

Abstract
A series of N6-(p-sulfophenyl)alkyl and N6sulfoalkyl derivatives of adenosine was synthesized, revealing that N6-(p-sulfophenyl)adenosine (10b) is a moderately potent (Ki vs [3H]PIA in rat cortical membranes was 74 nM) and A1-selective (120-fold) adenosine agonist, of exceptional aqueous solubility of >1.5 g/mL (≈3 M). Compound 10b was very potent in inhibiting synaptic potentials in gerbil hippocampal slices with an IC50 of 63 nM. At a dose of 0.1 mg/kg ip in rats, 10b inhibited lipolysis (a peripheral A1 effect) by 85% after 1 h. This in vivo effect was reversed using the peripherally selective A1-antagonist 1,3-dipropyl-8-[p-(carboxyethynyl)phenyl]xanthine (BW1433). The same dose of 10b in NIH Swiss mice (ip) was nearly inactive in locomotor depression, an effect that has been shown to be centrally mediated when elicited by lower doses of other potent adenosine agonists, such as N6-cyclohexyladenosine (CHA) (Nikodijevic et al. FEBS Lett. 1990, 261, 67). HPLC studies of biodistribution of a closely related and less potent homologue, N6-[4-(p-sulfophenyl)butyl]adenosine indicated that a 25 mg/kg ip dose in mice resulted in a plasma concentration after 30 min of 0.46 μg/mL and no detectable drug in the brain (detection limit <0.1% of plasma level). Although 10b at doses >0.1 mg/kg in mice depressed locomotor activity, this depression was unlike the effects of CHA and was reversible by BW1433. These data suggest that 10b is a potent adenosine agonist in vivo and shows poor CNS penetration.
Introduction
Adenosine agonists, such as N6-cyclohexyladenosine (CHA), cause intense behavioral effects at low doses.1–4 The locomotor depression elicited by peripherally administered N6-substituted adenosine analogs is usually interpreted as a central nervous system (CNS) effect. Evidence for central mediation of the depressant effect is that very low agonist doses administered directly into the brain are effective5 and that peripherally selective antagonists, such as (p-sulfophenyl)xanthine derivatives,2,6,7 are inactive in reversing this depression. The nonselective xanthines, caffeine and theophylline, which freely cross the blood-brain barrier, reverse adenosine-agonist-induced behavioral depression.
It has been shown that adenosine A1 agonists protect against ischemia-induced brain degeneration,8,9 possibly by modulating excitatory amino acid toxicity in the CNS. However, the multiple actions of adenosine,10,11 including both peripherally and/or centrally-mediated effects, complicate interpretation of these results.
As tools to aid in delineating central and peripheral adenosine agonist effects, we have synthesized a series of sulfonates related to N6-(phenylalkyl) and N6-alkyl derivatives of adenosine that are known potent agonists. The sulfonate group is completely charged at physiological pH and is expected to preclude passage of the derivative across the blood brain barrier, by analogy with (p-sulfophenyl)-theophylline and its analogues.6
Chemistry
The sulfoamine derivatives 3b and 6b–c were prepared according to the routes shown in Figure 1. For 3b, monosulfonation of the dibromoalkane 1 was followed by amination. Compounds 6b and 6c were obtained by amination of 4 and sulfonation of 5, respectively. Reaction of 6-chloropurine riboside, 7, with the appropriate sulfonated amine provided compounds 8b–12b (Figure 2).
Figure 1.
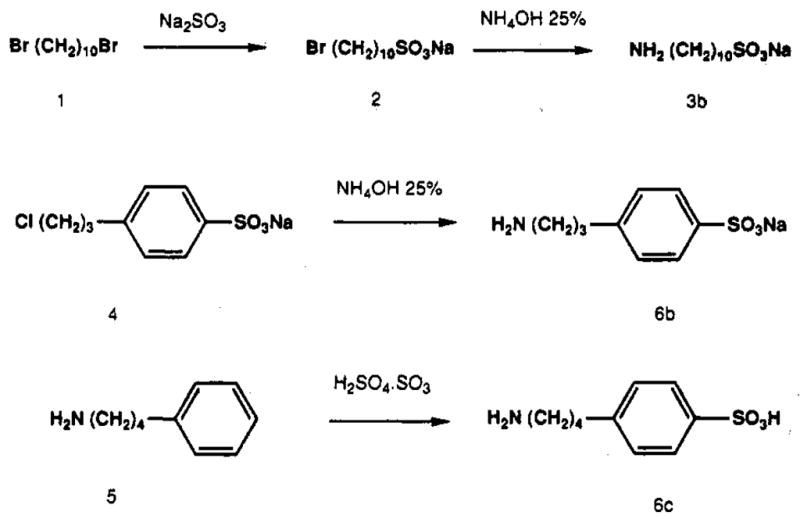
Synthesis of sulfoaralkylamine and sulfoalkylamine intermediates.
Figure 2.

Synthesis of N6-(sulfoaralkyl)- and N6-(sulfoalkyl)-adenosine analogs.
The sulfonate derivatives were considerably more hydrophilic than the corresponding hydrogen analogs. Compound 11b could be dissolved in neutral phosphate buffer (100 mM) at a concentration of 0.8 M. In comparison, a carboxylic acid derivative prepared in a previous study,13 N6-[p-(carboxymethyl)phenyl] adenosine, had a maximum aqueous solubility of 2.1 mM. Compound 10b was especially soluble, with an aqueous solubility of > 1.5 g/mL (≈3 M).
