

Abstract
Common inflammatome gene signatures as well as disease-specific signatures were identified by analyzing 12 expression profiling data sets derived from 9 different tissues isolated from 11 rodent inflammatory disease models. The inflammatome signature significantly overlaps with known drug targets and co-expressed gene modules linked to metabolic disorders and cancer. A large proportion of genes in this signature are tightly connected in tissue-specific Bayesian networks (BNs) built from multiple independent mouse and human cohorts. Both the inflammatome signature and the corresponding consensus BNs are highly enriched for immune response-related genes supported as causal for adiposity, adipokine, diabetes, aortic lesion, bone, muscle, and cholesterol traits, suggesting the causal nature of the inflammatome for a variety of diseases. Integration of this inflammatome signature with the BNs uncovered 151 key drivers that appeared to be more biologically important than the non-drivers in terms of their impact on disease phenotypes. The identification of this inflammatome signature, its network architecture, and key drivers not only highlights the shared etiology but also pinpoints potential targets for intervention of various common diseases.
Keywords: Bayesian network, co-expression network, inflammatome, inflammatory diseases, key regulators
Introduction
Inflammation is the response of an organism’s immune system to damage incurred by its cells, organs, and vascularized tissues by microbial pathogens, injurious chemicals, or physical insults. The initial stages of inflammation involve changes in local blood flow combined with the accumulation of various inflammatory cells (monocytes, neutrophils, eosinophils, lymphocytes, dendritic cells, and mast cells) at the site of tissue trauma. Foreign pathogens, cell debris caused by the inflammatory response, and the inflammatory cells themselves are then removed as tissue repair commences. Under normal circumstances, tissue function is restored. However, if this delicate balance between inflammation and resolution of the events leading to the inflammation is dysregulated, inflammation can lead to disease pathology (Medzhitov, 2008). As the mechanisms of more and more diseases have been elucidated over the past decade, it has become clear that most of the major chronic diseases previously not associated with inflammation, including atherosclerosis (Hansson and Libby, 2006; Aviram, 2011; Mizuno et al, 2011), obesity (Kanda et al, 2006; Heber and Carpenter, 2011; Kobayashi et al, 2011), diabetes (Muoio and Newgard, 2008; Das and Mukhopadhyay, 2011; Hess and Grant, 2011), osteoarthritis (Wieland et al, 2005; Rosenthal, 2011; Zivanovic et al, 2011), stroke (Nakase et al, 2008; Benbir et al, 2011; Drake et al, 2011; Zakai et al, 2011), sarcopenia (Jensen, 2008; Meng and Yu, 2010), and cancer (Mantovani et al, 2008; Bergman et al, 2011; Grange et al, 2011; Porta et al, 2011; Sansone and Bromberg, 2011), all have a clear inflammation component.
The connection between atherosclerosis and inflammation was established by studies in animals and humans, which showed that hypercholesterolemia causes focal activation of the endothelium in large and medium-sized arteries. The infiltration and retention of LDL in the arterial intima induce an inflammatory response in the artery wall (Pentikainen et al, 2000). The molecular link between inflammation and obesity was established based on the findings that TNFα is overexpressed in the adipose tissues of mouse models of obesity (Hotamisligil et al, 1993) as well as in the adipose and muscle tissues of obese humans (Hotamisligil et al, 1995). It soon became clear that obesity is characterized by a broad inflammatory response and that many inflammatory mediators exhibit patterns of expression and impact insulin action in a manner similar to that of TNFα during obesity, as observed in a range of organisms from mice to humans (Dandona et al, 2004). In addition, it has been shown in epidemiological studies that chronic inflammation could lead to various types of cancer. For example, Helicobacter pylori infection is associated with gastric cancer (Fox and Wang, 2007), hepatitis B or C virus infection is associated with hepatocellular carcinoma (Gurtsevitch, 2008), and inflammatory bowel disease is associated with colon cancer (Rhodes, 1996).
It is therefore not surprising to see that single anti-inflammatory agents can treat a variety of diseases. For example, glucocorticoids have been used to treat rheumatoid arthritis (RA), psoriasis, gout, Crohn’s disease, asthma, atopic dermatitis, and transplant rejection. Likewise, non-steroidal anti-inflammatory drugs (NSAIDs) such as Coxibs are used for alleviating RA, ankylosing spondylitis (AS), gout, acute/chronic pain, and cancer. More recently, anti-cytokine therapies, particularly anti-TNF therapies, have been broadly applied in RA, AS, Crohn’s disease, and psoriatic arthritis (Van Hauwermeiren et al, 2011). Novel therapeutic agents are being developed based on the assumption that several clinical indications can be treated by targeting common pathways (O'Neill, 2006).
The clearly demonstrated inflammatory nature of many common chronic diseases puts forward a hypothesis that a representative gene signature can be acquired from multiple disease models. In support of this, pathogen-induced host responses, autoimmune diseases, and lung inflammatory diseases have shared gene expression changes accessed by transcriptional profiling of blood or hematopoietic cells (Jenner and Young, 2005; Gilchrist et al, 2006; Nilsson et al, 2006; Pennings et al, 2008; Pankla et al, 2009; O'Hanlon et al, 2011). However, it is not yet clear whether (1) common signatures are shared across different tissue types and across different types of inflammatory diseases/conditions, (2) common signature genes have causal relationships with each disease, (3) common signatures have therapeutic potentials, (4) coherent and common gene–gene interaction networks and regulatory mechanisms underlie various disease states, and (5) there are disease-specific genes and processes in each disease model.
To address these questions, we selected 12 inflammation-related gene expression profiling data sets representing 9 different tissues isolated from 11 disease models. The disease models include asthma, emphysema, pulmonary fibrosis, lipopolysaccharide (LPS)-treated acute injury, inflammation and neuropathic pain, atherosclerosis, stroke, obesity, diabetes, and age-related sarcopenia. We derived a representative gene signature of 2483 genes across 12 disease model-tissue combinations as well as disease-specific signatures. The common gene signature was found to be significantly enriched for genes involved in inflammation and immune response, thus was termed as the ‘inflammatome’. The inflammatome signature was then compared with current known drug targets, candidate disease-associated genes from genome-wide association studies (GWAS), and co-expression network modules developed from independent mouse and human cohorts to assess the disease-causal nature and potential co-regulation patterns of the inflammatome signature. We also integrated the inflammatome signature with Bayesian networks (BNs) developed from independent mouse and human cohorts to derive consensus Bayesian subnetworks that delineate the relationships among the signature genes as well as key regulators of the signature based on the network topology. Experimental evidence was also provided to support the role of the key regulators identified.
Results
Rodent inflammatory models included in the analysis
The 12 rodent inflammatory model-tissue combinations include an ovalbumin (OVA)-challenged asthma model (lung), a high fat diet (HFD)-treated ApoE knockout (KO) atherosclerosis model (aorta), an IL-1β transgenic emphysema model (lung), a db/db diabetes model (adipose and islet), a TGFβ transgenic (Tg) pulmonary fibrosis model (lung), a CGN-induced inflammation pain model (skin), an LPS-treated acute injury model (liver), a Chung neuropathic pain model (dorsal root ganglia, DRG), an ob/ob obesity model (adipose), a middle cerebral artery occlusion (MCAO) stroke model (brain), and an age-related sarcopenia model (muscle) (Table I). The total data set derives from 11 rodent animal models and includes molecular profiling data from 9 different tissues.
Table 1. Twelve rodent inflammatory disease models and the number of cases and controls used in the current analysis.
| Disease | Model | Species | Tissue profiled | No. of Cases | No. of Controls | No. of total arrays |
|---|---|---|---|---|---|---|
| Asthma | OVA | Mouse | Lung | 5 | 4 | 9 |
| COPD | IL-1β Tg | Mouse | Lung | 5 | 3 | 8 |
| Fibrosis | TGFβ Tg | Mouse | Lung | 4 | 4 | 8 |
| Atherosclerosis | ApoE KO HFD | Mouse | Aorta | 3 | 3 | 6 |
| Diabetes | db/db | Mouse | Adipose | 3 | 3 | 6 |
| Diabetes | db/db | Mouse | Islet | 5 | 5 | 10 |
| Obesity | ob/ob | Mouse | Adipose | 3 | 3 | 6 |
| Multiple | LPS | Rat | Liver | 4 | 4 | 8 |
| Stroke | MCAO | Rat | Brain | 4 | 4 | 8 |
| Neuropathic pain | Chung | Rat | DRG | 4 | 4 | 8 |
| Inflammation pain | CGN | Rat | Skin | 4 | 5 | 9 |
| Sarcopenia | Aged versus Young | Rat | Muscle | 5 | 5 | 10 |
Identification and functional annotation of the ‘inflammatome’ signature
To identify a representative gene signature, we adopted a two-way ANOVA statistical analysis approach and adopted the Benjamini and Hochberg multiple testing correction scheme. For the two-way ANOVA, the two main factors applied were disease model and disease state. This analysis resulted in the identification of 2483 genes (Supplementary Table 1) that were differentially expressed between the disease and normal states (P<1.0e−9; Figure 1). The P-value threshold used for gene selection was relatively stringent to ensure the identification of a set of genes achieving strong consensus across all 12 data sets. We separated the gene signature into two k-means clusters of upregulated (1499 genes) and downregulated (984) genes. Overall, 41% (1026) of the signature genes were consistently upregulated (614 genes) or downregulated (412 genes) in at least 10 data sets and 119 genes upregulated (83 genes) or downregulated (36) across all 12 data sets. Selected cuts of upregulated and downregulated genes among the 12 models are summarized in Table II.
Figure 1.

A heat map of the inflammatome signature comprising 1499 upregulated and 984 downregulated genes. The rows represent the disease samples from the 12 data sets and the columns represent the 2483 signature genes that were grouped into two k-means clusters of upregulated and downregulated genes.
