

Abstract
Human leukocyte antigen HLA-B alleles have better protective activity against HIV-1 than HLA-A alleles, possibly due to differences in HLA-restricted HIV-1-specific CD8+ cytotoxic T lymphocyte (CTL) function, but the mechanism is unknown. HIV-1 negative regulatory factor (Nef) mediates down-regulation of surface expression of class I HLA (HLA-I) and may therefore impair immune recognition by CTL. Because of sequence differences in the cytoplasmic domains, HLA-A and -B are down-regulated by Nef but HLA-C and -E are not affected. However, the latter are expressed at low levels and are not of major importance in the CTL responses to HIV-1. Here, we compared the role of the cytoplasmic domains of HLA-A and -B in Nef-mediated escape from CTL. We found HLA-B cytoplasmic domains were more resistant to Nef-mediated down-regulation than HLA-A cytoplasmic domains and demonstrated that these differences affect CTL recognition of virus-infected cells in vitro. We propose that the relative resistance to Nef-mediated down-regulation by the cytoplasmic domains of HLA-B compared with HLA-A contributes to the better control of HIV-1 infection associated with HLA-B-restricted CTLs.
It is known that HLA-B types are more protective against HIV-1 (HIV-1) than HLA-A types (1, 2). This phenomenon is puzzling at first because HLA-A and -B are expressed at similar levels on leukocytes and have the same basic function, which is to present peptides to CD8+ CTL. The protection is not entirely explained by a confounding effect of a few highly protective HLA-B types such as B57, B58, B27, B51, and B81 (3). It is striking, however, that the most protective types are all B allotypes, findings made independently by conventional HLA typing and by genomic methods (4, 5). It has been shown that HLA-B-restricted HIV-1-specific T cells are more polyfunctional, have higher avidity receptors, and show greater clonal turnover, compared with HLA-A-restricted T cells (6, 7), but these properties could be secondary to the better control of the virus that these HLA types confer.
HIV-1 negative regulatory factor (Nef) is well known to down-regulate surface expression of HLA class I (8). However, because down-regulation of HLA class I is less complete than the down-regulation of CD4, also mediated by Nef but through a different mechanism, its importance has been uncertain. If the effect is big enough to diminish CTL responses, it is hard to reconcile with the often-rapid selection of virus escape mutants by CTL in vivo (9, 10). On the other hand, strong evidence that MHC down-regulation is important comes from Swigut et al. (11), who infected rhesus monkeys with a simian immunodeficiency virus (SIV) carrying hard-to-revert mutations in Nef that abrogated the down-regulation of MHC class I. Initially the animals controlled the challenge SIV more effectively than animals infected with wild-type virus, but then the virus repaired the mutations to restore full Nef activity and virus control was diminished. Therefore, the SIV-Nef effect on MHC class I must be important to virus virulence; it is likely that this is also true of HIV-1 Nef.
The obvious consequence of down-regulation of class I HLA would be to facilitate evasion of CTL attack. Similar strategies have evolved in many other persisting viruses, although through several different mechanisms (12–15). It is important to note, however, that virus-mediated down-regulation of HLA class I is compatible with strong antiviral CTL responses because the latter can be primed through cross-presentation of viral proteins taken up by uninfected dendritic cells (16). Thus, the abundant antiviral CTL usually present in HIV-1-infected patients may be severely limited in CTL ability to recognize infected cells by the actions of Nef.
It has previously been shown that HLA-C is resistant to down-regulation by HIV-1 Nef, unlike HLA-A and -B (17). However, because HLA-C is normally expressed at low levels (18), HLA-C-specific CTL may not be very important, although epitopes presented by HLA-C are sometimes present (10, 19, 20). On the other hand, retention HLA-C, even at low levels, may mean that it can still inhibit natural killer (NK) cells that have HLA-C-specific KIR2DL receptors (17).
Previous studies have shown that that HLA-A and -B molecules are both sensitive to Nef down-regulation, but it was briefly noted that HLA-A is more down-regulated than HLA-B (17). Differences between the HLA-A, -B, and -C molecules in their sensitivity to intracellular Nef are likely to reflect genetic polymorphism in the amino acid sequences of their cytoplasmic domains. Here we test the hypothesis that HLA-B-restricted CTL responses are stronger than HLA-A-restricted responses because of their differing susceptibility to Nef-mediated down-regulation. We generated transfected cells that expressed the extracellular dominance of HLA-B, complementing previous studies that have shown much more frequent and greater effect of HLA-B than HLA-A-restricted anti-HIV-1 specific T-cell responses (1, 5, 21, 22).