Biological Evaluation
Binding Assay
The analogs were tested in a radioligand binding assay for affinity at adenosine receptors in rat brain membranes. The compounds were assayed for affinity at rat A1 cortical receptors using [3H]-N6-(phenylisopropyl)adenosine17 and at rat A2 striatal receptors using [3H]CGS 21680 (Table I).12 The (sulfoalkyl)-adenosine derivative 8b was essentially inactive in binding to both A1 and A2 adenosine receptors. Compound 9b was comparable in potency and selectivity to the longer chain [(sulfophenyl)alkyl]adenosine derivatives. The Ki values for the [(sulfophenyl)alkyl]adenosine derivatives 10b–12b ranged from 74 to 610 nM at A1 receptors. The orders of both potency and selectivity at A1 receptors were 10b > 12b > 11b. The highest degree of A1 selectivity, 120-fold, was observed for compound 10b. Compound 11b was only slightly A1 selective. Except for 8b, the effect of adding a sulfonate group to an N6-adenosine derivative was to diminish the A1-receptor affinity by 20–60-fold. At A2 receptors, the differences in affinity between the sulfo and corresponding H analogues was less pronounced. Curiously, the very low affinity of N6-decyladenosine, 9a, at A2 receptors was enhanced 7-fold in the corresponding terminal sulfo derivative, 9b.
Table I.
Potency of N6-(Sulfoaralkyl)- and N6(Sulfoalkyl)adenosines in Competitive Binding Assays at Rat Brain A1 and A2 Adenosine Receptors and a Comparison with the Corresponding Unsulfonated Analoga,b

| |||||
|---|---|---|---|---|---|
| no. | R | A1 receptor Ki, nM | A2a receptor Ki, nM | affinity ratio A1/A2 | |
| 8a | ethyl | 4.9 | 8900 ± 770 | 1800 | |
| 8b | 2-sulfoethyl | 41% at 100 μM | 0% at 100 μM | >1 | |
| ratio | >20 000 | >12 | |||
| 9a | decyl | 21f | 60 000 ± 4500 | 2900 | |
| 9b | 10-sulfodecyld | 712 ± 31 | 8780 ± 3990 | 12 | |
| ratio | 34 | 0.14 | |||
| 10a | phenyl | 3.5 ± 0.5g | 663c | 190 | |
| 10b | p-sulfophenyl | 74 ± 8.5 | 8900 ±880 | 120 | |
| ratio | 21 | 13 | |||
| 11a | 3-phenylpropyl | 12 ± 2g | 455 ± 160 | 38 | |
| 11b | 3-(p-sulfophenyl)propyl | 610 ± 39d | 3840 ± 174e | 6.3 | |
| ratio | 51 | 8.4 | |||
| 12a | 4-phenylbutyl | 7.5 ± 1.5g | 4750 ± 283 | 630 | |
| 12b | 4-(p-sulfophenyl)butyl | 432 ± 18e | 11 300 ± 292 | 25 | |
| ratio | 58 | 2.4 | |||
Versus 1.0 nM [3H]PIA at A1 receptors and versus 5.0 nM [3H]CGS 21680 at A2 receptors, unless noted. Values are given as the average of two or three (±SEM) determinations performed in triplicate.
Triethylammonium salt tested, unless noted.
Versus [3H]NECA.
Sodium salt.
Ammonium salt.
Calf brain; data from ref 17
Data from ref 29.
Brain versus Plasma Concentrations
For HPLC studies of biodistribution of the sulfonated adenosine derivatives, a weakly potent derivative [N6-4-(p-sulfophenyl)butyl] adenosine, 12b) was chosen to allow larger doses to be injected for detection purposes. Thus, a relatively high plasma concentration could be achieved without complicating or detrimental biological effects. A 25 mg/kg ip dose of 12b in mice resulted in a plasma concentration of 0.46 μg/mL after 30 min and no detectable drug in the brain (detection limit <0.1% of plasma level). At this dose moderate locomotor depression of the mice was observed.
Electrophysiology
Adenosine derivatives are potent inhibitors of synaptic potentials in the hippocampal formation, an action mediated by A1 receptors.18 Several sulfonate derivatives and reference ligands were characterized for the ability to inhibit synaptic transmission using in vitro rat or gerbil hippocampal slices (Table II). Synaptic responses (field excitatory postsynaptic potential (fEPSPs)) were recorded in the CA1 region. All of the adenosine derivatives inhibited fEPSPs in a dose-dependent manner. The order of potency corresponded more roughly to affinity at A1 receptors. CGS 21680, an A2-selective agonist, was of very low potency. Compound 10b was very potent in inhibiting synaptic potentials in both rat and gerbil hippocampal slices with IC50 values of 74 and 63 nM, respectively, comparable in potency to R-PIA. Characteristic of R-PIA and other potent adenosine N6 derivatives, such as CHA (N6-cyclohexyladenosine) and ADAC (N6-[4-[[[4-[[[(2-aminoethyl)amino]-carbonyl]methyl]anilino]carbonyl]methyl]phenyl]-adenosine), is a slow onset of action and a slow washout. The onset of action of 10b was comparable to that of adenosine itself (very rapid), and the washout was also rapid. The A1-selective adenosine antagonist CPX (8-cyclopentyl-1,3-dipropylxanthine) blocked the inhibitory effects of this derivative (gerbil). The propyl derivative 11b was found to be approximately as potent as 2-chloroadenosine (2-CADO) in this assay with an IC50 value of 1.7 μM. Thus, this sulfonate derivative clearly acted as an agonist, in spite of its relatively low affinity at A1 receptors (Ki 610nM). This derivative had a slightly slower onset of action in this assay than adenosine itself, but a faster onset of action than N6-cyclopentyladenosine (CPA). The dose–response curves show that this sulfonate derivative is more potent than adenosine, yet considerably less potent than CPA.
Table II.
