

Abstract
High-risk human papillomaviruses (HPV), such as HPV-16, are etiologic agents of a variety of anogenital and oral malignancies, including nearly all cases of cervical cancer. Cervical cancers arising in transgenic mice that express HPV-16 E7 in an inducible manner require the continuous expression of E7 for their maintenance. However, in HPV-associated cancers in vivo, E6 and E7 invariably are co-expressed. In this study, we investigated whether cervical cancers rely on the continuous expression of E7 in the context of constitutively expressed E6. We placed the inducible HPV-16 E7 transgene onto a background in which HPV-16 E6 was constitutively expressed. In transgenic mice with high-grade cervical dysplastic lesions and cervical cancer, repressing the expression of E7 led to the regression of all cancers and the vast majority of high-grade dysplastic lesions. In addition, cervical cancers were occasionally observed in transgenic mice in which E7 was repressed and then re-expressed. Our findings therefore indicate that even in the presence of constitutively expressed E6, the continuous expression of E7 is required for the maintenance of cervical cancers and most precancerous lesions. These data have important implications for the potential clinical use of drugs designed to inhibit the expression and/or function of E7 to treat HPV-associated cancers.
Keywords: human papillomavirus, HPV, cervical cancer
Introduction
Human papillomaviruses (HPV) are small, double-stranded DNA viruses that infect the stratified squamous epithelium and cause benign proliferative lesions. The “high-risk” HPVs, which include HPV-16 and HPV-18, are causal agents of nearly all cervical cancers, a majority of other anogenital cancers, and a distinct subset of head and neck squamous cell carcinomas (1). HPV-16 encodes the E5, E6, and E7 oncogenes, and of the three, E7 is the most oncogenically potent in mucosal tissue (2, 3). Although the E7 oncoprotein is capable of binding to dozens of cellular proteins (4), its ability to bind to and degrade the tumor suppressor pRb is its best-characterized function (5–8). Similarly, the HPV-16 E6 oncoprotein can bind to a host of intracellular partners (9) but is best known for its ability to bind to and induce the degradation of the p53 tumor suppressor (10–13).
Many studies in vitro have suggested that the continuous expression of the papillomaviral oncogenes is required for the maintenance of the transformed phenotype of cell lines derived from HPV-positive cervical cancers (14–19). In vivo, the continuous expression of oncogenes is required for the maintenance of neoplastic lesions in the context of other types of cancers (20–31). To investigate whether the same was true for E7 in the context of cervical cancers, we previously generated Bi-L E7 transgenic mice that expressed HPV-16 E7 in an inducible manner. Bi-L E7 transgenic mice harbor a construct encoding HPV-16 E7 and firefly luciferase, the expression of which is driven from bi-directional minimal cytomegaloviral promoters under the control of a tetracycline response element. To induce the expression of these genes, we crossed Bi-L E7 mice to a line of mice that expresses the tetracycline transactivator (tTA) protein under the control of the keratin 5 (K5) promoter (K5-tTA mice) (32). In Bi-L E7/K5-tTA bitransgenic mice, we showed that E7 and luciferase were expressed in the epithelium and that their expression could be silenced by administering doxycycline. Bi-L E7/K5-tTA bitransgenic mice developed cervical cancers and widespread cervical dysplasia when treated chronically with estrogen for 6 months, and cancers and high-grade dysplasia both regressed completely within 1 month of administering doxycycline (33). These data indicated that E7 may be a relevant target for anti-cancer therapy and for the treatment of HPV-associated high-grade cervical dysplasia.
In cancers driven by the expression of two oncogenes or by the expression of an oncogene and the inactivation of a tumor suppressor gene, independence from the continuous expression of an oncogene can arise. For example, although the majority of mammary tumors in transgenic mice regressed once the expression of an inducible transgene encoding c-Myc was silenced, the presence of de novo mutations in Ras prevented the regression of a subset of the tumors (23). Likewise, experiments performed in mice expressing inducible Wnt-1 on a wild-type genetic background or in mice hemizygous for p53 (p53+/−) showed that the failure of mammary tumors to regress once the expression of Wnt-1 was silenced was correlated with a loss of heterozygosity at the p53 locus on the p53+/− background (27). These results have important clinical implications, evidenced by the common occurrence of de novo mutations in human oncogenes that permit their escape from targeted therapies (34–36).
