

Abstract
The neurotransmitter, glutamate interacts with glutamate receptor proteins leading to the activation of multiple signaling pathways. Dysfunction in the glutamatergic signaling pathway has been well established as a frequent player in diseases such as Schizophrenia, Alzheimer’s, and brain tumors (gliomas). Recently aberrant functioning of this pathway has also been shown in melanoma. In both glioma and melanoma cases glutamate secretion stimulates tumor growth, proliferation and survival through activation of the MAPK and PI3K/Akt pathways. Extracellular glutamate levels and glutamatergic signaling may in the future serve as biological markers for tumorigenicity and help in targeted therapy for melanoma.
Background
Glutamate is a major excitatory neurotransmitter of the central nervous system (CNS) that potentiates processes such as; neuronal precursor-cell proliferation, cellular homeostasis, synaptic transmission, growth, migration, invasion, survival as well as cell death (1-3). Extracellular glutamate binds to and activates multiple glutamate receptors of which there are two major types; the ionotropic- and the metabotropic-glutamate receptor family. The ionotropic family consists of N-methyl-D-aspartate receptors (NMDA), α-Amino -3-hydroxy-5-methyl-4-isoazolepropionic acid (AMPA), and Kainate receptors. The metabotropic receptor family consists of eight different subtypes (mGluR1-8 or GRM1-8) that are subdivided into three different groups based on sequence similarity and their coupling to small G-proteins. Recent findings have demonstrated expression of functional glutamate receptors in non-neuronal peripheral cells such as; skin, placenta, and colon (4, 5). Furthermore, genomic and proteomic studies have revealed glutamate signaling to be the target of dysregulation in several different cancer types including melanoma thus implicating it in cancer tumorigenesis (5-8). These findings make glutamate receptors and their downstream effector molecules compelling candidates for further investigation into their role in melanomagenesis as well as their investigation as potential therapeutic targets. It must be noted that developmentally, cells of the central nervous system (CNS) such as glia, astrocytes and neurons and melanocytes are derived from neural crest stem cells (9, 10). We propose, based on the developmental lineage, that glial and melanocytes share similar signaling pathways that are important for homeostasis, growth, proliferation and survival and thus may be subject to similar patterns of deregulation leading to disease.
The ionotropic glutamate receptors are composed of large complexes of multi-protein subunits creating ion channels in the cell plasma membranes that allow for influx or efflux of mono- or divalent cations (e.g., Ca2+) (11). Upon binding of glutamate, these ligand-gated channels change their conformation giving rise to ion permeability and oscillations that in many neuronal cells (cerebellar granule cells, neurons, astrocytes, and glial cells) are important for synaptic transmissions, cellular migration and survival (1, 12, 13).
The NMDA receptor plays a major role in many neuronal processes and disease etiologies such as learning, memory and neurodegeneration (i.e., Schizophrenia) (14). The NMDARs are heterotetrameric complexes consisting of; two NR1 (GRIN1) subunits and two NR2 (GRIN2 (A-D)) or a mixture or GRIN2 and GRIN3 subunits predominately expressed in neuronal cells. Upon binding of its cognate ligands, NMDAR permits Ca2+ influx resulting in increased intracellular calcium levels leading to activation of calcium-dependent signal transduction (12). Moderate levels of intracellular Ca2+ influx leads to calmodulin (CaM) and calmodulin-dependent kinase (CaMKs) activation triggering increased protein kinase A (PKA) activity. However, hyperactivation of NMDAR by glutamate leads to calcium-dependent cell death (excitotoxicity), a phenomenon observed in neurodegenerative diseases and brain tumors (15, 16). Recent studies have demonstrated the importance of functional NMDA receptors as GRIN2A and GRIN2B were shown to be mutated with decreased channel permeation in Schizophrenia patients (17). Furthermore, GRIN2A was discovered to be highly mutated in malignant melanoma patients utilizing a whole-exome screen of 14 tumor samples (18).
The AMPA and Kainate receptors are involved in synaptic transmissions in the mammalian CNS and are important for short-term and long-term synaptic plasticity. Both types of receptors have been implicated in neuronal diseases such as; seizures, epilepsy, or alzheimer’s (19). AMPA and Kainate receptors form hetero- or homomeric complexes found primarily in the pre- and/or postsynaptic regions of neuronal cells (12). Glutamate-mediated receptor activation allows for increased membrane permeability resulting in Ca2+ influx causing differential neuronal plasticity (short-term vs. long term) and signaling mechanisms culminating in variable pathway activation (13). AMPA and Kainate receptor dysfunction have been implicated as possible candidates in certain tumorigenic models (20), but to date very little research has been done on their role in melanomagenesis and thus in the interest of time and space will not be discussed any further in detail.