Table 2. A summary of number of consistent upregulated and downregulated genes in 12 disease models.
| Upregulated | Accumulated no. of gene | Downregulated | Accumulated no. of gene |
|---|---|---|---|
| All 12 models | 83 | All 12 models | 36 |
| ≥11 models | 303 | ≥11 models | 171 |
| ≥10 models | 614 | ≥10 models | 412 |
| ≥9 models | 939 | ≥9 models | 639 |
| ≥8 models | 1193 | ≥8 models | 810 |
| ≥7 models | 1357 | ≥7 models | 925 |
We then tested for enrichment of biological functions in the upregulated and downregulated inflammatome signature using GO biological process enrichment analysis (Table III). The majority of the biological processes enriched in the upregulated genes were associated with immune and inflammatory responses (e.g., inflammatory response, leukocyte activation, cytokine production, chemotaxis, TLR signaling pathway, and antigen presentation); other processes not directly involved in immune response were also included, such as mitotic cell cycle, induction of apoptosis, extracellular matrix remodeling, osteoclast differentiation, translation, and angiogenesis. Several protein families, such as caspase (Casp1, Casp3, Casp4, Casp7, and Casp8), cathepsin (Ctsb, Ctsc, Ctsd, Ctse, Ctsh, Ctsk, Ctsl, Ctss, and Ctsz), chemokine/chemokine receptor (Ccl1, Ccl2, Ccl3, Ccl5, Ccl6, Ccl7, Ccl9, Ccl11, Ccl12, Ccl17, Ccl20, Ccr1, Ccr5, Cxcl1, Cxcl2, Cxcl3, Cxcl5, Cxcl9, Cxcl10, Cxcl13, Cxcl16, and Cxcr3), complement/complement receptor (C1q, C1r, C3, C4b, C3ar1, and C5ar1), Fc receptors (Fcer1g, Fcgr1, Fcgr2b, Fcgr3, and Fcgr4), interleukin/interleukin receptor (Il1b, Il1r2, Il4ra, Il6, Il10, Il10ra, Il10rb, Il19, and Il33), matrix metallopeptidase (Mmp2, Mmp3, Mmp8, Mmp9, Mmp12, Mmp13, Mmp14, and Mmp19), minichromosome maintenance deficient (Mcm2, Mcm3, Mcm5, Mcm6, Mcm7, and Mcm10), and proteasome (Psma5, Psmb2, Psmb8, Psmb9, Psmb10, and Psme1) are upregulated in the majority (>9) of the 12 data sets. The downregulated genes were significantly enriched for pathways or processes associated with nerve impulse transmission, energy generation and all major metabolic processes involving amino acid, fatty acid, and carbohydrate metabolism (Table III).
Table 3. Annotation of upregulated (left panel) and downregulated (right panel) inflammatome signatures.
| Similar set: upregulated | Enrichment P | Overlap | Set | Similar set: downregulated | Enrichment P | Overlap | Set |
|---|---|---|---|---|---|---|---|
| All P-values from the Fisher’s Exact Test were Bonferroni-corrected to account for multiple testing. | |||||||
| Inflammatory response | 4.76E−61 | 208 | 704 | Transmission of nerve impulse | 3.32E-11 | 78 | 639 |
| Leukocyte activation | 2.13E−32 | 164 | 704 | Valine, leucine, and isoleucine degradation | 1.34E-08 | 18 | 42 |
| Regulation of immune response | 1.44E−25 | 84 | 260 | Carboxylic acid metabolic process | 4.03E-06 | 68 | 661 |
| Cytokine production | 6.10E−18 | 85 | 335 | Cofactor metabolic process | 1.30E-05 | 31 | 198 |
| Chemotaxis | 4.97E−16 | 74 | 284 | Generation precursor metabolites/energy | 9.18E-05 | 57 | 554 |
| Humoral immune response | 3.25E−14 | 69 | 271 | Fatty acid catabolic process | 0.000122 | 16 | 65 |
| Mitotic cell cycle | 7.64E−13 | 87 | 414 | Ubiquinone metabolism | 0.000562 | 11 | 46 |
| Induction of apoptosis | 1.74E−12 | 86 | 412 | Amino acid metabolic process | 0.000813 | 31 | 236 |
| TLR signaling pathway | 4.66E−12 | 21 | 47 | Fatty acid β-oxidation | 0.001857 | 13 | 52 |
| Phagocytosis | 2.74E−11 | 38 | 111 | Cellular catabolic process | 0.005883 | 64 | 736 |
| Innate immune response | 9.29E−11 | 48 | 173 | Electron transport | 0.007212 | 37 | 341 |
| ECM remodeling | 9.67E−11 | 22 | 59 | GPCRs in the regulation of muscle tone | 0.018483 | 14 | 103 |
| Osteoclast differentiation | 3.61E−10 | 31 | 82 | Tricarboxylic acid cycle | 0.026331 | 8 | 24 |
| Regulation of cell proliferation | 4.19E−10 | 112 | 662 | Fatty acid oxidation | 0.028396 | 14 | 75 |
| Antigen processing and presentation | 7.44E−10 | 32 | 89 | Fatty acid metabolic process | 0.034657 | 34 | 323 |
| Positive regulation of translation | 3.64E−09 | 45 | 170 | G-protein signaling_Rap1A regulation | 0.034694 | 9 | 45 |
| Cytokine production by Th17 cells | 6.33E−09 | 17 | 40 | Lipid catabolic process | 0.042439 | 23 | 180 |
| Angiogenesis | 9.41E−09 | 60 | 277 | Signal transduction_cAMP signaling | 0.053522 | 14 | 113 |
| Cell-cycle process | 2.79E−08 | 104 | 636 | Carbohydrate metabolic process | 0.075586 | 52 | 606 |
| Wound healing | 1.79E−07 | 57 | 274 | NF-AT signaling in cardiac hypertrophy | 0.107942 | 12 | 91 |
| Regulation of translation | 4.44E−07 | 66 | 349 | regulation of neurotransmitter levels | 0.124713 | 20 | 155 |
| Macrophage activation | 1.61E−06 | 20 | 49 | Long-term depression | 0.721233 | 13 | 76 |
| Interleukin-12 production | 1.70E−06 | 18 | 40 | Secretion | 0.786091 | 55 | 733 |
Identification of disease-specific signatures
We also identified disease-specific gene signatures, defined as genes that are statistically significantly differentially expressed between the disease and control group at a false discovery rate (FDR)<5% in only one of the rodent models. At an FDR<5%, 1175, 26, 1120, 284, 177, 28, 782, 1123, 1208, 476, 292, and 222 genes were identified as disease model-specific signatures for Apoe KO (aorta), db/db adipose, db/db islet, Chung neuropathic pain (DRG), CGN-induced pain (skin), IL-1β Tg emphysema model (lung), LPS-treated acute injury (liver), ob/ob (adipose), OVA-challenged asthma (lung), sarcopenia (muscle), MCAO stroke (brain), and pulmonary fibrosis model (lung), respectively (Supplementary Table 2). In average 576 genes show disease-specific differential expression, supporting the presence of disease-specific alternations in gene expression. However, compared with the ∼2500 genes in the inflammatome signature shared among the disease models, the disease specific signatures are much smaller. In addition, a GO biological process enrichment analysis only revealed enrichment of a limited set of functional categories (such as sensory perception, ion homeostasis, and neuron development) for four of the disease model-specific signatures (ApoE KO, db/db islet, ob/ob, and MCAO stroke) at a Bonferroni-corrected P<0.05 (Supplementary Table 3).
Although disease-specific signature genes can help identify disease-specific mechanisms, targets, and biomarkers, the smaller signature sizes limit our power to identify robust disease-specific signatures, as indicated by the poor coherence in the functionalities of the disease-specific signature genes in comparison with the common inflammatome signature. We therefore conducted a more in-depth analysis of the inflammatome signature in the subsequent sections.
Comparison of the common inflammatome signature with current drug targets and disease-associated genes
We next assessed the relevance of the rodent model-derived inflammatome signature for human disease. At the time this manuscript was prepared, there were 803 known drug target genes (including marketed drugs and drugs under development) according to GeneGO ( http://www.genego.com/). In addition, there were 3498 reported candidate genes adjacent to SNPs that have been annotated as being associated with various diseases or disease-related intermediate phenotypes in the NHGRI GWAS catalog ( http://www.genome.gov/gwastudies, accessed April 28, 2012). In all, 168 of the 803 drug target genes and 413 of the 3498 GWAS candidate genes were in the inflammatome signature (Supplementary Table 4), representing a significant enrichment of inflammatome genes in each of these gene sets (Fisher’s Exact Test P-values <5.1e−22 and 1.8e−5, respectively; fold enrichments=2.1 and 1.2, respectively). There are 66 overlapping genes among the 3 gene sets and the targeting of several of them (e.g., CD40, CHRM3, IL12A, and VEGFA) has been shown to produce a significant therapeutic effect on multiple diseases (Supplementary Table 4). Among the overlapping genes between the inflammatome and current drug target lists are Ppara and Prkaa2 (Ampk), with these genes downregulated in all 12 inflammatory disease models and agonists for these genes either in the market or under development (Narkar et al, 2008; Richter and Ruderman, 2009). This comparative analysis indicated that the inflammatome signature is significant enriched for current drug targets and candidate GWAS genes for common human diseases, supporting a causal relationship between the inflammatome genes and many human diseases, and it provides a rich source of new drug targets for consideration in many different disease areas.
Comparison of the common inflammatome signature with multiple mouse and human tissue gene co-expression networks
To explore the co-regulation pattern of the common inflammatome signature genes and cross-validate their connection to multiple disease phenotypes in independent studies, we performed an integrative analysis by linking co-expression network data obtained from multiple murine F2 genetic crosses and multiple human cohorts segregating different disease phenotypes. Gene co-expression network analysis (GCENA) has been increasingly used to identify gene subnetworks for prioritizing gene targets associated with a variety of common human diseases such as cancer and obesity (Gargalovic et al, 2006; Horvath et al, 2006; Chen et al, 2008; Emilsson et al, 2008). Networks generally provide a convenient framework for exploring the context within which single genes operate. Networks are simply graphical models composed of nodes and edges. In a gene co-expression network, the nodes represent genes and edges (links) between any two nodes indicate a relationship (a similar expression pattern) between the two corresponding genes. One important end product of GCENA is gene modules comprising highly interconnected sets of genes. It has been demonstrated that these types of modules are generally enriched for known biological pathways, for genes that associate with disease traits, and for genes that are linked to common genetic loci (Zhu et al, 2004, 2008; Zhang and Horvath, 2005; Gargalovic et al, 2006; Horvath et al, 2006; Lum et al, 2006; Chen et al, 2008; Emilsson et al, 2008; Schadt et al, 2008). In this way, one can identify those key groups of genes that sense perturbations from genetic loci, and subsequently define the intermediate steps that actually lead to disease states.
A gene co-expression network can be fully represented by a topological overlap matrix (TOM). Topological overlap between two genes not only reflects their more proximal interactions (e.g., two genes physically interacting or having correlated expression values), but also reflects the higher order interactions that these two genes may have with other genes in the network (Schadt et al, 2005). Following a previously described method of weighted GCNA (WGCNA; Zhang and Horvath, 2005), we constructed eleven (11) gene co-expression networks corresponding to 11 data sets derived from four human and one mouse gene expression studies as detailed in Table IV. The TOM plots of the networks are depicted in Figure 2. A complete description of the studies can be found in Materials and methods.