Because the papillomaviral E6 and E7 oncoproteins invariably are co-expressed in human cervical cancers (37), in this study we investigated whether their co-expression could eliminate the dependence of high-grade cervical dysplasia and cervical cancers on the continuous expression of E7. To do this, we placed the Bi-L E7 transgene onto a K14E6 background in which HPV-16 E6 is expressed constitutively in the stratified squamous epithelia (38). Due to the morbidity that arose in Bi-L E7/K5-tTA bitransgenic mice because of the high level of expression of E7 in the epidermis (33), we used K14-tTA mice (39), in which the expression of the tTA protein is controlled by the K14 rather than the K5 promoter, to induce the expression of luciferase and E7 from the Bi-L E7 transgene. Because K14E6 and K14E7 mice display only weak overt epidermal phenotypes, we reasoned that using the K14 promoter to drive the expression of tTA might reduce the expression of E7 in the epidermis of mice crossed with K14-tTA mice versus K5-tTA mice. Crossing Bi-L E7/K14E6 mice to K14-tTA mice yielded Bi-L E7/K14E6/K14-tTA triply transgenic mice in which we could repress the expression of E7 with doxycycline without affecting the expression of E6. Triply transgenic mice developed high-grade dysplastic cervical lesions and cervical cancers when treated chronically with estrogen, and despite the presence of E6, all cervical cancers and all but one high-grade dysplastic lesion regressed once we repressed the expression of E7. In triply transgenic mice in which E7 was repressed and then re-expressed, cervical cancers occasionally were observed, although whether those cancers represented a re-emergence of neoplastic disease or were cancers that never regressed is unclear. Our data suggest that targeting the expression and/or function of E7 alone holds promise for treating HPV-associated neoplastic disease, although the potential for the re-emergence of cancers after the initial regression of overt disease may necessitate long-term treatment with anti-E7 therapies.
Materials and Methods
For a full description of the Materials and Methods, see Supplementary Information.
Transgenic mice
K14E6 (38), K14E7 (40), Bi-L E7 (33), and K14-tTA (39) transgenic mice have been described previously.
Treatment with estrogen
To induce persistent estrus and to eliminate cycling through estrus, continuous-release pellets delivering 0.05 mg of 17β-estradiol (estrogen) per 60-d period (Innovative Research of America) were implanted s.c. into the shoulder fat pads of adult female virgin mice.
Treatment with doxycycline
Doxycline-containing chow (2 g/kg; Bio-Serv) was used to repress the expression of the Bi-L E7 transgene.
Procurement of female reproductive tracts and histologic analysis
One hour before sacrifice, mice were injected with 5-bromo-2′-deoxyuridine (BrdUrd; 12.5 mg/mL in PBS) at 10 μL/g body weight. Procured female reproductive tracts were fixed overnight at 4°C in 4% paraformaldehyde (w/v), embedded in paraffin, sectioned, and stained with H&E for histologic analysis of neoplastic and dysplastic disease.
Analysis of luciferase
Tissue lysates were analyzed for the activity of luciferase using the Luciferase Assay System (Promega Corp.) according to the manufacturer’s instructions.
Analysis of E7
A mouse anti-HPV-16 E7 antibody (Santa Cruz Biotechnology) was used to examine the expression of E7 by Western blot.
Analysis of E6
Arbor Vita’s test for HPV E6 applies the lateral flow format for detection of the E6 oncoprotein of HPV-16, 18, and 45 (41).
Analysis of p53
p53 specific (Santa Cruz Biotechnology) Westerns were performed on the same samples used for the analysis of E6 protein levels. Epithelial cell equivelents were assessed using an ab specific for keratin 14 (Abcam).
Immunohistochemistry
Mouse anti-BrdUrd (Calbiochem Immunochemicals), mouse anti-minichromosome maintenance protein 7 (MCM7; NeoMarkers Corp.), and mouse anti-p16 (Santa Cruz Biotechnology) were used for immunohistochemical analysis.
Quantification of BrdUrd
Eight visual fields at ×40 magnification were scored as either positive (brown) or negative (blue) for BrdUrd.
Results
Characterization of Bi-L E7/K14-tTA bitransgenic mice
As already noted, to perform the experiments described herein, for humane reasons we were unable to use Bi-L E7/K5-tTA mice used in our previous studies because of the severe overt epidermal phenotypes that they developed (33). To regulate the expression of the Bi-L E7 transgene, we instead used K14-tTA mice (39), which express the same tTA protein in the stratified squamous epithelium as K5-tTA mice but do so under the control of the K14 rather than the K5 promoter. Based on the location of the overt phenotypes in K14E6 and K14E7 mice (unpublished data), we suspected that the K14 promoter may drive the transcription of genes more weakly in the epidermis than does the K5 promoter, thereby precluding the onset of morbidity due to high-level epidermal expression of E7. Indeed, when we generated Bi-L E7/K14-tTA bitransgenic mice, their epidermis had only weak overt phenotypes that were not fully penetrant (data not shown), which was in stark contrast to the severe and fully penetrant epidermal phenotypes we observed in Bi-L E7/K5-tTA bitransgenic mice that we had used previously to evaluate the dependence of cervical cancers on the expression of E7 in the absence of E6 (33).