The metabotropic glutamate receptor family is composed of eight different subtypes called GRMs or mGluRs. mGluR subtypes are subdivided into three groups; I, II, and III based on sequence homology and intracellular effects potentiated by variable G-protein coupling (21). mGluRs are members for the G-protein coupled receptor (GPCR) superfamily. GPCRs are membrane bound proteins that mediate signal transduction upon ligand binding and subsequent activation of their cognate trimeric G-protein complex (α, β, and γ subunits). Activation of the secondary messenger proteins, GTP-bound Gα subunit and Gβγ dimer, allows for transit stimulation of signaling pathways important for cellular proliferation, growth, migration, survival, and calcium-mediated cellular homeostasis (21-23). Recently, eloquent investigations have implicated some of these receptors in both glioma and melanoma tumorigenesis through, pharmacological inhibition studies, gain of function mutations or overexpression experiments using transgenic mice (24-28).
Group I consists of mGluR1 (GRM1) and mGluR5 (GRM5). Group I-mGluRs are coupled to Gαq/Gα11 G-proteins that upon glutamate-mediated activation results in stimulation of PLCβ thus causing hydrolytic cleavage of phosphatidylinositol -4,5-bisphosphate (PIP2) forming inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). Release of these secondary messenger’s leads to increased calcium release from ER stores and activation of protein kinase C (PKC) resulting in downstream effector target stimulation.
Group II consists of mGluR2 (GRM2) and mGluR3 (GRM3) and are coupled to Gαi/o. Activation the Gαi/o-proteins cause Gαi/o-mediated inhibition of adenylyl cyclase (AC) causing reduced production of cyclic adenosine monophosphate (cAMP). The release of their cognate Gβγ subunit reduces certain ion channel properties and other downstream effector molecules (21). Group III contains the rest of the mGluRs (4, 6, 7, & 8). These receptors are also coupled to the Gαi/o proteins and potentiate or attenuate some of the same pathways that group II mGluRs target including the MAPK and PI3K/Akt signaling pathways (29, 30).
Glutamate receptors as novel drivers in cancer
Recent studies have demonstrated the role glutamate receptors play in neurodegenerative diseases and tumorigenesis (see Table 1). The implications that NMDA receptor functions as a conveyor of cellular signals important for cognitive learning and apoptosis have been well established in neuronal diseases and brain tumors (13, 31, 32). A recent genetic screen of Schizophrenia patients by Endele et al revealed novel somatic mutations in the genes encoding either GRIN2A or GRIN2B. They showed that mutation of these NMDA receptor channel proteins resulted in suppression of ion permeability or channel conductance. They proposed that reduced Ca2+-influx may lead to reduced activation of Ca2+-dependent signaling mechanisms which may be the underlying cause of the disease (17). However, the causative effect seen in tumors is not the lack of Ca2+ or inhibition of influx, but rather the uncontrolled ion permeability in neuronal cells leading to cell death resulting in dramatic changes in the architecture of the surrounding tissue within the brain (15, 16).
Table 1.
Glutamate receptors in various diseases and malignancies
| Glutamate receptor type | Subclass | Disease type | Genetic Alteration | Possible mechanism | References |
|---|---|---|---|---|---|
| iGluR | GRIN1 | Malignant melanoma | Somatic mutations | Not reported | 18 |
| GRIN2A/2B | Schizophrenia | Germline and de novo translocations | Lack of proper ion conductance | 17 | |
| GRIN2A | Malignant melanoma | Somatic mutations (mini hotspots) | Not reported | 18 | |
| GRIN3 | Malignant melanoma | Somatic mutations | Not reported | 18 | |
| GluR2 | Schizophrenia Alzheimer’s Glioblastoma Multiforme |
Downregulation of GluR2 subunit which is less permissive to Ca2+ ion influx. |
Deregulated AMPA receptor function | 21 | |
| mGluR | mGluR1 | Malignant melanoma | Possible amplification | Deregulated overexpression | 27 |
| Breast cancer | Not reported | Inhibition with antagonists resulted in decreased breast cancer cell growth in a xenograft model |
47 | ||
| mGluR2 | Glioma | Not reported | mGluR2 activity important for glioma growth in vitro/vivo |
26
25 |
|
| mGluR3 | Glioma | Not reported | mGluR3 activity important for glioma growth in vitro/vivo |
26
25 |
|
| Malignant melanoma | Somatic mutations (mini hotspots) | Activating mutations leading to hypersensitive to agonist stimulation of the MEK-MAPK pathway |
28 | ||
| mGluR4 | Colorectal carcinoma | Overexpression in tumor tissue | Not reported | 46 | |
| mGluR5 | Malignant melanoma | Overexpression | Increased activation of MAPK pathway | 51 | |
| mGluR8 | Malignant melanoma | Somatic mutations | Not reported | 28 |
iGluR, ionotropic glutamat receptor family; mGluR, metabotropic glutamate receptor family; GRIN1, N-methyl-D-aspartate receptors (NMDAR) subunit 1; GRIN2A, NMDAR subunit 2A; GRIN2B, NMDAR subunit 2B; GRIN3, NMDAR subunit 3; GluR2, alpha-Amino -3-hydroxy-5-methyl-4-isoazolepropionic acid receptor (AMPAR) subunit 2; mGluR1/2/3/4/5/8, metabotropic glutamate receptors-1/2/3/4/5/8.