Table 4. Data sets used for co-expression network analysis.
| Cohort | Specie | Tissue | Disease state |
|---|---|---|---|
| BxH ApoE−/− | Mouse | Adipose/muscle/liver | Diabetes and obesity |
| NKI | Human | Breast | Cancer |
| HCC | Human | Liver | Cancer |
| IFA | Human | Adipose | Normal |
| HLC | Human | Liver | Normal |
Figure 2.

Topological overlap matrix (TOM) plots of weighted, gene coexpression networks constructed from one mouse studies (A–F) and four human studies including IFA (G–H), NKI (I), HLC (J) and HCC (K). Each symmetric heat map with rows and columns as genes represents the network connection strength (as indicated by the different shades of color—from white signifying not significantly correlated to red signifying highly significantly correlated) between any pair of nodes (genes) in the corresponding network. The network connection strength is measured as the topological overlap between genes. The network modules highlighted as color block along the rows and columns (each color block represents a module) were identified via an average linkage hierarchical clustering algorithm using topological overlap as the dissimilarity metric. In each network, the module highlighted with a black box is most enriched with the inflammatome signature. (A) Mouse male adipose, (B) mouse male liver, (C) mouse male muscle, (D) mouse female adipose, (E) mouse female liver, (F) mouse female muscle, (G) mouse male adipose, (H) human female adipose, (I) human breast cancer, (J) human normal liver, (K) human cancer liver.
Each of the 11 gene co-expression networks includes at least one module that is significantly enriched for the inflammatome signature, with Fisher’s exact test P-values ranging from 7.6e−28 to 1.1e−203 as shown in Table V. For example, the blue module comprised of 1991 genes from the BxH ApoE−/− female adipose network, includes 672 of the inflammatome genes and this represents a 3.28-fold enrichment over what we would have expected by chance (P<e−203). Roughly one-third of the black module genes in the NKI breast cancer network overlap the inflammatome signature, representing a three-fold enrichment (P<2e−51). All these modules are significantly enriched for genes in the inflammatory and immune response pathways. On the other hand, the purple module in NKI network and the turquoise modules in the HCC network, both containing many typical cell cycle genes such as TOP2A, CHEK1, E2F1, and EZH2, are also significantly enriched for the inflammatome signature with Fisher's exact test P-values <2.6e−37 and <1.6e−31, respectively. Interestingly, the purple module of the NKI network is the most predictive of patient’s survival time (Cox model P-value<e−12) while the black module is moderately predictive of survival time as well. These results indicate that the inflammatome signature is highly co-regulated not only in multiple tissues (liver, adipose, muscle, and breast cancer tissue) but also in multiple disease states (cancer, obesity, and diabetes) in data sets completely independent of those from which the signature was derived.
Table 5. Network gene modules most enriched with the inflammatome signature.
| Cohort | Tissue | Gender | Module | Network size | Signature size | Signature in network | Module size | Signature in module | Fold enrichment | Enrichment P-value |
|---|---|---|---|---|---|---|---|---|---|---|
| BxH ApoE−/− | Adipose | Female | Blue | 21 936 | 2505 | 2258 | 1991 | 672 | 3.2789252 | 1.13E−203 |
| BxH ApoE−/− | Adipose | Male | Brown | 21 936 | 2505 | 2258 | 1604 | 597 | 3.6157922 | 2.58E−203 |
| BxH ApoE−/− | Muscle | Male | Blue | 21 836 | 2505 | 2249 | 2803 | 721 | 2.4974442 | 1.16E−143 |
| BxH ApoE−/− | Liver | Female | Red | 21 936 | 2505 | 2258 | 605 | 291 | 4.6727346 | 2.81E−129 |
| BxH ApoE−/− | Liver | Male | Yellow | 21 936 | 2505 | 2258 | 1206 | 395 | 3.1818763 | 1.29E−108 |
| BxH ApoE−/− | Muscle | Female | Turquoise | 21 842 | 2505 | 2250 | 4518 | 858 | 1.8435331 | 4.40E−91 |
| NKI | Breast (cancer) | All | Purple | 19 570 | 2276 | 1995 | 387 | 130 | 3.2951888 | 2.56E−37 |
| NKI | Breast (cancer) | All | Black | 19 570 | 2276 | 1995 | 644 | 201 | 3.0616681 | 1.97E−51 |
| HCC | Liver (cancer) | All | Turquoise | 14 878 | 2276 | 1835 | 2405 | 510 | 1.7193472 | 2.47E−42 |
| IFA | Adipose | Male | Turquoise | 5580 | 2276 | 824 | 1123 | 316 | 1.90552352 | 1.50E−40 |
| IFA | Adipose | Female | Turquoise | 5561 | 2276 | 842 | 1696 | 411 | 1.6005041 | 2.10E−34 |
| HLC | Liver | All | Yellow | 4408 | 2276 | 623 | 180 | 84 | 3.3018727 | 7.63E−28 |
We previously described the identification of macrophage-enriched metabolic network (MEMN) modules in mouse and human and showed that MEMNs are enriched for genes supported as causal for virtually all key metabolic disease traits associated with diabetes, obesity, and cardiovascular disease (CVD) (Chen et al, 2008; Emilsson et al, 2008; Yang et al, 2010). Since the enriched functional categories (i.e., inflammatory and immune responses) and tissue-specific gene expression patterns (macrophage- and leukocyte-enriched) are very similar between inflammatome and MEMNs, we performed a comparison among the inflammatome signature, mouse-derived MEMN, and human-derived MEMN gene sets and found highly significant overlaps between any two of these sets (Figure 3). This analysis indicated that the MEMNs, in addition to playing key roles in metabolic disease traits, could also be involved in additional diseases such as lung inflammation, sarcopenia, and pain. In fact, among the 420 overlapping genes between the inflammatome and MEMN sets, 119 were found to be in the Mouse Genome Informatics (MGI) database ( ftp://ftp.informatics.jax.org/pub/reports/index.html#pheno) and 68 displayed 770 unique phenotypes relevant to various disease areas ranging from metabolic, cardiovascular, morphological, neurological, and immune system disorders to cancer and embryonic lethality (Supplementary Table 5). Note that the likelihood that 57% of the 119 genes tested with mutant phenotypes is significantly higher than the overall rate of 28% of the 9116 genes tested in the MGI database with mutant phenotypes (proportion test P<2.9e−12). In addition, 127 of the 420 genes in the MEMN have been linked to 119 diseases/traits (P<2.5e−3; Supplementary Table 6) in public GWAS studies as cataloged by NHGRI (accessed April 28, 2012). Moreover, one of the overlapping genes, C3ar1, has been tested and shown to affect obesity (Schadt et al, 2005; Yang et al, 2009), diabetes (Mamane et al, 2009), atherosclerosis (Yang et al, 2010), and asthma (Hasegawa et al, 2004). These lines of evidence again strongly support the critical causal role of these genes in multiple diseases.
Figure 3.

A Venn diagram showing overlaps among the inflammatome, human macrophage-enriched metabolic network (MEMN), and mouse MEMN signatures. One-third of the inflammatome signature genes are in the human MEMN and the three signatures share 420 genes.
Employing BNs to infer regulatory relationships among the inflammatome signature genes
Neither expression signatures nor co-expression networks can provide causal relationships among genes. Probabilistic causal networks are one way to model such relationships, where causality in this context reflects a probabilistic belief that one node in the network affects the behavior of another either directly or indirectly. BNs (Zhu et al, 2004; Zhu et al, 2007; Zhu et al, 2008; Zhu et al, 2012) are one type of probabilistic causal networks that provide a natural framework for integrating highly dissimilar types of data. Unlike co-expression networks, which allow one to look at the overall gene–gene correlation structure at a high level, BNs are sparser but allow a more granular look at the relationships and directional predictions among genes or between genes and other traits such as disease. We have previously described a method to reconstruct probabilistic, causal networks by integrating genetic and gene expression data (Zhu et al, 2004, 2007, 2008, 2012, 92). Toward this end, we constructed 66 mouse BNs from 11 mouse crosses (Lum et al, 2006; Chen et al, 2008) and three human BNs from the previously described human studies (Icelandic Family Blood (IFA), Emilsson et al, 2008; Human Liver Cohort (HLC), Yang et al, 2010; and breast cancer, Tran et al, 2011). The 66 mouse BNs include 25 constructed from liver data, 23 from adipose data, and 18 from muscle in mouse studies. Each data set involves hundreds to thousands of samples that were leveraged for the network reconstructions. All mouse studies were carried out in the context of a genetic F2 intercross design, where different F2 crosses have different genetic perturbation architectures and different sets of causal reactive relationships that can be inferred. Thus, we constructed a network for each data set independently. Each BN was gender and tissue specific and was constructed using the genetic and gene expression data generated from each population (Zhu et al, 2004, 2007, 2008, 94). Our previous studies have shown that predictive BNs can be constructed based on genetic and gene expression data with over 100 samples (Zhu et al, 2004, 2007, 2008, 2012, 92). As the construction of BNs is an NP-hard problem, we used a Monte Carlo Markov Chain (MCMC) method to construct the networks. To construct the network, we simultaneously run 1000 MCMC chains to produce 1000 networks (Friedman et al, 2000; Zhu et al, 2007). Our final network then represents common features among these 1000 structures, minimizing any issues relating to overfitting of the data. For each of the three tissues, we derived a tissue-specific consensus BN as the union of all the BNs for the same tissue. The three mouse consensus BNs and three human BNs served as (1) the network framework representative of the inflammatome signature and (2) the sources for identifying the key regulators of the inflammatome signature via a modification of the key driver identification algorithm (Zhu et al, 2008, 2012; Tran et al, 2011).
The key driver algorithm works as follows. For each of the six causal BNs, we first derived a subnetwork comprised of the directed links between the inflammatome signature genes. The nodes and edges of the six inflammatome causal networks are listed in Supplementary Table 7. Then, for each gene in the subnetwork, we defined its downstream signature as the set of nodes in the subnetwork that could be reached by the gene following directed links throughout the 6-edge neighborhood of the node. A node was claimed as a candidate driver if the number of its downstream nodes was significantly enriched in the inflammatome subnetwork. A candidate driver became a global driver if it was not in the downstream of any other candidate driver; otherwise it was a local driver. The candidate drivers that had the number of out-links significantly greater than random were promoted as global drivers. Finally, the key and local drivers for the inflammatome signature were derived based upon the consistency of the status of the candidate drivers across the six causal networks. A driver was defined as a key driver of the inflammatome signature if (1) it was a global driver in at least two causal networks or (2) it was a global driver in one causal network and a local driver in at least one other network. A driver became a local driver if (1) it was a global driver in only one causal network but not a driver in any other network or (2) it was only a local driver in at least two causal networks.