To characterize the induction and repression of the Bi-L E7 transgene by the K14-tTA line of transgenic mice, we performed assays on epithelial tissues harvested from Bi-L E7/K14-tTA bitransgenic mice to detect the activity of luciferase, which serves as a reporter for expression from the Bi-L E7 transgene. In the dorsal skin, ears, and lower female reproductive tracts of Bi-L E7/K14-tTA bitransgenic mice, we observed a significant induction of luciferase over what was observed in nontransgenic mice (P < 0.03, two-sided Wilcoxon rank-sum test) (Fig. 1A). In agreement with the weak overt epidermal phenotypes, the induction of the Bi-L E7 transgene by the K14-tTA transgene was weakest in the dorsal skin. We observed no significant induction of luciferase in the epithelia of Bi-L E7 singly transgenic mice or in bitransgenic mice given doxycycline for as little as 3 days, indicating that there was no leaky expression of the transgene and that it could be repressed effectively with doxycycline.
Figure 1.

The expression of luciferase and E7 in the stratified squamous epithelia of Bi-L E7/K14-tTA bitransgenic mice is eliminated on treatment with doxycycline. A, luciferase assays done on lysates from the dorsal skin, ear, or lower female reproductive tracts of mice from the indicated genotypes and treatment groups. Columns, mean; bars, SD. *, P < 0.05 versus nontransgenic mice by a two-sided Wilcoxon rank-sum test, and n ≥ 3 for all groups of mice. B, Western blot to examine the expression of E7 in lysates from the lower female reproductive tract. To compare the level of expression of E7 in Bi-L E7/K14-tTA bitransgenic mice to that seen in K14E7 mice, equivalent amounts of lysates from three K14E7 mice were pooled, and the indicated amounts of this pool were analyzed in parallel with lysates from mice of the indicated genotypes. Detection of β-actin was used as a loading control.
We next examined the expression of E7 in Bi-L E7/K14-tTA bitransgenic mice by performing Western blots on lysates from the lower female reproductive tract (Fig. 1B). To determine the level of expression of E7 in bitransgenic mice relative to K14E7 mice, a line of mice that we have used extensively in the past to characterize the oncogenic properties of E7 in vivo (2, 3, 40), we loaded control lanes with known amounts of protein from lysates from the tracts of K14E7 mice. The level of E7 expression in Bi-L E7/K14-tTA mice was approximately 10–30% that in the K14E7 mice. This is less than the level of expression of E7 in the Bi-L E7/K5-tTA mice, which was approximately 50% that of K14 E7 (33). There was no detectable expression of E7 in bitransgenic mice given doxycycline or in singly transgenic mice. Thus, even though the level of induction of luciferase in bitransgenic mice was strongest in the lower female reproductive tract, the level of E7 expressed in Bi-L E7/K14-tTA bitransgenic mice was lower than that observed in K14E7 mice. Nevertheless, these data indicate that E7 and luciferase are expressed in the epithelia of Bi-L E7/K14-tTA bitransgenic mice and that this expression can be repressed within as little as 3 days following the administration of doxycycline.
Finally, we investigated whether the expression of E7 in the reproductive tracts of Bi-L E7/K14-tTA bitransgenic mice had detectable, acute effects on the epithelium. We first performed immunohistochemical staining for BrdUrd to measure the synthesis of DNA in endocervical sections from mice injected with the nucleotide analogue one hour before sacrifice (Fig. 2A) and quantified the results (Fig. 2B). Whereas suprabasal endocervical epithelial cells from nontransgenic mice only rarely replicate their DNA, in Bi-L E7/K14-tTA bitransgenic mice we observed a significant induction of the suprabasal synthesis of DNA (P < 1 × 10−6 versus nontransgenic mice, two-sided Wilcoxon rank-sum test). Interestingly, despite the lower level of expression of E7 in the reproductive tracts of bitransgenic versus K14E7 mice (Fig. 1B), the degree to which the induction of the suprabasal synthesis of DNA occurred in both genotypes was statistically indistinguishable. Not surprisingly, we detected no increase in the suprabasal synthesis of DNA in the endocervices of bitransgenic mice given doxycycline or in singly transgenic mice, in which we did not detect E7 (Fig. 1B).
Figure 2.

The endocervical epithelium of Bi-L E7/K14-tTA bitransgenic mice displays aberrant proliferation. A, immunohistochemical analysis of the incorporation of BrdUrd in sections of the endocervical epithelium from mice of the indicated genotypes. Brown nuclei represent cells positive for BrdUrd, and the black line delineates the basement membrane. B, quantification of nuclei positive for BrdUrd in sections shown in A. Eight visual fields at ×40 magnification were scored for nuclei positive or negative for BrdUrd. Columns, mean; bars, SD. *, P < 1 × 10−4 versus nontransgenic mice by a two-sided Wilcoxon rank-sum test, and n ≥ 3 for all groups of mice. C, immunohistochemical analysis of the expression of MCM7 in sections of the endocervical epithelium from mice of the indicated genotypes. Brown nuclei represent cells positive for MCM7, and the black line delineates the basement membrane.
