

Abstract
PCR ribotyping is the most commonly used Clostridium difficile genotyping method, but its utility is limited by lack of standardization. In this study, we analyzed four published whole genomes and tested an international collection of 21 well-characterized C. difficile ribotype 027 isolates as the basis for comparison of two capillary gel electrophoresis (CGE)-based ribotyping methods. There were unexpected differences between the 16S-23S rRNA intergenic spacer region (ISR) allelic profiles of the four ribotype 027 genomes, but six bands were identified in all four and a seventh in three genomes. All seven bands and another, not identified in any of the whole genomes, were found in all 21 isolates. We compared sequencer-based CGE (SCGE) with three different primer pairs to the Qiagen QIAxcel CGE (QCGE) platform. Deviations from individual reference/consensus band sizes were smaller for SCGE (0 to 0.2 bp) than for QCGE (4.2 to 9.5 bp). Compared with QCGE, SCGE more readily distinguished bands of similar length (more discriminatory), detected bands of larger size and lower intensity (more sensitive), and assigned band sizes more accurately and reproducibly, making it more suitable for standardization. Specifically, QCGE failed to identify the largest ISR amplicon. Based on several criteria, we recommend the primer set 16S-USA/23S-USA for use in a proposed standard SCGE method. Similar differences between SCGE and QCGE were found on testing of 14 isolates of four other C. difficile ribotypes. Based on our results, ISR profiles based on accurate sequencer-based band lengths would be preferable to agarose gel-based banding patterns for the assignment of ribotypes.
INTRODUCTION
Clostridium difficile is the most frequently identified cause of hospital-acquired diarrhea and antibiotic-associated pseudomembranous colitis. The emergence and international spread of the virulent ribotype 027/pulsed-field gel electrophoresis (PFGE) type NAP1/restriction endonuclease analysis (REA) type B1 has raised the importance of accurate strain typing in infection control. PCR ribotyping is a commonly used typing method for C. difficile that was first described in 1993 (3) and introduced as a routine method in 1996 (11). It is based on variations in sequence lengths (mainly) and copy numbers of 16S-23S rRNA intergenic spacer regions (ISRs) (2, 7, 16).
Conventional agarose gel-based PCR ribotyping is still commonly used but has a number of limitations. The lack of a standardized type strain collection has led to the development of different ribotyping nomenclatures (1, 7, 13, 14, 16), analysis of band sizes is somewhat subjective because of poor resolution, which makes interlaboratory comparison of results unreliable (7), and heteroduplex DNA artifacts are formed, as noticed by authors of previous studies (4, 6, 7). These limitations can make it difficult to distinguish closely related ribotypes with only one or two band differences, such as the newly described ribotypes 176, 198, and 244, which are closely related to and easily mistaken for ribotype 027 (18).
Indra et al. (7) described the use of sequencer-based capillary gel electrophoresis (SCGE) as an alternative to conventional gel electrophoresis, using 5′-end fluorescein-labeled primers and the AB 310 Genetic Analyzer (Applied Biosystems, Carlsbad, USA) with a 41-cm capillary loaded with a POP4 gel. This method was highly reproducible (the standard deviation of peak sizes was ±0.5 bp), independent of the reagents used, and had much higher discriminatory power than conventional agarose gel electrophoresis. More accurate assignment of band sizes makes interlaboratory comparisons of PCR ribotyping results more reliable (7). Recently, an automated commercial CGE method, QIAxcel (Qiagen, Hilden, Germany), which does not require the use of fluorescein-labeled primers, has become available for C. difficile PCR ribotyping (http://www.qiagen.com/literature/render.aspx?id=104730).
Considerable variation in patterns can occur within some individual PCR ribotypes (7), which sometimes makes reliable differentiation difficult, depending on the criteria used. In addition, several different primer pairs have been used for C. difficile PCR ribotyping (1, 13, 16), but their performance has not been compared.
The aim of this study was to analyze published whole-genome sequences in silico, using three different primer sets (“virtual ribotyping”) to accurately determine the numbers and theoretical sizes of ISR bands, as a solid basis for standardization of C. difficile PCR ribotyping and comparison of the accuracy and reproducibility of two different methods based on CGE. We used ribotype 027 as a convenient example, because of its international importance and because there are four published whole-genome sequences and we have access to an international panel of 21 well-characterized isolates (8).
