

Abstract
Alcoholism and acquired immune deficiency syndrome are associated with severe muscle wasting. This impairment in nitrogen balance arises from increased protein degradation and a decreased rate of protein synthesis. The regulation of protein synthesis is a complex process involving alterations in the phosphorylation state and protein-protein interaction of various components of the translation machinery and mammalian target of rapamycin (mTOR) complexes. This review describes mechanisms that regulate protein synthesis in cultured C2C12 myocytes following exposure to either alcohol or human immunodeficiency virus antiretroviral drugs. Particular attention is given to the upstream regulators of mTOR complexes and the downstream targets which play an important role in translation. Gaining a better understanding of these molecular mechanisms could have important implications for preventing changes in lean body mass in patients with catabolic conditions or illnesses.
Keywords: AMP-activated protein kinase/tuberous sclerosis complex 2/Ras homolog enriched in brain, Rag GTPases, Phospholipase D, Mitogen-activated protein kinase, Translation initiation, Elongation
INTRODUCTION
Alcoholism can cause diverse metabolic and functional changes in skeletal and cardiac muscle, and when consumed in excess, it has detrimental effects on essentially all organs. A hallmark of prolonged alcohol (EtOH) abuse is the loss of lean body mass, similar to that commonly observed in patients with acquired immune deficiency syndrome (AIDS), cancer and sepsis[1,2]. Muscle wasting during catabolic illness is linked to increased morbidity and mortality, with the loss of muscle arising from a decreased rate of protein synthesis and/or increased protein degradation[3].
Protein synthesis is a highly regulated process that includes amino acid transport, signal transduction events, transcription and translation. Ribosomal translation of mRNA is composed of three phases: (1) initiation, whereby methionyl-tRNA attaches to a 40S ribosomal subunit to form the 43S pre-initiation complex. This complex binds to mRNA, with subsequent binding of 60S to form an active 80S ribosome complex capable of translation; (2) elongation, during which tRNA-bound amino acids are incorporated in growing polypeptide chains; and (3) termination, where the completed protein is released from the ribosome[4-7].
EtOH adversely affects protein synthesis, both in animal isolated organs and in cell culture systems[8-11]. The mechanism responsible for the EtOH-induced inhibition of muscle protein synthesis is, in part, mediated through the mammalian target of rapamycin (mTOR). mTOR is a central regulator of cell growth, survival and metabolism under both catabolic and anabolic conditions[12-15]. The mTOR kinase exerts its effect as part of two biochemically and functionally distinct protein complexes termed mTOR complex (mTORC)1 and mTORC2[16,17]. In addition to mTOR, the proteins mLST8/GβL and Deptor (DEP- domain-containing mTOR-interacting protein)[18] are members of both mTORC1 and mTORC2. On the other hand, raptor (regulatory associated protein of mTOR) and proline rich Akt substrate of 40 kDa (PRAS40), are unique members of mTORC1, whereas Rictor (rapamycin insensitive companion of mTOR), stress-activated protein kinase (SAPK)-interacting protein 1 (Sin1) and PRR5/Protor (protein observed with rictor) are distinct components of mTORC2[17,19-21].
A number of signaling pathways are involved in enhancing or inhibiting mTORC1 activity. For example, stimulation of mTORC1 by growth factors is regulated via phosphoinositol-3 kinase (PI3K)/Akt and the mitogen-activated protein kinase (MAPK) pathways which are initiated by ligand binding to cell membrane-bound cognate receptors[14,22,23]. Recently, phospholipase D (PLD) and its metabolite phosphatidic acid (PA) have been shown to regulate mTORC1 activity[24-27]. Likewise, Rag GTPases appear to play an important role in amino acid signaling to mTORC1[28-33]. In each case, the activation of mTORC1 alters the phosphorylation state and the function of direct substrates, such as the eukaryotic initiation factor 4E binding protein 1 (4E-BP1) and ribosomal S6 kinase 1 (S6K1). As such, these changes are tightly coupled with increased protein synthesis.
In contrast to anabolic stimuli, environmental and nutritional stressors negatively affect mTORC1 through the metabolic controller AMP-activated protein kinase (AMPK)[34-38]. Despite progress in understanding how growth factors regulate mTORC1 signaling, it remains unclear how EtOH controls this pathway. In this review, we focus on the mechanism by which EtOH regulates mTORC1 as well as the downstream targets involved in the process of translation initiation and elongation.
REGULATION OF MTORC1 ACTIVITY BY ETOH ACTIVITY BY ETOH
The control of protein synthesis is a complex process involving alterations in the phosphorylation of various downstream signaling components of mTORC1 and mTORC2[7,39,40]. In the presence of EtOH, there is a decrease in mTORC1 activity towards its substrate proteins 4E-BP1 and S6K1[8,10]. Decreased 4E-BP1 phosphorylation redistributes eIF4E from the active eIF4E/eIF4G complex to the inactive eIF4E/4E-BP1 complex. This suppresses the binding of the active complex to the mRNA cap and inhibits translation initiation. Reduced S6K1 activity, on the other hand, affects several substrates including eukaryotic elongation factor 2 kinase (eEF2K), and protein phosphatase 2A (PP2A)[41]. As a consequence of these changes, there is an increase in eEF2 phosphorylation, thereby suppressing the elongation process. Conversely, EtOH stimulates mTORC2 activity as exemplified by increased S473 phosphorylation of Akt[40]. This, in turn, enhances the phosphorylation of PRAS40, as well as increasing its interaction with mTOR[10]. Collectively, these data indicate a regulatory link between these two mTOR complexes.
Based on current information, several upstream proteins play an important role in regulating mTORC1 activity in response to EtOH (Figure 1). AMPK phosphorylation and activation are increased by EtOH, thereby downregulating mTORC1 signaling. EtOH also affects the PI3K pathway that signals through the Akt kinase and PLD pathways. Finally, MAPK signaling has been shown to negatively mediate the effect of EtOH and regulate the activity of mTORC1.
Figure 1.