We also examined the expression of MCM7 (Fig. 2C), which is up-regulated in the endocervical epithelium of K14E7 and Bi-L E7/K5-tTA mice (33, 42) and is a marker for HPV-associated high-grade cervical dysplasia and cancer (43). In nontransgenic mice, the expression of MCM7 was restricted to the basal and parabasal cells of the endocervical epithelium. In Bi-L E7/K14-tTA bitransgenic mice, we observed the expression of MCM7 throughout the full thickness of the epithelium. The pattern of expression of MCM7 in singly transgenic mice and in bitransgenic mice given doxycycline was indistinguishable from the pattern observed in nontransgenic mice. Although the level of expression of E7 in the reproductive tracts of Bi-L E7/K14-tTA bitransgenic mice was lower than that observed in K14E7 mice (Fig. 1B), it still was sufficient to induce the expression of MCM7 in suprabasal epithelial cells.
Taken together, these data indicate that the expression of luciferase and E7 in Bi-L E7/K14-tTA bitransgenic mice is induced in multiple epithelial tissues, and the expression of E7 results in acute endocervical phenotypes indicative of aberrant proliferation. Furthermore, the expression of both E7 and luciferase can be repressed and the phenotypes of bitransgenic mice can be reversed with the administration of doxycycline.
Cervical cancers in Bi-L E7/K14E6/K14-tTA triply transgenic mice are dependent on the continuous expression of E7 for their maintenance
To induce cervical cancers in Bi-L E7/K14E6/K14-tTA triply transgenic mice, we treated them chronically with estrogen, which is a necessary co-factor for the development (2, 44) and maintenance (42) of cervical cancers in mice transgenic for E6 and E7. Previously, we found that 6 to 7 months of treatment with estrogen was sufficient to induce cervical cancers in K14E7 and Bi-L E7/K5-tTA mice (33), but based on the lower level of expression of E7 in the lower female reproductive tracts of Bi-L E7/K14-tTA mice versus K14E7 mice (Fig 1B), we extended the treatment of triply transgenic mice so that it ranged from 9 to 10 months. After the completion of the treatment, we sacrificed the mice, harvested and fixed their reproductive tracts, and had them embedded in paraffin and sectioned for analysis. We stained the sections with H&E and examined them histopathologically to score for the presence of dysplastic disease and cancer. Based on the most severe lesion observed in each mouse, we assigned them into the following categories: nondysplastic hyperplasia, low-grade dysplasia [cervical intraepithelial neoplasia 1 (CIN1)], mid-grade dysplasia (CIN2), high-grade dysplasia (CIN3), and cervical cancer. The results are summarized in Table 1.
Table 1.
Incidence of cervical disease in mice treated chronically with estrogen
| Genotype (n) | Estrogen (mo) | Doxycycline (mo) | Grade of cervical disease
|
||||
|---|---|---|---|---|---|---|---|
| Hyperplasia | CIN1 | CIN2 | CIN3 | Cervical cancer (%) | |||
| Nontransgenic (20) | 10 | - | 5 | 13 | 2 | ||
| K14-tTA (20) | 10 | - | 4 | 12 | 3 | 1 | |
| Bi-L E7 (19) | 10 | - | 11 | 6 | 1 | 1 (5) | |
| Bi-L E7/K14E6/K14-tTA (19) | 9 | - | 5 | 9 | 5 (26)*† | ||
| Bi-L E7/K14E6/K14-tTA (17) | 10 | - | 5 | 7 | 5 (29)*† | ||
| Bi-L E7/K14E6/K14-tTA (20) | 10 | 1 | 6 | 10 | 3 | 1 | |
| Bi-L E7/K14E6/K14-tTA (12) | 12 | +1/−2 | 3 | 5 | 2 | 2 (17) | |
NOTE: Mice of the indicated genotypes were treated with estrogen for the duration listed and either treated or not treated (−) with doxycycline for the indicated number of months. The number of mice placed into each category of cervical disease is indicated.
P < 1 × 10−6, when comparing the average severity of disease with that of nontransgenic mice by a two-sided Wilcoxon rank-sum test.
P < 0.03, when comparing the incidence of cancer with that of nontransgenic mice by a two-sided Fisher’s exact test.