Potency of Adenosine Analogs as Inhibitors of Synaptic Potentials in the Hippocampal Formation18 (synaptic responses (fEPSPs) were recorded in the CA1 region of in vivo hippocampal slices, in rat brain, unless noted)
In Vivo Lipolysis
Adenosine agonists cause inhibition of lipolysis via activation of A1 receptors in isolated adipocytes and likewise result in a lowering of serum glycerol levels in vivo.29 Three (sulfophenyl)alkyl adenosine derivatives (10b–12b) were examined as inhibitors of lipolysis in vivo in rats at doses of 0.1–1.0 mg/kg and were found to lower serum glycerol levels. The maximum effect was seen at 30–60 min post subcutaneous injection (Figure 3). The (sulfophenyl)propyl derivative 11b lowered the serum glycerol levels in rats at 60 min following injection by 30% and 34% at doses of 0.5 and 1.0 mg/kg, respectively (data not shown). Compound 10b was considerably more potent in the inhibition of lipolysis, with a dose of 0.1 mg/kg nearly completely suppressing serum glycerol levels. Compound 12b was the weakest of the three derivatives in the lipolysis assay. The antilipolytic effects elicited by adenosines agonists 10b–12b (Figure 3) were fully or nearly completely antagonized by the peripherally selective adenosine antagonist BW1433 (1,3-dipropyl-8-[p-(carboxyethynyl)phenyl]xanthine)23 at a dose of 4 mg/kg, administered simultaneously. BW1433 alone raised serum glycerol levels by a small amount, from 296 ± 20 μM before injection to 326 ± 15 μM at 60 min postinjection.
Figure 3.

Time course for the effects of sulfoadenosine derivatives on lipolytic activity in rats (n = 3 for each point). Serum glycerol levels were measured enzymatically.15 Key: animals injected with 10b at 0.1 mg/kg sc (open symbols) or 12b at 1.0 mg/kg (closed symbols) for adenosine agonist alone (squares) or combination of agonist and BW1433, 4.0 mg/kg sc (triangles). BW1433 alone raised serum glycerol levels by a small amount, from 296 ± 20 μM before injection to 326 ± 15 μM at 60 min postinjection.
Locomotor Activity
Adenosine agonists are potent locomotor depressants,1–3 and the onset of the effect is generally rapid. Several derivatives were examined in a behavioral assay, using a computerized animal activity monitor cage. The locomotor activity of mice, control and drug-treated, was followed over a 90-min time course, with measurements made in 10-min intervals (Figure 4A). A 0.1 mg/kg dose of 10b administered to NIH Swiss mice (ip) was only weakly active in locomotor depression (total distance traveled during 60 min was 82 ± 11% of vehicle control, n = 6). Doses of 0.3 and 1.0 mg/kg in mice produced locomotor activity over 60 min of 73 ± 7.8% (n = 20) and 11.6 ± 2.6% (n = 7) of control, respectively. This depression was quantitatively and qualitatively unlike the effects of N6-cyclohexyladenosine, for which the EC50 dose was found to be much lower (~0.1 mg/kg). At the higher doses of 10b, the animals did not respond to touch, there was no analgesia, and body posture was different from that of CHA. CHA-depressed animals respond to touch and exhibit a diminished response to painful stimulus, such as pressure applied to the tail. Over a period of 1 h, mice injected with 0.3 mg/kg 10b traveled 3900 ± 400 cm/60 min (total distance, n = 20) compared to control animals traveling 5300 ± 300 cm/60 min (n = 25). The depression elicited by a dose of 0.3 mg/kg 10b was completely reversed (over a 30-min period) by a 0.25 mg/ kg ip dose of CPX. The depression elicited by 0.3 mg/kg 10b was also reversed by a 4 mg/kg dose of BW1433 (Figure 4B). Mice receiving a combination of 10b and BW1433 traveled 5150 ± 700 cm/60 min (n = 12). BW1433 alone resulted in 5650 ± 700 cm/60 min traveled (n = 10). The butyl derivative, 12b, was shown to be inactive as a locomotor depressant in mice at a dose of 1 mg/kg (data not shown). However, at a 5 mg/kg dose of 12b pronounced locomotor depression was observed, and this effect disappeared after 60 min postinjection. Very high doses of the propyl derivative 11b (25 and 50 mg/kg ip) also produced moderate locomotor depression in rats during 1 h.
Figure 4.

(A) Effects of N6-(4-sulfophenyl)adenosine, 10b, on locomotor activity in mice.3 Key: vehicle-injected animals (open squares), 10b at 0.3 mg/kg ip (closed squares), 10b at 1.0 mg/kg ip (circles); n = 7–25. P values for control vs 0.3 mg/kg 10b were <0.01 at 10 min and <0.001 at 20 min. (B) Reversal by the adenosine antagonist BW1433 of the locomotor depression elicited by a 0.3 mg/kg dose of compound 10b. Key: vehicle-injected animals (open squares), 10b at 0.3 mg/kg ip (closed squares), 10b + BW1433, 4.0 mg/kg ip (triangles), BW1433 alone (circles); n = 10–25.
Unlike the depression elicited by 10b, the locomotor depression elicited by CHA was not reversed by BW1433, consistent with the primary central mode of action of CHA at this dose. Over a 30-min period, CHA alone (100 μg/kg, ip) produced locomotor activity of 64 ± 12% (n = 7) of control. CHA in combination with a 4 mg/kg dose of BW1433 elicited locomotor activity of 73 ± 6.4% (n = 10), relative to BW1433 alone.
Body Temperature
Either parenteral (ip) or central (icv) administration of the (sulfophenyl)propyl derivative, 11b, caused rectal temperature depression in rats (Figure 5). In comparison, vehicle administered by the same routes caused a small rise in temperature (by approximately 1°C). Thus, it appears that there are multiple components (both peripheral and central) to temperature depression by adenosine agonists. This observation is consistent with previous reports on hypothermic effects of adenosine analogues.10,21,22
Figure 5.

Effects of compound 11b on body temperature in rats: (a) by ip administration at the dose indicated or (b) by icv administration in a cannulated animal.