It is well established that glioma cells secrete glutamate to the extracellular milieu where glioma cells and neurons reside both in vitro or in vivo (15). High concentrations of glutamate cause neurons to become permeable to Ca2+ ions. Overstimulation of glutamate receptors cause increased intracellular levels of Ca2+ leading to apoptosis allowing glioma cells room to grow and spread (16). The limitations for growth that gliomas face, because of the non-expandable nature of the cranium, necessitates the damage and elimination of normal brain tissue. Recent research has demonstrated that at the periphery of glioma tumor growth one observes the highest levels of glutamate (found at micromolar levels which is ten times that of normal extracellular concentration seen in synapses) (33). It is this prolonged or hyperactivation of glutamate receptors (especially NMDA receptors) on neuron cells via this excess extracellular glutamate that leads to the phenomenon called excitotoxicity (15, 16). Furthermore, the normal protective mechanism that helps maintain excess levels of glutamate in the extracellular space surrounding cells which utilizes a Na2+-dependent transport system is diminished (34). The transporters responsible for this are collectively referred to as the excitatory amino acid transporters 1-4 (EAAT1-4) and are typically found on astrocytic cells. Recent studies using glioma patient samples as well as knockout studies in mice have clearly demonstrated a role these transporters play in neurodegenerative diseases and glioma (35, 36). Collectively, these findings illustrate the major role glutamate receptors and transporters play in cellular homeostasis of neurons, glial cells and astrocytes.
Recently, our lab was the first to demonstrate through the use of whole-exome sequencing the high prevalence of somatic mutations in GRIN2A in malignant melanoma (18). Whole-exome sequencing of 14 tumor and normal matched samples from treatment naïve patients with melanoma allowed the unexpected discovery that the gene encoding for NMDA receptor subunit ε−1 or GRIN2A to harbor 34 somatic mutations in 125 melanoma samples (25.2%). The mutations were distributed throughout the gene, with clustering of mutations at amino acids 371, 372 and 373 or amino acids 1073, 1074 and 1076 within important functional domains. We also observed three recurrent alterations at S278F, E371K, and E1175K as well as 5 nonsense mutations. Furthermore, another group recently published a whole-exome screen of 8 melanoma samples and found 2 additional somatic mutations in GRIN2A, suggesting that genetic alteration of this gene is important (37). We hypothesize, that GRIN2A is a novel tumor suppressor in melanoma. The high percent of somatic mutations that introduce a premature stop codon (in 15% of cases) would result in a truncated protein which may affect ion channel assembly and function. Preliminary functional assessment of mutant GRIN2A corroborates this hypothesis. This report by our group was the first to demonstrate that the glutamate signaling pathway is significantly altered in melanoma (Figure 1). We have since identified additional genes within the glutamate pathway to be somatically mutated in melanoma creating a broader picture of the genetic landscape important for deregulating this pathway. Our lab found GRIN1, GRIN3, PLCβ4, GRM3, PYK2, and ERBB4 to be highly mutated in melanoma. Interestingly, genetic findings in schizophrenia patients implicated alterations to some of the exact genes found mutated in melanoma (14, 17, 38).We hypothesize that mutation in GRIN1 and GRIN3 result in loss of function as proposed earlier for the somatic mutations observed in GRIN2A. In contrast, select mutations in GRM3, a metabotropic glutamate receptor, in melanoma resulted in activation of the GPCR signaling to MEK1/2 causing increased melanoma cell migration and anchorage-independent growth (27). PLCβ is directly downstream of both the NMDAR and GRM receptors and is stimulated in a calcium-dependent manner via CaMKs. Somatic mutations in PLCβ may thus result in aberrant hydrolysis of PIP2. Lastly, we showed that mutation of ErbB4 in melanoma cells causes constitutive kinase activity resulting in increased proliferation, transformation, and migration (39).