Applying this algorithm, we identified 151 key and 212 local drivers (Supplementary Table 8 for the driver list and Supplementary Table 9 for the downstream genes of the key drivers). The key driver genes are significantly enriched for genes in pathways like leukocyte activation (P=1.49e−22), regulation of immune response (P=3.55e−21), cytokine production (P=1.40e−18), and inflammatory response (P=1.45e−13). Notably, 55 of the 151 key drivers are global drivers in at least 2 causal networks and they represent the top key drivers. Figure 4 shows two causal networks with their key drivers highlighted.
Figure 4.

Inflammatome gene regulatory (Bayesian) networks and their predicted key drivers that are highlighted as large red nodes. The nodes in each network are the inflammatome signature genes and the directed links between them are derived from the causal networks reconstructed by integrating genetic and gene expression data in the corresponding cohort: (A) the human adipose IFA study; (B) the human liver HLC study. HCK, CD53, and TYROBP are the top drivers of both inflammatome subnetworks.
In order to address whether the drivers represent biologically more critical genes than non-driver genes, we downloaded the mutant phenotype data from the MGI. As shown in Table VI, compared with a frequency of 28.7% of genes in the phenotype database that showed observable altered phenotypes overall, the frequencies of genes with mutant phenotypes among the key drivers, local drivers, and non-driver genes were 63.6, 57.9, and 39.2%, respectively, representing significant enrichments for genes with mutant phenotypes among the key drivers and local drivers (Fisher’s Exact Test P=2e−12 and 1.7e−12, respectively). In addition, the frequencies of genes with mutant phenotypes in the key and local driver groups were significantly higher than that of the non-driver group (proportion test P=0.001 and 0.005, respectively). Thus, the key drivers identified indeed appear to be more biologically important than the non-drivers. Notably, 19 of the top 55 key drivers were tested in MGI and 73.7% (14) had mutant phenotypes.
Table 6. Comparison of mutant phenotypes between Bayesian network key drivers, local drivers, and non-drivers.
| Group | No. of genes | No. of gene tested in the MGI phenotype database | No. of genes with MGI phenotype(s) | % Tested genes with phenotype(s) |
|---|---|---|---|---|
| Top 55 key drivers | 55 | 19 | 14 | 73.7 |
| Key drivers | 151 | 44 | 28 | 63.6 |
| Local drivers | 212 | 57 | 33 | 57.9 |
| Non-drivers | 2098 | 609 | 239 | 39.2 |
The top five key drivers are Cd53, Hck, Tyrobp, Nckap1l, and Aif1. Cd53 has been suggested to have a role in multiple inflammatory diseases such as RA, atopic eczema, B-cell chronic lymphoproliferative disorders, and interstitial lung disease (Taylor et al, 2000; Barrena et al, 2005; Jockers and Novak, 2006; Pedersen-Lane et al, 2007); Hck (Hematopoietic cell kinase) has been associated with COPD and poor outcome of chronic myeloid leukemia (Zhang et al, 2007; Lee et al, 2008; Yanagisawa et al, 2009); Tyrobp has been implicated in presenile dementia with bone cysts and a cognitive disorder Nasu-Hakola disease (Paloneva et al, 2000; Thrash et al, 2009); Nckap1l (NCK-associated protein 1-like), expressed only in hematopoietic cells, is associated with the Wiskott-Aldrich Syndrome (WAS), a rare X-linked primary immunodeficiency (Snapper and Rosen, 1999); Aif1 (allograft inflammatory factor 1) has been associated with atherosclerosis, systemic sclerosis, Type 1 diabetes, Type 2 diabetes, and RA (Arvanitis et al, 2005; Alkassab et al, 2007; Harney et al, 2008; Zhang et al, 2008; Eike et al, 2009; Rouskas et al, 2012).
Other notable key drivers include C3ar1 and Alox5ap, two susceptibility genes for various diseases. C3ar1, which has been experimentally validated as a causal gene for obesity, diabetes, and atherosclerosis (Schadt et al, 2005; Mamane et al, 2009; Yang et al, 2009, 2010), and has been linked to asthma in human genetic association studies (Hasegawa et al, 2004), is a key driver in mouse adipose tissue. Alox5ap activates 5-lipoxygenase (encoded by Alox5) and has been linked to a spectrum of diseases including CVDs, stroke, asthma, arthritis, and cancers. In addition, Alox5ap mutants have been reported to affect metabolism, homeostasis, immune system, and skeleton phenotypes (Byrum et al, 1997), and an Alox5ap inhibitor, MK866, has been shown to have anti-atherosclerosis (Jawien et al, 2006) and anti-tumor activities (Mayburd et al, 2006). Interestingly, when C3ar1 or Alox5 genes are modified in KO mice (Mehrabian et al, 2005; Yang et al, 2009), the inflammatome network was significantly perturbed, as reflected by significant overlap of liver expression signatures of C3ar1 and Alox5 KO mice with the inflammatome network (P=7.65e−3 for C3ar1 signature and 1.03e−12 for Alox5 signature, respectively). The C3ar1 signature is enriched for inflammatory pathways such as complement pathway and IL-10 signaling as well as metabolic pathways such as fatty acid and amino acid metabolism. Similarly, the Alox5 signature is enriched for lipid/amino acid/fatty acid/steroid metabolic processes as well as inflammatory processes such as T cell/lymphocyte/leukocyte activation. These KO signatures not only support the regulatory role of the key drivers in modulating the inflammatome but also tie the inflammatory genes with other metabolic processes.
In addition, 32 key driver genes are candidate genes for 53 diseases/traits in human GWAS based on NHGRI GWAS catalog (accessed April 28, 2012; Supplementary Table 10), again supporting the notion that these key driver genes impact a variety of human diseases. Therefore, a majority of these genes have literature support for their critical role in mediating multiple disorders.
Comparison of inflammatome signature and key drivers with inflammatory signatures identified in previous studies
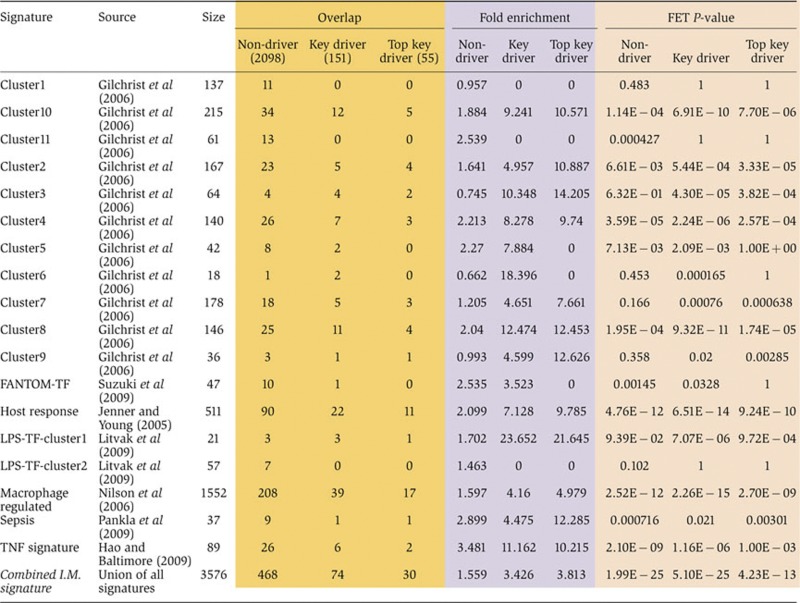
Previously, several studies investigated gene expression patterns in blood or various hematopoietic cell lineages from different inflammatory conditions/diseases and analyzed the potential regulatory transcription factors (Jenner and Young, 2005; Gilchrist et al, 2006; Nilsson et al, 2006; Hao and Baltimore, 2009; Litvak et al, 2009; Pankla et al, 2009; Suzuki et al, 2009). From these publications, we collected 18 inflammatory response gene signatures. These signatures were also combined into a super signature by taking the union over all sets. As shown in Table VII, the inflammatome signature and its key driver list significantly overlap with 15 and 16 of the 19 signatures, respectively. Among the 15 published signatures significantly overlapping the inflammatome signature, 14 have higher fold enrichment for the driver list than for the non-drivers of the inflammatome signature, suggesting a higher cross-study consistency among the key driver genes. Among all the enrichment tests, the driver list shows the highest likelihood of harboring two previously identified groups of macrophage-induced transcription factors, namely, Cluster 6 and LPS-TF-Cluster1 (Gilchrist et al, 2006; Litvak et al, 2009), with fold enrichments of 18.4 and 23.7, respectively. Both groups comprising many transcription factors regulated in the early phase of temporal activation of macrophages and likely control gene expression in the intermediate and late phases (Gilchrist et al, 2006; Litvak et al, 2009), again supporting the cross-study consistency as well as the regulatory power of the inferred drivers as opposed to the non-drivers in the inflammatome signature. In addition, the top 55 key drivers have higher likelihood than the 151 key drivers to be in 11 of the 16 signatures significantly enriched for the key drivers. This and the previous overlap analysis between the key drivers and the MGI phenotype database suggest the importance of the rank order of the predicted drivers.
Table 7. Enrichment test of our inflammatome signature and its drivers for the inflammatory signatures from the literature using Fisher's Exact Test (FET). The combined I.M. signature is a union of all the other signatures reported in this table.
|
Although there is a statistically significant over-representation of these previously identified signatures in the driver and inflammatome lists, 51% or 77 of the predicted key drivers and 74% or 1831 of the inflammatome signature genes from our study are novel, suggesting that this approach is complementary to all the previous studies.
Discussion
In the current analysis, we first identified a representative ‘inflammatome’ signature of roughly 2500 genes from 12 expression data sets covering 9 different tissues from 11 rodent inflammatory disease models. The inflammatome signature is highly enriched with current known drug target and GWAS genes, suggesting that it could be used as a reference gene set for new target identification. We also found that the inflammatome bears close resemblance to the previously identified MEMN, a gene module identified in both human and mouse adipose and liver tissues and enriched for causal genes involved in metabolic disorders (Chen et al, 2008; Emilsson et al, 2008). When mapping the 2500 inflammatome genes to previously constructed gene networks, we found that members of this gene set are highly connected in both Bayesian and gene co-expression networks across multiple tissues of multiple independent data sets. The identification of this ‘inflammatome’ gene signature extends the coverage of MEMN beyond adipose and liver in the metabolic disease to multiple diseases in numerous tissues. The fact that inflammatome signature also highly overlaps with network modules that predict cancer survival suggests that the importance of the signature can be extended to diseases that were not covered by the 11 models investigated here. It is also interesting to note that the inflammatome contains all proposed cancer therapeutic targets (e.g., Hif-1a, Vegf, M-csf, Stat3, Nfkb1, Nfkb2, RelB, Mmp12, and Tgfb) associated with inflammation-related genes proposed in a recent review article by Sica et al (2008).