Results
Polymorphism of the HLA-I Cytoplasmic Domain and Nef-Mediated Down-Regulation.
HIV-1 Nef-induced down-regulation of HLA-I is mediated by its interaction with the cytoplasmic domain of HLA-I and the clathrin adaptor protein complex-1 (AP-1) (23). To assess the role of the cytoplasmic domain of HLA-I allotypes in CTL-mediated HIV-1 control, we initially evaluated the polymorphism in the HLA class I exons 6,7, and 8 that encodes the carboxyl-terminal of HLA-I molecule (Table 1 and Table S1) according to available HLA-I sequences from the gene bank database. This region of HLA-I is relatively conserved compared with the external alpha-1 and alpha-2 domains (17). Whether polymorphisms in this domain, apart from those that regulate HLA-C expression, affect CTL responses to HIV-1 has not previously been examined. Examination of the international ImMunoGeneTics information system (IMGT)/HLA database (http://www.ebi.ac.uk/imgt/hla/) shows that for the HLA-A alleles there are seven cytoplasmic domain variants, compared with two for HLA-B and seven for HLA-C (Table 1 and Table S1). Six of the seven HLA-C, but none of the HLA-A and -B cytoplasmic regions have the Y321C and D328N variations, which are important in resisting Nef-mediated down-regulation (17, 23).
Table 1.
Aligned cytoplasmic domain sequences of cell lines generated representing HLA-A, -B, and -C alleles in the database
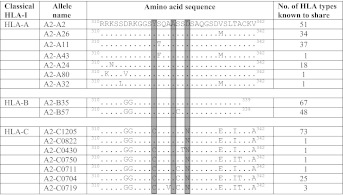 |
Aligned cytoplasmic domain sequences of cell lines generated representing HLA-A, -B, and -C alleles in the database. Dots represent residues that match with HLA-A0201 sequence. Highlighted amino acids are believed to be crucial residues that are important in Nef-mediated HLA-I down-regulation (17). The numbering corresponds to the residue number in HLA-Cw4.
We generated Jurkat T-cell lines stably expressing chimeric human HLA-I, molecules having HLA-A0201 extracellular and transmembrane domains fused to different cytoplasmic sequences of HLA-I identified from the IMGT/HLA database to examine the role of cytoplasmic domain polymorphism on differential HIV-1 Nef-mediated HLA-I down-regulation. Even though we selected Jurkat cells with the strongest 2% expression after transduction, it was clear that the expression of HLA-A2 chimeric molecules with HLA-C cytoplasmic domains, with the exception of A2-C1205, was at least 10-fold lower than chimeras containing HLA-A or HLA-B cytoplasmic domains (Fig. S1). Isoleucine at position 337, present in all C cytoplasmic domains, is thought to be responsible for accelerated internalization, which leads to a lower expression of HLA-C molecules on the surface (18). Expression levels of chimeric HLA-A2 molecules with HLA-A and HLA-B cytoplasmic domains were at a high level and similar. The cell lines were then transiently transfected either wild-type HIV-1SF2Nef or a HIV-1G2ANef mutant, both tagged with GFP. The G2A Nef is mutated at amino acid position 2 so that it is not myristoylated and is therefore defective in mediating HLA-I down-regulation (24). The level of surface HLA-A2 expression was then compared between cell populations gated on GFP positive cells (Fig. 1A). The HLA-A2 molecules with HLA-A cytoplasmic domains were more down-regulated by Nef than those with HLA-B domains (P < 0.01, Student’s t test; Fig. 1B). As expected, there was no down-regulation of HLA-A2 fused to any of the HLA-C cytoplasmic domains.
Fig. 1.

Differential down-regulation of HLA-A, -B, and -C alleles with Nef and HIV infection. (A) Representative flow cytometry demonstrating magnitude of down-regulation of HLA-A2-A, HLA-A2-B, and HLA-A2-C chimera measured 24 h after transiently transfecting with plasmids expressing HIV-1SF2Nef/GFP or control plasmid HIV-1G2ANef/GFP. (B) The fold down-regulation of A2 chimera in the presence of HIV-1SF2Nef/GFP as calculated as in methods. (C) Representative flow cytometry demonstrating magnitude of down-regulation of HLA-A2-A, HLA-A2-B, and HLA-A2-C chimera measured 6 d postinfection with HIV-IIIB virus. (D) The fold down-regulation of A2 chimera in infected cells, calculated by comparing MFI of A2 in p24 positive cells (described in detail in Materials and Methods). All data are representative of three independent experiments. Error bars, SEM, x = mean fold down-regulation of HLA-A2. P as calculated by Student's t test.