We found that 9 months of treatment with estrogen was sufficient to induce cervical cancers in 26% of Bi-L E7/K14E6/K14-tTA triply transgenic mice (P < 0.03 versus nontransgenic mice, two-sided Fisher’s exact test). The proportion of triply transgenic mice with cancer following a tenth month of treatment with estrogen was 29%, which did not represent a statistically significant increase over what we observed in mice treated for 9 months. All triply transgenic mice treated for 9 to 10 months with estrogen had at least CIN2 lesions in their reproductive tracts, with over 70% of both groups displaying CIN3 lesions or cervical cancers. The average severity of disease in the reproductive tracts of triply transgenic mice treated for 9 or 10 months with estrogen was significantly worse than that observed in nontransgenic mice (P < 1 × 10−6, two-sided Wilcoxon rank-sum test).
We next treated a subset of Bi-L E7/K14E6/K14-tTA triply transgenic mice with doxycycline for the final month of a 10-month treatment with estrogen to repress the expression of E7 (Table 1). After being treated for 1 month with doxycycline, none of the triply transgenic mice had any remaining cervical cancers. Furthermore, the majority of triply transgenic mice that received doxycycline had no or only low-grade dysplastic cervical lesions, and the average severity of cervical disease in these mice was indistinguishable from that of nontransgenic mice. One mouse did still have a high-grade dysplastic cervical lesion, however, which is in contrast to the complete absence of high-grade lesions observed previously with Bi-L E7/K5-tTA bitransgenic mice (33).
To confirm whether E6 was still expressed in the endocervical epithelium of triply transgenic mice treated with doxycycline, total cellular protein from the lower reproductive tracts of triply transgenic mice that had or had not been treated with doxycycline were subjected to a quantitative assay to detect the expression of the HPV-16 E6 protein (Fig. 3A). E6 was expressed in both doxycycline-treated and untreated mice; however, the expression of E6 was ~ 2-fold lower in mice treated with doxycycline. To assess whether this small reduction in E6 impaired its function we measured levels of p53 in the same tissues and found no increase in levels of 53, indicative of E6 retaining its ability to destabilize p53 (Fig. 3B).
Figure 3.

Expression of E6 in the lower reproductive tracts of Bi-L E7/K14E6/K14-tTA triply transgenic mice. A, lateral flow assays for the detection of high-risk HPV E6 proteins. Left, duplicate runs for CaSki cell lysates (positive control, top) and lysis buffer (negative control, bottom). The top and bottom panels to the right of the control strips represent duplicate runs of the assay for each of six experimental samples. Along the top, the total amount of cellular protein, in μg, used from each sample is displayed. Samples were taken from Bi-L E7/K14E6/K14-tTA mice treated for 10 months with estrogen that were (+Dox) or were not (No Dox) treated with doxycycline. Shown in the margins are the relative positions for detection of HPV-16 (16), HPV-18 (18) and HPV-45 (45) E6 proteins as well as an internal control (C) that accounts for any interference that lysates may have with the assay’s method for detecting bound protein. Note the signal in each assay that reflects the presence of HPV-16 E6. Under each sample is provided the estimate of amount of E6 per ug of sample based upon a standard curve established using a serial dilution of total protein lysate from the HPV-16-positive CaSki cervical cancer cell line. B. quantification of p53 levels in the same tissues evaluated for E6. To learn whether the 2 fold reduction in E6 led to an impairment in its overall function we analyzed levels of 53, a known target of E6 that is destabilized through E6’s recruitment of the ubiquitin ligase, E6AP (ube3a). No changes in the levels of p53 were noted. To account for the amount of epithelial cell content in the tissue samples, Westerns were reprobed for the epithelial specific marker, keratin 14 (K14).
We conclude that although high-grade dysplasia rarely may remain after the expression of E7 is repressed, cervical cancers still require the continuous expression of E7 for their maintenance even in the presence of the continuous expression of E6.
Cervical cancers are occasionally observed in Bi-L E7/K14E6/K14-tTA triply transgenic mice in which E7 is repressed and then re-expressed
We were interested in knowing whether the re-expression of E7 could lead to the reappearance of neoplastic disease even though cervical cancers had overtly regressed when its expression was repressed, similar to what has been observed in transgenic mice harboring inducible HER2/neu (26), MYC (28), or Wnt1 (27). To investigate this, we treated a group of Bi-L E7/K14E6/K14-tTA triply transgenic mice with doxycycline for 1 month, starting after 9 months of treatment with estrogen. After 1 month, we removed the doxycycline and allowed the mice to age an additional 2 months on estrogen, and we verified by the analysis of luciferase that expression from the Bi-L E7 transgene was re-induced after the removal of doxycycline (data not shown). After this regimen, 2 of 12 triply transgenic mice had cervical cancer (Table 1); however, this does not represent a statistically significant increase in the incidence of cervical cancer over the absence of cancers observed in nontransgenic mice (P ≈ 0.13, two-sided Fisher’s exact test).