In addition to helping define the network architecture of the inflammatome signature, the integrative network analysis also identified 151 key regulatory, that is, key driver, genes among the inflammatome genes. Multiple lines of evidence support the importance of these key drivers: (1) enrichment for genes which, when perturbed, cause phenotypic changes in KO mice, (2) experimental validation of a key driver C3ar1, and (3) 32 key drivers have been identified as candidate susceptibility genes for over 50 disease traits in GWAS studies. Among the 151 key drivers, Cd53, Hck, Tyrobp, Nckap1l, and Aif1 are highly consistent key drivers across majority of the causal networks and they have been linked to many inflammation-related disorders. The identification of these key drivers implies a critical role in numerous disease settings and they may serve as key biomarkers or drug targets.
To our knowledge, this is the largest systematic investigation of multiple tissues (9) of multiple disease models (11) with an inflammatory component. Compared with previous studies investigating inflammatory genes in blood or hematopoietic cell lineages in limited sets of inflammatory diseases or conditions, our study identified a significantly larger number of inflammatome genes and key regulators that are not only consistent in multiple tissues but are involved in a much broader spectrum of disease types. In addition, the integration of gene expression profiling data with knowledge-based databases and data-driven networks not only helps identify a common inflammatome signature and relates the signature to the diseases under investigation, but also helps uncover the network architecture and key genes that drive network and disease variance. Since the inflammatome was derived from multiple disease models and tissues, it points to a central role that infiltrating inflammatory cells such as macrophages play in all major disease areas. Several genes of macrophage origin, when perturbed, have been shown to impact multiple disease outcomes. It is conceivable that further mining and validation of the inflammatome signature, especially the key drivers identified, could result in additional high value targets that can be used for multiple inflammatory diseases rather than individual diseases.
It is important to note that the common inflammatome genes and their drivers do not necessarily undermine the significant value of disease-specific signatures for personalized medicine, as commonly practiced in today’s pharmaceutical industry and medical system. Our study indeed identified many disease-specific genes, supporting the existence of unique features in each disease type. Future studies that involve larger sample sizes in each disease model can help further our understanding of disease-specific processes. In light of a comprehensive understanding of the shared as well as the distinct molecular mechanisms, the most effective and personalized treatment of a particular disease could be the combination of a drug targeting the core processes common to multiple diseases and a drug that is more specific to each disease type.
Materials and methods
Rodent inflammatory disease models
As shown in Table I, 12 rodent inflammatory disease models are used in the current study and the tissues involved include lung, aorta, adipose, islet, liver, brain, DRG, skin, and muscle. Gene expression data for all disease models reported in the current study have been deposited to Gene Expression Omnibus under GSE31559.
OVA mouse model for asthma
Five male BALB/c mice (20–25g) are immunized with OVA 1 week apart. Two weeks after the first immunization, mice are subjected to 0.5 × PBS or 5% OVA (in 0.5 × PBS) aerosol challenge for 20 min via whole body exposure for 3 consecutive days. One day after the last aerosol challenge, mice are anesthetized with Ketamine/diazepam/sodium pentobarbital and surgically tracheotomized and jugular vein cannulated before attaching to a small animal ventilator (Flexivent) for invasive lung mechanical responses to intravenous methacholine. Mice are removed from the ventilator and whole lungs are harvested. Right versus left lungs are rinsed in PBS, blotted dry, and immediately frozen in liquid nitrogen for storage at −80°C. Right lung tissues were used for profiling. Total RNA was extracted from left lung using RNeasy Midi kit as described by the manufacturer (Qiagen, Valencia, CA, USA). Samples were treated with DNaseI on-column (Qiagen) for 30 min. RNA concentration was measured using a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, USA) and RNA integrity was determined with a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Samples displaying an RNA integrity number (RIN) >7.5 were used for profiling. Samples from four mice treated with PBS were combined to serve as reference pools and compared with individual lung samples of PBS- and OVA-treated animals. Microarray profiling was conducted as previously described (Lampe et al, 2004), using a mouse custom 44K array comprising 40 572 genes. The two-color microarrays were scanned using the Agilent scanner and proprietary image acquisition software. Rigorous image quality control (QC) was performed using proprietary software and the raw data warehoused in a proprietary database. Experimental QC was performed in MATLAB (Mathworks, Inc. Natick, MA, USA; http://www.mathworks.com). Expression data were loaded into the Resolver (Rosetta proprietary software database http://www.rosettabio.com/products/resolver/default.htm), for transformation normalization and error modeling. Fluor-reversed pairs for each sample were combined to give a single log-ratio and a P-value for technical variability for each biological sample compared with its appropriate control.
IL-1β Tg mouse model for COPD
Lung samples from three IL-1β Tg mice treated with water were used as the reference and compared with five individual lung samples from Tg Dox-treated lungs to obtain the IL-1β Tg-induced signatures. All lung samples were collected 7 days after Dox treatment when active airway inflammation was observed. RNA samples were prepared as described above in the mouse OVA model. Samples were amplified and labeled using a custom automated version of the RT/IVT protocol and reagents provided by Affymetrix. Hybridization, labeling, and scanning were completed following manufacturer’s recommendations (Affymetrix) and microarray profiling was performed by using a mouse Affymetrix custom array containing 44 000 probe sets.
TGFβ Tg mouse model for fibrosis
TGFβ Tg and WT littermate control mice were treated with water or doxycycline (Dox) for 14 days. At day 14, doxycycline-treated transgenic animals manifest both pronounced lung fibrosis and airway inflammation. These end points were measured before profiling samples to ensure that each Dox-treated animal displays a uniform response. Lung samples from four mice per group (TGFβ Tg and WT) were collected. Total RNA purification and microarray profiling were performed in the same way as described above in the mouse OVA model.
ApoE−/− HFD mouse model for atherosclerosis
Three C57BL/6 WT and three ApoE−/− mice (The Jackson Laboratory) were weaned at 4 weeks of age. For gene expression profiling studies, an atherogenic western HFD containing 21% fat and 0.15% cholesterol (88137; Harlan Teklad) was given to both WT and ApoE−/− animals for 16 weeks started when the animals were at 8 weeks of age.
RNA from whole aorta was extracted with Qiagen RNeasy kit (Cat# 74181) according to manufacturer’s recommended protocol. Reverse transcription (RT) was carried out with high capacity cDNA archive kit (Applied Biosystems, Cat# 4322171). For total RNA samples from whole aorta tissue, 500 ng RNA was used for each 100 μl RT reaction. Microarray profiling was performed in the same way as described above in the mouse OVA model.
dbdb mouse model for diabetes (adipose)
Epidydimal white adipose tissue (eWAT) was isolated from six db/db and six lean control (db/+) mice at 10 week of age. The db/db mice are obese, severely hyperglycemic, and only modestly hyperinsulinemic at 10 weeks of age. Total RNA purification and microarray profiling were performed in the same way as described above in the mouse OVA model.
dbdb mouse model for obesity (islet)
Pancreatic islets were isolated from the db/db and the lean control (db/+) mice at 9 weeks of age. The db/db mice are obese, severely hyperglycemic, and only modestly hyperinsulinemic, indicating the de-compensation of β-cell to the insulin resistance. Comparing gene expression profiles in db/db and db/+ islets at this time point allowed us to find genes involved in the β-cell dysfunction during the development of diabetes in this model. Islet RNA samples were purified and profiled from five mice per group essentially the same way as described above in the mouse OVA model.
ob/ob mouse model for obesity
eWAT from nine ob/ob and nine WT mice on C57Bl/6J background at 8 weeks of age was collected and three samples each from WT and ob/ob mice were combined to form three pooled groups each. RNA samples were purified and profiled essentially the same way as described above in the mouse OVA model.
LPS rat model for multiple inflammatory diseases
Four Sprague-Dawley rats received either an intraperitoneal injection of LPS of 3 mg/kg body weight (n=4) or sodium chloride (SC) 0.9% (n=4). Animals were killed 24 h after LPS or SC injection. Total RNA was extracted from liver using RNeasy Midi kit as described by the manufacturer (Qiagen). Samples were treated with DNaseI on-column (Qiagen) for 30 min. RNA concentration was measured using a NanoDrop ND-1000 (NanoDrop Technologies) and RNA integrity was determined with a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Samples displaying an RIN >7.5 were used for profiling. Samples from livers treated with SC were combined to serve as reference pools and compared with individual liver samples of SC- and LPS-treated animals. Microarray profiling was conducted as previously described (Lampe et al, 2004), using a rat Agilent custom array contains ∼44 000 probes.
Transient MCAO rat model for stroke
Brain tissues were collected from four transient MCAO (tMCAO) rats and four controls at 24 h. Selection of optimal time points was primarily based upon the time course of infarct formation (histological data). Brain tissue of 4 mm thick ipsilateral brain section (dissected into cortex and striatum) was collected but only cortex samples were used for expression profiling. RNA purification and microarray profiling were performed essentially the same way as described above for LPS rat model except microarray platform was using a rat custom Agilent array contains ∼25 000 probes.
Chung rat model for neuropathic pain
Rat DRG at 7 days following spinal nerve ligation that elicits neuropathic pain or sham operation was profiled. Four DRG samples per group each pooled from two animals were included in the analysis. RNA purification and microarray profiling were performed essentially the same way as described above for LPS rat model except microarray platform was using a rat custom Agilent array contains ∼25 000 probes.
Carrageenan rat model for inflammation pain
Four rats were treated with carrageenan (CGN) at 30 mg/kg body weight (mpk) and then compared with five control animals without CGN treatment. Three hours post-CGN paw edema and hyperalgesia to mechanical pressure were measured. Rats were euthanized 4 h post-CGN for skin tissue harvesting. Skin RNA samples were purified and QC in the same way as described above in the LPS rat model but microarray profiling was performed using a custom Affymetrix array.
Aged rat model for sarcopenia
Five 24-month-old male Sprague-Dawley rats were obtained and served as the aged group to compare with five 6-month-old male Sprague-Dawley rats which served as young controls. All animals were housed in a 12:12 h light cycle with ad lib food and water for at least a month (age at necropsy was 28 months and 7 months for old and young, respectively). Body composition measures were collected using a rodent magnetic resonance (MR) instrument and grip strength was assessed for eight animals in each group. Animals were euthanized with CO2 and exsanguination and the whole hind limb lateral gastrocnemius muscle from the left leg was dissected and snap frozen in liquid nitrogen. RNA samples were purified and QC in the same way as described above in the LPS rat model but microarray profiling was performed using a custom Affymetrix array.
Inflammatome signature identification
The inflammatome signature was identified by adopting a two-way ANOVA approach using two factors and their interaction term; ‘model’ (12 levels, 12 rodent models) and ‘disease status’ (2 levels, normal and disease samples). In all, 2483 genes with disease P-values of ⩽1.0e−9 (Benjamini–Hochberg corrected) were selected.