We then asked whether whole virus infection of the Jurkat cells with the laboratory strain HIV-IIIB down-regulated the chimeric HLA-A2 molecules in the same way (Fig. 1C). Again, we found that down-regulation of chimeric HLA-A2 molecules was determined by the cytoplasmic tail and again there was significantly less down-regulation of HLA-A2 with -B compared with A cytoplasmic domains (P < 0.05, Student’s t test ; Fig. 1D). HLA-A2 with C cytoplasmic domains was not down-regulated.
T Cells Recognize an HIV-1 Epitope Presented by HLA-A2-B Chimeras Better than HLA-A2-A Chimeras.
We then asked whether the different degree of Nef-mediated down-regulation of HLA-A2, dependent on the cytoplasmic domain, affected T-cell recognition, using the live virus IFN-γ enzyme-linked immunosorbent spot (ELISPOT) assay to measure CTL recognition of infected cells (25). Jurkat T-cell lines expressing HLA-A2 chimeras were infected with HIV-IIIB and cocultured overnight with an HLA-A2-restricted Gag p17 77–85 (amino acid sequence SLNYTVATL)–specific CTL clone (G10), which was 95% positive for epitope-specific T-cell receptors by HLA A2-epitope tetramer staining. The IFN-gamma-producing T cells were then counted in an ELISPOT assay at a ratio of 1:50 G10:infected cells to ensure maximal stimulation of the T cells (Fig. 2 A and B). We consistently observed more IFN-gamma-producing T cells [measured as spot-forming units (SFU)] when the clone was exposed to infected cells expressing HLA-A2 chimeras with HLA-B cytoplasmic domains compared with HLA-A domains, although HLA-A2-A26 was an exception (Fig. 2A).
Fig. 2.

Control of HIV infection by A2-restricted Gag-p17-specific CTL. (A) Results of live virus ELISPOT assay carried out with E:T = 400:20,000. All data are representative of three independent experiments. Error bars, SD, P as calculated by Student's t test. (B) Correlation between mean fluorescent intensity of A2 chimera of HIV-IIIB-infected cells and mean spot-forming units. (C) Representative flow-based viral suppression assay results of selected HIV-IIIB-infected chimeric cell lines cocultured with A2 Gag-restricted CTL clones. CD8+ cells were gated out. (D) Percentage suppression of HIV-1-infected cells (p24 positive cells) compared with cultures incubated without A2 Gag-specific clones.
Comparatively fewer SFU were observed when the clone was incubated with infected cells expressing HLA-A2-C, with the exception of the relatively highly expressed A2-C1205, which stimulated a strong response. The poor T-cell responses could be attributed to low expression of most of the HLA-A2-C chimeras on the surface of the cell lines. There was a highly significant correlation between mean fluorescent intensity of HLA-A2 staining or fold down-regulation of A2 chimera (HLA-A and -B) on HIV-1-infected cells and number of SFU (Fig. S2 and Table S4). Therefore, these experiments demonstrate that the degree of HLA-A2 down-regulation strongly influences recognition of virus-infected cells by this Gag-specific CTL clone.
We confirmed these results by measuring the disappearance of infected cells in the presence of the G10 T-cell clone in vitro (Fig. 2C). We found some p24 negative cells down-regulated CD4 and likely represent the infected cells at early stage when p24 expression is low. The escape by the infected cells with the HLA-A cytoplasmic domains could be titrated and was less marked at higher effector-to-target ratios (Fig. 2D). Complementary results were found when the expression of CD107a was measured on the G10 cells cocultured with infected cells. Cell-surface CD107a levels were notably higher when G10 was exposed to cell lines with the HLA-A2 chimeras that were not down-regulated by HIV-1 (Fig. S3).
HLA-B7 is More Resistant to Nef-Mediated Down-Regulation than HLA-A2 and -A3 in Cell Lines Naturally Expressing both Alleles.
To exclude the possibility that the results shown above are limited by the use of cells transfected with hybrid HLA molecules, we extended the study to cells expressing native HLA molecules. We used two cell lines, Jurkat and 293T cells. The number of cell lines that can be used for this kind of analysis is restricted by the availability of specific anti-HLA-I antibodies, but for Jurkat (HLA type HLA-A*3, HLA-B*7, and HLA-B*35) and for 293T (HLA type A2, A3, and B7), antibodies to HLA-A*2, A*3, and B*7 were accessible. On uninfected cells the level of expression of HLA-A and -B was very similar. However, after HIV-1 infection of Jurkat with HIV-IIIB or transient transfection of 293T with HIV-1SF2Nef-GFP, we observed that HLA-A2 and -A3 were always more down-regulated than HLA-B7 (Fig. 3 A–C).
