

Abstract
Aurora kinases are highly conserved, essential regulators of cell division. Two Aurora kinase isoforms, A and B (AURKA and AURKB), are expressed ubiquitously in mammals, whereas a third isoform, Aurora C (AURKC), is largely restricted to germ cells. Because AURKC is very similar to AURKB, based on sequence and functional analyses, why germ cells express AURKC is unclear. We report that Aurkc−/− females are subfertile, and that AURKB function declines as development progresses based on increasing severity of cytokinesis failure and arrested embryonic development. Furthermore, we find that neither Aurkb nor Aurkc is expressed after the one-cell stage, and that AURKC is more stable during maturation than AURKB using fluorescently tagged reporter proteins. In addition, Aurkc mRNA is recruited during maturation. Because maturation occurs in the absence of transcription, posttranscriptional regulation of Aurkc mRNA, coupled with the greater stability of AURKC protein, provides a means to ensure sufficient Aurora kinase activity, despite loss of AURKB, to support both meiotic and early embryonic cell divisions. These findings suggest a model for the presence of AURKC in oocytes: that AURKC compensates for loss of AURKB through differences in both message recruitment and protein stability.
Aurora kinases are highly conserved cell-cycle regulators with essential roles in chromosome segregation. There are three Aurora kinases in mammals: Aurora kinases A and B (AURKA or -B) are ubiquitously expressed and their functions have been extensively studied, whereas AURKC is largely limited to germ cells (1–3); many human cancer cell lines express AURKC (4) and some somatic tissues express AURKC at low levels (5–7). It is not clear, however, why germ cells require a third AURK. Because isoforms can have different functions, it is tempting to speculate that AURKC exists because its mitotic counterparts simply cannot execute unique features of meiosis.
One unique feature of meiosis is the generation of haploid gametes from diploid precursor cells by a reductional chromosome segregation during meiosis I (MI) followed by an equational division at meiosis II (MII) without an intervening round of DNA replication. In oocytes, another unique feature is that meiosis is not a continuous process because there is a growth period during a prolonged arrest at prophase I, followed by a cell division cycle during oocyte maturation, and a second arrest at metaphase of MII, until fertilization, which triggers completion of MII. Furthermore, proteins in the oocyte must support the first mitotic cell cycles of the embryo before zygotic genome activation. Despite these obvious differences, several observations suggest that AURKC may not have a specialized function. AURKB and AURKC are highly similar in sequence (61% identical), and AURKC can functionally compensate for loss of AURKB when ectopically expressed in somatic cells (8, 9). Furthermore, embryos that lack AURKB can develop to but not beyond the blastocyst, as long as AURKC is present, consistent with the idea that AURKB and -C have similar functions (10).
Given the sequence similarity and apparent redundant function, it is unclear why germ cells have a third AURK. Male mice lacking AURKC are subfertile because of postmeiotic defects, including abnormally condensed chromatin and abnormally shaped acrosomes, but females were not examined (11). Mutations in human AURKC cause meiotic arrest and formation of tetraploid sperm (12), suggesting an essential role in cytokinesis in male meiosis. Experiments in mouse oocytes using a chemical inhibitor of AURKB (ZM447439) do not address the function of AURKB because AURKC is also inhibited (13–17). Strategies using dominant-negative versions of AURKC are also difficult to interpret, because the mutant may also compete with AURKB (18). Overexpression studies have similar limitations because both kinases interact with inner centromere protein (INCENP) and these studies did not report expression levels of AURKB versus AURKC (19). Therefore, no experiments to date have directly addressed why oocytes contain a third AURK.
A hint as to the need for oocytes to express AURKC comes from comparisons of AURKB and AURKC sequences. AURKB contains N-terminal destruction motifs that AURKC lacks. In mitotic cell cycles, these motifs regulate AURKB destruction by the anaphase-promoting complex/cyclosome (APC/C) at cytokinesis, before G1 of the following cell cycle (20, 21). The CDH1 (FZR1 in mouse) regulator of the APC/C binds the KEN box (amino acids 4–9 in mouse) and both CDH1 and CDC20 bind the A-box (amino acids 26–29 in mouse). AURKB also contains four putative D-boxes, which AURKC also contains, the functions of which in regulating its stability are unclear (20, 21). Because there are two rounds of chromosome segregation without an intervening cell cycle in meiosis, if AURKB is degraded after MI (as it is during mitosis), there may be no opportunity to regenerate additional AURKB to support MII. Based on these sequence comparisons, we hypothesized that oocytes contain a third AURK because of differential regulation of AURKC protein levels relative to AURKB.
We demonstrate that female mice lacking AURKC are subfertile because of phenotypes that begin with mild chromosome misalignment causing arrest at MI and increase in severity during embryogenesis with cytokinesis failure. The progression of these phenotypes indicates a gradual loss of AURK activity during oocyte maturation and early development. Consistent with this model, we find that AURKB protein is less stable than AURKC during meiotic maturation. Moreover, we find that the Aurkc message is recruited for translation during oocyte maturation to ensure sufficient AURK activity during meiosis and embryonic development. Expression of AURKB in Aurkc−/− oocytes and embryos rescues the meiotic and cytokinesis defects, respectively, consistent with the hypothesis that AURKB can compensate for the loss of AURKC. Taking these data together, we propose that AURKC is an example of an isoform of a cell-cycle regulator that is recruited during meiotic maturation to support meiosis, fertilization and early embryonic cell division.
Results
AURKC Is Required for Normal Oocyte Maturation and Embryo Development.
AURKC expression is largely limited to germ cells, yet a role during oocyte meiotic maturation is not clear. We rederived cryopreserved Aurkc+/− embryos to generate mice lacking Aurkc (11) to determine the requirement of AURKC in female gametes. Oocytes from knockout mice do not express detectable Aurkc message or AURKC protein; the amount of Aurkb mRNA is unaffected in knockout mice (Fig. S1). The number of full-grown oocytes from superovulated, sexually mature female Aurkc−/− mice (34.6 ± 3.8, n = 10) was not statistically different from the numbers from control littermates (44.67 ± 6.2, n = 9). Nevertheless, our breeding trials revealed that they were subfertile, averaging two fewer pups per litter (Fig. 1A), but with a similar number of days between litters compared with control littermates (Fig. S2).
