

Abstract
We have shown previously that high-dose lipid amphotericin preparations are not more efficacious than lower doses in aspergillosis. We studied toxicity, drug concentrations and localization, and quantitative infection concurrently, using a 4-day model of central nervous system (CNS) aspergillosis to assess early events. Mice given Aspergillus fumigatus conidia intracerebrally, under a cyclophosphamide immunosuppressive regimen, were treated for 3 days (AmBisome at 3 or 10 mg/kg of body weight, Abelcet at 10 mg/kg, amphotericin B deoxycholate at 1 mg/kg, caspofungin at 5 mg/kg, or voriconazole at 40 mg/kg). Sampling 24 h after the last treatment showed that AmBisome at 3 but not at 10 mg/kg, as well as Abelcet, caspofungin, and voriconazole, reduced brain CFU. All regimens reduced renal infection. Minor renal tubular changes occurred with AmBisome or Abelcet therapy, whereas heart, lung, and brain showed no drug toxicity. Amphotericin B tissue and serum concentrations did not correlate with efficacy. Endothelial cell activation (ICAM-1 and P-selectin in cerebral capillaries) occurred during infection. Amphotericin B derived from AmBisome and Abelcet localized in activated endothelium and from Abelcet in intravascular monocytes. In 10-day studies dosing uninfected mice, minor renal tubular changes occurred after AmBisome or Abelcet at 1, 5, or 10 mg/kg with or without cyclophosphamide treatment; nephrosis occurred only with Abelcet in cyclophosphamide-treated mice. Hepatotoxicity occurred with AmBisome and Abelcet but was reduced in cyclophosphamide-treated mice. Marked CFU reduction by AmBisome at 3 mg/kg occurred in association with relatively more intense inflammation. Abelcet renal localization appears to be a precursor to late nephrotoxicity. Hepatotoxicity may contribute to high-dose Abelcet and AmBisome failures. Our novel observation of endothelial amphotericin localization during infection may contribute to amphotericin mechanism of efficacy.
INTRODUCTION
Central nervous system (CNS) aspergillosis is a common site of dissemination in invasive disease (12). Therapy for this manifestation of disease is, however, relatively ineffective, and there is a high risk of mortality. We have examined various potential therapies, including azoles, echinocandins, amphotericin B preparations, and combination therapy, for efficacy using a murine model of CNS aspergillosis (4–6, 21, 22, 36). Although in each of these studies one or more regimens have shown efficacy, none of the tested regimens has proven curative. Interestingly, lipid-carried amphotericin B preparations showed a flat dose-response curve. High dosages were not more efficacious than lower doses and in some instances could show deleterious effects (5, 6).
In the current study, we sought to further examine possible reasons for the observed efficacy profile of the lipid-carried amphotericin B preparations. We felt that several possibilities existed. The induction of nonlethal toxicity in the organs could influence drug efficacy, as could differences in accumulation or distribution of amphotericin B in the tissues. We also felt that severity of host response could influence treatment outcome. Thus, our aim was assessing comparative drug toxicity by histological methods and determining concentrations of amphotericin B in tissues and correlating these with drug efficacy. Although accumulation of amphotericin B in the tissues is important, we believe that where the drug is in the tissues in relationship to the infecting organisms is more important and thus examined amphotericin B localization in tissues by an immunohistochemistry method. Furthermore, we examined the expression of inflammatory markers, ICAM-1 and P-selectin, by the cerebral vascular endothelium to focalize the inflammatory response and compare with localization of amphotericin B. We chose to examine these various aspects early in the course of disease to reduce the confounding effects animals dying from infection might have, and we compared them with uninfected mice treated in the same manner.
MATERIALS AND METHODS
CNS model and therapy.