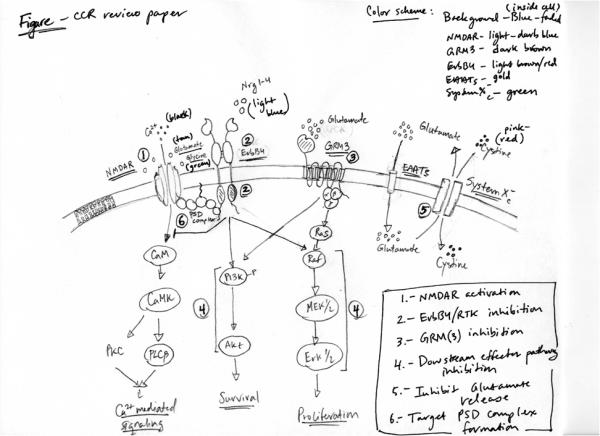
Figure 1. Glutamate signaling pathways in melanoma.

The genes that function in glutamate signaling are specified. Potential therapeutic intervention points are indicated by arrows or inhibition bars followed by a list of small molecule inhibitors or proposed monoclonal antibody . PSD complex, post synaptic density complex. NMDAR, N-methyl-D-aspartic acid (NMDA) receptor. RTK, receptor tyrosine kinase. GRM3, metabotropic glutamate receptor-3. EAAT, excitatory amino acid transporter. System Xc-, cystine-glutamate cotransporter protein.
Pathway analysis and statistical tests of our melanoma exomes allowed the identification of the glutamate signaling pathway to be significantly deregulated in melanoma (18), thus integrating the genes described above as somatically mutated into one single pathway. A similar scenario was described in previous reports by Hahn et al. where they demonstrated a direct link between the glutamate receptor (NMDAR) and receptor tyrosine kinase ErbB4 in Schizophrenic patients. Increased NRG1-induced ErbB4 activity was observed in postmortem tissue samples from Schizophrenic patients causing attenuation of the NMDA receptor function (32). In corroboration with these results, several recent reports have further demonstrated the link between glutamate receptor signaling via the NMDAR and the cellular components found in the Post Synaptic Density (PSD) complex consisting of; PSD-95, PSD-93, SAP-102/97, Pyk2, Fyn (40-44). The PSD complex, through direct biding with the C terminal tails in NMDAR2A (GRIN2A) and ErbB4 helps to localize this signalosome at the cellular/synaptic surface. To date most research on the PSD complex has been done in neuronal derived samples. However, we hypothesize that NMDAR and ErbB4 mediate downstream signal transduction via a cross-talk mechanisms using a form of the PSD complex present in melanoma cells important for tumorigenesis (Figure 1). In addition to the role of ionotropic glutamate receptor signaling in disease, support for a role in signaling through metabotropic glutamate receptors in both neuronal and peripheral cells has also been established (1-5). Activation of mGluR1, mGluR3, mGluR4 and mGluR5 receptors supports proliferation and survival of neural stem cells in fetal brain and neural regions of the adult brain, gliomas, breast cancer, colorectal carcinoma and melanoma tumors through stimulation of the MAPK and PI3K/Akt signaling pathways (16, 23-28, 30, 31). The reports by Chen and colleagues on glutamate signaling and melanoma have helped bring to light the importance that mGluRs play in the development of this disease. They first established the link using an insertional mutant mouse, TG3 that developed spontaneous melanoma on hair-less regions of the mice (26). They demonstrated that the insertion fell within intron 3 of grm1gene in these mice resulting in ectopic expression of mGluR1. To confirm the effects of ectopic expression was directly responsible for melanoma development, Chen and colleagues used a melanocyte specific promoter to drive the expression of mGluR1 in transgenic mice and observed the same phenotype. A similar effect was observed in the hyperproliferation of melanocytes leading to transformation and malignant melanoma. Furthermore, they have found that mGluR1 positive human melanoma and stably expressing mGluR1 melan-a cells secrete more extracellular glutamate than normal melanocytes or control melan-a cells (45, 46). They hypothesize that aberrant secretion of glutamate leads to activation of the mGluR1-dependent signaling pathways responsible for their aggressive tumorigenic phenotypes observed in vitro and in vivo.