Fig. 1.

Loss of AURKC leads to meiotic abnormalities. (A) Results of fertility trials. The average number of pups born to females of the indicated genotype over an 8-mo breeding trial is shown. (B) GV-intact oocytes were isolated from mice of the indicated genotype and matured in vitro for 8 h before fixation at MI. Percent of oocytes with misaligned chromosomes was plotted for each mouse analyzed. (C–F) GV-intact oocytes were isolated from mice of the indicated genotype and matured in vitro to determine incidence (B) and timing (C) of polar body extrusion (PBE). Cells were fixed when controls (WT) had reached MII and processed for immunocytochemistry to detect chromosomes and spindles. The percentage of oocytes that contained abnormal chromosome configurations at either MI or MII was determined (E), and representative images are shown (F). Graphs represent mean (± SEM) from at least 30 oocytes from three independent experiments. (Scale bars, 5 μm.) One-way ANOVA was used to analyze the data in B–D. *P < 0.05, **P < 0.01, ***P < 0.001; WT, wild-type; HET, heterozygous; KO, knockout.
To determine if AURKC is required for oocyte meiotic maturation, we first matured oocytes to metaphase of MI and, after fixation and staining, examined chromosome alignment. Although the overall percentage of MI oocytes from knockout mice with chromosome misalignment was not strikingly different from wild-type controls, we observed a greater variation from mouse-to-mouse in oocytes from knockout females (Fig. 1B). Next, we monitored the time of polar body emission and found that significantly more oocytes from AURKC knockout mice arrested in MI, as indicated by failure to extrude a polar body (Fig. 1C). Furthermore, the oocytes that failed to extrude a polar body did not undergo cytokinesis, whereas those oocytes that did extrude a polar body were delayed by 1 h entering anaphase I compared with controls (Fig. 1D). These oocytes were then fixed and analyzed by immunocytochemistry, and a significant portion of those from Aurkc−/− mice displayed abnormal chromosome alignment regardless of whether they arrested at MI or progressed to MII (Fig. 1 D–F). We did not observe, however, any increase in aneuploidy incidence (1 of 35 aneuploid eggs in knockouts and 0 of 12 aneuploid eggs in controls). This finding is consistent with oocytes containing misaligned chromosomes arresting at metaphase of MI.
Because mice lacking Aurkb develop to the blastocyst stage (10), it is likely that AURKC also functions during early embryonic cell divisions. To test this hypothesis, we isolated one-cell embryos from wild-type, heterozygous, or knockout females that were mated to wild-type males and allowed the embryos to develop in vitro. Significantly fewer embryos from Aurkc−/− females cleaved to the two-cell stage (Fig. 2A). Moreover, we found that significant numbers of one-cell embryos attempted and failed to complete cytokinesis (Fig. 2 B and C), and we frequently observed prolonged membrane ruffling or blebbing (Fig. S3). Cytokinesis failure is a well-established phenotype of AURKB inhibition or mutation (22–25), and we used this phenotype to compare AURKB function in MI vs. the one-cell stage in oocytes and embryos from Aurkc−/− females. The cytokinesis failure phenotype is much more severe at the one-cell stage, indicating that AURKB activity declines as development progresses (Fig. 2D).
Fig. 2.

Embryonic development is compromised in Aurkc−/− mice. (A–F) Female mice of the indicated genotype were mated to wild-type males and one-cell embryos were isolated and cultured in vitro. (A) The percentage of one-cell embryos that cleaved to the two-cell embryonic stage. (B–D) Embryos were imaged live by DIC every 5–7 min. In B, the percentage of embryos with abnormal cytokinesis was plotted for each mouse. C contains representative images of a wild-type embryo undergoing normal cytokinesis (Upper) and a KO embryo failing cytokinesis (Lower). The time stamp is h:min after hCG injection. (Scale bar, 5 μm.) (D) The percentage of normal cytokinesis events from Fig. 1C (MI) and panel B (1C) were compared. (E and F) Embryo development was monitored at the indicated times after hCG and mating. These experiments were conducted four times with at least two mice per genotype each time, and the data are expressed as the mean ± SEM. (G and H) mRNA levels were measured by quantitative RT-PCR at the indicated stages. Data were normalized against a probe that detects exogenously added Gfp message. Mean (± SEM) from three independent experiments are shown. One-way ANOVA was used to analyze the data. *P < 0.05; **P < 0.01; ***P < 0.001. BL, blastocyst; frag, fragmented; GV, full grown GV-intact oocyte; Inc, meiotically incompetent oocyte; Mor, morula; 1C, one-cell embryo; 2C, two-cell embryo; 4C, four-cell embryo; 8C, eight-cell embryo.
Consistent with a gradual decline in AURKB activity, development of embryos from Aurkc−/− mothers became progressively worse with extended culture periods. Sixty-four hours after mating, ∼35% of the control embryos developed to the eight-cell embryonic stage compared with only ∼15% of the embryos from the knockout mothers (Fig. 2E). Some of the latter embryos were fragmented, with many arrested at the two-cell stage. At 113 h, when nearly 75% of control embryos developed to the blastocyst stage, only ∼25% of embryos from the knockout mothers were blastocysts (Fig. 2F). Those embryos from the knockout mothers that did develop to the blastocyst stage appeared indistinguishable from control embryos. At this time point, most of the embryos that had lagged behind in development had fragmented. These findings indicate that the subfertility of Aurkc−/− females was because of perturbations that begin during meiotic maturation (chromosome misalignment and cell-cycle arrest) and become more pronounced during embryonic development (failure in cytokinesis).
AURKC Is More Stable than AURKB.
Mice lacking AURKB develop up to the blastocyst stage of embryonic development because AURKC is sufficient for supporting the early mitotic divisions (10). In contrast, AURKB function declines between MI and the one-cell stage, as indicated by increasing cytokinesis failure (Fig. 2D), and AURKB does not support development to the blastocyst stage (Fig. 2 E and F). AURKB is unstable during mitosis because it is targeted for ubiquitin-mediated proteolysis after M phase during each round of the cell cycle (20, 21). AURKC, however, lacks destruction motifs that AURKB contains, suggesting that it could be more stable. Moreover, quantitative real-time PCR demonstrated that although oocytes (meiotically incompetent and full-grown oocytes) and eggs (MII) contained both Aurkb and Aurkc mRNA, as previously described (13, 14, 18), these messages declined by the two-cell stage (Fig. 2 G and H). Taken together, these data support the model that the amount of AURKB declines during meiosis and early embryonic mitosis and cannot be resynthesized until the blastocyst stage, whereas the amount of AURKC is maintained during preimplantation development.