Discussion
Adenosine agonists have been proposed to be cerebro-protective agents.8,9 CHA, a potent A1-selective agonist had a protective effect on the ischemic brains in gerbils and rats, as judged from both histological evidence8,9 and survival rates.9 A proposed mechanism for this phenomenon is interference with the glutamic acid pathways, either presynaptically to inhibit the release of glutamic acid or postsynaptically to interfere with its “excitotoxic” effect through reduction of calcium influx.
It was suggested that other effects of adenosine, such as temperature depression, cardiovascular depression, or CNS depression, may complicate interpretation of the phenomenon of cerebroprotection. Hypothermia alone has been demonstrated to be neuroprotective in the rat model.23
In general, there is uncertainty about the central vs peripheral nature of some of the physiological effects of adenosine. For example, temperature regulation by adenosine receptor ligands appears to have both central and peripheral components.10,21,22 Even the question of whether some of the more potent adenosine agonists such as R-PIA enter the CNS altogether when administered peripherally has been subject to debate.24
We have synthesized a series of charged derivatives of adenosine as putative peripheral agonists. These sulfonate derivatives may prove useful in determining if the cerebroprotective effect of adenosine agonists and other adenosine effects are due to the central and/or peripheral activation of adenosine receptors.
It was desirable to introduce a sulfonate group, which would be permanently charged at physiological pH, on an N6-substituent, the position of greatest flexibility of substitution among A1-selective analogs. However, negatively charged groups at the N6-position are known to diminish affinity at adenosine receptors.13 Several carboxylate-bearing derivatives have been reported to have affinities roughly two orders of magnitude less than the corresponding hydrogen-bearing derivatives. For example, N6-[p-(carboxymethyl)phenyl] adenosine and N6-p-tolyladenosine have Ki values at A1 receptors of 210 and 2.5 nM, respectively.13 Likewise, in the case of xanthines acting as adenosine antagonists, the presence of anionic groups on 8-phenyl substituents leads to diminished affinity.25 Nevertheless, p-(sulfophenyl)theophylline has found wide use,2,9,26 and its lower potency is acceptable due to its selective biodisposition and favorable aqueous solubility. Both 8-SPT (8-(p-sulfophenyl)theophylline) and the more potent carboxylate xanthine derivative BW143320 appear to be peripherally-selective adenosine antagonists. BW1433 is inactive in reversing the CNS-mediated effects of adenosine agonists in previous studies (S. Daluge, unpublished results) and in this study. BW1433 administered ip in rats as the sodium salt (30 mg/kg, suspension in 0.5% methylcellulose) has a half-life of 2.4 h (P. Savina and R. Blum, unpublished results). Similarly, the present agonist derivatives are considerably less potent in binding assays than the analogous uncharged derivatives (Table I). It appears that a sulfonate group diminishes affinity independent of the size of the N6-substituent, suggesting an unfavorable interaction with negatively charged membrane components, e.g. lipids. The lower potency of some of these sulfonated agonists may be tolerated for in vivo studies taking into consideration selective biodisposition, as indicated in the HPLC studies.
The hydrophilicity of the sulfoadenosine derivatives is consistent with the low pKa of an aryl sulfonic acid group (0–1) compared to that of a carboxylic acid (3–5).30 Thus, the proportion of the analog that exists in charged form at physiological pH would be much greater for a sulfonic acid than for a carboxylic acid. Also, the π-value of a sulfonate group (−4.76) suggests a somewhat greater tendency to partition into an aqueous phase than the value for a carboxylate (−4.36).31 The log P value for R-PIA was reported to be ~2.0.32 Applying the correlation analysis for changing substituents predicts a log P value of 0.7 for N6-phenyladenosine. Thus, the sulfo derivatives 10b and 12b have predicted log P values of −4.1 and −2.1, respectively. These negative log P values are consistent with the observed low brain penetration.
A primary neuromodulatory action of adenosine is to inhibit excitatory synaptic responses in the brain. In the hippocampus, this effect has been shown to be mediated by A1 receptors.18,27 Both 10b and the propyl derivative 11b applied directly to hippocampal slices were found to be more potent in an electrophysiological model than would be predicted from their potencies in a binding assay relative to other agonists. In fact, the IC50 for inhibition of fEPSPs by 10b was identical to its Ki value at A1 receptors (Table II). The effects of the sulfo adenosine analogues and of adenosine itself are qualitatively different from those of CHA, ADAC, and other N6 derivatives16,18 in rapidity of onset and washout. The relatively high activity in the hippocampal slice assay might relate to its lack of diffusion into fatty structures or not being taken up into cells; thus it is fully available in the medium to activate A1 receptors. For instance, uptake is sufficient to alter the apparent potency of adenosine and its analogs in slices (for a discussion see ref 33). Thus, compound 10b represents a unique type of adenosine agonist which exhibits both high in vivo potency and rapid reversibility.
Locomotor depression by uncharged and relatively hydrophobic adenosine agonists such as CHA is centrally mediated.2–4 At a dose of 0.1 mg/kg (highly active in the inhibition of lipolysis in vivo) 10b was nearly inactive in locomotor depression, although higher doses depressed behavioral activity. Thus, at lower doses the action of 10b (e.g. inhibition of lipolysis) is purely peripheral. At higher doses 10b preferentially activates A1 receptors in the peripheral nervous system, as suggested by the reversibility of the locomotor effects by BW1433. The precise mechanism for locomotor depression at higher doses is yet unknown, but may relate to cardiac depression or vasodilatation. It is a result of activation of A1 receptors, since it is reversible by both BW1433 and the potent and highly A1-selective antagonist CPX, but the tissue location of the relevant receptors is undetermined.