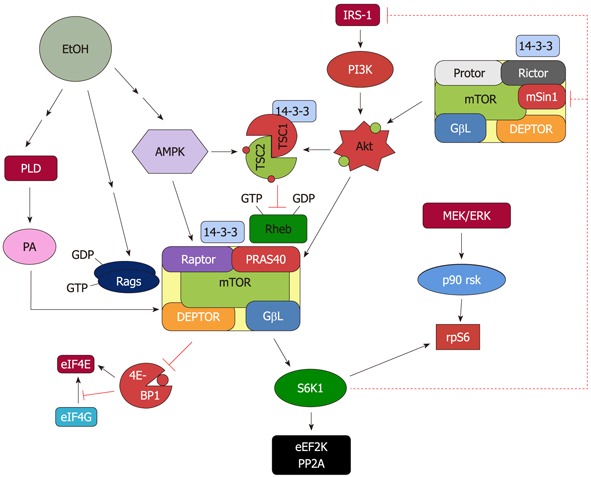
Proposed model for the regulation of mammalian target of rapamycin complex 1 and mammalian target of rapamycin complex 2 in response to EtOH. EtOH decreases the activity of mammalian target of rapamycin complex (mTORC)1 toward its substrates 4E binding protein 1 (4E-BP1) and S6 kinase 1 (S6K1). This process is mediated via multiple signaling routes including AMP-activated protein kinase (AMPK), phosphoinositol-3 kinase (PI3K)/Akt, phospholipase D (PLD) and Rag GTPases. The role each of these upstream regulators plays in affecting mTORC1 function is discussed in the text. The decrease in S6K1 phosphorylation with EtOH signals to mTORC2 as part of a feedback loop. As such, EtOH increases mTORC2 activity toward Akt, which then enhances phosphorylation of its substrate proline rich Akt substrate of 40 kDa (PRAS40).
UPSTREAM REGULATORS OF MTORC1
AMPK pathway
AMPK plays an important role in the process of cellular energy homeostasis. Activation of AMPK leads to the upregulation of ATP production pathways; conversely, this suppresses energy consuming processes such as protein synthesis[42]. Stimulation of AMPK is known to have a protective effect in cells. For example, in cardiomyocyctes, AMPK activation constrains hypertrophic growth[34]. Likewise, this kinase appears to play a role in protecting against hypoxic injury[38]. In contrast to muscle, AMPK activity is reduced in liver and cultured hepatoma cells following EtOH treatment[43-45], and this may explain a number of adverse effects associated with this drug. Along these lines, upregulation of AMPK with the pharmacological agent 5-aminoimidazole-4-carboxamide-1-β-D ribonucleoside (AICAR) prevents the development of alcohol-induced fatty liver in animals[46]. A more detailed examination of the differences between muscle and liver responses to EtOH is beyond the scope of the current review.
Recently, we reported that EtOH increases the phosphorylation of AMPK in C2C12 myocytes[10,41]. This increase was associated with enhanced AMPK activity, as determined via an in vitro kinase assay utilizing immunoisolated AMPK and the substrates acetyl-CoA carboxylase and raptor. The impairment of mTOR signaling by EtOH is mediated by a variety of protein modifications that are facilitated by AMPK. These include enhanced phosphorylation levels for the mTORC1 component raptor, the GTPase-activator protein (GAP) tuberous sclerosis complex 2 (TSC2)[47] and the elongation factor eEF2 (for more details see below).
Raptor functions as a scaffold protein in mTORC1 and it recruits mTORC1 substrates. The importance of raptor in mTOR signaling has been highlighted using deletion and knockdown approaches, both of which diminish mTORC1 activity[48-50]. Alteration of the phosphorylation state of various mTORC1 components can affect the activity of this complex. These modifications also affect the protein-protein interactions that occur within mTORC1, as well as with other cytosolic proteins. Indeed it has been reported that activation of AMPK under energy stress increases the phosphorylation of raptor at S792 and enhances the interaction of this protein with the cytosolic anchor protein14-3-3[36]. The 14-3-3 protein regulates the activity of binding partners by inducing changes in their catalytic activity, either through a conformational change, a masking of the catalytic region of the protein, or the translocation of the protein to another site, such as from the nucleus to the cytoplasm[51-54]. As such, existing protein-protein interactions are disrupted. EtOH increases raptor phosphorylation at this residue, concurrent with an increased interaction with 14-3-3. Furthermore, this drug increases the binding of mTOR with other mTORC1 components in C2C12 myocytes[10]. Hence, it is possible that the increased association of raptor with mTOR and 14-3-3 is partially responsible for the decreased mTOR activity, as assessed by its effect on 4E-BP1 and S6K1.
AMPK regulates mTOR, at least in part, via its action on TSC2[36,55]. Following activation, TSC2 localizes to the endosome/lysosome membrane where it serves as a GAP for the small G-protein Ras homolog enriched in brain (Rheb)[56,57]. When TSC2 is active, it converts Rheb from the GTP form to Rheb-GDP, thereby blocking the ability of Rheb to activate mTOR signaling. The activity of Rheb is also dependent upon its ability to bind to mTOR[57-60]. Interestingly, Rheb binds to mTOR in either its active or inactive state[61]. However, the GTP form of Rheb is required for the activation of mTOR signaling, thereby enhancing the ability of mTOR to regulate its substrates.
The function of TSC2 can be either stimulated or inhibited following phosphorylation, and this is dependent upon the site targeted. Phosphorylation of TSC2 at S1345 by AMPK enhances the activity of this protein[55]. In contrast, phosphorylation of TSC2 (T1462, S939) by PI3/Akt inhibits the function of TSC2 and reduces its suppressive effect toward Rheb[56,62-64]. It should be noted that phosphorylation of TSC2 does not directly affect the GAP activity of this protein, suggesting that other mechanisms are responsible for its function. Indeed, it has been proposed that phosphorylation of TSC2 at S939 by Akt increases binding of TSC2 with 14-3-3[65]. As such, this may sequester TSC2 in the cytosol, thereby preventing the interaction of this protein with Rheb.
Under conditions of energy starvation, there is an increase in phosphorylation of TSC2 at S1345 and T1227. The importance of these sites has been demonstrated in studies in which mutations of S1337, S1341 and S1345 inhibited the stress-induced activation of TSC2, thereby decreasing its ability to regulate Rheb[55]. Similar to the effects of energy deprivation, EtOH increases the phosphorylation of TSC2 at S1387, and this may enhance the ability of TSC2 to negatively regulate Rheb[66]. However, it is unknown whether EtOH alters the nucleotide state of Rheb. Nevertheless, if EtOH increases the amount of Rheb in the GDP form, then this change may account for the observed decrease in protein synthesis. Regardless, EtOH has been shown to decrease the extent of the Rheb-mTOR interaction. Again, this does not necessarily signify a decrease in the GTP form of Rheb, because both Rheb-GTP and Rheb-GDP bind with equal efficiency to mTOR[61]. However, the overall decrease in the Rheb-mTOR interaction would be expected to decrease mTORC1 activity.
The MEK/ERK signaling pathway also mediates mTOR signaling via its effect on TSC2. Upon activation, ERK phosphorylates TSC2 at residue S664, thereby repressing the GAP activity of this protein towards Rheb[67,68]. This is comparable to the inhibition of TSC2 produced by insulin activation of the PI3K/Akt pathway. Owing to the fact that AMPK, ERK and Akt phosphorylate TSC2 at different sites, it appears that these pathways act independently in regulating the function of TSC2. At present it is unknown whether EtOH signals to TSC2 via the MEK/ERK pathway, although EtOH has been reported to decrease MEK/ERK and p90RSK signaling[69]. In contrast to insulin, EtOH does not alter TSC2 phosphorylation at the site (T1462) which is targeted by Akt[10]. Thus, these data suggest that EtOH signals to TSC2/Rheb via a PI3K/Akt/TSC2 independent pathway.