To test whether AURKB and AURKC are differentially stable, we used a live-cell imaging assay. We comicroinjected Aurkb-mCherry and Aurkc-Gfp cRNAs into germinal vesicle (GV) oocytes, then inhibited translation by adding cycloheximide, and measured mCherry and GFP intensities over time as reporters for the abundance of AURKB and -C. Because long-term incubation in cycloheximide inhibits major meiotic cell-cycle transitions (26, 27), we monitored the destruction of AURKB and C during three shorter intervals: from GV to MI (GV-MI), from MI to MII (MI-MII), and in activated eggs that are exiting the MII arrest. Although both AURKC-GFP and AURKB-mCherry are progressively degraded (Fig. 3 A–C), AURKC-GFP is more stable than AURKB-mCherry in each interval. Overall, ∼30% of AURKC-GFP was degraded between the GV stage and the two-cell embryonic stage compared with ∼70% of AURKB-mCherry (Fig. 3D). To confirm that degradation was not specific to the fluorophore, we compared AURKB-GFP to AURKB-mCherry and found identical degradation kinetics (Fig. S4A). As a photo-bleaching control, nondegradable cyclin B-GFP fluorescence was constant under identical conditions (Fig. S4B). Taken together, these results show that AURKB and -C gradually decline during meiotic maturation but AURKC is more stable.
Fig. 3.

AURKC is more stable than AURKB during meiotic maturation. (A–C) GV-intact oocytes were coinjected with the indicated cRNAs and matured to the indicated stage. In each graph, the first time point is 1 h after cycloheximide addition, and fluorescent images were obtained at the indicated times. Below are representative images from each time course. Data represent mean (± SEM) from at least 30 oocytes from two independent experiments. (Scale bars, 5 μm.) (D) Merge of the data from A–C.
There are conflicting reports regarding whether oocytes contain AURKB. Some studies have reported localization of endogenous AURKB and exogenous AURKB-GFP to centromeres and midbodies in mouse oocytes (17), whereas others have failed to do so (18). This discrepancy may be because of differences in the abundance of AURKB in the different mouse strains used coupled with poor reagents for detection. Aurkb and Aurkc mRNA are detectable by quantitative real-time PCR, and immunocytochemistry on maturing oocytes and developing embryos showed that AURKC localized to metaphase chromatin, and was undetectable by the blastocyst stage (Fig. S5). Even though Aurkc message was virtually absent in the early embryonic stages (Fig. 2 G and H), the protein still remained in the embryo (Fig. S5). We failed to reliably detect AURKB protein with commercially available reagents, but given the presence of Aurkb message and that Aurkc−/− mice are viable, it is likely that oocytes and early embryos contain AURKB protein. Furthermore, immunocytochemistry of human oocytes also demonstrates that both AURKB and AURKC are present (28).
AURKB Degradation Does Not Depend on Its N Terminus.
Because the N terminus of AURKB contains destruction motifs (Fig. 4A) that are recognized by the APC/C during mitosis, we asked if AURKB and AURKC degradation is proteasome-dependent. Treatment with the proteasome inhibitor MG132 reduced degradation of both AURKs (Fig. 4B and Fig. S6). To test whether the N-terminal motifs in AURKB could explain the difference in kinetics of proteasome-dependent destruction between AURKB and -C, we tested the effects of mutating the KEN and A-boxes and deleting the first 93 amino acids of AURKB-GFP. In both cases the degradation kinetics were not affected (Fig. 4 C and D and Fig. S7 A and B). Furthermore, a chimeric version of AURKC that contained the first 90 amino acids of AURKB was degraded with similar kinetics as wild-type AURKC (Fig. S7C). Taken together, these data strongly suggest that the difference in degradation kinetics between AURKB and C, although proteasome-dependent, cannot be explained by the destruction motifs in AURKB’s N terminus.
Fig. 4.

AURKB stability does not depend upon its N terminus. (A) Schematic representation of AURKB and AURKC. KEN, A-, and D-boxes are indicated, and the conserved regions are shaded in light gray. (B–D) GV-intact oocytes were coinjected with the indicated cRNAs and matured to the indicated stage. Cycloheximide was added 1 h before the first time point and fluorescent images were obtained at the indicated times. Data represent mean (± SEM) from at least 30 oocytes from two independent experiments. MG132 was added as indicated to inhibit the proteasome (B).
Aurkc Is a Maternally Recruited Message.
AURKC protein appears to increase between MI and MII when immunofluorescent signals are compared (Fig. S5). Because oocyte meiotic maturation takes place in the absence of transcription, protein levels are regulated by mRNA recruitment and mRNA degradation; recruitment is regulated by cytoplasmic polyadenylation element (CPE) and DazL binding sequences in 3′UTRs (29, 30). Aurkc contains a conserved CPE (UUUUAU) in its 3′ UTR that is in close proximity (11 nucleotides) to its hexanucleotide polyadenylation sequence (HEX) (Fig. 5A) and three putative DAZL binding sites (U2–10C/GU2–10), suggesting that the increase in AURKC protein is caused by recruitment of Aurkc mRNA during meiotic maturation. The Aurkb 3′ UTR, however, also contains a CPE adjacent to the HEX and a second CPE further 5′ to the HEX. To test whether Aurkb and Aurkc mRNAs are recruited during meiotic maturation, we fused the 3′ UTRs of these kinases to a firefly luciferase (Luc) reporter (Fig. 5A). Following injection and assay for luciferase activity, we found that the 3′ UTR of Aurkc recruited Luc mRNA ∼10-fold following maturation (Fig. 5B), compared with ∼threefold for the Aurkb 3′ UTR. Recruitment of Luc mRNA significantly depended upon the CPE in Aurkc because it was reduced to ∼2.5-fold when two nucleotides of the Aurkc CPE had been mutated (Fig. 5). These data indicate that AURKC protein levels increase during meiotic maturation because its message is recruited for translation in a CPE-dependent manner.