The role of mGluR3 in glioma tumorigenesis has been well delineated using functional studies and pharmacological antagonists specific for individual receptors (24, 25). mGluR3 expression is ubiquitous in glioma cells and gliomblastomas. Treatment of glioma cells using specific antagonists for group II mGluRs, LY341495 caused reduced cellular growth in vitro and in vivo via suppression of MAPK and Akt pathways. Recently, we revealed, through exon-capture sequencing of the GPCR superfamily in malignant melanoma, that GRM3 is highly mutated (27). GRM3 or mGluR3 is a group II metabotropic glutamate receptor that is a 7-transmembrane (7-TM) receptor protein which binds glutamate as a ligand resulting in shift from an inactive to an active state. As described earlier, ligand binding induces activation of the cognate small G-protein via a conformational change thus signaling to downstream effector molecules. The identified mutations were located throughout the coding region of the gene and affected the extracellular domains as well as the 7-TM domain with two mini-hotspots (M547K and E807K) located proximal to the transmembrane domains. We discovered that four of the non-synonymous changes in the 7-TM domain (G561E, S610L, E767K, and E870K) resulted in increased activation of the MEK1/2 kinase, increased migration and anchorage independence of melanoma cells. Furthermore, melanoma cells harboring mutant forms of GRM3 exhibited reduced survival in the presence of the MEK1/2 inhibitor, AZD-6244 and were sensitive to shRNA-mediated depletion compared to wild type expressing cells in in vitro and in vivo assays. Expression of mGluRs is restricted to certain tissues and cell types in the CNS demonstrating the essential role they play in neurological processes (21). Hoogduijn et al. showed that human melanocytes lack expression of most forms of mGluRs but express certain forms of NMDAR and AMPAR subunits demonstrating a possible role for glutamate signaling in melanocyte homeostasis (4). The absence of mGluR1, mGluR2/3, and mGluR4, 6-8 on melanocytes has been corroborated by others , however work by Nicolletti and colleagues demonstrated expression of functional mGluR5 receptors in cultured human melanocytes (47). They showed mGluR5-dependent cell proliferation and survival using specific group I mGluR agonists and antagaonists. Interestingly, a recent report by Choi et al., implicated ectopic expression of mGluR5 in the development of melanoma in mice (48). They generated transgenic mice using two forms of mGluR5 (WT and S901A) to delineate the effects of PKC-dependent phosphorylation on mGluR5 plays on CaM binding and subsequent receptor function in Ca2+ oscillations in cells. PKC phosphorylation on S901 in mGluR5 inhibits CaM binding and mGluR5-dependent Ca2+ oscillations. They discovered that one founder mouse (mGluR5 S901A) exhibited pronounced formation of melanoma tumors and increased muscle and bone metastases. Using a melanocyte specific promoter to drive mGluR5 expression they observed an increased incidence in mice with melanoma with a 100% penetrance in the progeny for both WT and S901A mice. MAPK was activated in mice expressing mGluR5 under the TRP1 promoter suggesting increased presence of cellular signaling important for proliferation and growth.
Clinical-Translational Advances
One of the most important advances for the melanoma field has been the validation of c-KIT and BRAF as successful therapeutic targets in melanoma patients (49-55). However, despite these successes it is important to search for additional melanoma drug targets as not all patients harbor c-KIT or BRAF mutations and even patients that harbor these mutations and show response to the inhibitors, develop resistance to the drug (56-59).
Our genomic screening and functional studies have demonstrated the dysregulation of the glutamate receptor signaling pathway consisting of ionotropic- and metabotropic-glutamate receptors in melanoma (18, 27). Figure 1 depicts the glutamate receptor signaling pathways and genes known to be affected by genetic alteration in melanoma. There are several nodes in the glutamate receptor pathway that could be utilized for clinic applications highlighted in Figure 1 and described below: (1) targeted activation of NMDAR using specific agonists such as Ibotenic acid or homoquinolinic acid or glutamate (glutamate analogs-NMDA) would increase the intracellular levels of Ca2+ and potentially induce calcium-mediated cell death, (2) inhibition of ErbB4 signaling in melanoma cells harboring mutant forms of ErbB4 using small molecule inhibitors such as lapatinib, erolotinib, or neratinib or monoclonal antibodies (mAb) to the ErbB4 ectodomain to inhibit ligand induced dimerization and transactivation. This approach is supported by treatment of melanoma cells expressing mutant forms of ErbB4 showing sensitivity to low doses of lapatinib leading to suppression of Akt activation and increased apoptosis (39), (3) use of mGluR antagonists or mGluR3 specific antagonists (BAY 36-7620, LY341495) to inhibit glutamate signaling via this GPCR. As seen in treatment of glioma cells, using LY341495 resulted in reduced cellular growth via suppression of MAPK and Akt pathways (24). The only caveat to utilizing this approach is that prolonged suppression of mGluR3 causes reduced expression of glutamate transporters and potentially reduced proliferation of glioma stem cells (60, 61), (4) targeting downstream effector molecules that are nodal points in cellular signaling for malignant melanoma will allow for better combinatorial therapeutic approaches. Melanoma cells with aberrant glutamate receptor signaling exhibit hyperactive PI3K/Akt and MEK/MAPK pathways (27, 39); thus using inhibitors to PI3K (BKM120, PX866,or GSK2126458), Akt (Perifosine, MK-2206,or GSK2141795), Raf (PLX4032, PLX4720, or PLX4732)MEK (AZD-6244, GSK1120212) or MAPK (AEZS-131) in combination with one of the above mentioned therapies (ErbB4 inhibitor or GRM3 antagonists) to attenuate proliferative and survival signals resulting in increased and specific tumor cell apoptosis, may be suggested (5) secretion suppression of excess glutamate to the extracellular milieu. Using Riluzole, a FDA approved drug for treatment of Amytropic Lateral Sclerosis (ALS) to treat mGluR1 expressing cells showed reduced secreted glutamate levels resulting in decreased melanoma cell proliferation compared normal melanocytes lacking mGluR1 expression (46, 62). Furthermore, Chen and colleagues recently completed a phase 0 clinical trial treating late stage malignant melanoma patients with Riluzole with 33% of the patients exhibiting significant clinical response (63). Importantly, patient samples exhibited reduced MAPK and Akt activation in the presence of Riluzole demonstrating the efficacy and importance of targeting glutamate availability to tumors.