A murine model of CNS aspergillosis was used in these studies. Five-week-old male CD-1 mice (Charles River, Hollister, CA) were used. Two days prior to infection, all mice were dosed intraperitoneally with 200 mg of cyclophosphamide/kg of body weight; additional doses were given every 5 days thereafter. Prior to infection, Aspergillus fumigatus strain 10AF from long-term storage was grown, and conidia were collected as previously described (11, 18). Infection was initiated by direct intracerebral inoculation of 3.26 × 106 conidia of A. fumigatus strain 10AF as previously described (3, 5, 6). All animal studies were done under an approved protocol and following the guidelines of the Institutional Animal Care and Use Committee of the California Institute for Medical Research.
Treatments began 1 day after infection and were administered for 3 days. AmBisome (AmBi; Gilead Sciences), Abelcet (ABLC; Enzon), deoxycholate-formulated amphotericin B (AMBd), and 5% dextrose water (D5W) were prepared per the manufacturers' instructions and given intravenously. Voriconazole (VCZ; Pfizer) was administered orally in 4% polyethylene glycol 400 (PEG 400) (5, 6); mice receiving VCZ were provided 50% grapefruit juice in lieu of normal drinking water (5, 6). Caspofungin (CAS; Merck) was given subcutaneously. Groups received AmBi at 3 or 10 mg/kg of body weight, ABLC at 10 mg/kg, AMBd at 1 mg/kg, CAS at 5 mg/kg, or VCZ at 40 mg/kg. Doses and routes of administration were selected from previous efficacy observations (3, 5, 6, 21, 22, 36). Additional groups of uninfected mice were also studied, with the cyclophosphamide treatment and all drug treatments given on the same schedule as in the infection part of the study.
Sample collection.
All mice were preassigned for CFU, pharmacology, histopathology, or immunohistochemistry studies to eliminate possible sampling bias.
One day after the last treatment (day 4 postinfection), surviving mice were anesthetized using inhaled isoflurane and exsanguinated for serum collection. After euthanasia, CFU determinations for drug efficacy were done on brain and kidneys as previously described by homogenization of the organ and plating of serially diluted samples (5, 6, 18). For those mice preassigned for histopathology and pharmacologic studies, the brain was removed intact. For histopathology studies, tissue samples were fixed in 10% neutral buffered formalin (NBF). A second sample of the tissue was frozen at −20°C for determination of amphotericin B concentration. Serum samples were also submitted for serum chemistry analysis.
Amphotericin B determination.
The concentration of amphotericin B in the serum or tissues (lung, liver, heart, brain, kidneys) was determined by bioassay using Paecilomyces variotii as the indicator organism (8, 18, 19, 39). Tissue samples were homogenized and extracted in 100% methanol and clarified by centrifugation (13,000 × g for 5 min), and aliquots were taken to dryness using a SpeedVac (Savant Instruments, Inc., Farmingdale, NY). After extraction and drying, each sample was reconstituted with pooled human serum for bioassay. Serum samples were extracted with 100% methanol (2 ml per 0.1 ml serum) and taken to dryness using a SpeedVac (Savant), and residue material was suspended in pooled human serum; these samples were then assayed directly. Unextracted serum was also assayed. The lower limit of the amphotericin B bioassay was 0.031 μg/ml; limits of detection for tissues are indicated in each figure.
Histopathology and immunohistochemistry.