In conclusion, N6-(p-sulfophenyl)adenosine (10b) is a potent A1 adenosine agonist in receptor binding, in inhibitory electrophysiological effects in hippocampal slices, and in inhibition of lipolysis in vivo. A close homologue, 12b, did not cross the blood-brain barrier, even at very high doses. Compound 10b is even more polar than 12b, and thus is expected to be similarly excluded from the brain. 10b, 11b, and 12b depressed locomotor activity only at high doses, but this depression appeared to be related to the peripheral effects of adenosine. A peripherally selective A1 adenosine agonist might have utility in treating cardiac ischemia,28 without producing CNS side effects.
Experimental Section
Chemistry
New compounds were characterized (and resonances assigned) by 300-MHz proton nuclear magnetic resonance mass spectroscopy using a Varian GEMINI-300 FT-NMR spectrometer. Unless noted, chemical shifts are expressed as ppm downfield from tetramethylsilane. Synthetic intermediates were characterized by chemical-ionization mass spectrometry (NH3) and adenosine derivatives by fast atom bombardment mass spectrometry (JEOL SX102). C, H, N, and S analyses were carried out by Atlantic Microlabs (Norcross, GA), and ±0.4% was acceptable. 6-Chloropurine riboside, 7, taurine, 3a, and sulfanilic acid, 6a, were purchased from Aldrich (St. Louis, MO) and p-(3-chloropropyl)benzenesulfonate, 4, was obtained from Schweizerhall, Inc. (South Plainfield, NJ). N6-Phenyladenosine and 2-chloroadenosine were purchased from Research Biochemicals, Inc. (Natick, MA). Compounds 11a and 12a were the gift of Dr. Ray Olsson (University of South Florida, Tampa, FL). Compounds 11a and 12a were the gift of Dr. Ad IJzerman (Center for Bio-pharmaceutical Sciences, Leiden, The Netherlands). BW1433 was the gift of Dr. S. Daluge (Burroughs Wellcome Co., Research Triangle Park, NC). Analytical TLC plates and silica gel (230–400 mesh) were purchased from VWR (Bridgeport, NJ). Silica gel 100 C18 reversed-phase was obtained from Fluka (Ronkonoma, NY).
Sodium 10-Aminodecanesulfonate (3b)
A mixture of sodium sulfite (1.4 g, 1.1 mmol), 1,10-dibromodecane (1) (7.5 mL, 3.3 mmol), 25 mL of ethanol, and 20 mL of water was refluxed for 6 h. The upper phase (1,10-dibromodecane) was removed, and the lower aqueous phase was washed with chloroform three times. The aqueous phase was concentrated by rotary evaporation at 50 °C. The solid residue was dissolved in 10 mL of a 25% solution of ammonium hydroxide and heated at 100 °C in a sealed container for 3 h. Sodium 10-aminodecanesulfonate (3b) separated upon cooling as a white crystalline solid. This material was isolated by filtration and washed successively with cold water and ether. MS (FAB [pos]/glycerol matrix): m/z238 MH2+. 1H NMR δ (D2O): 1.3 (m, 12 H), 1.65 (m, 4 H, CH2CH2-NH2 + CH2CH2SO3), 3.28 and 3.29 (2 t, 4 H, CH2NH2 + CH2-SO3).
Sodium p-(3-Aminopropyl)benzenesulfonate (6b)
p-(3-Chloropropyl) benzenesulfonate (4) (6 g, 23.4 mmol) was dissolved in 28 mL of a 25% solution of ammonium hydroxide and heated at 100 °C in a closed vessel for 3 h. Sodium p-(3-aminopropyl)-benzenesulfonate (6b) separated upon cooling as a white crystalline solid. This material was isolated by filtration and washed successively with cold water and ether (3.65 g, 66%). MS (FAB [neg]/glycerol matrix): m/z 214 M−. 1H NMR δ (DMSO-d6): 1.85 (m, 2 H, CH2CH2C6H3), 2.65 and 2.80 (2 t,4 H, CH2NH2 + CH2C6H3), 7.15 (d, 2 H, arom), 7.50 (d, 2 H, arom).
p-(4-Aminobutyl)benzenesulfonic Acid (6c)
4-Phenyl-butylamine (5) (15 g, 0.1 mol) was added dropwise at 0 °C to a mixture of concentrated sulfuric acid (26 mL) and fuming sulfuric acid (15 mL). After stirring at room temperature for 1 h, the mixture was refluxed for an additional hour. After cooling, the solution was then poured into 1L of dioxane. The solid obtained was filtered, washed several times with dioxane, and dried (18 g,78%). MS (FAB [pos]/glycerol matrix): m/z 230MH2+. Anal. C10H15NO3S (C, H, N, S).
N6-[(p-Sulfophenyl)alkyl]-,N6-(ω-Sulfoalkyl)-,or N6-(p-Sulfophenyl)adenosine, Triethylammonium, Ammonium, or Sodium Salts (General Procedure for 8b–12b)
6-Chloropurine riboside (7) (1 g, 3.5 mmol) and the appropriate sulfoamine (4.2 mmol) were suspended in 40 mL of n-butyl alcohol, and a 3-fold excess of triethylamine (1.6 mL) was added. For preparation of compounds 9b and 11b, the sodium salts of the sulfo amines 3b and 4 were used. The mixture was heated at 120 °C for 24 h. The reaction mixture was concentrated to a syrup and after dilution with chloroform loaded onto a 15 × 4.5 cm column of silica gel (flash type). It then was eluted with a gradient of methanol in ethyl acetate (20–50%) to remove the traces of unreacted material, followed by a mixture of chloroform-methanol (7:3, by volume) to elute the product. For compound 12b, 10% concentrated NH4OH was included in the elution solvent, the ammonium salt of the sulfo adenosine derivative was obtained. For compounds 9b–12b, the solid obtained after evaporation was crystallized from a mixture of water and ethanol (roughly 1:50, by volume) that was placed under an ether atmosphere overnight. Compound 8b was purified by low-pressure reversed-phase chromatography on C18 silica (eluant: water/methanol, 8:2, by volume). Yields ranged between 42% and 63%. Analysis: (C, H, N, S) for 8b (+ 1.5H2O, Et3N salt), 10b (Et3N salt), and 12b (+ 1 H2O, NH4 salt); (C, H, N) for 11b (+ 1.5H2O, sodium salt); (C, H) for 9b (+ 1.5H2O, sodium salt), N calcd 13.05, found 12.33. 1H NMR for 10b δ (DMSO-d6): 1.15 (t, 9 H, CH3CH2), 3.10 (m, 6 H, CH2CH3), 3.60 and 3.70 (m, 2 H, 5′-H), 3.98 (m, 1 H, 4′-H), 4.18 (m, 1 H, 3′-H), 4.64 (m, 1 H, 2′-H), 5.20 (d, 1 H, OH), 5.26 (1,1 H, OH), 5.50 (d, 1 H, OH), 5.96 (d, 1 H, l′-H J1′,2′ = 6.4 Hz), 7.55 and 7.90 (d, 4 H, arom), 8.40 and 8.55 (s, 2 H, 2-H and 8-H), 10.0 (s, 1 H, NH). ε305 for 10b in methanol (λmax) 33 100. MS (FAB [neg]/glycerol matrix): m/z 422 M−. Mp for 10b: 155–165 °C.