Disease-specific signature identification
One-way ANOVA was used to identify differential expression signatures in individual rodent models. FDR was estimated using Q-value approach to control for multiple testing and statistical cutoff was set to FDR<5%. Genes that were only significant in one model were defined as disease-specific genes for the particular model.
Mouse BXH cross study
In a previously described cross between C57BL6/J (B6) and C3H/HeJ (C3H) on an Apoe−/− background (referred to here as the BXH cross) (Wang et al, 2006), muscle, liver, and adipose tissues were extracted from 334 F2 animals in the B3H cross and profiled on an Agilent custom murine gene expression microarray17. All F2 animals were genotyped at >1300 single-nucleotide polymorphism markers and clinically characterized with respect to obesity-, diabetes-, and atherosclerosis-related traits.
Human study populations
For the NKI breast cancer cohort (van de Vijver et al, 2002), about 300 cancer breast samples were collected and profiled on the Agilent Human 25K platform comprising 24 496 non-control oligonucletoide probes. For details about tissue collection and RNA/DNA isolation, RNA sample preparation, microarray hybridization, and expression analysis, see the original paper (van de Vijver et al, 2002). The gene expression data were adjusted for estrogen and progesterone receptor (ER/PR) status as well as age to avoid their influence.
For the IFA cohort, 673 Icelandic subjects ranging in age from 18 to 85 years were recruited and of these 553 formed 124 dense three-generation pedigrees. Individuals were recruited so as to maximize the density for any of the given pedigrees. All participants in the IFA were scored for various clinical traits related to obesity, including height, weight, waist circumference, hip circumference, and percentage body fat (PBF) measured by bioimpedance. For details about tissue collection and RNA/DNA isolation, RNA sample preparation, microarray hybridization, and expression analysis, see our previous paper (Emilsson et al, 2008).
For the HLC, a total of 427 liver samples (1–2 g) were acquired from Caucasian individuals from three independent liver collections at tissue resource centers at Vanderbilt University, the University of Pittsburgh, and Merck Research Laboratories. The Vanderbilt samples (N=504) included both postmortem tissue and surgical resections from organ donors and were obtained from the Nashville Regional Organ Procurement Agency (Nashville, TN), the National Disease Research Interchange (Philadelphia, PA), and the Cooperative Human Tissue Network (University of Pennsylvania, Ohio State University, and University of Alabama at Birmingham). The Pittsburgh samples were normal postmortem human liver and were obtained through the Liver Tissue Procurement and Distribution System (Dr. Stephen Strom, University of Pittsburgh, Pittsburgh, PA). The University of Pittsburgh samples (N=211) were all postmortem, as were the Merck samples (N=65) collected by the Drug Metabolism Department and reported previously1. All samples were stored frozen at −80°C from collection until processing for RNA and DNA; some samples had been stored for over a decade before being processed for this study. Demographic data varied across centers for these samples and were missing in many cases. In cases where age, gender, or ethnicity data were not available in the patient records, we imputed it from the gene expression and/or genotype data (see below). For more details about tissue collection and RNA/DNA isolation, RNA sample preparation, microarray hybridization, and expression analysis, see our previous paper (Schadt et al, 2008).
For the human hepatocellular carcinoma (HCC) cohort, ∼250 matched tumor and adjacent normal samples were collected from HCC patients during surgical resection and assess (Burchard et al, 2010). Demographic and pathologic parameters for the 272 ethnic Chinese HCC patients who received curative surgery and used in this study are shown (see Supplementary Table 1). Half of the patients (51.1%) suffered from tumor recurrence during the follow-up period. The primary end points measured were overall survival, DFS, and tumor stage (pTNM). Flash frozen tissue was placed in a chilled milling tube along with a stainless steel bead, dipped in a liquid nitrogen bath and loaded onto the QIAGEN TissueLyser for milling (30 Hz in 30 s intervals). Multiple cycles of milling were sometimes required to achieve complete pulverization of the tissue to a fine powder. After milling, the tissue powder was recovered and rapidly manually split for extraction DNA and RNA while all the time maintaining sub-zero temperatures. Samples were amplified and labeled using a custom automated version of the RT/IVT protocol and reagents provided by Affymetrix. Hybridization, labeling, and scanning were completed following manufacturer’s recommendations (Affymetrix). Sample amplification, labeling, and microarray processing were performed by the Rosetta Inpharmatics Gene Expression Laboratory in Seattle, WA.
Gene co-expression network analysis
The weighted network analysis begins with a matrix of the Pearson correlations between all gene pairs, then converts the correlation matrix into an adjacency matrix using a power function f(x)=xˆβ. The parameter β of the power function is determined in such a way that the resulting adjacency matrix (i.e., the weighted co-expression network) is approximately scale free. To measure how well a network satisfies a scale-free topology, we use the fitting index (Zhang and Horvath, 2005) (i.e., the model fitting index R2 of the linear model that regresses log(p(k)) on log(k) where k is connectivity and p(k) is the frequency distribution of connectivity). The fitting index of a perfect scale-free network is 1. For each data set, we select the smallest β that leads to an approximately scale-free network. The distribution p(k) of the resulting network approximates a power law: 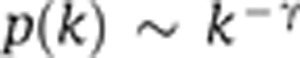 .
.
To explore the modular structures of the co-expression network, the adjacency matrix is further transformed into a TOM (Zhang and Horvath, 2005). As the topological overlap between two genes reflects not only their direct interaction, but also their indirect interactions through all the other genes in the network, previous studies (Ravasz et al, 2002; Zhang and Horvath, 2005) have shown that topological overlap leads to more cohesive and biologically meaningful modules.
To identify modules of highly co-regulated genes, we used average linkage hierarchical clustering to group genes based on the topological overlap of their connectivity, followed by a dynamic cut-tree algorithm to dynamically cut clustering dendrogram branches into gene modules (Langfelder et al, 2007). To distinguish between modules, each module was assigned a unique color identifier, with the remaining, poorly connected genes colored gray. The whole procedure leads to an ordered TOM heat map in which the rows and the columns represent genes in a symmetric manner, and the color intensity represents the interaction strength between genes. This connectivity map highlights that genes in a transcriptional network fall into distinct network modules, where genes within a given module are more interconnected with each other (blocks along the diagonal of the matrix) than with genes in other modules. There are a couple of network connectivity measures, but one particularly important one is the within module connectivity (k.in). The k.in of a gene was determined by taking the sum of its connection strengths (co-expression similarity) with all other genes in the module that to which the gene belonged.
Construction of BNs
Recent progress on reconstruction of gene regulatory networks has shown that integration of multiple sources of genetic data can lead to gene causal networks predictive of complex phenotypes (Zhu et al, 2004, 2008; Mehrabian et al, 2005; Schadt et al, 2005; Kulp and Jagalur, 2006; Lum et al, 2006). Among a variety of approaches, BNs have shown superior performance. BN is a probabilistic representation of the gene regulatory network. In all, 6312, 6349, 6268, and 5802 differentially regulated genes for NKI (van 't Veer et al, 2002), Wang (Wang et al, 2005), Miller (Miller et al, 2005), and Christos (Sotiriou et al, 2006), respectively, were provided as input into a BN reconstruction software program based on a previously described algorithm (Zhu et al, 2004). Searching optimal BN structures given a data set is an NP-hard problem. We employed an MCMC method to do local search of optimal structures. As the method is stochastic, the resulted structure will be different from each run. In our process, 1000 BNs were reconstructed using different random seeds to start the stochastic reconstruction process. For each seed, 15 × n2 iterations of MCMC were run on average, where n is the number of nodes. The Bayesian Information Criterion (BIC) scores were used as the optimization criteria. From the resulting set of 1000 networks generated by this process, edges that appeared in >30% of the networks were used to define a consensus network. The 30% cutoff threshold for edge inclusion is based on our previous simulation study (Zhu et al, 2007), where 30% cutoff yields the best tradeoff between recall rate and precision. The consensus network resulted by averaging may not be a BN (a directed acyclic graph) any more. To make the consensus network structure into a directed acyclic graph, edges in this consensus network were removed if and only if (1) the edge was involved in a loop and (2) the edge was the most weakly supported of all edges making up the loop.
Assessing biological importance of BN key drivers via mutant phenotypes
The mutant phenotype data were downloaded from the MGI database ( ftp://ftp.informatics.jax.org/pub/reports/index.html#pheno) and genes within mutant alleles in the database were annotated based on the key driver analysis results. The frequency of genes with mutant phenotypes in each of the three groups, namely, key drivers, local drivers, and non-drivers, was calculated. One-sided Fisher's exact was used to test enrichment of genes that showed mutant phenotypes in each group compared with all genes with mutant alleles in the database. One-sided proportion test was used to compare the frequency of genes with mutant phenotypes between key drivers, local drivers, and non-drivers. Significance level was set to P<0.05.
Identification of liver gene expression signatures of C3ar1 and Alox5 KO mice
The gene expression profiling experiments for C3ar1 and Alox5 have been described previously (Mehrabian et al, 2005; Yang et al, 2009). Briefly, the liver tissues of three C3ar1 KO, four littermate C3ar1 WT mice, five Alox5 KO, and five littermate Alox5 WT mice were profiled with Rosetta/Merck Mouse TOE 75k Array 1 microarray (GPL3562) manufactured by Agilent Technologies (Palo Alto, CA). The array consists of 23 574 non-control oligonucleotides extracted from mouse Unigene clusters and combined with RefSeq sequences and RIKEN full-length cDNA clones. The expression data have been deposited to GEO under an accession number GSE31559, respectively. A two-sided Student’s T-test was used to identify signature genes with significant differences between KO animals and the WT control mice.
Supplementary Material
Acknowledgments
We thank Amit Kulkarni for assistance with data upload.
Author contributions: IW, BZ, and XY conceived the framework, analyzed the data, and wrote the manuscript, with critical input from EES. SS, JZ, CZ, MP, YH, CN, JY, PYL, JL, EES, HD, and CR participated in data analysis. DS, MAC, HH, CMT, EA, GO, MJL, RT, CC, UB, and HAW provided rodent animal modes. All authors read, edited, and approved the final manuscript.
Footnotes
IW, CZ, DS, MAC, HH, CMT, EA, GO, RT, JY, CC, HAW, HD, and CR are current employees at Merck and own Merck stocks. Merck Sharp and Dohme is a corporation involved in the research, development, manufacturing, and commercialization of ethical pharmaceuticals. This includes developing drugs targeting inflammation, cancer, and atherosclerosis. The remaining authors declare that they have no conflict of interest.