PLD
PLD is a lipid metabolizing enzyme that plays an important role in epidermal growth factor-mediated cellular proliferation[25]. Recent work suggests a role for PLD as an upstream regulator of the mTOR pathway, perhaps being linked to Rheb[24,70]. The putative connection between these proteins is based on the observation that Rheb binds to PLD under in vitro conditions and stimulates its activity. As noted above, Rheb is activated by the AMPK/TSC2 pathway, suggesting that AMPK also plays a role in regulating PLD. From a mechanistic standpoint, PLD acts to hydrolyze phosphatidylcholine into choline and PA, with PA functioning as a second messenger. PA also regulates the mTOR pathway in mitogenesis and in the mechanical stimulation of muscle growth. Although the mode of PA action in regulating mTORC1 is uncertain, it has been posited to interact with mTOR, thereby stabilizing the interaction of mTOR to the other components in mTORC1 and mTORC2[26]. On the other hand, it has also been suggested that PA regulates mTOR via an indirect mechanism. As such, the PA metabolite LPA (lysophosphatidic acid) has been proposed to increase mTOR activity via the activation of the MEK/ERK pathway[27].
EtOH and 1-butanol appear to effect mTOR signaling via their negative impact on PLD activity[25]. Although the mechanisms have not been established, EtOH has been shown to affect AMPK/TSC2/Rheb[66]. Because Rheb is upstream mediator of PLD[24], the inactivation of Rheb by EtOH may downregulate the production of PA. In addition to these signaling events, a second mechanism has also been proposed to explain the decrease in PA levels following EtOH exposure. Ethanol completes with water as a substrate for PLD, and as such, this results in the preferred production of phosphatidylethanol[71]. Although the effects of phosphatidylethanol on mTORC1 signaling are unknown, the decrease in PA levels should negatively impact mTORC1 activity. Accordingly, butanol decreases serum-stimulated phosphorylation of S6K1 and 4E-BP1, and this effect can be counteracted via the addition of PA[70]. Likewise, the adverse effects of EtOH on differentiation and mTOR signaling for neural stem precursor cells are negated following the addition of PA[25]. In agreement with these studies, we observed that PA addition counteracts the suppressive effect of EtOH in C2C12 myocytes.
Rag GTPases
The Rag proteins are a family of four related small GTPases which have been implicated in amino acid signaling to mTORC1[28,29,31]. Rag proteins form heterodimers, whereby a single RagA or RagB protein interacts with either RagC or RagD. The function of the Rag complexes are controlled by the nucleotide loading state of RagA or RagB, as evidenced by over-expression of the constitutively active form of GTP-bound Rag heterodimer[30]. Recent studies reported an increased interaction of Rag proteins with raptor and mTOR following exposure to leucine. This Rag-mTORC1 complex is translocated to endosomal/lysosomal membranes where it associates with the ragulator, a trimeric complex consisting of p14, p18 and MP1 proteins[72]. The association of mTORC1 with lysosomal membranes promotes mTORC1 activation by increasing its co-localization with Rheb.
Rag proteins may be an important target for the action of stressors. Along these lines, EtOH has been shown to cause leucine resistance under both in vivo and in vitro circumstances[73]. Indeed, we showed that EtOH attenuates the association of Rag proteins with raptor in C2C12 myocytes. This decreased interaction also reduces the binding of Rheb and mTOR. Thus, it may account for the decline in mTORC1 activity. In addition to the effects on protein-protein interactions, EtOH may affect the nucleotide state of Rag proteins. Along these lines, the suppressive effect of EtOH on mTORC1 activity can be overcome by over-expressing GTP-bound Rag proteins. Thus, these data suggest that a change from active to inactive Rag heterodimer plays an important role in mediating the negative effects of EtOH[66].
THE MTORC1 DOWNSTREAM TARGETS
S6K1 and 4E-BP1
Various signaling pathways converge on mTORC1, thereby affecting its activity towards downstream targets including 4E-BP1 and S6K1. The translation repressor protein 4E-BP1 binds to eIF4E and inhibits the eIF4E-eIF4G interaction. This prevents the formation of the active eIF4E-eIF4G complex which plays an important role in the cap-dependent translation initiation process. Hyperphosphorylation of 4E-BP1 in response to insulin and insulin-like growth factor (IGF-1) releases eIF4E from 4E-BP1, and increases its binding with eIF4G[74]. On the other hand, hypophosphorylation of 4E-BP1 under catabolic conditions, such as observed in sepsis and burn, or after treatment with human immunodeficiency virus (HIV) antiretroviral drugs increases its binding with eIF4E and blocks the formation of the initiation complex[75-78]. EtOH also decreases the phosphorylation of 4E-BP1 both in muscle and in C2C12 myocytes, thereby disrupting the formation of active eIF4E-eIF4G initiation complex[69,79]. Thus, this treatment appears to diminish the binding of mRNA with the ribosome and limit protein synthesis.
S6K1 plays an important role in protein synthesis in skeletal muscle. This is evidenced by data from genetic studies in which S6K1 null mice are smaller than their wild-type littermates and exhibit muscle atrophy. Likewise, exposure to rapamycin or S6K1 deletion causes similar reductions in myocyte size[80]. Activation of S6K1 by growth factors and mitogenic stimuli is regulated through serial phosphorylation. In contrast, both in vivo and in vitro studies of muscle and C2C12 myocytes show that EtOH inhibits the ability of S6K1 to regulate downstream targets such as ribosomal protein S6 (rpS6)[8,69,81]. Whereas the function of rpS6 is not fully understood, S6K1 also phosphorylates several substrate proteins involved in translation initiation including eIF4B, PDCD4, as well as a kinase and phosphatase that control the elongation process (eEF2K, PP2A). In addition, S6K1 regulates rictor and insulin receptor substrate-1 as part of the mTORC1-dependent feedback loop with mTORC2[82,83].
eEF2K, PP2A and eEF2
Translation elongation utilizes a set of proteins termed eukaryotic elongation factors (eEFs). These proteins, which include eEF1A, eEF1B and eEF2 mediate ribosomal translocation[84]. Phosphorylation of eEF2 is a prime regulator of this protein, with increased phosphorylation resulting in reduced activity, along with decreases in the rate of protein synthesis[85]. One of the upstream regulators of eEF2 is eEF2K, a Ca2+/calmodulin kinase III[86,87]. The activity of eEF2K can be mediated via a number of signaling pathways, including mTOR/S6K1 and the SAPK 2a/p38. These finding have been confirmed via studies using the inhibitors rapamycin and SB 203580 which block the ability of eEF2K to regulate eEF2[88-90]. The activation of this kinase is regulated through single or multisite phosphorylation when cells are exposed to various stimuli. As such, there is increased phosphorylation of eEF2K at the Ser 366 residue following exposure to insulin, IGF-I or neurotrophic factors. Phosphorylation of eEF2K by insulin inhibits its activity, with phosphorylation levels being inversely related to the rate of elongation. On the other hand, phosphorylation of eEF2K at other sites in response to stress conditions increases its activity, thereby resulting in the inactivation of eEF2[84]. Thus, this contributes to the overall decrease in peptide chain elongation and protein synthesis. Together, these results indicate that phosphorylation of eEF2K can regulate eEF2 either positively or negatively, depending on the stimuli and the particular residue that is phosphorylated.