MATERIALS AND METHODS
C. difficile reference panel and DNA preparation.
An international panel of 21 previously well-characterized PCR ribotype 027 isolates was provided by Brandi Limbago, Centers for Disease Control and Prevention, Atlanta, GA (8). All isolates were retrieved from storage by subculture on blood agar plates (Columbia II agar base supplemented with 5% horse blood) and incubated at 37°C under anaerobic conditions for 48 h. One to five colonies were picked from the plates, suspended in 200 μl of molecular biology-grade water, and heated at 100°C for 15 min. The suspension was centrifuged for 5 min at 16,800 × g, and the supernatant was used immediately for PCR or stored at −20°C.
Another 14 C. difficile isolates, belonging to ribotypes 001 (seven isolates), 002 (three isolates), 014, and 015 (two isolates each) from the same well-characterized reference set (8), were also used in subsequent testing and processed as described above.
Amplification of the intergenic sequence region.
The primers used in this study, their combinations, and their labeling are shown in Table 1. We chose three of the six published pairs of primers to evaluate their performance and to test the reliability and comparability of two CGE methods. The primer pairs chosen for SCGE were two that have been most commonly used internationally, namely, 16S-UK/23S-UK (16) and 16S-USA/23S-USA (1, 7), and another pair, 16S-UK/23S-AU (13, 16), which produce a relatively long amplicon. The forward primers (16S-UK and 16S-USA) were carboxyfluorescein (FAM)-labeled at the 5′ end. Primers used for QCGE (16S-UK/23S-AU) were unlabeled (Table 1).
Table 1.
Published primers for amplification of C. difficile ISRs
| Primer | Gene target | GenBank accession no. | Sequence (5′–3′)e | Tm (°C) |
|---|---|---|---|---|
| 16S-USAa,d | 16S rRNA gene | FN545816 | 12293 GTGCGGCTGGATCACCTCCT 12312 | 71.0 |
| 16S-UKb,d | 16S rRNA gene | FN545816 | 12256 CTGGGGTGAAGTCGTAACAAGG 12277 | 66.6 |
| 16S-AUc | 16S rRNA gene | FN545816 | 12297 GGCTGGATCACCTCCTT 12313 | 60.2 |
| 23S-USAa,d | 23S rRNA gene | FN545816 | 12621 CCCTGCACCCTTAATAACTTGACC 12598 | 67.1 |
| 23S-UKb | 23S rRNA gene | FN545816 | 12617 GCgCCCTTtgTAgCTTGACC 12598 | 67.9 |
| 23S-AUc,d | 23S rRNA gene | FN545816 | 12638 TAGTGCCAAGGCATCCGCCCT 12618 | 73.6 |
Stubbs et al. (16). Of note, reverse primer 23S-UK has a 4-nucleotide mismatch (lowercase) compared to 23S rRNA genes of four fully sequenced C. difficile PCR ribotype 027 genomes (Table 2).
Sadeghifard et al. (13).
For SCGE, primer pairs 16S-USA/23S-USA, 16S-UK/23S-UK, and 16S-UK/23S-AU were used, with forward primers 16S-UK and 16S-USA being 5′-FAM labeled; for QCGE, nonlabeled conventional primer pair 16S-UK/23S-AU was used.
Numbers indicate sequence positions in corresponding GenBank sequences. Theoretical size differences between PCR products amplified by different primer pairs versus 16S-USA/23S-USA were 32 bp for 16S-UK/23S-UK, 13 bp for 16S-AU/23S-AU, and 53 bp for 16S-UK/23S-AU.
The 25-μl PCR mixture was prepared as follows: 2 μl template DNA, 0.5 μM forward and reverse primers (Sigma-Aldrich, Sydney, Australia), 2.5 mM deoxynucleoside triphosphates (Roche, Castle Hill, Australia), 10× PCR buffer (Qiagen, Doncaster, Victoria, Australia), 0.5 U Qiagen HotStar Taq polymerase and molecular biology-grade H2O (Eppendorf, North Ryde, Australia). The PCR was performed as follows: 95°C for 15 min, 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, followed by a further extension at 72°C for 10 min.
Sequencer-based CGE PCR ribotyping.
The AB 3730xl DNA analyzer (Applied Biosystems) with a 48-capillary, 50-cm POP7 gel was used for PCR fragment analysis. PCR products were diluted 1:30 with molecular biology-grade H2O (Eppendorf) to a final volume of 30 μl before loading. Sample injection was at 1.6 kV over 15 s with a total running time of 6,200 s at 15 kV run voltage. A 20- to 1,200-bp LIZ 1,200 ladder (Chimerx) was used as an internal marker for each sample. The size of each peak was determined using GeneMapper 4.0 software (Applied Biosystems).