Biological Methods
Receptor Binding
Rat cerebral cortical membranes and striatal membranes were prepared12,13 and treated with adenosine deaminase (0.5 units/mL) for 20 min at 37 °C prior to radioligand binding studies or incorporation studies. Solid samples of the adenosine derivatives were dissolved in DMSO and stored at −20 °C. The stock solutions were diluted with DMSO to a concentration of ≤0.1 mM prior to adding to the aqueous medium. The final concentration of DMSO in the assay medium was generally ≤2%.
Inhibition of binding of 1 nM [3H]-N6-(phenylisopropyl)-adenosine (Dupont NEN, Boston, MA) to A1 receptors in rat cerebral cortex membranes was measured as described.17 Membranes (~100 μg of protein, 5 mg wet weight, per tube) were incubated for 1.5 h at 37 °C in a total volume of 2 mL of 50 mM Tris hydrochloride, at pH 7.4. Adenosine deaminase was present (2 IU/mL) during the incubation with radioligand. Bound and free radioligand were separated by addition of 4 mL of a buffer containing 50 mM Tris-HCl, at pH 7.4 at 5 °C followed by vacuum filtration using a Brandel Cell Harvester (Brandel, Gaithersburg, MD) and a Whatman GF/B glass fiber filter with additional washes totaling 12 mL of buffer. Nonspecific binding was determined with 10 μM 2-chloroadenosine.
Inhibition of binding of 5 nM [3H]CGS 21680 [4-[2-[[6-amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-y]amino]-ethyl] benzenepropanoic acid] was carried out as described12 using 20 μM 2-chloroadenosine to determine nonspecific binding. Membranes (~80 μg of protein, 5 mg wet weight, per tube) were incubated for 1 h at 25 °C in a total volume of 1 mL. Adenosine deaminase was present (3 IU/mL) during the incubation with radioligand. Filtration was carried out using a Brandel Cell Harvester, as above.
At least seven different concentrations spanning 4 orders of magnitude, adjusted appropriately for the IC50 of each compound, were used. IC50 values, computer-generated using a nonlinear regression formula on the GraphPAD program (Institute for Scientific Information), were converted to apparent Ki values using KD values12,13 of 1.0 and 14 nM for [3H]PIA and [3H]CGS 21680 binding, respectively, and the Cheng-Prusoff equation.14
HPLC Studies of Tissue Extracts
Mice (7 months old, CR:CFW strain, average weight 40 g) were injected ip with a solution of the adenosine derivative (dissolved in 0.5 mL of 20% aqueous DMSO). After 30 min, 200 μL of pentobarbital (70 mg/ mL) were injected ip, and 500 μL of blood was collected by cardiac puncture (right auricle) into a syringe containing 50 μL of EDTA (5 mM) and frozen on dry ice. The brains were removed and placed on dry ice.
Tissue Preparation
Blood (200 μL) was mixed with 600 μL of acetonitrile at 4 °C and vortexed for 30 s. After centrifuging on the microfuge for 2 min, the supernatant was removed and evaporated under nitrogen, and the residue was redissolved in acetonitrile containing 0.1% trifluoroacetic acid (HPLC solvent A, 60 μL). A chopped whole brain was ground (tissue grinder or dounce homogenizer) with 500 μL of acetonitrile, vortex-mixed in a glass tube, and centrifuged 10 min at no. 5 speed (500 × G) on a clinical centrifuge. The supernatant was removed and evaporated under nitrogen. The residue was redissolved in 50 μL of HPLC solvent A.
HPLC Conditions
Plasma and brain extracts were analyzed for the appropriate adenosine derivative (compared to standard) by reversed-phase HPLC. A C-18 column (Rainin, 0.4 × 25 cm, 5 μ, 300-Å pore size, 2.3 mL/min) was used, with a mobile phase consisting of mixtures of solvent A and water containing 0.1% trifluoroacetic acid, using a gradient of 5–10% (20 min), followed by 10–15% (2 min), and 15–20% (18 min). Detection was by UV absorption at 254 nm. The retention time of compound 12b was 30 min.
Locomotor Activity
Individual adult male mice of the NIH (Swiss) strain between 5 and 6 weeks of age weighing at least 25 g were studied in a Digiscan activity monitor (Omnitech Electronics Inc., Columbus, OH) with computerized data analysis (ILAM software).2,3 Different animals were used for each experiment. Adenosine and xanthine analogs were dissolved in a 20:80 v/v mixture of Alkamuls EL-620 (formerly Emulphor, Rhône-Poulenc, Cranbury, NJ) and phosphate buffered saline and injected ip in a volume of 5 mL per kg body weight. Where appropriate, the antagonist was injected 5 min prior to injection of the agonist. Animals were placed in the cage immediately after the final injection, and monitoring was begun after 5 min. Total distance traveled was used as a measure of locomotor activity. Activity was monitored for up to 90 min, with data collected in 10-min intervals. Control values for vehicle-injected animals were determined for each experiment.