To assess whether there was re-expression of E7 in these two cancers, we performed immunohistochemical analysis for MCM7 and p16 (Fig. 4), which are biomarkers linked to the expression of E7 (43). Both biomarkers were expressed throughout the cancers arising in the triply transgenic mice never treated with doxycycline. In the epithelia of triply transgenic mice treated for 1 month with doxycycline, the expression of MCM7 was restricted to the basal and parabasal compartments and p16 was detected only weakly. In the cancers observed in the triply transgenic mice treated with doxycycline for 1 month and then allowed to age an additional 2 months without doxycycline, we found abundant expression of both MCM7 and p16, indicating that E7 was re-expressed in these cancers.
Figure 4.
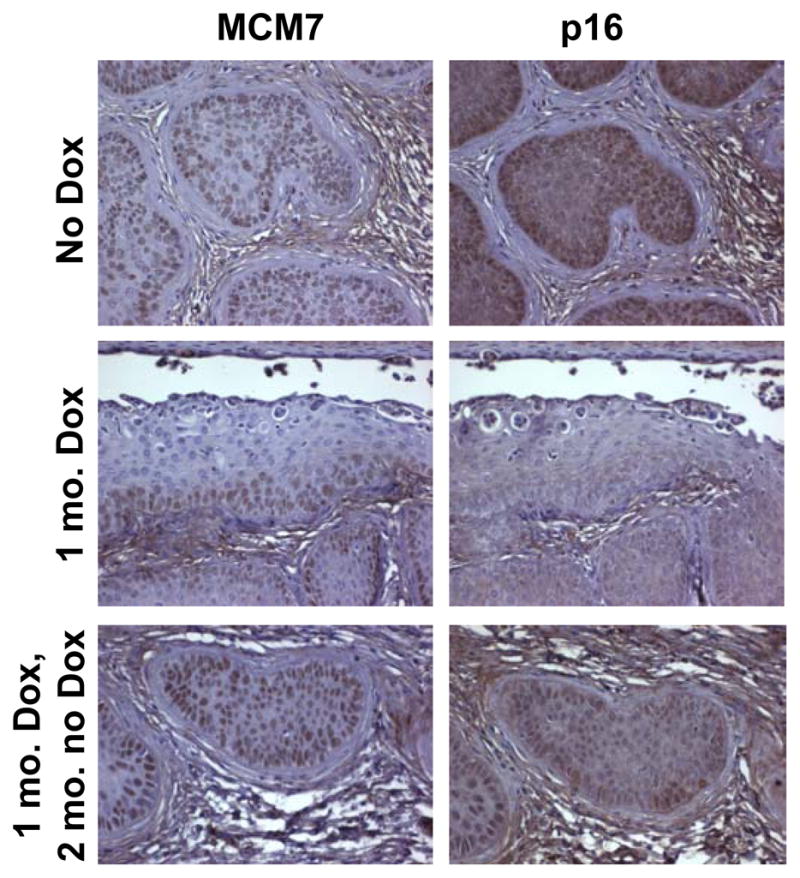
Expression of E7-specific biomarkers in Bi-L E7/K14E6/K14-tTA triply transgenic mice treated chronically with estrogen. Shown are representative microscopic images at ×40 magnification of tissue sections from triply transgenic mice stained with antibodies to either MCM7 (left) or p16 (right). Top, cervical cancers from mice treated for 10 months with estrogen that were not treated with doxycycline. Middle, endocervical epithelia from mice treated for 10 months with estrogen that were treated during the final month with doxycycline. Bottom, cervical cancers from mice treated for 10 months with estrogen that were treated with doxycycline during the 10th month, then taken off of doxycycline and maintained on estrogen for an additional 2 months.
Our data from Bi-L E7/K14E6/K14-tTA triply transgenic mice show that even in the presence of the constitutive expression of E6, established cervical cancers and high-grade dysplastic lesions still require the continuous expression of E7 for their maintenance, although we observed cervical cancers in two triply transgenic mice in which E7 had been repressed and then re-expressed. Whether those cancers represented a re-emergence of neoplastic disease or were cancers that never regressed, however, is unclear.
Discussion
Co-expression of E6 and E7 does not decrease the dependence of cervical cancers on the continuous presence of E7
It has been shown by others that when two oncogenes are co-expressed to induce murine tumors, repressing the expression of both often leads to the more complete regression of tumors than repressing either one alone (45, 46). In our mice, however, all cervical cancers in Bi-L E7/K14E6/K14-tTA triply transgenic mice regressed once we repressed only the expression of E7 (Table 1), despite the constitutive expression of the E6 oncogene (Fig. 3). Our results parallel those observed in cell lines derived from HPV-positive cervical cancers, which require the continuous expression of both E6 and E7 to maintain their growth (14).