QIAxcel-based CGE PCR ribotyping.
QCGE was performed according to the manufacturers' instructions (Qiagen). Briefly, 10 μl of PCR product was loaded per sample, and analysis was performed with a QX DNA high-resolution cartridge, using the OM500 method and QX 15-bp/1-kb alignment markers. Fragment sizes were calculated using the BioCalculator (Qiagen).
ISR sequences from C. difficile PCR ribotype 027 whole-genome sequences.
Publicly available whole-genome sequences of four C. difficile PCR ribotype 027 isolates in GenBank were downloaded into BioManager (https://bm.angis.org.au/) and analyzed by “virtual ribotyping” as reference standards for ISR lengths (Table 2). The strains used were 2007855 (GenBank accession no. FN665654), CD196 (GenBank accession no. FN538970), BI1 (GenBank accession no. FN668941), and R20291 (GenBank accession no. FN545816). It has recently been suggested that R20291 should be regarded as the reference type strain of C. difficile ribotype 027 (18).
Table 2.
ISR sizes amplified from whole-genome sequences of four C. difficile PCR ribotype 027 isolates by different primer sets
| Strain, GenBank accession no. | ISR lengths (bp) amplified with different primer pairsa,e |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a | b | Rareb | c | d | Rareb | e | f | gc | h | |
| R20291, FN545816 | 235/267/288 | 268/300/321 | NP | 330/362/383 | 332/364/385 | NP | 371/403/424 | 428/460/481 | NP | 552/583/605 |
| BI1, FN668941 | 235/267/288 | 268/300/321 | NP | 330/362/383 | 332/364/385 | NP | 371/403/424 | 428/460/481 | NP | 552/583/605 |
| CD196, FN538970 | 235/267/288 | 268/300/321 | NP | 330/362/383 | 332/364/385 | 362/394/415 | 371/403/424 | 428/460/481 | NP | 552/583/605 |
| 2007855, FN665654 | 235/267/288 | 268/300/321 | 310/342/363 | 330/362/383 | 332/364/385 | NP | 371/403/424 | NPd | NP | 552/583/605 |
The three numbers (separated by slashes) listed in this table are ISR (16S-23S rRNA intergenic spacer region) lengths calculated with primer pairs (i) 16S-USA/23S-USA, (ii) 16S-UK/23S-UK, and (iii) 16S-UK/23S-AU; they represent correlated peaks in SCGE-based and bands in QCGE-based PCR ribotyping (see Fig. 1 and 2). NP, not present.
These two peaks were only found in one of four genome sequences analyzed and were not detected in any of the 21 PCR ribotype 027 C. difficile reference strains tested.
Whether peak g, detected in the present study in reference panel isolates but not whole genomes (see Table 2) and by Valiente et al. (18), is an artifact (4) or a fault in whole-genome sequence analysis requires further investigation.
An ISR of 428 bp was not found in the genome sequence of strain 2007855.
RESULTS
Analysis of C. difficile ribotype 027 whole-genome sequences.
In silico analyses of the four publicly available ribotype 027 whole-genome sequences using the primer pair 16S-USA/23S-USA showed that the number of ISR copies in different genomes varied between 8 and 10 each and produced bands of nine different lengths: 235 bp (band a), 268 bp (b), 310 bp, 330 bp (c), 332 bp (d), 362 bp, 371 bp (e), 428 bp (f), and 552 bp (h). Six bands (a, b, c, d, e, and h) were found in all four genomes, and band f was found in three. ISR bands of 310 bp and 362 bp in length were found in only one genome sequence each, namely, 2007855 and CD196, respectively (Table 2), and neither was identified by either SCGE or QCGE PCR ribotyping in the panel of reference strains tested (see below). Despite these differences, this bioinformatic analysis provided size reference data for the seven bands that were present in all or most of the genomes.
Comparison of whole-genome band patterns with those of reference panel isolates.
The results of SCGE-based PCR ribotyping using three different primer pairs, are summarized in Table 3; Fig. 1 shows the electropherogram of strain UK6 as an example. Eight bands were identified with all three primer pairs in all 21 reference strains, including peak h, although its signal was quite weak.
Table 3.