Fig. 5.

Aurkc is a maternally recruited message. (A) Schematic representation of the Firefly luciferase (Luc) fusions to the Aurkb and Aurkc 3′ UTRs. Putative CPE and HEX motifs are underlined and the number of nucleotides between the motifs are indicated in the gray boxes. In Aurkc, the two nucleotides in the CPE that were mutated are in bold. Note that the lengths are not to scale. (B) GV-intact oocytes were coinjected with Luc fused to the indicated 3′ UTR and Renilla luciferase as an injection volume control. Luminescence was measured and quantified as the fold-difference in MII compared with GV. Data represent mean (± SEM) from 10 oocytes from two independent experiments. mut, mutated.
AURKB Can Compensate for Loss of AURKC in Vivo.
Mitotic cells depleted for AURKB are rescued by ectopic expression of AURKC, suggesting that they can carry out the same functions (8, 9). Our data indicate that AURKB protein levels decline during oocyte maturation and embryonic development (Figs. 1–4). To determine if AURKB is functionally equivalent to AURKC and if increasing the amount of AURK activity in knockout oocytes and embryos can compensate for loss of AURKC, we matured oocytes from Aurkc−/− mice that were microinjected with cRNA encoding mCherry, Aurkc-mCherry, or Aurkb-mCherry; oocytes from wild-type mice injected with mCherry served as controls. Expression of either Aurkc- or Aurkb-mCherry rescued both the MI arrest phenotype in Aurkc−/− oocytes (Fig. 6A) and the cytokinesis phenotype in one-cell embryos from Aurkc−/− mothers (Fig. 6C). Furthermore, AURKB was no longer restricted to centromeres at MI in Aurkc−/− oocytes, as it was in wild-type cells, but it adopted a chromatin localization that is typical of AURKC (Fig. 6B). Similarly, AURKB-GFP mimicked AURKC centromere localization in Aurkc−/− eggs, whereas AURKB was not detected at centromeres in wild-type MII eggs (Fig. 6B). Our findings that AURKB rescued the Aurkc−/− polar body extrusion and one-cell cytokinesis defects and adopted the subcellular localization of AURKC indicate that AURKB can compensate for the loss of AURKC during meiosis and support the model that AURKB activity declines during meiotic maturation and embryonic development.
Fig. 6.
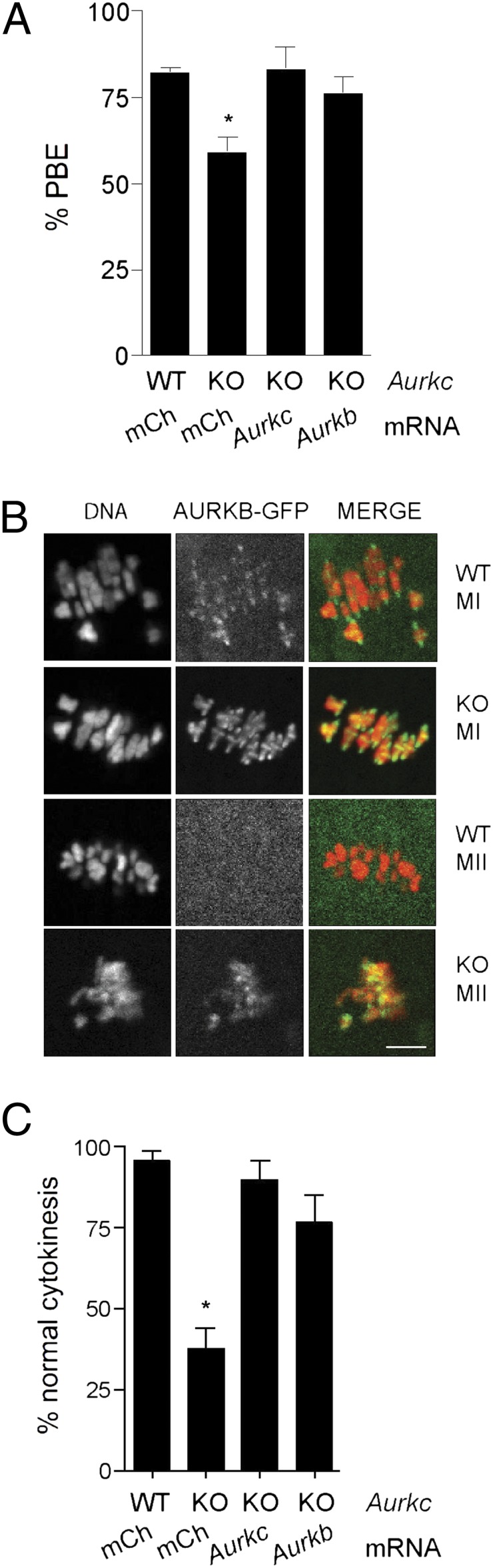
Ectopic expression of AURKB in oocytes and embryos from Aurkc KO mice rescues their defects. (A and C) GV-intact oocytes or one-cell embryos from a single mouse of the indicated genotype were subdivided and microinjected with the indicated cRNAs and matured to MII to determine incidence of PBE or developed to the two-cell stage to monitor cytokinesis. These experiments were repeated three times with two to four KO mice per experiment. (B) Wild-type or Aurkc KO oocytes were microinjected with Aurkb-Gfp cRNA, matured to MI and MII, and imaged live. Shown are representative images. (Scale bar, 5 μm.) One-way ANOVA was used to analyze the data. *P < 0.05; mCh, mCherry.