We have previously reported that increased eEF2 phosphorylation in response to EtOH is not mediated via eEF2K[41] (Figure 2). This conclusion is based on the observation that EtOH decreased both the phosphorylation (Ser 366) and activity of eEF2K, with this latter result being verified using an in vitro assay with purified eEF2 as substrate. This decrease in eEF2K activity was consistent with the reduced activity of its upstream regulators mTOR and S6K1. Furthermore, treatment of C2C12 myocytes with an eEF2K inhibitor (rottlerin) did not suppress the EtOH-induced increased in eEF2 phosphorylation, further confirming the lack of a role for this kinase.
Figure 2.
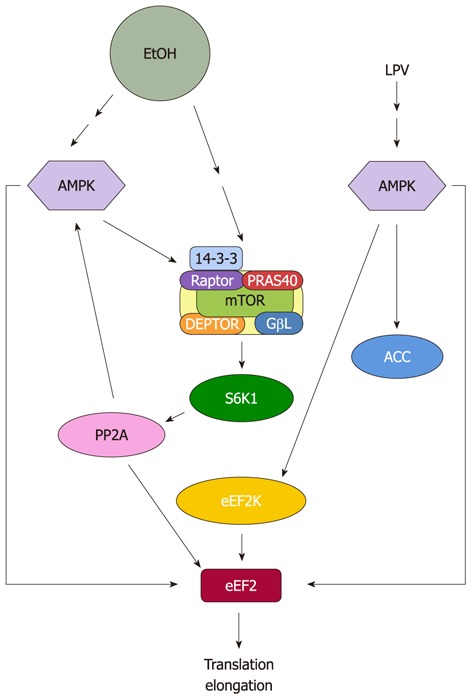
Proposed model for regulation of eukaryotic elongation factor 2 by EtOH and human immunodeficiency virus antiretroviral drug lopinavir. EtOH and lopinavir (LPV) increase eukaryotic elongation factor 2 (eEF2) phosphorylation and inactivate this protein. This process is directly mediated by increased AMP-activated protein kinase (AMPK) activity. EtOH reduces eEF2 function owing to decreased protein phosphatase 2A (PP2A) activity, while LPV regulates this process by increasing eEF2 kinase (eEF2K) activity. mTOR: Mammalian target of rapamycin.
Because eEF2K does not appear to be responsible for EtOH-induced eEF2 phosphorylation, it is possible that this activity is controlled by another S6K1 target, namely the phosphatase PP2A. Indeed, a recent study showed that EtOH decreases PP2A level in heart muscle[91]. Likewise, incubation of C2C12 myocytes with EtOH reduced the in vitro activity of PP2A, as judged using immunoisolated eEF2 substrate. Therefore, inactivation of eEF2 can be directly mediated by PP2A, at least in response to EtOH[41]. The role for PP2A in this process has been further verified by experiments using rapamycin and the MEK inhibitor PD98059. These compounds were shown to increase PP2A activity, and as such, this may account for the ability of these agents to block the effects of EtOH on eEF2 phosphorylation.
eEF2 can also be regulated independent of mTORC1/S6K1 signaling, with this occurring via the action of AMPK. Along these lines, EtOH directly increases the activity of this kinase towards eEF2[41]. From a mechanistic standpoint, the increased phosphorylation and activity of AMPK may be the result of increased upstream activation and/or a decreased PP2A activity. For instance, EtOH has also been shown to reduce the in vitro activity of PP2A towards AMPK. Thus, suppression of PP2A can indirectly affect eEF2 phosphorylation, via the reduced action of this phosphatase on AMPK.
Rictor and mTORC2
mTORC2 is a protein complex regulating a number of ATP kinase family members including Akt, protein kinase Cα and serum-and glucocorticoid-induced protein kinase 1[92-95]. In contrast to mTORC1, the function of mTORC2 is not well characterized, although it has been reported to play an important role in regulating the actin cytoskeleton[96]. One of the core subunits of mTORC2 is rictor, and its importance has been established by studies in which the deletion of this gene is embryonically lethal in mice[50]. Recent studies demonstrate that over-expression of rictor in gliomas leads to hyperactivation of mTORC2. As such, this promotes tumor cells proliferation[97]. Conversely, down regulation of rictor inhibits the ability of mTORC2 to regulate its target Akt at S473[39,98]. This downregulation also increases S6K1 and 4EBP1 phosphorylation in mesangial cells, thereby increasing protein synthesis[39]. Recently, we showed that shRNA knockdown (KD) of rictor is capable of stimulating protein synthesis in C2C12 myocytes. This increase was correlated with decreased rictor phosphorylation and binding with 14-3-3. In addition, rictor KD increased S6K1 and PP2A activities, as well as decreasing Akt and eEF2 phosphorylation[10]. Hence, the observed increased in protein synthesis in C2C12 myocytes with rictor KD suggests a possible regulatory link between the action of mTORC1 and mTORC2 following EtOH exposure.
The activity of mTORC2 can be regulated by changes in the phosphorylation state of component proteins, as well as through alterations in protein-protein interactions. It is noteworthy that recent studies indicate the phosphorylation of rictor at T1135 by the mTORC1-dependent kinase S6K1 is not essential for mTORC2 assembly and activity[83,99,100]. On the other hand, studies have shown that increased phosphorylation of rictor at this site in response to growth factors correlates with enhanced binding of 14-3-3[82,83]. In C2C12 myocytes treated with EtOH, there is a reduction in S6K1 activity along with a decreased phosphorylation of rictor at T1135. In addition, EtOH decreases the association of rictor with the negative regulators 14-3-3 and Deptor. These alterations appear to be responsible for the increased activity of mTORC2, as assessed by enhanced S473 Akt phosphorylation. These results have been confirmed under in vitro conditions, where EtOH stimulates mTORC2 activity towards the purified substrate Akt[10]. It should be noted that elevated Akt phosphorylation in C2C12 myocytes is not necessarily due to the action of mTORC2. Instead, this may be the result of an EtOH-induced reduction in PP2A activity[41]. However, in contrast to C2C12 myocytes, a recent study reported that EtOH increases PP2A activity in heart muscle, while simultaneously decreasing Akt phosphorylation[101]. Thus, the effects of EtOH may be cell-type specific.