Fig. 3.

Relative resistance to Nef-mediated down-regulation by native HLA-B leads to strong effector response in CD8+ T cells observed with HLA-B alleles. (A) Dot plots and representative histogram demonstrating down-regulation of HLA-A3 and HLA-B7 in HIV-1SF2Nef-GFP transfected Jurkat cells (n = 3). (B) Dot plots and representative histogram showing down-regulation of HLA-A3 and HLA-B7 in HIV-IIIB-infected Jurkat cells (n = 3). (C) Dot plots and representative histogram showing down-regulation of HLA-A2 and HLA-B7 in HIV-1SF2Nef-GFP expressing 293T cells (n = 3). Magnitude of HLA-A/B down-regulation was measured 6 d postinfection or 24 h postelectroporation. (D) Live virus IFN-γ ELISPOT assay was performed with pNL4-3 or M20A pNL4-3 virus-infected MT2 cells at different infectivity ratios. These were cocultured either with HLA-A24 Nef and HLA-B51 Gag-restricted CTL clones at E:T= 400:20,000 overnight. SFU were normalized by comparing SFU obtained for each clone with mean SFU obtained for MT2 cells pulsed with relevant peptide. All data are representative of three independent experiments. Error bars, SEM, P as calculated by Student's t test.
Effect of HIV-1Nef on HLA-B- and HLA-A-Restricted T-Cell Responses.
Finally we compared HLA-A- and HLA-B-restricted T-cell recognition of HIV-1 in the same cell. MT2 cells, which express both HLA-A24 and HLA-B51, were infected with wild-type HIV-1 (NL4-3) or Nef-mutant HIV-1 (M20A NL4-3). The latter expresses a Nef protein with a mutation at position 20 (M20A), which selectively abolishes HLA down-regulation (26). HLA-A24-restricted Nef 134–142-specific and B51-restricted Gag 327–335-specific CTL clones were used as effector T cells, with an HLA B8-restricted Nef-specific CTL clone as a negative control. T-cell recognition of virus was measured using a live virus ELISPOT assay (Fig. 3D). The T-cell response for each clone incubated with virus-infected MT2 cells at a ratio of T-cell clone to infected cells of 1:50, was compared with the response obtained with the Nef-mutant HIV-1-infected cells. We repeatedly found no significant difference in the magnitude of the IFN-γ response by HLA B51-restricted CTLs when incubated with wild-type HIV-1-infected cells compared with M20A pNL4-3 virus-infected target cells. In contrast, the HLA A24-specific CTLs responded to MT2 cells infected with Nef mutant virus significantly more effectively than to cells infected with the wild-type virus.
Discussion
It is widely held that HLA-B alleles are superior in function to HLA-A in control of HIV-1 (1, 2). All of the confirmed strongly protective HLA alleles are B (4, 5), but these do not entirely account for the association between HLA-B-restricted T-cell responses and slower disease progression; the studies by Kiepiela et al. (1) and Pereyra et al. (5) show that B allotypes have a greater effect, which can either be positive or be negative. It is also clear that HLA-B-restricted responses are much more frequent that HLA-A-restricted responses by a ratio of over 2 to 1. It has been assumed that the stronger and functionally superior HLA-B-restricted CTL responses are determined entirely by the nature of the CTL epitopes (6, 7). It has also been suggested that the protective HLA-B27 and -B57 molecules select a more broadly responding T-cell receptor repertoire in the thymus because of amino acid sequence constraints in the peptides they bind (27). These findings, however, only give part of the explanation. It is unlikely that the T-cell receptor repertoire hypothesis would extend across all HLA-B alleles and the T-cell functional arguments do not explain why those functions are better in the first place.
Here, we demonstrate that the cytoplasmic domain of HLA-B molecules avoids reduced CTL cell recognition of HIV-1-infected cells by resisting Nef-mediated down-regulation of HLA class I molecules. Although that resistance is incomplete, it appears sufficient to enable strong and effective CTL responses to be maintained and that this could lead to efficient control of virus replication and lead to more effective elimination of infected cells. Preliminary experiments (not shown) indicate that the GG motif in the cytoplasmic domain shared by HLA-B and -C but not -A, together with shortened cytoplasmic tail, are needed to abrogate the down-regulation.