Discussion
Here, we report that female mice lacking Aurkc are subfertile. Oocytes from knockout mice have a higher incidence of chromosome misalignment and arrest at MI, and one-cell embryos often fail in cytokinesis (Figs. 1 and 2). These phenotypes are hallmarks of cells that lack Aurora kinase activity (22, 24, 25) and worsen as development continues (Fig. 2D). Interestingly, the phenotypic severity (both in vivo and in vitro) varies between mice (Figs. 1F and 2B, and Fig. S8B). These differences could be explained by the degree of compensation by AURKB. In some cases AURKB levels may fall below a threshold necessary to support maturation or embryo development, and therefore viability is compromised. It is unlikely that the six additional pseudogene copies of Aurkc (31) or putative N-terminal splice variants (6) compensate for loss of AURKB because we did not detect their presence in oocytes from knockout mice (Fig. S1A) using a Taqman probe that is 97–100% identical in sequence to these regions, nor did we detect AURKC protein by immunocytochemistry with an antibody that recognizes the C terminus (Fig. S1C). Our in vitro maturation and development data demonstrate that the requirement for AURKC in vitro appears more severe than its requirement in vivo (compare Figs. 1 B–F and 2 A–F with Fig. 1A). This difference may be because of hormonal priming for the in vitro experiments versus natural matings in the in vivo fertility trials. Alternatively, the amount of AURKB may be less following culture in vitro, and therefore it is unable to compensate as well as in vivo. When we isolated in vivo developed blastocysts from hormonally primed mice, we found fewer in the knockout females compared with wild-type, favoring the first model (Fig. S8). Unfortunately, testing the second model is technically not feasible because of limitations in our ability to quantify the amount of AURKB protein.
Our findings that AURKB and AURKC stability and recruitment differ during meiosis (Figs. 3, 4C, and 5B) provide an explanation for the existence of AURKC in oocytes. AURKB is a critical regulator of mitotic cell division, but it is not stable during meiotic maturation, and it is not transcribed again until the blastocyst stage (Figs. 2G and 3). In contrast, AURKC protein likely increases during meiotic maturation, due to its greater stability and CPE-mediated recruitment of Aurkc mRNA (Fig. 5B and Fig. S4). Although AURKB degradation depends upon the proteasome and the N terminus of AURKB contains destruction elements that AURKC lacks, the N terminus does not appear to regulate AURKB stability during meiosis as it does in mitosis (Fig. 4). Because both exogenous proteins gradually decline during meiosis, both may be regulated via their C-terminal D-boxes (Fig. 4A), but AURKB degradation is more pronounced (Fig. 3D).
The discovery of these regulatory mechanisms, combined with evidence that embryos can develop to the blastocyst stage in the absence of AURKB (10), suggests that oocytes express a third AURK to compensate for the inherent instability of AURKB. Notably, expression of a nondegradable AURKB in somatic cells leads to aneuploidy, indicating that its degradation is important for mitotic cell cycles (20). Consistent with others studies, our data support the hypothesis that AURKB and AURKC have similar functions (8–10). There may be subtle functional differences between AURKB and -C, however, that are not revealed in our assays, and oocytes lacking both AURKB and AURKC will be a powerful tool for answering this question.
AURKC is an example of a cell-cycle regulator isoform that is recruited during meiotic maturation to support meiosis, fertilization, and early embryonic cell division. Other examples include WEE1B kinase and IP3 receptor, which contribute to exit from MII (32, 33), and CDC6 and ORC6L, which allow DNA replication following fertilization (34, 35). We propose that these recruited mRNAs comprise a strategy to switch from a program of oocyte growth without cell division, during the prolonged cell-cycle arrest, to meiotic and mitotic divisions without growth (i.e., early embryonic cleavage stages).
Materials and Methods
Oocyte and Embryo Collection, Culture, and Microinjection.
For experiments in Figs. 1–4, fully grown GV-intact oocytes from equine CG-primed (44–48 h before collection) 6-wk-old female CF-1 mice (Harlan) (Figs. 1–4) or from the indicated Aurkc genotype (Figs. 5–6) were obtained as previously described (35). Meiotic resumption was inhibited by addition of 2.5 μM milrinone (Sigma) to the collection, culture, or microinjection medium (36). Oocytes were cultured in Chatot, Ziomek, and Bavister (CZB) medium in an atmosphere of 5% CO2 in air at 37 °C and were collected and microinjected in MEM/PVP (polyvinylpyrrolidone, 3 g/L) (37). To inhibit the proteasome, MG132 (Sigma) was added to the culture medium to a final concentration of 20 μM. To activate MII eggs, the eggs were placed in Ca2+/Mg2+-free CZB plus 10 mM SrCl2 and 5 mg/mL cytochalasin B (Sigma C2743) 18 h after maturation. After 3 h, the eggs were washed free of SrCl2. Oocytes were microinjected with 250 ng/μL of Aurkb/c cRNAs, as previously described (35). Injected oocytes were held for 2–12 h before induction of maturation. For maturation experiments, oocytes were washed and cultured in milrinone-free CZB medium.
In Figs. 2 and 6, primed mice of the indicated Aurkc genotype (Fig. 6) were administered human CG (hCG) and mated to B6D2F1/J males (Jackson Laboratories). In Fig. 6, one-cell embryos were isolated 20 h post-hCG from the ampullae and cultured in KSOM + amino acids (Millipore) in an atmosphere of 5% CO2 in air at 37 °C. In Fig. S5, embryos of the indicated stage were collected as previously described and immediately frozen (38). All animal experiments were approved by the institutional animal use and care committee and were consistent with the National Institutes of Health (NIH) guidelines.
Generation and Genotyping of Aurkc−/− Mice.
Generation of Aurkc−/− was described previously (11). Lexicon Pharmaceuticals transferred cryopreserved Aurkc+/− embryos into pseudopregnant females and the resulting pups were sent to the University of Pennsylvania for further breeding. For genotyping, the copy number of Neo was quantified by real-time PCR per the manufacturer’s protocol. Briefly, tails were digested in 400 μL of lysis buffer (125 mM NaCl, 40 mM Tris, pH 7.5, 50 mM EDTA, pH 8, 1% (vol/vol) sarkosyl, 5 mM DTT, and 50 μM spermidine) with 6 μL of Proteinase K (Sigma #P4850) for 2 h at 65 °C. After dilution of 1:30 in water, the lysates were boiled for 5 min to denature Proteinase K. Two miroliters of the diluted DNA was added to each reaction. Primers to detect Neo (F: 5′ CTCCTGCCGAGAAAGTATCCA-3′; R: GGTCGAATGGGCAGGTAG-3′) were used at a final concentration of 300 nM and primers to detect Csk (for sample normalization) (F 5′-CTGGCCATCCGGTACAGAAT-3′; R 5′-TGCAGAAGGGAAGGTCTTGCT-3′) were used at a final concentration of 100 nM. The TAMRA-quenched Neo probe (ABI) was conjugated to 6-fluorescein amidite and used at a final concentration of 100 nM and the TAMRA-quenched Csk probe was conjugated to VIC and used at a final concentration of 100 nM. The comparative Ct method was used to calculate the Neo copy number.