The notion of glutamate addiction in melanoma is corroborated by the findings that human melanoma cells secrete elevated levels of glutamate and express reduced levels of the glutamate transporter proteins EAAT2/EAAT4 ((45, 46) and our unpublished data). The excitatory amino acid transporter-2/4 proteins are normally expressed on neuronal cells to help in reducing the excitotoxicity effect of excess glutamate released by glial cells and astrocytes in the CNS (36). Evidence suggests that high-grade level glioblastomas have reduced expression of EAAT-2 and potentially other EAATs (64). Overexpression of EAAT-2 in glioblastomas resulted in decreased growth and tumorigenesis in vitro and in vivo. In our lab we observed that a panel of malignant melanoma cell lines exhibited reduced transcript levels of both EAAT-2 and EAAT-4, but no difference in expression levels of EAAT-1, EAAT-2 or EAAT-5 (unpublished data). Lastly, another potential target pertaining to the glutamate pathway is the (6) PSD complex. It has recently been reported by Tymianski and colleagues that uncoupling the PSD complex in neuronal cells using a PSD-95 inhibitor prevented stroke damage due to neurotoxicity (65). This approach may be promising in melanoma as both ErbB4 and GRM3 may utilize PSD complexes for proximal localization and regulation of downstream molecules.
Evidence presented in this review suggests that the glutamatergic signaling pathways play key roles in melanoma tumorigenesis. Targeting these pathways directly in conjunction with their downstream effector molecules may prove effective in treating melanoma patients.
Figure 2.
Acknowledgments
Grant Support – This work was supported by the Intramural Research Programs of the National Human Genome Research Institute at the National Institutes of Health.
Footnotes
Disclosure of Potential Conflicts of Interest – No potential conflicts of interest
References
- 1.Komuro H, Rakic P. Modulation of neuronal migration by NMDA receptors. Science (New York, NY. 1993;260:95–7. doi: 10.1126/science.8096653. [DOI] [PubMed] [Google Scholar]
- 2.Nacher J, McEwen BS. The role of N-methyl-D-asparate receptors in neurogenesis. Hippocampus. 2006;16:267–70. doi: 10.1002/hipo.20160. [DOI] [PubMed] [Google Scholar]
- 3.Schlett K. Glutamate as a modulator of embryonic and adult neurogenesis. Curr Top Med Chem. 2006;6:949–60. doi: 10.2174/156802606777323665. [DOI] [PubMed] [Google Scholar]
- 4.Hoogduijn MJ, Hitchcock IS, Smit NP, Gillbro JM, Schallreuter KU, Genever PG. Glutamate receptors on human melanocytes regulate the expression of MiTF. Pigment cell research / sponsored by the European Society for Pigment Cell Research and the International Pigment Cell Society. 2006;19:58–67. doi: 10.1111/j.1600-0749.2005.00284.x. [DOI] [PubMed] [Google Scholar]
- 5.Kim MS, Chang X, Nagpal JK, Yamashita K, Baek JH, Dasgupta S, et al. The N-methyl-D-aspartate receptor type 2A is frequently methylated in human colorectal carcinoma and suppresses cell growth. Oncogene. 2008;27:2045–54. doi: 10.1038/sj.onc.1210842. [DOI] [PubMed] [Google Scholar]
- 6.Kaderlik KR, Minchin RF, Mulder GJ, Ilett KF, Daugaard-Jenson M, Teitel CH, et al. Metabolic activation pathway for the formation of DNA adducts of the carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in rat extrahepatic tissues. Carcinogenesis. 1994;15:1703–9. doi: 10.1093/carcin/15.8.1703. [DOI] [PubMed] [Google Scholar]
- 7.Stepulak A, Luksch H, Gebhardt C, Uckermann O, Marzahn J, Sifringer M, et al. Expression of glutamate receptor subunits in human cancers. Histochem Cell Biol. 2009;132:435–45. doi: 10.1007/s00418-009-0613-1. [DOI] [PubMed] [Google Scholar]
- 8.Tamura H, Suzuki M, Moriya Y, Hoshino H, Okamoto T, Yoshida S, et al. Aberrant methylation of N-methyl-D-aspartate receptor type 2B (NMDAR2B) in non-small cell carcinoma. BMC Cancer. 2011;11:220. doi: 10.1186/1471-2407-11-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adameyko I, Lallemend F. Glial versus melanocyte cell fate choice: Schwann cell precursors as a cellular origin of melanocytes. Cell Mol Life Sci. 2010;67:3037–55. doi: 10.1007/s00018-010-0390-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sommer L. Generation of melanocytes from neural crest cells. Pigment cell & melanoma research. 2011;24:411–21. doi: 10.1111/j.1755-148X.2011.00834.x. [DOI] [PubMed] [Google Scholar]
- 11.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, et al. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science (New York, NY. 1992;256:1217–21. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 12.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–96. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–42. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- 14.Lang UE, Puls I, Muller DJ, Strutz-Seebohm N, Gallinat J. Molecular mechanisms of schizophrenia. Cell Physiol Biochem. 2007;20:687–702. doi: 10.1159/000110430. [DOI] [PubMed] [Google Scholar]