Formalin-fixed tissues were trimmed, processed, and embedded in paraffin, and approximately 5-μm sections were prepared and stained with hematoxylin and eosin (H&E). Gridley-stained sections (for organism visualization) were also prepared from brain from all infected mice and from liver, lung, heart, and kidney from some. Five sections from the CNS tissues were examined; 2 cerebrum, 1 midbrain, 1 cerebellum, and 1 brainstem. All brain sections (infected and uninfected animals) were stained immunohistochemically for amphotericin B, ICAM-1, and P-selectin. Primary antibodies against amphotericin B (affinity-purified rabbit anti-amphotericin B antibody, customized antibody produced by Antibodies Inc., Davis, CA), mouse ICAM-1 (biotinylated goat anti-mouse ICAM-1 antibody; R&D Systems, Minneapolis, MN), and mouse P-selectin (goat anti-mouse P-selectin; Santa Cruz Biotechnology, Santa Cruz, CA) were used. Immunohistochemistry to detect amphotericin B was performed as previously described (37). To detect ICAM-1 and P-selectin, tissues were placed in citrate buffer (0.01 mol/liter citrate buffer, pH 6) and heated for 15 min in a pressure cooker for antigen retrieval. Endogenous peroxidase activity was blocked with 3% hydrogen peroxidase (Sigma, St. Louis, MO) for 10 min at room temperature. A protein block (0.5% casein, 1% bovine serum albumin, and 1.5% goat serum) designed to reduce nonspecific binding was used. Following the protein block, sections were incubated with the primary antibody dilutions (1:25 dilution for biotinylated goat anti-mouse ICAM-1 and 1:50 for goat anti-mouse P-selectin) for 1 h at room temperature. Biotinylated secondary antibody (1:500 dilution of donkey anti-goat IgG, heavy and lights chains; Jackson ImmunoResearch, West Grove, PA) was applied to P-selectin-stained slides for 30 min. All slides were reacted for 30 min with ABC Elite reagent (Vector Laboratories, Burlingame, CA), and diamino benzidine chromogen substrate (Fast DAB tablets, Sigma-Aldrich, St. Louis, MO) was applied for 10 min as a substrate for the peroxidase reaction. All slides were counterstained with hematoxylin (Richard-Allan Scientific, Kalamazoo, MI) in saturated lithium carbonate (Sigma), dehydrated through alcohols, and cleared in xylene (EMD, Gibbstown, NJ), and coverslips were applied. Sections incubated with isotype-matched irrelevant antibodies served as negative controls in each technique. Appropriate positive controls were included in all experiments.
For the amphotericin B studies, amphotericin B deoxycholate (X-Gen Pharmaceuticals, Northport, NY) prepared as a 20-μg/ml solution in water and parathyroid hormone-related protein (nonspecific background control) (Sigma) at a 20-μg/ml solution in water were spotted onto UV-activated resin slides (Instrumedics, Inc., St. Louis, MO) and UV adhered to the slide prior to fixation. The slides were air dried and immunostained as described for the cryosections. These slides served as positive and negative controls, respectively. Kupffer cells containing abundant phagocytized intracytoplasmic amphotericin B (mouse liver from an uninfected animal that received a total cumulative AmBi dose of 225 mg/kg) were used as an additional positive control for all immunohistochemistry localization of amphotericin B. Mouse kidney was used for the positive control of the inflammatory marker ICAM-1, since vascular endothelium in the mouse kidney constitutively expresses ICAM-1. Because circulating platelets, as well as megakaryocytes in the spleen, constitutively express P-selectin, these tissue elements were used as positive controls for P-selectin staining.
All slides were evaluated microscopically by a board-certified veterinary pathologist without reference to the study group. Each slide was examined for the presence and intensity (strength) of labeling, as well as the distribution and frequency (relative density) of positive cells or positive tissue elements for each antibody. The intensity was graded on a semiquantitative, 1+ (minimal) to 4+ (marked), basis.
Meningoencephalitis grading scheme.
Fungal meningoencephalitis was graded in severity using the following semiquantitative scoring system: 1+ (minimal) was given to animals with one to two foci of inflammation, necrosis, and hemorrhage identified in the cerebrum only; 2+ (mild) was given to animals with >2 sites of inflammation, necrosis, and hemorrhage in the cerebrum only; 3+ (moderate) was given to animals with multiple sites of inflammation, necrosis, and hemorrhage in the cerebrum plus similar lesions in one or more of the midbrain, brainstem, or cerebellum; 4+ (marked) was given to animals with multifocal sites of inflammation, necrosis, and hemorrhage in the cerebrum plus similar lesions in one or more of the midbrain, brainstem, or cerebellum and extensive inflammation and fungal invasion throughout the ventricular system. The average lesion severity score was calculated for each group.
Subacute toxicity study.