Activation of Akt has been reported to change the phosphorylation status of its two downstream targets TSC2 and PRAS40[60,102]. However, EtOH does not affect the phosphorylation of the TSC2 residue (T1462) that is targeted by PI3K/Akt[10]. Instead, EtOH increases the phosphorylation of TSC2 at S1387, and this is most likely due to the action of AMPK. On the other hand, the activation of Akt by EtOH increases phosphorylation of the PRAS40 protein[10]. Although the function of PRAS40 is not well established, it has been postulated to act as a negative regulator of mTORC1. For example, multiple lines of evidence indicate that insulin increases PRAS40 phosphorylation at T246, whereas AICAR decreases phosphorylation at this site. Phosphorylation of PRAS40 by insulin enhances binding of this protein with 14-3-3, and this appears to be crucial for relieving the inhibitory effect of PRAS40 on mTORC1 activity[10,103-105]. In contrast, others have reported that phosphorylation of PRAS40 and its binding to 14-3-3 is not required for the activity of mTORC1[106,107]. EtOH increases the phosphorylation of PRAS40 in C2C12 myocytes, and this appears to be in conflict with the known negative effect of this agent. Furthermore, this increased phosphorylation was not associated with a change in the PRAS40 binding with 14-3-3[10]. Instead, EtOH increased the interaction between PRAS40 and mTOR, consistent with reports in which stressors, such as amino acid deprivation or treatment with 2-deoxyglucose, also increased this binding[104]. Thus, it appears that PRAS40 phosphorylation per se, does not necessarily affect its action. Instead, protein-protein interactions seem to play a more important role.
PROTEIN SYNTHESIS SIGNALING AND HIV DRUGS
AIDS is associated with severe muscle wasting, as well as adverse affects on protein, lipid, and carbohydrate metabolism[108]. Currently, a number of anti-retroviral drugs are available for the treatment of HIV infection. These agents are classified according to their mechanism of action as HIV-1 proteinase inhibitors (PIs) (e.g., lopinavir, indinavir, nelfinavir), nucleoside reverse transcriptase inhibitors (e.g., nevirapine), and non-nucleoside reverse transcriptase inhibitors (e.g., zidovudine). A combination of drug regiments is used as part of the highly active anti-retroviral therapy[109]. Paradoxically, treatment with HIV drugs also causes metabolic disorders such as changes in protein metabolism, hormones and peripheral lipodystrophy[78,110], even though it markedly reduces morbidity and mortality in AIDS patients[111].
The mechanisms by which HIV drugs affect protein synthesis are not well defined. However, we have shown that a number of these agents impair protein synthesis in animals and C2C12 myocytes[77,78,112,113]. The mode of action for these agents is less well characterized than the mechanism mediating the effects of EtOH. However, HIV drugs and EtOH have been shown to affect many of the same targets, including AMPK and MAPK signaling, as well as key proteins involved in initiation and elongation processes. Nevertheless, there are notable differences regarding the effects of these drugs. For example, various classes of HIV antiretroviral agents decrease the phosphorylation of 4E-BP1 in muscle and C2C12 myocytes[113]. In contrast, the HIV PI indinavir suppresses 4E-BP1 and mTOR phosphorylation in animals, but it has no effect on these parameters in C2C12 myocytes[77,78]. Likewise, the PI lopinavir does not alter the phosphorylation of mTOR in meningioma cells[114]. Indinavir disrupts the formation of the active eIF4E-eIF4G initiation complex and thus diminishes the binding of mRNA with the ribosome. Indinavir also reduces the phosphorylation of S6K1 and its downstream target rpS6 in myocytes. Hence, changes in the phosphorylation state of proteins involved in the initiation process may, at least in part, account for the observed decrease in protein synthesis. Because the adverse effects of indinavir on protein synthesis are accompanied by a decline in MEK/ERK/p90RSK and p38 MAPK/Mnk1 signaling, it is possible that these pathways mediate the protein metabolic effects of this drug[69,77].
Previously, we reported that treatment of C2C12 myocytes with indinavir or lopinavir increases phosphorylation of eEF2 at T56[69,112] (Figure 2). This was correlated with an enhanced phosphorylation of eEF2K at S366. In addition, there is an increased activity of this protein, as demonstrated using an in vitro assay with purified eEF2 as the substrate. The most probable explanation for increased eEF2K activity is via the action of AMPK. Indeed, lopinavir increases the phosphorylation of AMPK at T172. In addition, there is increased in vitro kinase activity of AMPK, as determined by its ability to directly phosphorylate the purified eEF2K substrate[112]. Finally, AMPK can directly regulate eEF2 phosphorylation in response to lopinavir, with this being independent of the action of eEF2K.
In summary, the effects of lopinavir on eEF2 phosphorylation appear to be mediated via both the direct and indirect action of AMPK. The mTORC1 pathway may play some role in the process, but at present, this has not been examined. On the other hand, the effects of EtOH on eEF2 are regulated through both mTORC1 and AMPK signaling.
CONCLUSION
It is well established that EtOH adversely affects muscle protein synthesis, with this occurring both in animal models and in cells culture. The initial site of action for EtOH is unknown, and specific receptors or membrane related effectors have not been identified. To date, the impact of this drug is best characterized for signaling mediators that converge on mTORC1. As such, this controls downstream effectors which are central in initiation- and elongation- related processes.
AMPK appears to be a prime mediator for the negative effects of EtOH. This kinase directly phosphorylates the mTORC1 component raptor, and this action is thought to increase the interaction of raptor with 14-3-3. AMPK also phosphorylates TSC2 at a site which induces the GAP activity of this protein. This change alters the nucleotide state of Rheb, and/or the Rheb-mTOR interaction, thereby inhibiting its activity towards mTORC1. This, in turn, impacts the mTOR downstream regulatory proteins that are involved in control of translation initiation. The inhibition of Rheb may also affect mTORC1 activity via an indirect or secondary mechanism involving a decrease in PLD activity and a reduction of PA. The elongation process represents another target of AMPK. Increased AMPK activity by EtOH directly inactivates eEF2. Furthermore, reductions in eEF2 activity by EtOH can be mediated by PP2A, owing to the decrease in mTOR/S6K1 activity.