ISR sizes for C. difficile PCR ribotype 027 calculated from whole-genome sequences in silico and from reference strains by SCGE and QCGE
| Peaks/bandsa | ISR size (bp) as amplified by indicated primer pair and methodb |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 16S-USA/23S-USA |
16S-UK/23S-UK |
16S-UK/23S-AU |
|||||||||||||||
| GBc | SCGE |
GBc | SCGE |
GBc | SCGE |
QCGE |
|||||||||||
| Avg ± SD | Range | GB avg diffi | Avg ± SD | Range | GB avg diff | Diffd | Avg ± SD | Range | GB avg diff | Diffe | Avg ± SD | Range | GB avg diff | ||||
| a | 235 | 231.4 ± 0.0 | 231.4–231.5 | 3.6 | 267 | 265.2 ± 0.1 | 265.1–265.3 | 1.8 | 33.8 | 288 | 284.8 ± 0.1 | 284.6–284.9 | 3.2 | 53.4 | 279.4 ± 4.2 | 273.7–286.9 | 8.6 |
| b | 268 | 264.3 ± 0.1 | 264.2–264.4 | 3.7 | 300 | 298.0 ± 0.1 | 297.9–298.1 | 2.0 | 33.7 | 321 | 317.7 ± 0.1 | 317.6–317.9 | 3.3 | 53.4 | 309.3 ± 5.1 | 302.4–319.0 | 11.7 |
| c | 330 | 325.5 ± 0.1 | 325.3–325.6 | 4.5 | 362 | 359.0 ± 0.3 | 358.4–359.4 | 3.0 | 33.5 | 383 | 378.0 ± 0.1 | 378.2–378.7 | 5.0 | 52.5 | 375.1 ± 4.2f | 369.8–383.1f | NDf |
| d | 332 | 327.7 ± 0.1 | 327.5–327.8 | 4.3 | 364 | 361.3 ± 0.2 | 360.6–361.6 | 2.7 | 33.6 | 385 | 380.6 ± 0.1 | 380.3–380.7 | 4.4 | 52.9 | |||
| e | 371 | 366.0 ± 0.1 | 365.7–366.1 | 5.0 | 403 | 398.7 ± 0.3 | 398.3–399.1 | 4.3 | 32.7 | 424 | 418.8 ± 0.1 | 418.5–419.0 | 5.2 | 52.8 | 412.6 ± 5.1 | 406.9–421.9 | 11.4 |
| f | 428 | 424.1 ± 0.1 | 424.0–424.3 | 3.9 | 460 | 457.8 ± 0.2 | 457.6–458.1 | 2.2 | 33.7 | 481 | 477.0 ± 0.1 | 476.7–477.2 | 4.0 | 52.9 | 478.7 ± 6.5 | 470.9–488.7 | 2.3 |
| gg | NP | 446.0 ± 0.1 | 445.8–446.2 | NP | 479.7 ± 0.2 | 479.4–480.0 | 33.7 | NP | 499.0 ± 0.1 | 498.8–499.2 | 53.0 | 510.2 ± 9.5 | 498.9–525.3 | ||||
| hh | 552 | 546.8 ± 0.2 | 556.5–547.0 | 5.2 | 584 | 580.7 ± 0.3 | 580.3–581.1 | 3.3 | 33.9 | 605 | 599.7 ± 0.2 | 599.4–599.9 | 5.3 | 52.9 | |||
Designations representing peaks in SCGE-based PCR ribotyping and bands in QCGE-based PCR ribotyping (see Fig. 1 and 2).
Abbreviations: ISR, 16S-23S rRNA intergenic spacer regions; GB, GenBank; SCGE, sequencer-based CGE; QCGE, QIAxcel-based CGE; NP, not present.
Four publicly available C. difficile PCR ribotype 027 whole-genome sequences in GenBank (see Table 2 for details) were used for in silico analysis: strains R20291 (GenBank accession no FN545816); BI1 (GenBank accession no. FN668941); CD196 (GenBank accession no. FN538970); and 2007855 (GenBank accession no. FN665654).
ISR length difference between PCR products amplified by 16S-UK/23S-UK and 16S-USA/23S-USA.
ISR length difference between PCR products amplified by 16S-UK/23S-AU and 16S-USA/23S-USA.
QCGE cannot distinguish bands c and d (2-bp difference); therefore, a single c/d band was read (Fig. 2), and the value “GB avg diff” was not calculated (ND, not done).
All 21 C. difficile PCR ribotype 027 reference strains tested in this study showed an extra amplicon peak/band g in both QCGE and SCGE which was not present in any of the GenBank whole-genome sequences.