Lipolysis Assay
The following protocol was used for determination of adenosine agonist effects on in vivo lipolysis. Sprague-Dawley rats (200–250 g) were fasted overnight. A blood sample was taken from the tail vein 10 s before the subcutaneous injection of the drug (adenosine agonist or simultaneous combination of agonist and antagonist) in vehicle (DMSO). The volume injected was 0.1 mL/100 g body weight. Additional blood samples were taken from the tail vein at 30, 60, and 90 min postinjection and were kept on ice and centrifuged (14000g) within 30 min. A perchloric acid extract (3 %) of plasma was neutralized with a solution containing 3 M potassium hydroxide, 0.5 M 3-morpholinopropanesulfonic acid, and 0.1 M EDTA and then assayed enzymatically for glycerol as described.15 Each data point was the average for three animals.
Electrophysiology
Sprague-Dawley rats (200–300 g) and gerbils were sacrificed by decapitation under deep anesthesia with pentobarbital. Hippocampal slices were prepared as previously described.16 Extracellular recordings of field excitatory postsynaptic potentials (fEPSPs) were recorded in the stratum radiatum of area CA1 in response to stimulation of the combined Schaffer collateral and commissural afferents. Slices were superfused continuously for 30–40 min with artificial cerebrospinal fluid (ACSF),16 and drugs were tested by addition to the ACSF. The amplitudes of the fEPSPs in control and drug-treated slices were measured on-line with a computer (PC) and plotted over the time course of each experiment. The IC50 value was the concentration of an adenosine agonist that inhibited the fEPSP by 50%.
Temperature Depression
An adenosine analogue was administered to Sprague-Dawley male (250 g) rats, by injection either ip (dissolved in saline) or icv (dissolved in ACSF). For icv administration, a 22-gauge cannula was implanted stereotaxically under pentobarbital anesthesia. Drug was administered 3–7 days post cannula implant in 10 μL of solution over a 10-min period. Rectal temperature was recorded at 30-min intervals for as long as 3 h.
Acknowledgments
K.A.J., M.M., and K.S.L. wish to acknowledge the generous financial support of Cortex Pharmaceuticals, Inc., Irvine, CA, and NSF Grant No. RNS-8901154 to K.S.L.
References
- 1.Snyder SH, Katims JS, Annau Z, Bruns RF, Daly JW. Adenosine receptors and the actions of the methylxanthines. Proc Natl Acad Sci USA. 1981;78:3260–3264. doi: 10.1073/pnas.78.5.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikodijevic O, Daly JW, Jacobson KA. Characterization of the locomotor depression produced by an A2-selective adenosine agonist. FEBS Lett. 1990;261:67–70. doi: 10.1016/0014-5793(90)80638-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikodijevic O, Sarges R, Daly JW, Jacobson KA. Characterization of A1- and A2-adenosine receptor mediated components in locomotor depression elicited by adenosine agonists. J Pharm Exp Ther. 1991;259:286–294. [PMC free article] [PubMed] [Google Scholar]
- 4.Durcan MJ, Morgan PF. NECA-induced hypomotility in mice: Evidence for a predominantly central site of action. Pharmacol Biochem Behav. 1989;32:487–490. doi: 10.1016/0091-3057(89)90185-8. [DOI] [PubMed] [Google Scholar]
- 5.Barraco RA, Bryant SD. Depression of locomotor activity following bilateral injections of adenosine analogs into the striatum of mice. Med Sci Res. 1987;15:421–422. [Google Scholar]
- 6.Daly JW, Padgett W, Shamim MT, Butts Lamb P, Waters J. 1, 3-Dialkyl-8-(p-sulfophenyl)xanthines: potent water-soluble antagonists for A1-A2-adenosine receptors. J Med Chem. 1985;28:487–492. doi: 10.1021/jm00382a018. [DOI] [PubMed] [Google Scholar]
- 7.Baumgold J, Nikodijevic O, Jacobson KA. Penetration of adenosine antagonists into mouse brain as determined by ex vivo binding. Biochem Pharmacol. 1992;43:889–894. doi: 10.1016/0006-2952(92)90257-j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans MC, Swan JH, Meldrum BS. An adenosine analogue, 2-chloroadenosine, protects against long term development of ischaemic cell loss in the rat hippocampus. Neurosci Lett. 1987;83:287–92. doi: 10.1016/0304-3940(87)90101-7. [DOI] [PubMed] [Google Scholar]
- 9.von Lubitz DKJE, Marangos PJ. Cerebral ischemia in gerbils: Postischemic administration of cyclohexyl adenosine and 8-sulfophenyltheophylline. J Mol Neurosci. 1990;2:53–59. doi: 10.1007/BF02896926. [DOI] [PubMed] [Google Scholar]
- 10.Seale T, Abla KA, Shamim MT, Carney JM, Daly JW. 3,7-Dimethyl-1-propargylxanthine: a potent and selective in vivo antagonist of adenosine analogs. Life Sci. 1988;43:1671–1684. doi: 10.1016/0024-3205(88)90478-x. [DOI] [PubMed] [Google Scholar]
- 11.Bruns RF. Role of adenosine in energy supply/demand balance. Nucleosides Nucleotides. 1991;10:931–943. [Google Scholar]