Amino acid signaling pathways also appear to be targeted by EtOH. EtOH decreases the Rag GTPase-raptor interaction and this may account for reduced binding of mTORC1 and Rheb and the associated decrease in mTORC1 activity. EtOH also increases the activity of the mTORC2 complex toward Akt. Decreases in mTORC1 activity and S6K1 phosphorylation are associated with feedback regulation of this complex. As such, enhanced mTORC2 activity may be due to increased levels of rictor as well as reduced interactions between rictor and the inhibitory proteins 14-3-3 and Deptor.
A number of issues remain to be resolved regarding the effects of EtOH and HIV drugs have on protein synthesis. Because AMPK appears to be a prime mediator of this process, the function of this kinase should be the subject of intensive research. Likewise, there is an emerging role for PLD and Rag GTPase signaling as targets of EtOH. Therefore, it is important to gain a better understanding of the mechanisms that regulate each of these modulators of mTOR complexes. As such, this may lead to the identification of factors that can counteract the negative effects of EtOH and HIV antiretroviral agents on muscle protein synthesis.
ACKNOWLEDGMENTS
We thank Dr. C Randell Brown for critical review of the manuscript.
Footnotes
Supported by National Institute of Health Grants R37 AA-011290 and DK-072909
Peer reviewer: Jae Youl Cho, Professor, Department of Genetic Engineering, Sungkyunkwan University, Chuncheon-dong 300, Suwon 440-746, South Korea
S- Editor Cheng JX L- Editor A E- Editor Zheng XM
References
- 1.Tisdale MJ. Biology of cachexia. J Natl Cancer Inst. 1997;89:1763–1773. doi: 10.1093/jnci/89.23.1763. [DOI] [PubMed] [Google Scholar]
- 2.Evans WJ. Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am J Clin Nutr. 2010;91:1123S–1127S. doi: 10.3945/ajcn.2010.28608A. [DOI] [PubMed] [Google Scholar]
- 3.Hasselgren PO. Catabolic response to stress and injury: implications for regulation. World J Surg. 2000;24:1452–1459. doi: 10.1007/s002680010262. [DOI] [PubMed] [Google Scholar]
- 4.Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 5.Lang CH, Kimball SR, Frost RA, Vary TC. Alcohol myopathy: impairment of protein synthesis and translation initiation. Int J Biochem Cell Biol. 2001;33:457–473. doi: 10.1016/s1357-2725(00)00081-9. [DOI] [PubMed] [Google Scholar]
- 6.Ryazanov AG, Rudkin BB, Spirin AS. Regulation of protein synthesis at the elongation stage. New insights into the control of gene expression in eukaryotes. FEBS Lett. 1991;285:170–175. doi: 10.1016/0014-5793(91)80798-8. [DOI] [PubMed] [Google Scholar]
- 7.Karinch AM, Martin JH, Vary TC. Acute and chronic ethanol consumption differentially impact pathways limiting hepatic protein synthesis. Am J Physiol Endocrinol Metab. 2008;295:E3–E9. doi: 10.1152/ajpendo.00026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vary TC, Lynch CJ, Lang CH. Effects of chronic alcohol consumption on regulation of myocardial protein synthesis. Am J Physiol Heart Circ Physiol. 2001;281:H1242–H1251. doi: 10.1152/ajpheart.2001.281.3.H1242. [DOI] [PubMed] [Google Scholar]
- 9.Lang CH, Frost RA, Kumar V, Wu D, Vary TC. Impaired protein synthesis induced by acute alcohol intoxication is associated with changes in eIF4E in muscle and eIF2B in liver. Alcohol Clin Exp Res. 2000;24:322–331. [PubMed] [Google Scholar]
- 10.Hong-Brown LQ, Brown CR, Kazi AA, Huber DS, Pruznak AM, Lang CH. Alcohol and PRAS40 knockdown decrease mTOR activity and protein synthesis via AMPK signaling and changes in mTORC1 interaction. J Cell Biochem. 2010;109:1172–1184. doi: 10.1002/jcb.22496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong-Brown LQ, Frost RA, Lang CH. Alcohol impairs protein synthesis and degradation in cultured skeletal muscle cells. Alcohol Clin Exp Res. 2001;25:1373–1382. [PubMed] [Google Scholar]
- 12.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 15.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 17.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 18.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16:1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Pearce LR, Huang X, Boudeau J, Pawłowski R, Wullschleger S, Deak M, Ibrahim AF, Gourlay R, Magnuson MA, Alessi DR. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405:513–522. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woo SY, Kim DH, Jun CB, Kim YM, Haar EV, Lee SI, Hegg JW, Bandhakavi S, Griffin TJ, Kim DH. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor beta expression and signaling. J Biol Chem. 2007;282:25604–25612. doi: 10.1074/jbc.M704343200. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Proud CG. mTORC1 signaling: what we still don’t know. J Mol Cell Biol. 2011;3:206–220. doi: 10.1093/jmcb/mjq038. [DOI] [PubMed] [Google Scholar]
- 23.Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab. 2009;296:E592–E602. doi: 10.1152/ajpendo.90645.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y, Fang Y, Yoon MS, Zhang C, Roccio M, Zwartkruis FJ, Armstrong M, Brown HA, Chen J. Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc Natl Acad Sci USA. 2008;105:8286–8291. doi: 10.1073/pnas.0712268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujita Y, Hiroyama M, Sanbe A, Yamauchi J, Murase S, Tanoue A. ETOH inhibits embryonic neural stem/precursor cell proliferation via PLD signaling. Biochem Biophys Res Commun. 2008;370:169–173. doi: 10.1016/j.bbrc.2008.03.060. [DOI] [PubMed] [Google Scholar]
- 26.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol. 2009;29:1411–1420. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winter JN, Fox TE, Kester M, Jefferson LS, Kimball SR. Phosphatidic acid mediates activation of mTORC1 through the ERK signaling pathway. Am J Physiol Cell Physiol. 2010;299:C335–C344. doi: 10.1152/ajpcell.00039.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim E. Mechanisms of amino acid sensing in mTOR signaling pathway. Nutr Res Pract. 2009;3:64–71. doi: 10.4162/nrp.2009.3.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sancak Y, Sabatini DM. Rag proteins regulate amino-acid-induced mTORC1 signalling. Biochem Soc Trans. 2009;37:289–290. doi: 10.1042/BST0370289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dennis MD, Baum JI, Kimball SR, Jefferson LS. Mechanisms involved in the coordinate regulation of mTORC1 by insulin and amino acids. J Biol Chem. 2011;286:8287–8296. doi: 10.1074/jbc.M110.209171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Proud CG. Nutrient control of TORC1, a cell-cycle regulator. Trends Cell Biol. 2009;19:260–267. doi: 10.1016/j.tcb.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 34.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771–32779. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- 35.Crozier SJ, Vary TC, Kimball SR, Jefferson LS. Cellular energy status modulates translational control mechanisms in ischemic-reperfused rat hearts. Am J Physiol Heart Circ Physiol. 2005;289:H1242–H1250. doi: 10.1152/ajpheart.00859.2004. [DOI] [PubMed] [Google Scholar]