Our findings therefore explain how CTL could exert better control over HIV-1 infection when they are restricted by HLA-B compared with HLA-A. Consistently, HLA-B-restricted T cells recognized HIV-1 infected cells better than HLA-A-restricted T cells. HLA-C molecules are resistant to Nef-mediated down-regulation (17, 23) but the baseline level of expression of HLA-C was repeatedly low, with the exception of A2-C1205, despite sorting the top 2% expressing cells in three independent transductions for each cell line. However, HLA-A2-C chimeras were all sufficiently expressed to be able to determine accurately whether or not Nef or HIV-1 caused down-regulation. The levels of natural HLA-C molecules on the cell surface are known to be lower than HLA-A and -B (28), and this has been attributed to either faster mRNA turnover (29), inefficient β2m association (30), low availability of specific peptides (31), or faster protein internalization in which Ile337 in the cytoplasmic domain of HLA-C plays a key role (18). Our findings with the A2-C chimeras clearly implicate the cytoplasmic domain in determining low C expression. The higher expression of A2-C1205 does not influence this study of Nef-mediated down-regulation, but is of some interest. It does not correlate with any known polymorphism in natural HLA-C expression levels (32) and is being further investigated. Generally, T-cell recognition of the HLA-A2-C chimeric molecules was weak and there was a very strong correlation between the level of HLA expression after exposure to Nef and CTL recognition (Fig. 2 B and C).
The results obtained with the transfected HLA-A2 chimeric molecules were reproduced with native HLA-A and HLA-B alleles in two cell lines that are susceptible to HIV-1 infection. Although this type of experiment is difficult to extend because of the limited availability of HIV-1-susceptible cell lines with HLA types matched to available antibodies, it is likely that the results seen here can be generalized. Previously it has been shown that functional outcomes upon antigen engagement depend on the strength of the stimulus (6, 33) and by antigen concentration (34). We might expect therefore to see more polyfunctional responses in CD8 + T cells that respond to epitopes presented by HLA types that are not down-regulated by HIV-1. Our observation that IFN-γ response in ELISPOT assays were greater, and that there was more degranulation and IFN-γ secretion in cocultured HLA-B-specific CTL clones compared with HLA-A-specific CTL clones supports this idea.
There is no doubt about the contribution of the extracellular domains of HLA-I to HIV-1 control by selecting the viral peptide epitopes to which CTL respond. HIV-Nef has probably evolved to down-regulate HLA-I to enable the virus to escape cell-mediated immune responses (8, 35). That this function is of great importance is implied by experiments in macaques, where a hard-to-correct mutation of the Nef sequence to abrogate this function in challenge SIV was repaired in vivo in infected macaques as the infection progressed (11). Why the cytoplasmic domains are polymorphic in the first place is unknown. It might reflect some ancestral interactions with a Neflike viral protein.
In summary, this study describes a mechanism by which polymorphism in cytoplasmic domain can lead to better control of HIV-1. We have shown that CTLs are able to elicit a good response against epitopes presented by HLA-B alleles because of the relative resistance of the latter to Nef-mediated down-regulation. Our data provide further insight into HIV-1 pathogenesis and provide an immune mechanism that helps explain of previous data that show better HIV-1 control by HLA-B alleles compared with HLA-A.
Materials and Methods
Construction of HLA-A2 Chimera.
Primer pairs as specified below (Table S2) were used to alter HLA-A2 cytoplasmic tail sequences as detailed by PCR mutagenesis. An HLA-A0201 wild-type construct inserted into a pcDNA3.1 vector (Clontech) acted as template. The forward primer introduced a BamHI restriction site. For A2-A2, A2-A26, A2-A11, and A2-A43, the reverse primer introduced both desired point mutation and NotI restriction site to the gene terminus. For A2-A24, A2-A80, A2-A32, A2-B35, and A2-B57 the desired mutation was introduced in one round of PCR, the product purified by gel extraction, and then extended with primer A2R2Not/B57NotR/B35NotR to introduce the second restriction site (Table S3). PCR was performed using the proofreading DNA polymerase Pfu (Stratagene).
Replication incompetent recombinant lentivirus were generated using the three plasmid system by cotransfection of 293T cells with calcium phosphate. A DNA mix composed of 1.5-μg HLA-A2 chimeric sequence inserted lentiviral vector, 1-μg envelop plasmid; pMD.G expressing the vesicular stomatitis virus G-envelope protein, and 1-μg packaging plasmid pCMVΛR8.91, containing the HIV-1 gag/pol, tat, and rev genes required for efficient lentivirus production were used.