Cloning, Mutagenesis, and in Vitro Synthesis of cRNA.
Generation of H2B-mCherry and nondegradable cyclin B-GFP were described previously (39, 40). To generate Aurkb and Aurkc-GFP and -mCherry, mouse Aurk sequences were amplified and cloned into pIVT-GFP or -mCherry (41). Truncated AURKB and chimeric AURKB/C constructs were also generated by PCR and cloned into pIVT-GFP. The Aurkb-KEN/A box mutant was generated by site-directed mutagenesis using the QuikChange Multisite Mutagenesis (Agilent Technologies) kit per the manufacturer’s instructions. The KEN box (amino acids 4–9) was changed to AAN and the A-box (amino acids 26–29; QRVL) was changed to QAVA. The 3′ UTRs of Aurkb and Aurkc were PCR-amplified from full-length clones obtained from Open Biosystems and cloned into pIVT containing firefly luciferase. Renilla luciferase was generated as described previously (34). The CPE (TTTTAT) in the 3′ UTR of Aurkc was mutated to TTGGAT using QuikChange.
Linearized DNA for in vitro transcription was achieved by NdeI digest for all Gfp- and mCherry-containing constructs. EcoRI digestion was used for luciferase-containing constructs. After purification of the digests (Qiagen PCR Cleanup) complementary RNAs (cRNAs) were generated using the mMessage mMachine T7 kit (Ambion) according to the manufacturer’s instructions. The cRNA was purified using the RNAeasy kit (Qiagen) and eluted in 30 μL of RNase-free water.
Real-Time PCR.
Fifty oocytes or embryos at the indicated stage were isolated from CF-1 mice and frozen before processing. After thawing on ice, 2 ng of Gfp mRNA was added to each sample. Next, total RNA from the mixtures were purified using the PicoPure RNA isolation kit (Arcturus) per the manufacturer’s protocol. After purification, cDNA was generated by reverse transcription using random hexamers and SuperScript II, as previously described (35). Taqman probes specific for Aurkb (Mm01718146_g1) and Aurkc (Mm03039428_g1) (Applied Biosystems) were used for gene expression detection and the comparative Ct method was used to determine the difference in expression levels between stages. Data were acquired using an ABI Prism 7000 (Applied Biosystems).
Luciferase Assay.
The cRNAs of firefly luciferase fused to the 3′ UTR of Aurkb or Aurkc (200 ng/μL) and Renilla luciferase (25 ng/μL) were coinjected into GV-intact oocytes from CF-1 mice, as described above, and incubated in vitro for at least 2 h in milrinone-containing medium. After incubation, one-half of the injected oocytes were matured to MII for 17 h. The other half was maintained at the GV stage. After washing in PBS + PVP, single oocytes were collected and lysed in Passive Lysis Buffer (12 μL/oocyte) for 15 min at room temperature with shaking followed by incubation on ice until processing with the Dual Luciferase reporter assay system (Promega). The manufacturer’s instructions were followed except that 10 μL of sample and 50 μL of Luciferase Assay Reagent II and of Stop and Glo Reagent were used. Signal intensities were obtained using a Monolight 2010 luminometer (Analytical Luminescence Laboratory). Background fluorescence was subtracted by measuring signals in uninjected oocytes and firefly luciferase activities were normalized to that of Renilla luciferase and expressed as the fold-recruitment in MII eggs compared with GV oocytes.
Immunocytochemistry.
Following meiotic maturation or embryo development to the indicated stage, oocytes and embryos were fixed in PBS containing 3.7% paraformaldehyde for 1 h at room temperature and transferred to blocking buffer [PBS + 0.3% (wt/vol) BSA + 0.01% (vol/vol) Tween-20] for storage overnight at 4 °C. After 15 min of permeabilization in PBS containing 0.1% (vol/vol) Triton X-100 and 0.3% (wt/vol) BSA, cells were incubated in rabbit anti-AURKC antibody (Bethyl A400-023A; epitope BL1217) at 1:30 or a polyclonal anti–β-tubulin antibody conjugated to Alexa 488 (Cell Signaling 3623) at 1:75 in blocking buffer for 1 h at room temperature in a humidified chamber. After washing, secondary Alexa Fluor 594 anti-rabbit antibody (Invitrogen A11012) was diluted 1:200 in blocking solution and the samples incubated for 1 h at room temperature. After a final wash in blocking buffer containing Sytox Green (Invitrogen S7020; 1:5,000), cells processed to detect AURKC were mounted in VectaShield (Vector Laboratories). Cells processed to detect the spindle were mounted in VectaShield containing 3 μg/mL propidium iodide. Ploidy analysis was acquired as described previously (40, 42). Images were collected with a spinning disk confocal using a 100× 1.4 NA oil immersion objective and processed using ImageJ software (NIH), as previously described (43).
Live-Cell Imaging.
To monitor destruction of the exogenous AURKs, GV oocytes were comicroinjected with the indicated cRNAs. Following injection, oocytes were maintained at the GV stage for 16–17 h (for the GV-MI time course), 10–11 h (for the MI-MII time course), or 1 h (for the MII activation time course) before maturation. To inhibit translation, oocytes were placed in 10 ng/μL cycloheximide (Sigma C7698) 1 h before live imaging. Similar, but not as robust as effect, was observed when cycloheximide was omitted. For live imaging, each oocyte or activated egg was transferred into an individual drop of CZB medium or Ca2+/Mg2+-free CZB containing SrCl2 and cytochalasin B, respectively. All drops contained cycloheximide and were under oil in a FluoroDish (World Precision Instruments). Differential interference contrast (DIC), GFP, and mCherry image acquisition was started at 0 (GV-MI), 6 (MI-MII), and 18 h (MII activation) after maturation on a Leica DM6000 microscope with a 40× 1.25 NA oil immersion objective and a charge-coupled device camera (Orca-AG; Hamamatsu Photonics) controlled by Metamorph Software. The microscope stage was heated to 37 °C and 5% CO2 was maintained using a microenvironment chamber (PeCon) and an airstream incubator (ASI 400, Nevtek). Images of individual cells were acquired every 30–60 min during the indicated intervals. For each oocyte, fluorescence was calculated at each time point as a fraction of the fluorescence at the first time point.