- 36.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12:1419–1423. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- 38.Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–9575. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das F, Ghosh-Choudhury N, Mahimainathan L, Venkatesan B, Feliers D, Riley DJ, Kasinath BS, Choudhury GG. Raptor-rictor axis in TGFbeta-induced protein synthesis. Cell Signal. 2008;20:409–423. doi: 10.1016/j.cellsig.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 40.Hong-Brown LQ, Brown CR, Navaratnarajah M, Huber DS, Lang CH. Alcohol-induced modulation of rictor and mTORC2 activity in C2C12 myoblasts. Alcohol Clin Exp Res. 2011;35:1445–1453. doi: 10.1111/j.1530-0277.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong-Brown LQ, Brown CR, Huber DS, Lang CH. Alcohol regulates eukaryotic elongation factor 2 phosphorylation via an AMP-activated protein kinase-dependent mechanism in C2C12 skeletal myocytes. J Biol Chem. 2007;282:3702–3712. doi: 10.1074/jbc.M606593200. [DOI] [PubMed] [Google Scholar]
- 42.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116:1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–1808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 44.García-Villafranca J, Guillén A, Castro J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie. 2008;90:460–466. doi: 10.1016/j.biochi.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 45.Liangpunsakul S, Sozio MS, Shin E, Zhao Z, Xu Y, Ross RA, Zeng Y, Crabb DW. Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am J Physiol Gastrointest Liver Physiol. 2010;298:G1004–G1012. doi: 10.1152/ajpgi.00482.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomita K, Tamiya G, Ando S, Kitamura N, Koizumi H, Kato S, Horie Y, Kaneko T, Azuma T, Nagata H, et al. AICAR, an AMPK activator, has protective effects on alcohol-induced fatty liver in rats. Alcohol Clin Exp Res. 2005;29:240S–245S. doi: 10.1097/01.alc.0000191126.11479.69. [DOI] [PubMed] [Google Scholar]
- 47.Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cell Signal. 2009;21:827–835. doi: 10.1016/j.cellsig.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 48.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 49.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 50.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Bridges D, Moorhead GB. 14-3-3 proteins: a number of functions for a numbered protein. Sci STKE. 2005;2005:re10. doi: 10.1126/stke.2962005re10. [DOI] [PubMed] [Google Scholar]
- 52.Yaffe MB. How do 14-3-3 proteins work?-- Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 2002;513:53–57. doi: 10.1016/s0014-5793(01)03288-4. [DOI] [PubMed] [Google Scholar]
- 53.Chen S, Synowsky S, Tinti M, MacKintosh C. The capture of phosphoproteins by 14-3-3 proteins mediates actions of insulin. Trends Endocrinol Metab. 2011;22:429–436. doi: 10.1016/j.tem.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 54.Klein DC, Ganguly S, Coon SL, Shi Q, Gaildrat P, Morin F, Weller JL, Obsil T, Hickman A, Dyda F. 14-3-3 proteins in pineal photoneuroendocrine transduction: how many roles? J Neuroendocrinol. 2003;15:370–377. doi: 10.1046/j.1365-2826.2003.01000.x. [DOI] [PubMed] [Google Scholar]
- 55.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 56.Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- 57.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 59.Long X, Ortiz-Vega S, Lin Y, Avruch J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J Biol Chem. 2005;280:23433–23436. doi: 10.1074/jbc.C500169200. [DOI] [PubMed] [Google Scholar]
- 60.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 62.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 63.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 64.Zhang HH, Huang J, Düvel K, Boback B, Wu S, Squillace RM, Wu CL, Manning BD. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One. 2009;4:e6189. doi: 10.1371/journal.pone.0006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, Guo R, Johnson CL, Kiguchi K, Walker CL. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol. 2006;173:279–289. doi: 10.1083/jcb.200507119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hong-Brown LQ, Brown CR, Kazi AA, Navaratnarajah M, Lang CH. Rag GTPases and AMPK/TSC2/Rheb mediate the differential regulation of mTORC1 signaling in response to alcohol and leucine. Am J Physiol Cell Physiol. 2012;302:C1557–C1565. doi: 10.1152/ajpcell.00407.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 68.Ma L, Teruya-Feldstein J, Bonner P, Bernardi R, Franz DN, Witte D, Cordon-Cardo C, Pandolfi PP. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 2007;67:7106–7112. doi: 10.1158/0008-5472.CAN-06-4798. [DOI] [PubMed] [Google Scholar]
- 69.Hong-Brown LQ, Brown CR, Huber DS, Lang CH. Alcohol and indinavir adversely affect protein synthesis and phosphorylation of MAPK and mTOR signaling pathways in C2C12 myocytes. Alcohol Clin Exp Res. 2006;30:1297–1307. doi: 10.1111/j.1530-0277.2006.00157.x. [DOI] [PubMed] [Google Scholar]
- 70.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 71.Gustavsson L. ESBRA 1994 Award Lecture. Phosphatidylethanol formation: specific effects of ethanol mediated via phospholipase D. Alcohol Alcohol. 1995;30:391–406. [PubMed] [Google Scholar]
- 72.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lang CH, Frost RA, Deshpande N, Kumar V, Vary TC, Jefferson LS, Kimball SR. Alcohol impairs leucine-mediated phosphorylation of 4E-BP1, S6K1, eIF4G, and mTOR in skeletal muscle. Am J Physiol Endocrinol Metab. 2003;285:E1205–E1215. doi: 10.1152/ajpendo.00177.2003. [DOI] [PubMed] [Google Scholar]
- 74.Rapley J, Oshiro N, Ortiz-Vega S, Avruch J. The mechanism of insulin-stimulated 4E-BP protein binding to mammalian target of rapamycin (mTOR) complex 1 and its contribution to mTOR complex 1 signaling. J Biol Chem. 2011;286:38043–38053. doi: 10.1074/jbc.M111.245449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lang CH, Frost RA, Bronson SK, Lynch CJ, Vary TC. Skeletal muscle protein balance in mTOR heterozygous mice in response to inflammation and leucine. Am J Physiol Endocrinol Metab. 2010;298:E1283–E1294. doi: 10.1152/ajpendo.00676.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lang CH, Frost RA, Vary TC. Regulation of muscle protein synthesis during sepsis and inflammation. Am J Physiol Endocrinol Metab. 2007;293:E453–E459. doi: 10.1152/ajpendo.00204.2007. [DOI] [PubMed] [Google Scholar]