Supernatant was collected at 24, 48, and 72 h posttransfection, and stored at −80 °C. The virus was later thawed and concentrated by ultracentrifugation at 111,000 × g at 4 °C for 1.5 h under sterile conditions. The pellet was resuspended in 200 μL RPMI 1640 and aliquoted for storage at −80 °C.
Jurkat cell sub cell line (JJK) cells in the exponential growth phase were infected with the packaged virus. One aliquot of lentivirus was used to infect cells. The virus aliquot was added to cells and returned to incubation for 2 h at room temperature. Cells were mixed every 0.5 h before adding 1 mL of R10 and returning to 24-well plates for culturing. After 5 d, cells were checked for HLA-A2 expression and the top 2% positive cells were selected by flow-cytometric sorting.
Cell Culture.
Jurkat cells expressing CD4+, MT2, and C8166 were grown in RPMI supplemented with 10% (vol/vol) FCS, glutamine, and penicillin/streptomycin. The 293T cells were grown in Dulbelco Modified Medium supplemented with 10% FCS glutamine and penicillin/streptomycin.
Antibodies, Plasmids, and Viruses.
The following antibodies were used: Mouse antihuman HLA-A2 conjugated to PE (AbD Serotec) or Alexa Flour 647 (AbD Serotec), mouse antihuman HLA-ABC Alexa Flour®647 (AbD Serotec), anti-CD4/APC (Dako), anti-HLA Class-I A3 conjugated to biotin (AbCam), APC Donkey antimouse IgG (eBiosciences), purified anti-HLA Class-I B7 (AbD Serotec), Kc57-RD1 (Beckman Coulter), antihuman CD8 conjugated to APC H7(BD), and antihuman CD107a conjugated PE (BD).
Plasmids encoding wild-type or mutant Nef–GFP fusion proteins were created as described (36). HIV-IIIB virus was used for initial viral assays. The virus stocks were generated by infecting 1 × 107 C8166 cells with stock HIV-IIIB virus. In summary the cells were spun down in 15-mL falcon tubes and mixed with one vial of concentrated HIV-IIIB virus stock and incubated at 37 °C in 5% CO2 for 1.5 h. At the end of the incubation period the cells were washed twice with RPMI and resuspended in R10 and returned to the incubator. The supernatant was harvested on D4, D7, and D10. The virus concentration was measured using the commercial kit p24 capture ELISA according to manufacturer instructions (Immunodiagnostics).
Construction of Virus Containing M20A Mutation.
M20A pNL4-3 was generated with PCR mutagenesis using pNL4-3 template with Quick Change II XL Site directed mutagenesis kit (Stratagene) (37). Briefly mutagenesis PCR was performed with primers introducing the mutation according to manufacturer’s instructions. The PCR product was digested with DpnI enzyme to remove methylated DNA and then transformed into XL1-Gold Ultra competent cells and grown under Ampicillin selection. The generated mutation was confirmed by sequencing. Plasmid DNA was extracted using Maxi prep kit (Qiagen). Viruses were generated by tranfecting 293T cells with Lipofectamine 2000 (Invitrogen) according to manufacturer instructions.
HLA Down-Regulation Assay/Flow Cytometry.
The 4–6 × 106 JJK were spun down and washed with RPMI1640, then resuspended in 0.4-mL RPMI1640. Cells were gently mixed with 30-ug plasmid DNA (HIV-1SF2Nef and/or HIV-1G2ANef) and transferred to a 4-mm electroporation cuvette (BioRad). The cuvette was placed in the holder of the electroporation apparatus (BTX Electro cell manipulator 600; BTX) and shocked once at 260 V, 1070 uF, 72 Ω. Soon after, electroporation cells were transferred to 2-mL prewarmed R10 in a 6-well plate. The cells are returned to incubator at 37 °C, 5% CO2. Cells are analyzed using flow-cytometric assay after 24 h.
Transduced and transfected cells were stained for surface molecules using a panel of antibodies after 24 h. The panel included monoclonal antibodies against HLA-A2, MHC-I, CD4, and HLA-A3. Cells were incubated with the pretitrated antibodies on ice for 20 min and washed with 2 mL of FACS wash buffer [PBS + 2% FCS (FCS) +0.01% sodium azide]. If a secondary antibody was used, the staining steps were repeated. Intracellular staining was carried out with HIV-1 p24 specific antibody (Kc57-RD1), a marker of HIV-1-infected cells. Washed cells were fixed with fixing reagent before performing analysis using Dako CyAn flow cytometer with summit software.