Our observation that the dependence of cancers on a single oncogene is not reduced by the presence of a second oncogene is similar to results observed in transgenic mice harboring an inducible construct encoding murine oncogenic K-Ras4bG12D (24). Pulmonary adenocarcinomas that developed in these mice universally regressed once the expression of K-Ras4bG12D was repressed; furthermore, even when this experiment was repeated in the absence of the p53 or p16INK4a tumor suppressive genes, which permitted the more rapid and invasive growth of the adenocarcinomas, all malignancies still regressed completely once K-Ras4bG12D was silenced. Similarly, even on a p53−/−background, nearly all mammary tumors induced by Wnt-1 depended on its continuous expression for their maintenance (27). In contrast, other studies have shown that somatic oncogenic mutations in K-Ras2 (23) or p53 (27) can render cancers independent of the continuous expression of the initiating oncogene. One key difference between these sets of studies is that the former—those in which the cancers universally or nearly universally regressed—used transgenic mice harboring germline mutations in tumor suppressive genes, and the latter—those in which a substantial fraction of cancers persisted—involved somatic oncogenic mutations, although one notable exception is that the germline knockout of p19ARF can decrease somewhat the likelihood of mammary tumors regressing once Wnt1 is silenced (47). In general, the likelihood of regression may not depend on the number of oncogenic mutations present as much as it does on the genetic context in which the cancers develop and on the timing of such mutations. In the case of HPV-associated carcinogenesis, additional oncogenic mutations develop in the context of constitutive expression of both E6 and E7, and presumably mutations that provide the greatest selective advantage in growth do not overlap with the mechanisms by which E6 and E7 drive carcinogenesis. For this reason, the co-expression of E6 with E7 may not reduce the dependence of cervical cancers on the continuous expression of E7.
Cervical cancers may re-emerge after the re-expression of E7
Several previous studies have shown that even though overt cancers may regress once an oncogene is silenced, a residual subpopulation of neoplastic cells may remain (26–28). In Bi-L E7/K14E6/K14-tTA triply transgenic mice in which we reactivated the expression of E7 for 2 months after it had been repressed for a month, two of twelve had cervical cancer (Table 1), but this result was not statistically significant when compared to the absence of cancers in nontransgenic mice. It is therefore unclear whether those two cancers represented a re-emergence of neoplastic disease or were cancers that never regressed once the expression of E7 was repressed. If the cervical cancers re-emerged, two months is insufficient for them to arise de novo in transgenic mice expressing E6 and E7 (48); therefore, the rapidity with which they re-emerged once E7 was re-expressed in triply transgenic mice indicates that that the cancers may have arisen from residual neoplastic cells that were either isolated and undetectable by histopathology or that were part of a persisting low-grade dysplastic lesion. Alternatively, non-neoplastic cells that already had acquired most of the genetic changes required for carcinogenesis may have driven the re-emergence of cancers once E7 was re-expressed. These data suggest that any potential anti-cancer therapy that targets the expression and/or function of E7 may have to be administered long-term to prevent the re-emergence of disease.
Supplementary Material
Acknowledgments
We thank Dr. John Wysolmerski for providing the K14-tTA mice.
Grant Support
This study was supported by grants from the NIH (CA022443 and CA098428). Histology-related services were supported in part by the University of Wisconsin Carbone Cancer Center (NIH grant CA014520).
References
- 1.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 2.Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862–71. [PubMed] [Google Scholar]
- 3.Strati K, Lambert PF. Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer Res. 2007;67:11585–93. doi: 10.1158/0008-5472.CAN-07-3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLaughlin-Drubin ME, Munger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384:335–44. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4. [PubMed] [Google Scholar]
- 6.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 7.Balsitis S, Dick F, Lee D, Farrell L, Hyde RK, Griep AE, et al. Examination of the pRb-dependent and pRb-independent functions of E7 in vivo. J Virol. 2005;79:11392–402. doi: 10.1128/JVI.79.17.11392-11402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balsitis S, Dick F, Dyson N, Lambert PF. Critical roles for non-pRb targets of human papillomavirus type 16 E7 in cervical carcinogenesis. Cancer Res. 2006;66:9393–400. doi: 10.1158/0008-5472.CAN-06-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tungteakkhun SS, Duerksen-Hughes PJ. Cellular binding partners of the human papillomavirus E6 protein. Arch Virol. 2008;153:397–408. doi: 10.1007/s00705-007-0022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 11.Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–9. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 12.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. Embo J. 1991;10:4129–35. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 14.DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol. 2003;77:1551–63. doi: 10.1128/JVI.77.2.1551-1563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A. 2000;97:12513–8. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodwin EC, Yang E, Lee CJ, Lee HW, DiMaio D, Hwang ES. Rapid induction of senescence in human cervical carcinoma cells. Proc Natl Acad Sci U S A. 2000;97:10978–83. doi: 10.1073/pnas.97.20.10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang ES, Naeger LK, DiMaio D. Activation of the endogenous p53 growth inhibitory pathway in HeLa cervical carcinoma cells by expression of the bovine papillomavirus E2 gene. Oncogene. 1996;12:795–803. [PubMed] [Google Scholar]