To monitor meiotic timing and timing of one-cell cleavage, oocytes and embryos from the indicated genotypes were matured in drops of CZB or KSOM + AA, respectively, under oil in a FluoroDish. DIC images were acquired every 10 min on the microscope described above using a 20× 0.4 NA objective to monitor the timing of GVBD and polar body extrusion or every 5 min to capture cytokinesis in one-cell embryos.
Fertility Trials.
Wild-type and knockout Aurkc female mice of sexual maturity (6 wk) were continually mated to wild-type B6D2 (Jackson Laboratories) male mice of known fertility for 8 mo. Cages were checked daily for the presence and number of pups.
Statistical Analysis.
One-way ANOVA and Student’s t test, as indicated in figure legends, were used to evaluate the differences between groups using Prism software (GraphPad Software). P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
The authors thank Fabian Cardenas and Danielle Young for assistance with mouse genotyping, Kristy Shuda for generation of the Aurora kinase isoforms-GFP constructs, and Paula Stein and Sarah Kimmins for helpful discussions. This work was supported by National Institutes of Health Grants HD 055822 (to K.S.) and HD 058730 (to R.M.S. and M.A.L.), and a Searle Scholar Award (to M.A.L.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
See Author Summary on page 13150 (volume 109, number 33).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1120517109/-/DCSupplemental.
References
- 1.Gopalan G, Chan CS, Donovan PJ. A novel mammalian, mitotic spindle-associated kinase is related to yeast and fly chromosome segregation regulators. J Cell Biol. 1997;138:643–656. doi: 10.1083/jcb.138.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yanai A, Arama E, Kilfin G, Motro B. ayk1, a novel mammalian gene related to Drosophila aurora centrosome separation kinase, is specifically expressed during meiosis. Oncogene. 1997;14:2943–2950. doi: 10.1038/sj.onc.1201144. [DOI] [PubMed] [Google Scholar]
- 3.Tseng TC, Chen SH, Hsu YP, Tang TK. Protein kinase profile of sperm and eggs: Cloning and characterization of two novel testis-specific protein kinases (AIE1, AIE2) related to yeast and fly chromosome segregation regulators. DNA Cell Biol. 1998;17:823–833. doi: 10.1089/dna.1998.17.823. [DOI] [PubMed] [Google Scholar]
- 4.Baldini E, et al. Aurora kinases are expressed in medullary thyroid carcinoma (MTC) and their inhibition suppresses in vitro growth and tumorigenicity of the MTC derived cell line TT. BMC Cancer. 2011;11:411. doi: 10.1186/1471-2407-11-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price DM, Kanyo R, Steinberg N, Chik CL, Ho AK. Nocturnal activation of aurora C in rat pineal gland: Its role in the norepinephrine-induced phosphorylation of histone H3 and gene expression. Endocrinology. 2009;150:2334–2341. doi: 10.1210/en.2008-1507. [DOI] [PubMed] [Google Scholar]
- 6.Yan X, et al. Cloning and characterization of a novel human Aurora C splicing variant. Biochem Biophys Res Commun. 2005;328:353–361. doi: 10.1016/j.bbrc.2004.12.168. [DOI] [PubMed] [Google Scholar]
- 7.Yan X, et al. Aurora C is directly associated with Survivin and required for cytokinesis. Genes Cells. 2005;10:617–626. doi: 10.1111/j.1365-2443.2005.00863.x. [DOI] [PubMed] [Google Scholar]
- 8.Slattery SD, Mancini MA, Brinkley BR, Hall RM. Aurora-C kinase supports mitotic progression in the absence of Aurora-B. Cell Cycle. 2009;8:2984–2994. [PubMed] [Google Scholar]
- 9.Sasai K, et al. Aurora-C kinase is a novel chromosomal passenger protein that can complement Aurora-B kinase function in mitotic cells. Cell Motil Cytoskeleton. 2004;59:249–263. doi: 10.1002/cm.20039. [DOI] [PubMed] [Google Scholar]
- 10.Fernández-Miranda G, et al. Genetic disruption of aurora B uncovers an essential role for aurora C during early mammalian development. Development. 2011;138:2661–2672. doi: 10.1242/dev.066381. [DOI] [PubMed] [Google Scholar]
- 11.Kimmins S, et al. Differential functions of the Aurora-B and Aurora-C kinases in mammalian spermatogenesis. Mol Endocrinol. 2007;21:726–739. doi: 10.1210/me.2006-0332. [DOI] [PubMed] [Google Scholar]
- 12.Ben Khelifa M, et al. A new AURKC mutation causing macrozoospermia: Implications for human spermatogenesis and clinical diagnosis. Mol Hum Reprod. 2011;17:762–768. doi: 10.1093/molehr/gar050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shuda K, Schindler K, Ma J, Schultz RM, Donovan PJ. Aurora kinase B modulates chromosome alignment in mouse oocytes. Mol Reprod Dev. 2009;76:1094–1105. doi: 10.1002/mrd.21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swain JE, Ding J, Wu J, Smith GD. Regulation of spindle and chromatin dynamics during early and late stages of oocyte maturation by aurora kinases. Mol Hum Reprod. 2008;14:291–299. doi: 10.1093/molehr/gan015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lane SI, Chang HY, Jennings PC, Jones KT. The Aurora kinase inhibitor ZM447439 accelerates first meiosis in mouse oocytes by overriding the spindle assembly checkpoint. Reproduction. 2010;140:521–530. doi: 10.1530/REP-10-0223. [DOI] [PubMed] [Google Scholar]
- 16.Gautschi O, et al. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14:1639–1648. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 17.Vogt E, Kipp A, Eichenlaub-Ritter U. Aurora kinase B, epigenetic state of centromeric heterochromatin and chiasma resolution in oocytes. Reprod Biomed Online. 2009;19:352–368. doi: 10.1016/s1472-6483(10)60169-1. [DOI] [PubMed] [Google Scholar]