- 77.Hong-Brown LQ, Brown CR, Lang CH. Indinavir impairs protein synthesis and phosphorylations of MAPKs in mouse C2C12 myocytes. Am J Physiol Cell Physiol. 2004;287:C1482–C1492. doi: 10.1152/ajpcell.00038.2004. [DOI] [PubMed] [Google Scholar]
- 78.Hong-Brown LQ, Pruznak AM, Frost RA, Vary TC, Lang CH. Indinavir alters regulators of protein anabolism and catabolism in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;289:E382–E390. doi: 10.1152/ajpendo.00591.2004. [DOI] [PubMed] [Google Scholar]
- 79.Lang CH, Frost RA, Kumar V, Vary TC. Impaired myocardial protein synthesis induced by acute alcohol intoxication is associated with changes in eIF4F. Am J Physiol Endocrinol Metab. 2000;279:E1029–E1038. doi: 10.1152/ajpendo.2000.279.5.E1029. [DOI] [PubMed] [Google Scholar]
- 80.Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, Sonenberg N, Kelly PA, Sotiropoulos A, Pende M. Atrophy of S6K1(-/-) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol. 2005;7:286–294. doi: 10.1038/ncb1231. [DOI] [PubMed] [Google Scholar]
- 81.Kumar V, Frost RA, Lang CH. Alcohol impairs insulin and IGF-I stimulation of S6K1 but not 4E-BP1 in skeletal muscle. Am J Physiol Endocrinol Metab. 2002;283:E917–E928. doi: 10.1152/ajpendo.00181.2002. [DOI] [PubMed] [Google Scholar]
- 82.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29:5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Treins C, Warne PH, Magnuson MA, Pende M, Downward J. Rictor is a novel target of p70 S6 kinase-1. Oncogene. 2010;29:1003–1016. doi: 10.1038/onc.2009.401. [DOI] [PubMed] [Google Scholar]
- 84.Browne GJ, Proud CG. Regulation of peptide-chain elongation in mammalian cells. Eur J Biochem. 2002;269:5360–5368. doi: 10.1046/j.1432-1033.2002.03290.x. [DOI] [PubMed] [Google Scholar]
- 85.Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature. 1988;334:170–173. doi: 10.1038/334170a0. [DOI] [PubMed] [Google Scholar]
- 86.Hait WN, Ward MD, Trakht IN, Ryazanov AG. Elongation factor-2 kinase: immunological evidence for the existence of tissue-specific isoforms. FEBS Lett. 1996;397:55–60. doi: 10.1016/s0014-5793(96)01140-4. [DOI] [PubMed] [Google Scholar]
- 87.Ryazanov AG, Ward MD, Mendola CE, Pavur KS, Dorovkov MV, Wiedmann M, Erdjument-Bromage H, Tempst P, Parmer TG, Prostko CR, et al. Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc Natl Acad Sci USA. 1997;94:4884–4889. doi: 10.1073/pnas.94.10.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Knebel A, Haydon CE, Morrice N, Cohen P. Stress-induced regulation of eukaryotic elongation factor 2 kinase by SB 203580-sensitive and -insensitive pathways. Biochem J. 2002;367:525–532. doi: 10.1042/BJ20020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Knebel A, Morrice N, Cohen P. A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38delta. EMBO J. 2001;20:4360–4369. doi: 10.1093/emboj/20.16.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20:4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guo R, Scott GI, Ren J. Involvement of AMPK in alcohol dehydrogenase accentuated myocardial dysfunction following acute ethanol challenge in mice. PLoS One. 2010;5:e11268. doi: 10.1371/journal.pone.0011268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 95.García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 96.Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 97.Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, Gera J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007;67:11712–11720. doi: 10.1158/0008-5472.CAN-07-2223. [DOI] [PubMed] [Google Scholar]
- 98.Kumar A, Lawrence JC, Jung DY, Ko HJ, Keller SR, Kim JK, Magnuson MA, Harris TE. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. 2010;59:1397–1406. doi: 10.2337/db09-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boulbes D, Chen CH, Shaikenov T, Agarwal NK, Peterson TR, Addona TA, Keshishian H, Carr SA, Magnuson MA, Sabatini DM, et al. Rictor phosphorylation on the Thr-1135 site does not require mammalian target of rapamycin complex 2. Mol Cancer Res. 2010;8:896–906. doi: 10.1158/1541-7786.MCR-09-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–921. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ma H, Yu L, Byra EA, Hu N, Kitagawa K, Nakayama KI, Kawamoto T, Ren J. Aldehyde dehydrogenase 2 knockout accentuates ethanol-induced cardiac depression: role of protein phosphatases. J Mol Cell Cardiol. 2010;49:322–329. doi: 10.1016/j.yjmcc.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang H, Zhang Q, Wen Q, Zheng Y, Philip L, Jiang H, Lin J, Zheng W. Proline-rich Akt substrate of 40kDa (PRAS40): a novel downstream target of PI3k/Akt signaling pathway. Cell Signal. 2012;24:17–24. doi: 10.1016/j.cellsig.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 103.Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem. 2003;278:10189–10194. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- 104.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 105.Wang L, Harris TE, Lawrence JC. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J Biol Chem. 2008;283:15619–15627. doi: 10.1074/jbc.M800723200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 107.Fonseca BD, Smith EM, Lee VH, MacKintosh C, Proud CG. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem. 2007;282:24514–24524. doi: 10.1074/jbc.M704406200. [DOI] [PubMed] [Google Scholar]
- 108.Coodley GO, Loveless MO, Merrill TM. The HIV wasting syndrome: a review. J Acquir Immune Defic Syndr. 1994;7:681–694. [PubMed] [Google Scholar]
- 109.Jordan R, Gold L, Cummins C, Hyde C. Systematic review and meta-analysis of evidence for increasing numbers of drugs in antiretroviral combination therapy. BMJ. 2002;324:757. doi: 10.1136/bmj.324.7340.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carr A, Samaras K, Burton S, Law M, Freund J, Chisholm DJ, Cooper DA. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12:F51–F58. doi: 10.1097/00002030-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 111.Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 112.Hong-Brown LQ, Brown CR, Huber DS, Lang CH. Lopinavir impairs protein synthesis and induces eEF2 phosphorylation via the activation of AMP-activated protein kinase. J Cell Biochem. 2008;105:814–823. doi: 10.1002/jcb.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hong-Brown LQ, Brown CR, Lang CH. HIV antiretroviral agents inhibit protein synthesis and decrease ribosomal protein S6 and 4EBP1 phosphorylation in C2C12 myocytes. AIDS Res Hum Retroviruses. 2005;21:854–862. doi: 10.1089/aid.2005.21.854. [DOI] [PubMed] [Google Scholar]
- 114.Johnson MD, O’Connell M, Pilcher W. Lopinavir inhibits meningioma cell proliferation by Akt independent mechanism. J Neurooncol. 2011;101:441–448. doi: 10.1007/s11060-010-0281-y. [DOI] [PubMed] [Google Scholar]