- 18.Hwang ES, Riese DJ, 2nd, Settleman J, Nilson LA, Honig J, Flynn S, et al. Inhibition of cervical carcinoma cell line proliferation by the introduction of a bovine papillomavirus regulatory gene. J Virol. 1993;67:3720–9. doi: 10.1128/jvi.67.7.3720-3729.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Psyrri A, DeFilippis RA, Edwards AP, Yates KE, Manuelidis L, DiMaio D. Role of the retinoblastoma pathway in senescence triggered by repression of the human papillomavirus E7 protein in cervical carcinoma cells. Cancer Res. 2004;64:3079–86. doi: 10.1158/0008-5472.can-03-3739. [DOI] [PubMed] [Google Scholar]
- 20.Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–72. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 21.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 22.Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- 23.D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235–9. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 24.Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–62. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102–4. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- 26.Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, Dugan KD, et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2:451–61. doi: 10.1016/s1535-6108(02)00212-x. [DOI] [PubMed] [Google Scholar]
- 27.Gunther EJ, Moody SE, Belka GK, Hahn KT, Innocent N, Dugan KD, et al. Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumorigenesis. Genes Dev. 2003;17:488–501. doi: 10.1101/gad.1051603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–7. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 29.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–9. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 30.Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell. 2004;6:577–86. doi: 10.1016/j.ccr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 31.Holmen SL, Williams BO. Essential role for Ras signaling in glioblastoma maintenance. Cancer Res. 2005;65:8250–5. doi: 10.1158/0008-5472.CAN-05-1173. [DOI] [PubMed] [Google Scholar]
- 32.Diamond I, Owolabi T, Marco M, Lam C, Glick A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol. 2000;115:788–94. doi: 10.1046/j.1523-1747.2000.00144.x. [DOI] [PubMed] [Google Scholar]
- 33.Jabbar SF, Abrams L, Glick A, Lambert PF. Persistence of high-grade cervical dysplasia and cervical cancer requires the continuous expression of the human papillomavirus type 16 E7 oncogene. Cancer Res. 2009;69:4407–14. doi: 10.1158/0008-5472.CAN-09-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorre ME, Sawyers CL. Molecular mechanisms of resistance to STI571 in chronic myeloid leukemia. Curr Opin Hematol. 2002;9:303–7. doi: 10.1097/00062752-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 37.Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol. 1987;61:962–71. doi: 10.1128/jvi.61.4.962-971.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J Virol. 1999;73:5887–93. doi: 10.1128/jvi.73.7.5887-5893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dunbar ME, Dann P, Brown CW, Van Houton J, Dreyer B, Philbrick WP, et al. Temporally regulated overexpression of parathyroid hormone-related protein in the mammary gland reveals distinct fetal and pubertal phenotypes. J Endocrinol. 2001;171:403–16. doi: 10.1677/joe.0.1710403. [DOI] [PubMed] [Google Scholar]
- 40.Herber R, Liem A, Pitot H, Lambert PF. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol. 1996;70:1873–81. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schweizer J, Lu PS, Mahoney CW, Berard-Bergery M, Ho M, Ramasamy V, et al. Feasibility study of a human papillomavirus E6 oncoprotein test for diagnosis of cervical precancer and cancer. J Clin Microbiol. 2010;48:4646–8. doi: 10.1128/JCM.01315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brake T, Lambert PF. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc Natl Acad Sci U S A. 2005;102:2490–5. doi: 10.1073/pnas.0409883102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brake T, Connor JP, Petereit DG, Lambert PF. Comparative analysis of cervical cancer in women and in a human papillomavirus-transgenic mouse model: identification of minichromosome maintenance protein 7 as an informative biomarker for human cervical cancer. Cancer Res. 2003;63:8173–80. [PubMed] [Google Scholar]
- 44.Shai A, Brake T, Somoza C, Lambert PF. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res. 2007;67:1626–35. doi: 10.1158/0008-5472.CAN-06-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Podsypanina K, Politi K, Beverly LJ, Varmus HE. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc Natl Acad Sci U S A. 2008;105:5242–7. doi: 10.1073/pnas.0801197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J, et al. Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS ONE. 2008;3:e2125. doi: 10.1371/journal.pone.0002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Debies MT, Gestl SA, Mathers JL, Mikse OR, Leonard TL, Moody SE, et al. Tumor escape in a Wnt1-dependent mouse breast cancer model is enabled by p19Arf/p53 pathway lesions but not p16 Ink4a loss. J Clin Invest. 2008;118:51–63. doi: 10.1172/JCI33320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung SH, Lambert PF. Prevention and treatment of cervical cancer in mice using estrogen receptor antagonists. Proc Natl Acad Sci U S A. 2009;106:19467–72. doi: 10.1073/pnas.0911436106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.