The largest band, h, was not detectable by QCGE due to decreased signal strength of larger amplicons.
Difference between GB and the average ISR sizes measured by SCGE.
Fig 1.

SCGE-based PCR ribotyping results for C. difficile reference strain UK6 (PCR ribotype 027 [8]). (A, B, and C) SCGE-based PCR ribotyping amplified by primer pairs 16S-USA/23S-USA, 16S-UK/23S-UK, and 16S-UK/23S-AU, respectively. The sizes of peaks a to h are shown in Table 3. SCGE was performed on an AB 3730xl DNA analyzer (Applied Biosystems) with the 20- to 1,200-bp LIZ 1200 ladder as the internal size control.
SCGE distinguished peaks c and d with length differences as small as 2 bp (Table 3 and Fig. 1). The standard deviations for each band length ranged from 0 to ±0.2 bp, ±0.1 to ±0.3 bp, and ±0.1 to ±0.2 bp for primer pairs 16S-USA/23S-USA, 16S-UK/23S-UK, and 16S-UK/23S-AU, respectively, reflecting the high discriminatory power and reproducibility of SCGE.
The band patterns produced by the three primer sets for all 21 ribotype 027 isolates were identical. The additional ISR bands found in genomes of strain 2007855 and strain CD196 (Table 2) were not detected in these well-characterized isolates. However, an additional ISR of ∼450 bp (band g), not present in any of the whole-genome sequences, was identified in all 21 PCR ribotype 027 reference strains tested (Table 3, Fig. 1) regardless of the primer pairs used.
Evaluation of QCGE PCR ribotyping.
Gel-view results of QCGE-based PCR ribotyping for all 21 reference strains are shown in Fig. 2, and the ISR band sizes assigned by this method in Table 3. Six bands (a, b, c/d, e, f, and g) were identified. QCGE could not distinguish between ISRs of 330 bp and 332 bp owing to a manufacturer-specified limit of discrimination of 3 to 5 bp for fragments in the range of 100 to 500 bp separated with the high-resolution cartridge. Thus, combined c/d bands (Fig. 2) were identified. QCGE also failed to detect the largest ISR amplicon (band h, 605 bp) (Table 3), which was identified by both genome analysis and SCGE in all reference isolates. This was possibly due to competition in PCR amplification, causing a decrease in the band signal strength with increasing amplicon size, which QCGE was not sensitive enough to capture. Therefore, QCGE would not be able to distinguish ribotype 027 from the closely related ribotype 176, which differs only by the absence of band h (10, 18).
Fig 2.

QCGE-based PCR ribotyping of 21 well-characterized C. difficile PCR ribotype 027 reference strains (8) amplified by primer pair 16S-UK/23S-AU. Lanes 1 to 21: strains UK6, UK7, UK8, UK9, UK10, NL1, NL2, NL3, NL4, NL5, CA2, CA3, CA4, CA5, CA6, US31, US35, US38, US39, US41, and US42. Sizes of bands a to g are listed in Table 3. Note that QCGE cannot distinguish bands c and d, which differ in length by only 2 bp; therefore, a single c/d band was read.
The standard deviations for different band sizes assigned by QCGE ranged from ±4.2 to ±9.5 bp, and the maximum size difference for corresponding bands in different isolates from 3.2 to 26.4 bp (Table 3). The ISR lengths for corresponding bands differed by 2.3 to 11.7 bp when average QCGE sizes were compared to those measured by genome sequences. As the reproducibility, discriminatory power, and sensitivity of QCGE were relatively poor and do not meet the requirements for accurate and reliable C. difficile PCR ribotyping, it could only be recommended for preliminary screening of isolates in an outbreak investigation.
Comparability of three primer sets.
All three primer pairs used for SCGE-based PCR ribotyping generated identical band patterns for 21 C. difficile ribotype 027 isolates tested, with accurate size calling. The theoretical size difference between bands produced by the 16S-USA/23S-USA and 16S-UK/23S-AU primer sets was 53 bp (Table 1), and the observed size differences, ranging from 52.5 to 53.4 bp, were very similar (Table 3). For the 16S-USA/23S-USA and 16S-UK/23S-UK primer sets, the theoretical band size difference was 32 bp (Table 1), and the observed difference was 1 or 2 bp greater at 33.5 to 33.9 bp (Table 3). In addition, there were differences in the lengths measured using different primers compared with the theoretical lengths based on genome sequence analysis; the differences were 3.6 to 5.2 bp for 16S-USA/23S-USA, 1.8 to 4.3 bp for 16S-UK/23S-UK, and 3.2 to 5.3 bp for primer pair 16S-UK/23S-AU (see Table 3 for details). The reasons for these minor length differences are unclear but may include the accuracy of internal markers used for SCGE.