References
- Alkassab F, Gourh P, Tank FK, McNearney T, Fischbach M, Ahn C, Arnett FC, Mayes MD (2007) An allograft inflammatory factor 1 (AIF1) single nucleotide polymorphism (SNP) is associated with anticentromere antibody positive systemic sclerosis. Rheumatology (Oxford) 46: 1248–1251 [DOI] [PubMed] [Google Scholar]
- Arvanitis DA, Flouris GA, Spandidos DA (2005) Genomic rearrangements on VCAM1, SELE, APEG1 and AIF1 loci in atherosclerosis. J Cell Mol Med 9: 153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviram M (2011) Atherosclerosis: cell biology and lipoproteins - inflammation and oxidative stress in atherogenesis: protective role for paraoxonases. Curr Opin Lipidol 22: 243–244 [DOI] [PubMed] [Google Scholar]
- Barrena S, Almeida J, Yunta M, López A, Fernández-Mosteirín N, Giralt M, Romero M, Perdiquer L, Delgado M, Orfao A, Lazo PA (2005) Aberrant expression of tetraspanin molecules in B-cell chronic lymphoproliferative disorders and its correlation with normal B-cell maturation. Leukemia 19: 1376–1383 [DOI] [PubMed] [Google Scholar]
- Benbir G, Ince B, Kumral E, Ongen Z, Kultursay H, Tokgozoglu L, Oto A, Tuzun H ARDA Investigators. (2011) Antihypertensive drugs and inflammation in acute ischemic stroke as a predictor factor of future cardiovascular mortality. Inflammation 35: 65–73 [DOI] [PubMed] [Google Scholar]
- Bergman M, Djaldetti M, Salman H, Bessler H (2011) Inflammation and colorectal cancer: does aspirin affect the interaction between cancer and immune cells? Inflammation 34: 22–28 [DOI] [PubMed] [Google Scholar]
- Burchard J, Zhang C, AM LIU, Poon RT, Lee NP, Wong KF, Sham PC, Lam BY, Feruson MD, Tokiwa G, Smirth R, Leeson B, Beard R, Lamb JR, Lim L, Mao M, Dai H, Luk JM (2010) MicroRNA-122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol Syst Biol 6: 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrum RS, Goulet JL, Griffiths RJ, Koller BH (1997) Role of the 5-lipoxygenase-activating protein (FLAP) in murine acute inflammatory responses. J Exp Med 185: 1065–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, Leonardson A, Castellini LW, Wang S, Champy M-F, Zhang B, Emilsson V, Doss S, Ghazalpour A, Horvath S, Drake T et al. (2008) Variations in DNA elucidate molecular networks that cause disease. Nature 452: 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandona P, Aljada A, Bandyopadhyay A (2004) Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 25: 4–7 [DOI] [PubMed] [Google Scholar]
- Das A, Mukhopadhyay S (2011) The evil axis of obesity, inflammation and type-2 diabetes. Endocr Metab Immune Disord Drug Targets 11: 23–31 [DOI] [PubMed] [Google Scholar]
- Drake C, Boutin H, JOnes MS, Denes A, McColl BW, Selvarajah JR, Hulme S, Georgiou RF, Hinz R, Gerhard A, Vail A, Prenant C, Julyan P, Maory R, Brown G, Simgova A, Herholz K, Kassio9u M, Crossman D, Francis S et al. (2011) Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain Behav Immun 25: 1113–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eike MC, Olsson M, Undlien DE, Dahl-Jørgensen K, Joner G, Rønningen KS, Thorsby E, Lie BA (2009) Genetic variants of the HLA-A, HLA-B and AIF1 loci show independent associations with type 1 diabetes in Norwegian families.. Genes Immun 10: 141–150 [DOI] [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, Mouy M, Steinthorsdottir V, Eiriksdottir GH, Bjornsdottir G, Reynisdottir I, Gudbjartsson D, Lelgadottir A, Jonasdottir A, Onasdottir A, Styrkarsdottir U et al. (2008) Genetics of gene expression and its effect on disease. Nature 452: 423–428 [DOI] [PubMed] [Google Scholar]
- Fox JG, Wang TC (2007) Inflammation, atrophy, and gastric cancer. J Clin Invest 117: 60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N, Linial M, Machman I, Pe'er D (2000) Using Bayesian networks to analyze expression data. J Comput Biol 7: 601–620 [DOI] [PubMed] [Google Scholar]
- Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang W-P, He A, Truong A, Patel S, Nelson SF, Horvath S, Berliner JA, Kirchgessner TG, Lusis AJ (2006) Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA 103: 12741–12746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Kennedy K, Hai T, Bolouri H, Aderem A (2006) Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441: 173–178 [DOI] [PubMed] [Google Scholar]
- Grange JM, Krone B, Mastrangelo G (2011) Infection, inflammation and cancer. Int J Cancer 128: 2240–2241 [DOI] [PubMed] [Google Scholar]
- Gurtsevitch VE (2008) Human oncogenic viruses: hepatitis B and hepatitis C viruses and their role in hepatocarcinogenesis. Biochemistry (Mosc) 73: 504–513 [DOI] [PubMed] [Google Scholar]
- Hansson GK, Libby P (2006) The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol 6: 508–519 [DOI] [PubMed] [Google Scholar]
- Hao S, Baltimore D (2009) The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol 10: 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harney SM, Vilariño-Guell C, Adamopoulos IE, Sims AM, Lawrence RW, Cardon LR, Newton JL, Meisel C, Pointon JJ, Darke C, Athanasou N, Wordsworth BP, Brown MA (2008) Fine mapping of the MHC Class III region demonstrates association of AIF1 and rheumatoid arthritis. Rheumatology (Oxford) 47: 1761–1767 [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Tamari M, Shao C, Shimizu M, Takahashi N, Mao XQ, Yamasaki A, Kamada F, Doi S, Fujiwara H, Miyatake A, Fujita K, Tamura G, Matsubara Y, Shirakawa T, Suzuki Y (2004) Variations in the C3, C3a receptor, and C5 genes affect susceptibility to bronchial asthma. Hum Genet 115: 295–301 [DOI] [PubMed] [Google Scholar]
- Heber D, Carpenter CL (2011) Addictive genes and the relationship to obesity and inflammation. Mol Neurobiol 44: 160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess K, Grant PJ (2011) Inflammation and thrombosis in diabetes. Thromb Haemost Suppl 1: S43–S54 [DOI] [PubMed] [Google Scholar]
- Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, Lee Y, Scheck AC, Liau LM, Wu H, Geschwind DH, Febbo PG, Kornblum HI, Cloughesy TF, Nelson SF, Mischel PS (2006) Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci USA 103: 17402–17407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95: 2409–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259: 87–91 [DOI] [PubMed] [Google Scholar]
- Jawien J, Gajda M, Rudling M, Mateuszuk L, Olszanecki R, Guzik TJ, Cichocki T, Cholpicki S, Korbut R (2006) Inhibition of five lipoxygenase activating protein (FLAP) by MK-886 decreases atherosclerosis in apoE/LDLR-double knockout mice. Eur J Clin Invest 36: 141–146 [DOI] [PubMed] [Google Scholar]
- Jenner RG, Young RA (2005) Insights into host responses against pathogens from transcriptional profiling. Nat Rev Microbiol 3: 281–294 [DOI] [PubMed] [Google Scholar]
- Jensen GL (2008) Inflammation: roles in aging and sarcopenia. JPEN J Parenter Enteral Nutr 32: 656–659 [DOI] [PubMed] [Google Scholar]
- Jockers JJ, Novak N (2006) Different expression of adhesion molecules and tetraspanins of monocytes of patients with atopic eczema. Allergy 61: 1419–1422 [DOI] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasaz K-i, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashir K, Kasuga M (2006) MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116: 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Ueki K, Okazaki Y, Iwane A, Kubota N, Ohsugi M, Awazawa M, Kobayashi M, Sasako T, Kaneko K, Suzuki M, Nishikawa Y, Hara K, Yoshimura K, Koshima I, Goyama S, Murakami K, Sasaki J, Nagai R, Kurokawa M et al. (2011) Blockade of class IB phosphoinositide-3 kinase ameliorates obesity-induced inflammation and insulin resistance. Proc Natl Acad Sci USA 108: 5753–5758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp DC, Jagalur M (2006) Causal inference of regulator-target pairs by gene mapping of expression phenotypes. BMC Genomics 7: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe JW, Stepaniants SB, Mao M, Radich JP, Dai H, Linsley PS, Sh Friend, Potter JD (2004) Signatures of environmental exposures using peripheral leukocyte gene expression: tobacco smoke. Cancer Epidemiol Biomarkers Prev 13: 445–453 [PubMed] [Google Scholar]
- Langfelder P, Zhang B, Horvath S (2008) Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut library for R. Bioinformatics 24: 719–720 [DOI] [PubMed] [Google Scholar]
- Lee F, Fandi A, Voi M (2008) Overcoming kinase resistance in chronic myeloid leukemia. Int J Biochem Cell Biol 40: 334–343 [DOI] [PubMed] [Google Scholar]
- Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, Lampano AE, Nykter M, Shmulevich Aderem A. (2009) Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat Immunol 10: 437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum PY, Chen Y, Zhu J, Lamb J, Melmed S, Wang S, Drake TA, Lusis AJ, Schadt EE (2006) Elucidating the murine brain transcriptional network in a segregating mouse population to identify core functional modules for obesity and diabetes. J Neurochem 97(Suppl 1): 50–62 [DOI] [PubMed] [Google Scholar]
- Mamane Y, Chung Chan C, Lavallee G, Morin N, Xu LJ, Huang J, Gordon R, Thomas W, Lamb J, Schadt EE, Kennedy BP, Mancini JA (2009) The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes 58: 2006–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454: 436–444 [DOI] [PubMed] [Google Scholar]
- Mayburd AL, Martlinez A, Sackett D, Liu H, Shih J, Tauler J, Avis I, Mulslhine JL (2006) Ingenuity network-assisted transcription profiling: Identification of a new pharmacologic mechanism for MK886. Clin Cancer Res 12: 1820–1827 [DOI] [PubMed] [Google Scholar]
- Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454: 428–435 [DOI] [PubMed] [Google Scholar]
- Mehrabian M, Allayee H, Stockton J, Lum PY, Drake TA, Castellani LW, Suh M, Armour C, Edwards S, Lamb J, Lusis AJ, Schadt EE (2005) Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat Genet 37: 1224–1233 [DOI] [PubMed] [Google Scholar]
- Meng SJ, Yu LJ (2010) Oxidative stress, molecular inflammation and sarcopenia. Int J Mol Sci 11: 1509–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LD, Smeds J, Geroge J, Vega VB, Bergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J (2005) An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA 102: 13550–13555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Jacob RF, Mason RP (2011) Inflammation and the development of atherosclerosis: effects of lipid-lowering therapy. J Atheroscler Thromb 18: 351–358 [DOI] [PubMed] [Google Scholar]