- 15.Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer research. 1999;59:4383–91. [PubMed] [Google Scholar]
- 16.Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001;7:1010–5. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- 17.Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nature genetics. 2010;42:1021–6. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- 18.Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nature genetics. 2011;43:442–6. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mellor IR. The AMPA receptor as a therapeutic target: current perspectives and emerging possibilities. Future Med Chem. 2010;2:877–91. doi: 10.4155/fmc.10.27. [DOI] [PubMed] [Google Scholar]
- 20.Luksch H, Uckermann O, Stepulak A, Hendruschk S, Marzahn J, Bastian S, et al. Silencing of selected glutamate receptor subunits modulates cancer growth. Anticancer Res. 2011;31:3181–92. [PubMed] [Google Scholar]
- 21.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermans E, Challiss RA. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. The Biochemical journal. 2001;359:465–84. doi: 10.1042/0264-6021:3590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 24.D’Onofrio M, Arcella A, Bruno V, Ngomba RT, Battaglia G, Lombari V, et al. Pharmacological blockade of mGlu2/3 metabotropic glutamate receptors reduces cell proliferation in cultured human glioma cells. J Neurochem. 2003;84:1288–95. doi: 10.1046/j.1471-4159.2003.01633.x. [DOI] [PubMed] [Google Scholar]
- 25.Arcella A, Carpinelli G, Battaglia G, D’Onofrio M, Santoro F, Ngomba RT, et al. Pharmacological blockade of group II metabotropic glutamate receptors reduces the growth of glioma cells in vivo. Neuro Oncol. 2005;7:236–45. doi: 10.1215/S1152851704000961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nature genetics. 2003;34:108–12. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- 27.Prickett TD, Wei X, Cardenas-Navia I, Teer JK, Lin JC, Walia V, et al. Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nature genetics. 2011;43:1119–26. doi: 10.1038/ng.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi KY, Chang K, Pickel JM, Badger JD, 2nd, Roche KW. Expression of the metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15219–24. doi: 10.1073/pnas.1107304108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciccarelli R, D’Alimonte I, Ballerini P, D’Auro M, Nargi E, Buccella S, et al. Molecular signalling mediating the protective effect of A1 adenosine and mGlu3 metabotropic glutamate receptor activation against apoptosis by oxygen/glucose deprivation in cultured astrocytes. Mol Pharmacol. 2007;71:1369–80. doi: 10.1124/mol.106.031617. [DOI] [PubMed] [Google Scholar]
- 30.Iacovelli L, Bruno V, Salvatore L, Melchiorri D, Gradini R, Caricasole A, et al. Native group-III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phosphatidylinositol-3-kinase pathways. J Neurochem. 2002;82:216–23. doi: 10.1046/j.1471-4159.2002.00929.x. [DOI] [PubMed] [Google Scholar]
- 31.Rzeski W, Turski L, Ikonomidou C. Glutamate antagonists limit tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6372–7. doi: 10.1073/pnas.091113598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–8. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- 33.Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J Neurochem. 2008;105:287–95. doi: 10.1111/j.1471-4159.2008.05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 35.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–86. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 36.de Groot JF, Liu TJ, Fuller G, Yung WK. The excitatory amino acid transporter-2 induces apoptosis and decreases glioma growth in vitro and in vivo. Cancer research. 2005;65:1934–40. doi: 10.1158/0008-5472.CAN-04-3626. [DOI] [PubMed] [Google Scholar]