- 18.Yang KT, et al. Aurora-C kinase deficiency causes cytokinesis failure in meiosis I and production of large polyploid oocytes in mice. Mol Biol Cell. 2010;21:2371–2383. doi: 10.1091/mbc.E10-02-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharif B, et al. The chromosome passenger complex is required for fidelity of chromosome transmission and cytokinesis in meiosis of mouse oocytes. J Cell Sci. 2010;123:4292–4300. doi: 10.1242/jcs.067447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen HG, Chinnappan D, Urano T, Ravid K. Mechanism of Aurora-B degradation and its dependency on intact KEN and A-boxes: Identification of an aneuploidy-promoting property. Mol Cell Biol. 2005;25:4977–4992. doi: 10.1128/MCB.25.12.4977-4992.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart S, Fang G. Destruction box-dependent degradation of aurora B is mediated by the anaphase-promoting complex/cyclosome and Cdh1. Cancer Res. 2005;65:8730–8735. doi: 10.1158/0008-5472.CAN-05-1500. [DOI] [PubMed] [Google Scholar]
- 22.Yabe T, et al. The maternal-effect gene cellular island encodes aurora B kinase and is essential for furrow formation in the early zebrafish embryo. PLoS Genet. 2009;5:e1000518. doi: 10.1371/journal.pgen.1000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Severson AF, Hamill DR, Carter JC, Schumacher J, Bowerman B. The aurora-related kinase AIR-2 recruits ZEN-4/CeMKLP1 to the mitotic spindle at metaphase and is required for cytokinesis. Curr Biol. 2000;10:1162–1171. doi: 10.1016/s0960-9822(00)00715-6. [DOI] [PubMed] [Google Scholar]
- 24.Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol. 2001;152:669–682. doi: 10.1083/jcb.152.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hauf S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golbus MS, Stein MP. Qualitative patterns of protein synthesis in the mouse oocyte. J Exp Zool. 1976;198:337–342. doi: 10.1002/jez.1401980306. [DOI] [PubMed] [Google Scholar]
- 27.Downs SM. Protein synthesis inhibitors prevent both spontaneous and hormone-dependent maturation of isolated mouse oocytes. Mol Reprod Dev. 1990;27:235–243. doi: 10.1002/mrd.1080270309. [DOI] [PubMed] [Google Scholar]
- 28.Avo Santos M, et al. A role for Aurora C in the chromosomal passenger complex during human preimplantation embryo development. Hum Reprod. 2011;26:1868–1881. doi: 10.1093/humrep/der111. [DOI] [PubMed] [Google Scholar]
- 29.Richter JD. CPEB: A life in translation. Trends Biochem Sci. 2007;32:279–285. doi: 10.1016/j.tibs.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, et al. Genome-wide analysis of translation reveals a critical role for deleted in azoospermia-like (Dazl) at the oocyte-to-zygote transition. Genes Dev. 2011;25:755–766. doi: 10.1101/gad.2028911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu HM, Chuang CK, Lee MJ, Tseng TC, Tang TK. Genomic organization, expression, and chromosome localization of a third aurora-related kinase gene, Aie1. DNA Cell Biol. 2000;19:679–688. doi: 10.1089/10445490050199063. [DOI] [PubMed] [Google Scholar]
- 32.Xu Z, Williams CJ, Kopf GS, Schultz RM. Maturation-associated increase in IP3 receptor type 1: Role in conferring increased IP3 sensitivity and Ca2+ oscillatory behavior in mouse eggs. Dev Biol. 2003;254:163–171. doi: 10.1016/s0012-1606(02)00049-0. [DOI] [PubMed] [Google Scholar]
- 33.Oh JS, Susor A, Conti M. Protein tyrosine kinase Wee1B is essential for metaphase II exit in mouse oocytes. Science. 2011;332:462–465. doi: 10.1126/science.1199211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murai S, Stein P, Buffone MG, Yamashita S, Schultz RM. Recruitment of Orc6l, a dormant maternal mRNA in mouse oocytes, is essential for DNA replication in 1-cell embryos. Dev Biol. 2010;341:205–212. doi: 10.1016/j.ydbio.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anger M, Stein P, Schultz RM. CDC6 requirement for spindle formation during maturation of mouse oocytes. Biol Reprod. 2005;72:188–194. doi: 10.1095/biolreprod.104.035451. [DOI] [PubMed] [Google Scholar]
- 36.Tsafriri A, Chun SY, Zhang R, Hsueh AJ, Conti M. Oocyte maturation involves compartmentalization and opposing changes of cAMP levels in follicular somatic and germ cells: Studies using selective phosphodiesterase inhibitors. Dev Biol. 1996;178:393–402. doi: 10.1006/dbio.1996.0226. [DOI] [PubMed] [Google Scholar]
- 37.Chatot CL, Ziomek CA, Bavister BD, Lewis JL, Torres I. An improved culture medium supports development of random-bred 1-cell mouse embryos in vitro. J Reprod Fertil. 1989;86:679–688. doi: 10.1530/jrf.0.0860679. [DOI] [PubMed] [Google Scholar]
- 38.Ma P, Schultz RM. Histone deacetylase 1 (HDAC1) regulates histone acetylation, development, and gene expression in preimplantation mouse embryos. Dev Biol. 2008;319:110–120. doi: 10.1016/j.ydbio.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schindler K, Schultz RM. CDC14B acts through FZR1 (CDH1) to prevent meiotic maturation of mouse oocytes. Biol Reprod. 2009;80:795–803. doi: 10.1095/biolreprod.108.074906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duncan FE, Chiang T, Schultz RM, Lampson MA. Evidence that a defective spindle assembly checkpoint is not the primary cause of maternal age-associated aneuploidy in mouse eggs. Biol Reprod. 2009;81:768–776. doi: 10.1095/biolreprod.109.077909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Igarashi H, Knott JG, Schultz RM, Williams CJ. Alterations of PLCbeta1 in mouse eggs change calcium oscillatory behavior following fertilization. Dev Biol. 2007;312:321–330. doi: 10.1016/j.ydbio.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stein P, Schindler K. Mouse oocyte microinjection, maturation and ploidy assessment. J Vis Exp. 2011;53:e2851. doi: 10.3791/2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiang T, Duncan FE, Schindler K, Schultz RM, Lampson MA. Evidence that weakened centromere cohesion is a leading cause of age-related aneuploidy in oocytes. Curr Biol. 2010;20:1522–1528. doi: 10.1016/j.cub.2010.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