- Muoio DM, Newgard CB (2008) Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9: 193–205 [DOI] [PubMed] [Google Scholar]
- Nakase T, Yamazaki T, Ogura N, Suzuki A, Nagata K (2008) The impact of inflammation on the pathogenesis and prognosis of ischemic stroke. J Neurol Sci 271: 104–109 [DOI] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM (2008) AMPK and PPARdelta agonists are exercise mimetics. Cell 134: 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson R, Bajic VB, Suzuki H, Di Bernardo D, Björkegren J, Katayama S, Reid JF, Sweet MJ, Gariboldi M, Carninci P, Hayashizaki Y, Hume DA, Tegner J, Ravasi T (2006) Transcriptional network dynamics in macrophage activation. Genomics 88: 133–142 [DOI] [PubMed] [Google Scholar]
- O'Hanlon TP, Rider LG, Gan L, Fannin R, Paules RS, Umbach DM, Weinberg CR, Shah RR, Mav D, Gourley MF, Miller FW (2011) Gene expression profiles from discordant monozygotic twins suggest that molecular pathways are shared among multiple systemic autoimmune diseases. Arthritis Res Ther 13: R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA (2006) Targeting signal transduction as a strategy to treat inflammatory diseases. Nat Rev Drug Discov 5: 549–563 [DOI] [PubMed] [Google Scholar]
- Paloneva J, Kestila M, Wu J, Salminen A, Böhling T, Ruotsalainen V, Hakola P, Bakker AB, Phillips JH, Pekkarinen P, Lanier LL, Timonen T, Peltonen L (2000) Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet 25: 357–361 [DOI] [PubMed] [Google Scholar]
- Pankla R, Buddhisa S, Berry M, Blankenship DM, Bancroft GJ, Banchereau J, Lertmemongkolchai G, Chaussabel D (2009) Genomic transcriptional profiling identifies a candidate blood biomarker signature for the diagnosis of septicemic melioidosis. Genome Biol 10: R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen-Lane JH, Zurier RB, Lawrence DA (2007) Analysis of the thiol status of peripheral blood leukocytes in rheumatoid arthritis patients. J Leukoc Biol 81: 934–941 [DOI] [PubMed] [Google Scholar]
- Pennings JLA, Kimman TG, Janssen R (2008) Identification of a common gene expression response in different lung inflammatory diseases in rodents and macaques. PLoS ONE 3: e2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentikainen MO, Oorni K, Ala-Korpela M, Kovanen PT (2000) Modified LDL - trigger of atherosclerosis and inflammation in the arterial intima. J Intern Med 247: 359–370 [DOI] [PubMed] [Google Scholar]
- Porta C, Riboldi E, Sica A (2011) Mechanisms linking pathogens-associated inflammation and cancer. Cancer Lett 305: 250–262 [DOI] [PubMed] [Google Scholar]
- Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási A-L (2002) Hierarchical organization of modularity in metabolic networks. Science 297: 1551–1555 [DOI] [PubMed] [Google Scholar]
- Rhodes JM (1996) Unifying hypothesis for inflammatory bowel disease and associated colon cancer: sticking the pieces together with sugar. Lancet 347: 40–44 [DOI] [PubMed] [Google Scholar]
- Richter EA, Ruderman NB (2009) AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J 418: 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal AK (2011) Crystals, inflammation, and osteoarthritis. Curr Opin Rheumatol 23: 170–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskas K, Kouvatsi A, Paletas K, Papazoglou D, Tsapas A, Lobbens S, Vatin V, Durand E, Labrune Y, Delplanque J, Meyre D, Froquel P (2012) Common variants in FTO, MC4R, TMEM18, PRL, AIF1, and PCSK1 show evidence of association with adult obesity in the Greek population. Obesity (Silver Spring) 20: 389–395 [DOI] [PubMed] [Google Scholar]
- Sansone P, Bromberg J (2011) Environment, inflammation, and cancer. Curr Opin Genet Dev 21: 80–85 [DOI] [PubMed] [Google Scholar]
- Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, Guhathakurta D, Sieberts SK, Monks S, Reitman M, Zhang C, Lum PY, Leonardson A, Thieringer R, Metzger JM, Yang L, Castle J, Zhu H, Kash SF, Drake TA, Sachs A et al. (2005) An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet 37: 710–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, Zhu J, Millstein J, Sieberts S, Lamb J, GuhaThakurta D, Derry J, Storey JD, Avila-Campillo I, Kruger MJ, Johnson JM et al. (2008) Mapping the genetic architecture of gene expression in human liver. PLoS Biol 6: e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Allavena P, Mantovani A (2008) Cancer related inflammation: the macrophage connection. Cancer Lett 267: 204–215 [DOI] [PubMed] [Google Scholar]
- Snapper SB, Rosen FS (1999) The Wiskott-Aldrich syndrome protein (WASP): roles in signaling and cytoskeletal organization. Annu Rev Immunol 17: 905–929 [DOI] [PubMed] [Google Scholar]
- Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, Desmedt C, Larsimont D, Cardoso F, Peterse H, Kuyten D, Buyse M, Van de Vijver MJ, Bergh J, Piccart M, Delorernzi M (2006) Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst 98: 262–272 [DOI] [PubMed] [Google Scholar]
- Suzuki H, Forrest AR, van Nimwegen E, Daub CO, Balwierz PJ, Irvine KM, Lassmann T, Ravasi T, Hasegawa Y, de Hoon MJ, Katayama S, Schroder K, Carninci P, Tomaru Y, Kanamori-Katayama M, Kubosaki A, Akalin A, Ando Y, Arner E, Asada M et al. (2009) The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat Genet 41: 553–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor ML, Noble PW, White B, Wise R, Liu MC, Bochner BS (2000) Extensive surface phenotyping of alveolar macrophages in interstitial lung disease. Clin Immunol 94: 33–41 [DOI] [PubMed] [Google Scholar]
- Thrash JC, Torbett BE, Carson MJ (2009) Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu-Hakola disease. Neurochem Res 34: 38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran LM, Zhang B, Zhang Z, Zhang C, Xie T, Lamb JR, Dai H, Schadt EE, Zhu J (2011) Inferring causal genomic alterations in breast cancer using gene expression data. BMC Syst Biol 5: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van 't Veer LJ, Dai H, van de Jijver MJ, He YD, Hart AAM, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415: 530–536 [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH et al. (2002) A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347: 1999–2009 [DOI] [PubMed] [Google Scholar]
- Van Hauwermeiren F, Vandenbroucke RE, Libert C (2011) Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev 22: 311–319 [DOI] [PubMed] [Google Scholar]
- Wang S, Yehya N, Schadt EE, Wang H, Drake TA, Lusis AJ (2006) Genetic and genomic analysis of a fat mass trait with complex inheritance reveals marked sex specificity. PLoS Genet 2: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timemrmans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA (2005) Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 365: 671–679 [DOI] [PubMed] [Google Scholar]
- Wieland HA, Michaelis M, Kirschbaum BJ, Rudolphi KA (2005) Osteoarthritis - an untreatable disease? Nat Rev Drug Discov 4: 331–344 [DOI] [PubMed] [Google Scholar]
- Yanagisawa S, Sugiura H, Yokoyama T, Yamagata T, Ichikawa T, Akamatsu K, Koarai A, Hirano t, Nakanishi M, Matsunaga K, Minakata Y, Ichinose M (2009) The possible role of hematopoietic cell kinase in the pathophysiology of COPD. Chest 135: 94–101 [DOI] [PubMed] [Google Scholar]
- Yang X, Deignan JL, Qi H, Zhu J, Qian S, Zhong J, Torosyan G, Majid S, Falkard B, Kleinhanz RR, Karlsson J, Castellani LW, Mumick S, Wang K, Xie T, Coon M, Zhang C, Estrada-Smith D, Farer CR, Wang SS et al. (2009) Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet 41: 415–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Peterson L, Thieringer R, Deignan JL, Wang X, Zhu J, Wang S, Zhong H, Stepaniants S, Beaulaurier J, Wang IM, Rosa R, Cumiskev AM, Luo JM, Luo Q, Shah K, Xiao J, Nickle D, Plump A, Schadt EE et al. (2010) Identification and validation of genes affecting aortic lesions in mice. J Clin Invest 120: 2414–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhang B, Molony C, Chudin E, Hao K, Zhu J, Gaedigk A, Suver C, Zhong H, Leeder JS, Guengerich FP, Strom SC, Schuetz E, Rushmore TH, Ulrich RG, Slatter JG, Schadt EE, Kasarskis A, Lum PY (2010) Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res 10: 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakai NA, Lange L, Longstreth WT Jr, O'Meara ES, Kelley JL, Fornage M, Nikerson D, Cushman M, Reiner AP (2011) Association of coagulation-related and inflammation-related genes and factor VIIc levels with stroke: the Cardiovascular Health Study. J Thromb Haemost 9: 267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S (2005) A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4: Article17 [DOI] [PubMed] [Google Scholar]
- Zhang X, Mahmudi-Azer S, Connett JE, Anthonisen NR, He JQ, Paré PD, Sandford AJ (2007) Association of Hck genetic polymorphisms with gene expression and COPD. Hum Genet 120: 681–690 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Pointon JJ, Wordsworth BP (2008) Comment on: An allograft inflammatory factor 1 (AIF1) single nucleotide polymorphism (SNP) is associated with anticentromere antibody positive systemic sclerosis. Rheumatology (Oxford) 47: 556–557author reply 557 [DOI] [PubMed] [Google Scholar]
- Zhu J, Lum PY, Lamb J, GuhaThakurta D, Edwards SW, Thieringer R, Berger JP, Wu MS, Thompson J, Sachs AB, Schadt EE (2004) An integrative genomics approach to the reconstruction of gene networks in segregating populations. Cytogenet Genome Res 105: 363–374 [DOI] [PubMed] [Google Scholar]
- Zhu J, Sova P, Xu Q, Dombek KM, Xu EY, Vu H, Tu Z, Brem RB, Bumgarner RE, Schadt EE (2012) Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol 10: e1001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Wiener MC, Zhang C, Fridman A, Minch E, Lum PY, Sachs JR, Schadt EE (2007) Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLoS Comput Biol 3: e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang B, Schadt EE (2008) A systems biology approach to drug discovery. Adv Genet 60: 603–635 [DOI] [PubMed] [Google Scholar]
- Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, Bumgarner RE, Schadt EE (2008) Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet 40: 854–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanovic S, Rackov LP, Zivanovic A, Jevtic M, Nikolic S, Kocic S (2011) Cartilage Oligomeric Matrix Protein - inflammation biomarker in knee osteoarthritis. Bosn J Basic Med Sci 11: 27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.