- 37.Stark MS, Woods SL, Gartside MG, Bonazzi VF, Dutton-Regester K, Aoude LG, et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nature genetics. 2011;44:165–9. doi: 10.1038/ng.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giegling I, Genius J, Benninghoff J, Rujescu D. Genetic findings in schizophrenia patients related to alterations in the intracellular Ca-homeostasis. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:1375–80. doi: 10.1016/j.pnpbp.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 39.Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nature genetics. 2009;41:1127–32. doi: 10.1038/ng.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia RA, Vasudevan K, Buonanno A. The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3596–601. doi: 10.1073/pnas.070042497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delint-Ramirez I, Fernandez E, Bayes A, Kicsi E, Komiyama NH, Grant SG. In vivo composition of NMDA receptor signaling complexes differs between membrane subdomains and is modulated by PSD-95 and PSD-93. J Neurosci. 2010;30:8162–70. doi: 10.1523/JNEUROSCI.1792-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bjarnadottir M, Misner DL, Haverfield-Gross S, Bruun S, Helgason VG, Stefansson H, et al. Neuregulin1 (NRG1) signaling through Fyn modulates NMDA receptor phosphorylation: differential synaptic function in NRG1+/− knock-outs compared with wild-type mice. J Neurosci. 2007;27:4519–29. doi: 10.1523/JNEUROSCI.4314-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geddes AE, Huang XF, Newell KA. Reciprocal signalling between NR2 subunits of the NMDA receptor and neuregulin1 and their role in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:896–904. doi: 10.1016/j.pnpbp.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 44.Pitcher GM, Kalia LV, Ng D, Goodfellow NM, Yee KT, Lambe EK, et al. Schizophrenia susceptibility pathway neuregulin 1-ErbB4 suppresses Src upregulation of NMDA receptors. Nat Med. 2011;17:470–8. doi: 10.1038/nm.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin SS, Namkoong J, Wall BA, Gleason R, Lee HJ, Chen S. Oncogenic activities of metabotropic glutamate receptor 1 (Grm1) in melanocyte transformation. Pigment cell & melanoma research. 2008;21:368–78. doi: 10.1111/j.1755-148X.2008.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Namkoong J, Shin SS, Lee HJ, Marin YE, Wall BA, Goydos JS, et al. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer research. 2007;67:2298–305. doi: 10.1158/0008-5472.CAN-06-3665. [DOI] [PubMed] [Google Scholar]
- 47.Frati C, Marchese C, Fisichella G, Copani A, Nasca MR, Storto M, et al. Expression of functional mGlu5 metabotropic glutamate receptors in human melanocytes. Journal of cellular physiology. 2000;183:364–72. doi: 10.1002/(SICI)1097-4652(200006)183:3<364::AID-JCP9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 48.Choi KY, Chang K, Pickel JM, Badger JD, 2nd, Roche KW. Expression of the metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15219–24. doi: 10.1073/pnas.1107304108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, et al. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment cell & melanoma research. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327–34. doi: 10.1001/jama.2011.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flaherty KT, Fisher DE. New strategies in metastatic melanoma: oncogene-defined taxonomy leads to therapeutic advances. Clin Cancer Res. 2011;17:4922–8. doi: 10.1158/1078-0432.CCR-10-2612. [DOI] [PubMed] [Google Scholar]
- 55.Jiang X, Zhou J, Yuen NK, Corless CL, Heinrich MC, Fletcher JA, et al. Imatinib targeting of KIT-mutant oncoprotein in melanoma. Clin Cancer Res. 2008;14:7726–32. doi: 10.1158/1078-0432.CCR-08-1144. [DOI] [PubMed] [Google Scholar]
- 56.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer cell. 2010;18:683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alcala AM, Flaherty KT. BRAF inhibitors for the treatment of metastatic melanoma: clinical trials and mechanisms of resistance. Clin Cancer Res. 2012;18:33–9. doi: 10.1158/1078-0432.CCR-11-0997. [DOI] [PubMed] [Google Scholar]
- 60.Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, et al. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci. 2003;17:2106–18. doi: 10.1046/j.1460-9568.2003.02657.x. [DOI] [PubMed] [Google Scholar]
- 61.Nicoletti F, Arcella A, Iacovelli L, Battaglia G, Giangaspero F, Melchiorri D. Metabotropic glutamate receptors: new targets for the control of tumor growth? Trends in pharmacological sciences. 2007;28:206–13. doi: 10.1016/j.tips.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 62.Le MN, Chan JL, Rosenberg SA, Nabatian AS, Merrigan KT, Cohen-Solal KA, et al. The glutamate release inhibitor Riluzole decreases migration, invasion, and proliferation of melanoma cells. The Journal of investigative dermatology. 2010;130:2240–9. doi: 10.1038/jid.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yip D, Le MN, Chan JL, Lee JH, Mehnert JA, Yudd A, et al. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin Cancer Res. 2009;15:3896–902. doi: 10.1158/1078-0432.CCR-08-3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Groot J, Sontheimer H. Glutamate and the biology of gliomas. Glia. 2011;59:1181–9. doi: 10.1002/glia.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature. 2012;483:213–7. doi: 10.1038/nature10841. [DOI] [PubMed] [Google Scholar]