Fold down-regulation was calculated as follows:
Fold down-regulation of A2 chimera on the surface = MFI of A2 of uninfected/untransfected cell ÷ MFI of A2 of HIV-infected/Nef-transfected cells.
Live Virus ELISPOT Assay.
The production of IFN-γ when target cells were cocultured with CTLs was measured on D6 of infection (the highest infection rate of Jurkat cells is achieved on D6). The cells were incubated at E:T = 400:20,000 after correcting for infection rate. In brief, the ELISPOT plates are coated with 5 ug of mAB for at least 2 h at room temperature or overnight at 4 °C, then washed six times with PBS and blocked with R10 at 37 °C for 2 h. The cells were incubated at above concentration in R10 overnight (12–14 h) before developing according to manufacturer’s instructions. The plates are read using ELISPOT reader. ELISPOT assays using virus-infected cells were carried out in the level-3 containment laboratory. Uninfected cells pulsed with relevant peptide, PHA and/or PMA + Ionomycin are used as positive control. HLA-mismatched clones incubated with infected target cells, target cells alone, and media alone were used as negative controls.
CD8+ T-Cell Degranulation Assay.
To examine whether CTL degranulation depends on the amount of HLA-I allele present on the surface of the CD4+ T cells infected with viral variants, we monitored the level of CD107a upregulation on CTLs. Jurkat and MT2 cells infected in vitro with respective HIV-1 viruses for 4–6 d were used as targets. The level of degranulation was assessed as the proportion of CD107a+ cells among the CTLs. CD8 + T cells from clones that were 14 d old were cocultured in the presence of CD4+ T cells expressing same HLA-I alleles with Monensin at 0.7 μl ml−1, Brefeldin A at 10μg ml−1, and CD107a-PE for 6 h. Cells were washed and stained with CD3-PerCP, CD8-APC-H7, and CD4-FiTC (BD Biosciences) and dead cell stain (violet) (Invitrogen) and flow cytometric analysis was performed.
Statistical Analysis.
One- or two-tailed unpaired t test was used for comparison of data based on experimental hypothesis. SPSS version 17 was used for most of the statistical calculations and Graph Pad Prism was used for drawing figures.
Supplementary Material
Acknowledgments
BB7.2 purified antibody and W6/32 antibody were kind gifts of Mr. Alistair Waugh, Medical Research Council Human Immunology Unit. We thank Dr. Craig Waugh of Weatherall Institute of Molecular Medicine FACS facility for helping with cell sorting and Weatherall Institute of Molecular Medicine HIU sequencing facility for help with sequencing. This work was supported by Commonwealth Scholarship Commission; the Faculty of Medicine, Colombo, Sri Lanka; and Medical Research Council, United Kingdom.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1204199109/-/DCSupplemental.
References
- 1.Kiepiela P, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–775. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 2.Goulder PJ, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8:619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephens HA. HIV-1 diversity versus HLA class I polymorphism. Trends Immunol. 2005;26:41–47. doi: 10.1016/j.it.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Carrington M, O’Brien SJ. The influence of HLA genotype on AIDS. Annu Rev Med. 2003;54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 5.Pereyra F, et al. International HIV Controllers Study The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almeida JR, et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med. 2007;204:2473–2485. doi: 10.1084/jem.20070784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harari A, et al. Skewed association of polyfunctional antigen-specific CD8 T cell populations with HLA-B genotype. Proc Natl Acad Sci USA. 2007;104:16233–16238. doi: 10.1073/pnas.0707570104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz O, Maréchal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 9.Borrow P, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 10.Goonetilleke N, et al. CHAVI Clinical Core B The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swigut T, et al. Impact of Nef-mediated downregulation of major histocompatibility complex class I on immune response to simian immunodeficiency virus. J Virol. 2004;78:13335–13344. doi: 10.1128/JVI.78.23.13335-13344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahn K, et al. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity. 1997;6:613–621. doi: 10.1016/s1074-7613(00)80349-0. [DOI] [PubMed] [Google Scholar]