Comparison of SCGE and QCGE for other ribotypes.
The results of testing 14 C. difficile isolates of other ribotypes by SCGE and QCGE were similar to those for ribotype 027. SCGE assigned band sizes more accurately and reproducibly (deviations from reference/consensus band sizes for individual bands ranged from 0.1 to 0.3 bp) than QCGE (band size deviations were 0.4 to 7.2 bp). SCGE detected bands of >500 bp in all ribotypes tested, none of which was detected by QCGE. The high discriminatory power of SCGE allowed the identification of four different banding patterns for the seven ribotype 001 isolates, which were consistent regardless of the primer set used. One pattern (A; represented by four isolates) showed five low-density bands, ranging in size (produced by the 16S-USA/23S-USA primers) from 484 ± 0.2 bp to 549.0 ± 0.3 bp; the other patterns showed different combinations of four of these five bands (Fig. 3). Pattern A was consistent with the ribotype 001 pattern reported by Indra et al. (6). None of these five bands (all >500 bp) was identified by QCGE, which therefore did not distinguish the four patterns.
Fig 3.
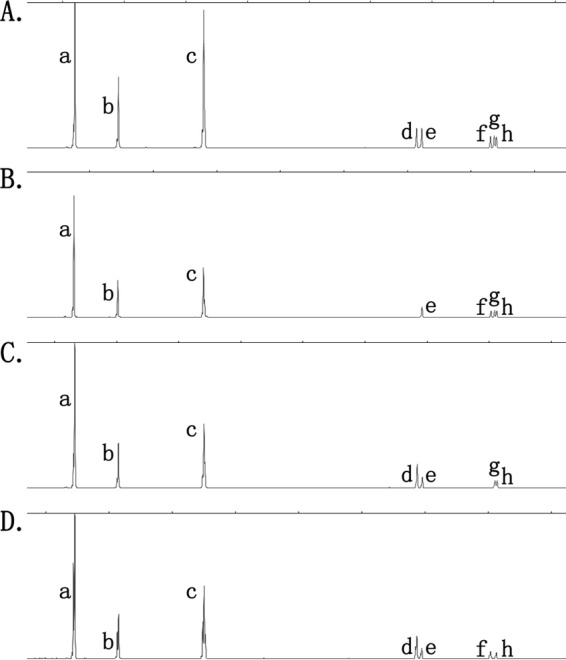
Four different band patterns identified from seven well-characterized C. difficile isolates previously assigned to ribotype 001. Pattern A, four isolates identified; patterns B, C, and D, one isolate each identified. Pattern A matches that described for ribotype 001 by Indra et al. (6).
DISCUSSION
Ideally, any microbial strain typing system should be standardized and calibrated against an objective gold standard. This would be particularly desirable for conventional C. difficile ribotyping based on agarose gel electrophoresis, which can be difficult to interpret because of variation in band strength and poor reproducibility (4, 7). C. difficile ribotyping based on CGE is more discriminatory, accurate, and reproducible (7). These characteristics have allowed the recognition of variability within what were previously regarded as single ribotypes, such as 027 (18). There is, therefore, some urgency to develop a system for standardization of methods and ribotype nomenclature.
We aimed to use C. difficile PCR ribotype 027 whole-genome sequences to provide reference data to evaluate the size-calling accuracy and comparability of C. difficile PCR ribotyping methods. However, in silico analyses of the four ribotype 027 whole-genome sequences showed that the number of ISR copies in different genomes varied and produced bands of different lengths. Stabler et al. (15) previously reported significant differences between the whole-genome sequences of two C. difficile ribotype 027 strains (CD196 and R20291), which suggests that the ISR profile differences between the strains that we identified are due to genuine genomic differences between strains rather than differences in methods of genome analysis (12, 17). The results raise the question as to whether the two strains (CD196 and 2007855) with one extra band each and/or the one with one extra and one missing band (2007855) should be classified as different ribotypes, similar to the new ribotypes closely related to ribotype 027 reported by Valiente et al. (18), based on the genome R20291-FN545816ISR profile (BI1-FN66894 has the same ISR-length profile) as the 027 reference standard.