- 12.Jarvis MF, Schutz R, Hutchinson AJ, Do E, Sills MA, Williams M. [3H]CGS 21680 anA2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J Pharmacol Exp Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 13.Jacobson KA, Kirk KL, Padgett WL, Daly JW. Functionalized congeners of adenosine: preparation of analogues with high affinity for Al-adenosine receptors. J Med Chem. 1985;28:1341–1345. doi: 10.1021/jm00147a039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 15.Eggstein M, Kuhlman E. Triglycerides and glycerol. In: Bermeyer HU, editor. Methods of Enzymatic Analysis. Academic Press; New York: 1974. pp. 1825–1831. [Google Scholar]
- 16.Lee K, Oliver M, Schottler F, Lynch G. Electron microscopic studies of brain slices: the effects of high-frequency stimulation on dentritic ultrastructure. In: Kerkut GA, Wheal HV, editors. Electrophysiology of Isolated Mammalian CNS Preparations. Academic Press; London: 1981. pp. 189–211. [Google Scholar]
- 17.Van Galen PJM, IJzerman AP, Soudijn W. Adenosine derivatives with N6-alkyl, -alkylamine, or -alkyladenosine substituents as probes for the A1 adenosine receptor. FEES Lett. 1987;223:197–201. doi: 10.1016/0014-5793(87)80535-5. [DOI] [PubMed] [Google Scholar]
- 18.Reddington M, Lee K, Schubert P. An A1-adenosine receptor characterized by [3H]cyclohexyladenosine binding mediates the depression of evoked potentials in the rat hippocampal slice preparation. Neurosci Lett. 1982;28:275–279. doi: 10.1016/0304-3940(82)90070-2. [DOI] [PubMed] [Google Scholar]
- 19.Vannucci SJ, Klim CM, Martin LF, LaNoue KF. A1-Adenosine receptor mediated inhibition of adipocyte adenylate cyclase and lipolysis in Zucker rats. Am J Physiol. 1989;257:E871–E878. doi: 10.1152/ajpendo.1989.257.6.E871. [DOI] [PubMed] [Google Scholar]
- 20.Clemo HF, Bourassa A, Linden J, Belardinelli L. Antagonism of the effects of adenosine and hypoxia on atrioventricular conduction time by two novel alkylxanthines: correlation with binding to adenosine A1 receptors. J Pharmacol Exp Ther. 1987;242:478–484. [PubMed] [Google Scholar]
- 21.Ticho SR, Radulovacki M. Role of adenosine in sleep and temperature regulation in the preoptic area of rats. Pharmacol Biochem Behav. 1991;40:33–40. doi: 10.1016/0091-3057(91)90317-u. [DOI] [PubMed] [Google Scholar]
- 22.Lee TF, Li DJ, Jacobson KA, Wang LCH. Improvement of cold tolerance by selective A1 adenosine receptor antagonists in rats. Pharmacol Biochem Behav. 1990;37:107–112. doi: 10.1016/0091-3057(90)90049-n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Busto R, Dietrich WD, Globus MY, Ginsberg MD. Postischemic moderate hypothermia inhibits CA1 hippocampal ischemic neuronal injury. Neurosci Lett. 1989;101:299–304. doi: 10.1016/0304-3940(89)90549-1. [DOI] [PubMed] [Google Scholar]
- 24.Brodie MS, Lee K, Fredholm BB, Stahle L, Dunwiddie TV. Central versus peripheral mediation of responses to adenosine receptor agonists: evidence against a central mode of action. Brain Res. 1987;415:323–330. doi: 10.1016/0006-8993(87)90214-9. [DOI] [PubMed] [Google Scholar]
- 25.Van Galen PJM, Stiles GL, Michaels G, Jacobson KA. Adenosine A1 and A2 receptors: Structure-function relationships. Med Res Rev. 1992;12:423–471. doi: 10.1002/med.2610120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evoniuk G, von Borstel RW, Wurtman RJ. Antagonism of the cardiovascular effects of adenosine by caffeine or 8- (p-sulphophenyl)-theophylline. J Pharmacol Exp Ther. 1987;2:428–432. [PubMed] [Google Scholar]
- 27.Dunwiddie T, Fredholm B. Adenosine receptors mediating inhibitory electrophysiological responses in rat hippocampus are different from receptors mediating cyclic AMP accumulation. Naunyn-Schmiedeberg’s Arch Pharmacol. 1984;326:294–301. doi: 10.1007/BF00501433. [DOI] [PubMed] [Google Scholar]
- 28.Thornton JD, Liu GS, Olsson RA, Downey JM. Intravenous pretreatment with A1-selective adenosine analogues protects the heart against infarction. Circulation. 1992;86:659–665. doi: 10.1161/01.cir.85.2.659. [DOI] [PubMed] [Google Scholar]
- 29.Ukena D, Olsson RA, Daly JW. Definition of subclasses of adenosine receptors associated with adenylate cyclase: interaction of adenosine analogs with inhibitory A1 receptors and stimulatory A2 receptors. Can J Physiol Pharmacol. 1987;66:365–376. doi: 10.1139/y87-063. [DOI] [PubMed] [Google Scholar]
- 30.Sturm K, Muschaweck R, Hropot M. 5-Sulfamoylorthanilic acids, a sulfonamide series with salidiuretic activity. J Med Chem. 1983;26:1174–1187. doi: 10.1021/jm00362a017. [DOI] [PubMed] [Google Scholar]
- 31.Hansch C, Leo A. Substituent constants for correlation analysis in chemistry and biology. Wiley-Interscience; New York: 1979. [Google Scholar]
- 32.Bridges AJ, Bruns RF, Heffner TG. Central nervous system actions of adenosine agonists and antagonists. Annu Rep Med Chem. 1988;23:39–48. [Google Scholar]
- 33.Dunwiddie T, Worth T, Olsson R. Adenosine analogs mediating depressant effects on synaptic transmission in rat hippocampus: Structure-activity relationships for the N6-subregion. Naunyn-Schmiedberg’s Arch Pharmacol. 1986;334:77–85. doi: 10.1007/BF00498743. [DOI] [PubMed] [Google Scholar]