- 13.Hill A, et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 14.Jones TR, et al. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc Natl Acad Sci USA. 1996;93:11327–11333. doi: 10.1073/pnas.93.21.11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Machold RP, Wiertz EJ, Jones TR, Ploegh HL. The HCMV gene products US11 and US2 differ in their ability to attack allelic forms of murine major histocompatibility complex (MHC) class I heavy chains. J Exp Med. 1997;185:363–366. doi: 10.1084/jem.185.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bevan MJ. Cross-priming. Nat Immunol. 2006;7:363–365. doi: 10.1038/ni0406-363. [DOI] [PubMed] [Google Scholar]
- 17.Cohen GB, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10:661–671. doi: 10.1016/s1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 18.Schaefer MR, et al. A novel trafficking signal within the HLA-C cytoplasmic tail allows regulated expression upon differentiation of macrophages. J Immunol. 2008;180:7804–7817. doi: 10.4049/jimmunol.180.12.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makadzange AT, et al. Characterization of an HLA-C-restricted CTL response in chronic HIV infection. Eur J Immunol. 2010;40:1036–1041. doi: 10.1002/eji.200939634. [DOI] [PubMed] [Google Scholar]
- 20.Mkhwanazi N, et al. Immunodominant HIV-1-specific HLA-B- and HLA-C-restricted CD8+ T cells do not differ in polyfunctionality. Virology. 2010;405:483–491. doi: 10.1016/j.virol.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dean M, Carrington M, O’Brien SJ. Balanced polymorphism selected by genetic versus infectious human disease. Annu Rev Genomics Hum Genet. 2002;3:263–292. doi: 10.1146/annurev.genom.3.022502.103149. [DOI] [PubMed] [Google Scholar]
- 22.Dong T, et al. Extensive HLA-driven viral diversity following a narrow-source HIV-1 outbreak in rural China. Blood. 2011;118(1):98–106. doi: 10.1182/blood-2010-06-291963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 24.Le Gall S, et al. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity. 1998;8:483–495. doi: 10.1016/s1074-7613(00)80553-1. [DOI] [PubMed] [Google Scholar]
- 25.Geyer M, Munte CE, Schorr J, Kellner R, Kalbitzer HR. Structure of the anchor-domain of myristoylated and non-myristoylated HIV-1 Nef protein. J Mol Biol. 1999;289:123–138. doi: 10.1006/jmbi.1999.2740. [DOI] [PubMed] [Google Scholar]
- 26.Ranasinghe SR, et al. The antiviral efficacy of HIV-specific CD8+ T-cells to a conserved epitope is heavily dependent on the infecting HIV-1 isolate. PLoS Pathog. 2011;7:e1001341. doi: 10.1371/journal.ppat.1001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blagoveshchenskaya AD, Thomas L, Feliciangeli SF, Hung CH, Thomas G. HIV-1 Nef downregulates MHC-I by a PACS-1- and PI3K-regulated ARF6 endocytic pathway. Cell. 2002;111:853–866. doi: 10.1016/s0092-8674(02)01162-5. [DOI] [PubMed] [Google Scholar]
- 28.Kosmrlj A, et al. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature. 2010;465:350–354. doi: 10.1038/nature08997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zemmour J, Parham P. Distinctive polymorphism at the HLA-C locus: Implications for the expression of HLA-C. J Exp Med. 1992;176:937–950. doi: 10.1084/jem.176.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCutcheon JA, Gumperz J, Smith KD, Lutz CT, Parham P. Low HLA-C expression at cell surfaces correlates with increased turnover of heavy chain mRNA. J Exp Med. 1995;181:2085–2095. doi: 10.1084/jem.181.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neefjes JJ, Ploegh HL. Allele and locus-specific differences in cell surface expression and the association of HLA class I heavy chain with beta 2-microglobulin: Differential effects of inhibition of glycosylation on class I subunit association. Eur J Immunol. 1988;18:801–810. doi: 10.1002/eji.1830180522. [DOI] [PubMed] [Google Scholar]
- 32.Falk K, et al. Allele-specific peptide ligand motifs of HLA-C molecules. Proc Natl Acad Sci USA. 1993;90:12005–12009. doi: 10.1073/pnas.90.24.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas R, et al. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat Genet. 2009;41:1290–1294. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 35.Valitutti S, Müller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med. 1996;183:1917–1921. doi: 10.1084/jem.183.4.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drakesmith H, et al. HIV-1 Nef down-regulates the hemochromatosis protein HFE, manipulating cellular iron homeostasis. Proc Natl Acad Sci USA. 2005;102:11017–11022. doi: 10.1073/pnas.0504823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, et al. Multilayered defense in HLA- B51-associated HIV viral control. J Immunol. 2011;187(2):684–691. doi: 10.4049/jimmunol.1100316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.