The 21 well-characterized C. difficile 027 isolates used in this study as a reference panel were part of a geographically diverse collection which had been previously subjected to seven typing methods in several laboratories (8). Our comparison of whole-genome band patterns with those of reference panel isolates tested by SCGE indicates that the PCR ribotype 027 strains in this international collection unambiguously belong to the same ribotype, as identified by SCGE, and that any of the three primer pairs produces satisfactory results. The absence of any of the eight peaks a to h or the detection of additional peaks in an isolate should prompt consideration that such an isolate may need to be assigned to another ribotype, such as the newly reported ribotypes 176, 198, and 244, which have apparently evolved from the 027 lineage (10, 18), or a new ribotype. Band g has been identified in previous studies (7, 18) (Table 2), as well as in the present study (Table 3). However, its absence from all of the published ribotype 027 whole genomes indicates that further investigation is required to determine whether it is an artifact (4) or a fault in genome sequence analysis (12).
This study focused on a single ribotype for which multiple whole-genome sequences are available. However, a limited comparison of SCGE and QCGE, using a small set of well-characterized isolates belonging to four other ribotypes, also showed that SCGE produces more reproducible band sizes and can identify large, low-intensity bands not detected by QCGE. As a result, SCGE identified variant banding patterns of ribotype 001, based on different combinations of five large but low-intensity bands that were not identified by QCGE. These results suggest that the benefits of SCGE for accurate identification of ribotype 027 and closely related (or variant) ribotypes are applicable to other ribotypes and should assist in the identification of new ones.
SCGE for C. difficile PCR ribotyping was first described by Indra et al. in 2008 (7). The use of fluorescein-labeled primers increases the sensitivity and, also, the cost of the method. SCGE requires access to a DNA sequencer, which may cause delay if PCR products are referred to an external sequencing facility, and the cost of consumables per isolate is greater ($6 to $7, including primers, labels, PCR reagents, and SCGE, including a component for equipment cost) than for QCGE (see below). SCGE requires the use of a more expensive instrument. We used the AB 3730xl DNA analyzer (purchase cost, ∼$480,000, compared with QIAxcel at ∼$55,000). However, the former has many other applications in a large reference or sequencing laboratory.
The use of QCGE for C. difficile PCR ribotyping was first reported by the manufacturer around 2009 (http://www.qiagen.com/literature/render.aspx?id=104730). QIAxcel is an automated electrophoresis platform that can deal with up to 96 samples per run with high efficiency and a turnaround time of 0.2 to 1.5 h depending on the number of specimens. As the method does not require fluorescein-labeled primers, it is simple to perform and potentially suitable for a clinical laboratory (4, 5, 9). The results can be exported in either electropherogram (similar to the SCGE result shown in Fig. 1) or gel-view format (as shown in Fig. 2). The major costs are for setup of the QIAxcel system hardware and BioCalculator analysis software and for consumables (cartridges), making the cost per sample around $3 to $4. However, compared with SCGE, QCGE has limited sensitivity and discriminatory power and cannot clearly distinguish closely related ribotypes, e.g., ribotypes 027 and 176, as shown in the present study.
Although all three primer pairs produced satisfactory results for C. difficile ribotype 027, we would not recommend 16S-UK/23S-UK primers because of the 4-bp mismatch of the 23S-UK reverse primer. The 16S-UK/23S-AU and 16S-USA/23S-USA sets are equally satisfactory, but the latter was used by Indra et al. (7) for initial evaluation of the SCGE method, and it produces relatively small amplicons which make comparison between runs easier.
Our aim was to develop a gold standard for C. difficile ribotype 027. Based on our bioinformatics and benchtop analyses, we recommend R20291 as the 027 reference strain, as suggested by Valiente et al. (18), and SCGE incorporating primer set 16S-USA/23S-USA as the reference method for C. difficile PCR ribotyping. This study was limited by the fact that only one PCR ribotype was tested. However, its strength was the use of an international set of well-characterized isolates, which allowed assessment of the consistency of patterns and reproducibility of the methods. Further studies of additional ribotypes will be needed in future. Nevertheless, this study has provided a solid reference for the evaluation, validation, and standardization of SCGE PCR ribotyping.
ACKNOWLEDGMENTS
We acknowledge Ying-Chun Xu from the Clinical Laboratory Department, Beijing Union Medical College Hospital, Chinese Academy of Medical Sciences, China, for providing a special grant from National Science Research Mega project against Major Infectious Diseases in China, grant 2009ZX10004-206.
The authors have declared no conflict of interest.
Footnotes
Published ahead of print 12 June 2012
REFERENCES
- 1. Bidet P, Barbut F, Lalande V, Burghoffer B, Petit JC. 1999. Development of a new PCR-ribotyping method for Clostridium difficile based on ribosomal RNA gene sequencing. FEMS Microbiol. Lett. 175:261–266 [DOI] [PubMed] [Google Scholar]
- 2. Bidet P, et al. 2000. Comparison of PCR-ribotyping, arbitrarily primed PCR, and pulsed-field gel electrophoresis for typing Clostridium difficile. J. Clin. Microbiol. 38:2484–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gurtler V. 1993. Typing of Clostridium difficile strains by PCR-amplification of variable length 16S-23S rDNA spacer regions. J. Gen. Microbiol. 139:3089–3097 [DOI] [PubMed] [Google Scholar]
- 4. Hoffmann M, et al. 2010. PCR-based method for targeting 16S–23S rRNA intergenic spacer regions among Vibrio species. BMC Microbiol. 10:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hughes-Stamm SR, Ashton KJ, van Daal A. 2011. Assessment of DNA degradation and the genotyping success of highly degraded samples. Int. J. Legal Med. 125:341–348 [DOI] [PubMed] [Google Scholar]
- 6. Indra A, et al. 2010. Mechanisms behind variation in the Clostridium difficile 16S–23S rRNA intergenic spacer region. J. Med. Microbiol. 59:1317–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Indra A, et al. 2008. Characterization of Clostridium difficile isolates using capillary gel electrophoresis-based PCR ribotyping. J. Med. Microbiol. 57:1377–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Killgore G, et al. 2008. Comparison of seven techniques for typing international epidemic strains of Clostridium difficile: restriction endonuclease analysis, pulsed-field gel electrophoresis, PCR-ribotyping, multilocus sequence typing, multilocus variable-number tandem-repeat analysis, amplified fragment length polymorphism, and surface layer protein A gene sequence typing. J. Clin. Microbiol. 46:431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McMurray CL, Hardy KJ, Hawkey PM. 2010. Rapid, automated epidemiological typing of methicillin-resistant Staphylococcus aureus. J. Microbiol. Methods 80:109–111 [DOI] [PubMed] [Google Scholar]
- 10. Nyc O, Pituch H, Matejkova J, Obuch-Woszczatynski P, Kuijper EJ. 2011. Clostridium difficile PCR ribotype 176 in the Czech Republic and Poland. Lancet 377:1407. [DOI] [PubMed] [Google Scholar]
- 11. O'Neill GL, Ogunsola FT, Brazier JS, Duerden BI. 1996. Modification of a PCR ribotyping method for application as a routine typing scheme for Clostridium difficile. Anaerobe 2:205–209 [Google Scholar]
- 12. Pignatelli M, Moya A. 2011. Evaluating the fidelity of de novo short read metagenomic assembly using simulated data. PLoS One 6:e19984 doi:10.1371/journal.pone.0019984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sadeghifard N, Gurtler V, Beer M, Seviour RJ. 2006. The mosaic nature of intergenic 16S–23S rRNA spacer regions suggests rRNA operon copy number variation in Clostridium difficile strains. Appl. Environ. Microbiol. 72:7311–7323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spigaglia P, Mastrantonio P. 2004. Comparative analysis of Clostridium difficile clinical isolates belonging to different genetic lineages and time periods. J. Med. Microbiol. 53:1129–1136 [DOI] [PubMed] [Google Scholar]
- 15. Stabler RA, et al. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 10:R102 doi:10.1186/gb-2009-10-9-r102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stubbs SL, Brazier JS, O'Neill GL, Duerden BI. 1999. PCR targeted to the 16S–23S rRNA gene intergenic spacer region of Clostridium difficile and construction of a library consisting of 116 different PCR ribotypes. J. Clin. Microbiol. 37:461–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suzuki S, Ono N, Furusawa C, Ying BW, Yomo T. 2011. Comparison of sequence reads obtained from three next-generation sequencing platforms. PLoS One 6:e19534 doi:10.1371/journal.pone.0019534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Valiente E, Dawson LF, Cairns MD, Stabler RA, Wren BW. 2012. Emergence of new PCR ribotypes from the hypervirulent Clostridium difficile 027 lineage. J. Med. Microbiol. 61:49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]