Five-week-old male CD-1 mice were used, with groups of 5 mice for each treatment arm. Mice were either dosed with cyclophosphamide using the schedule and dose described above or received no suppressive pretreatment. Mice were not infected.
Treatment with antifungals began 3 days after the first cyclophosphamide treatment. Mice were given one of the following regimens: no treatment; AMBd at 1 mg/kg; AmBi at 1, 5, or 10 mg/kg; or ABLC at 1, 5, or 10 mg/kg. AMBd, AmBi, and ABLC were given in D5W intravenously. Treatments were given for 10 consecutive days. Four days after the cessation of therapy, mice were anesthetized and exsanguinated for serum collection. The liver, kidney, and brain were placed in 10% NBF and processed to HE slides for histological examination. Sera were sent to Charles River Laboratories, Pathology Associates—Maryland (Frederick, MD), for determination of serum chemistries using a Research Chem 01 panel. This panel includes tests for the determination of cholesterol, alkaline phosphatase, alanine amino transferase, aspartate amino transferase, creatinine phosphokinase, bilirubin, blood urea nitrogen (BUN), creatinine, albumin, total protein, globulin, phosphorus, potassium, chloride, sodium, glucose, and bicarbonate.
Statistics.
For statistical purposes, to account for occasional missing data points due to death from infection, an arbitrary value of log10 6 was assigned as the CFU in the organ, ensuring that death was ranked more severely than survival with a lower CFU burden (23, 34). This assignment assumes that death is due to a high burden of infection and does not address death possibly being due to drug toxicity. CFU values were compared using a Mann-Whitney U test. Mean lesions scores were compared using a Mann-Whitney U test.
RESULTS
Drug efficacy.
Because the study was designed to use CFU in the organs as the parameter for therapeutic efficacy, the model was ended 1 day after the third treatment, on day 4 of infection. This was done to have a significant number of animals survive the infection. In spite of this, 4 of 10 D5W-treated mice and 2 of 10 mice given 1 mg/kg AMBd succumbed to infection on day 4 prior to sampling; no organs were taken from the dead mice. All mice in the other treatment regimens survived to day 4 and there were no statistical differences in survival among the various treatment groups. Thus, CFU remaining in the organs was used as the parameter of drug efficacy.
Brain.
Comparison of fungal burdens in the brain showed that ABLC at 10 mg/kg, AmBi at 3 mg/kg, CAS, and VCZ were efficacious with lower CFU than the diluent control D5W treatment (P = 0.04 to 0.009, depending on comparison), whereas AmBi at 10 mg/kg and AMBd at 1 mg/kg were not (P > 0.05) (Fig. 1). Among the various treatment groups, AmBi at 3 mg/kg was superior to all other regimens (P = 0.04 to 0.0001, depending on comparison) except CAS (P > 0.05). CAS was statistically more efficacious than AMBd at 1 mg/kg (P = 0.009) but equivalent to the other drug treatments (P > 0.05).
Fig 1.

CFU of A. fumigatus recovered from the brain and kidney of mice on day 4 of infection. Mice were given 3 days of treatment, and all surviving mice were euthanatized on day 4 for CFU determination. A value of 6 indicates that the animal died of infection. Abbreviations: D5W, 5% dextrose water; ABLC 10, Abelcet at 10 mg/kg; AmBi 3, AmBisome at 3 mg/kg; AmBi 10, AmBisome at 10 mg/kg; AMBd 1, deoxycholate amphotericin B at 1 mg/kg; Caspo 5, caspofungin at 5 mg/kg; VCZ 40, voriconazole at 40 mg/kg. Each group had 10 mice.
Kidney.
All drug regimens were efficacious in reducing fungal burden recovered from the kidneys compared to that of D5W treatment (P = 0.05 to 0.002, depending on comparison) (Fig. 1). However, all drug treatments were equivalent to each other (P > 0.05).
Of note, AmBi at 10 mg/kg was not superior to AmBi at 3 mg/kg, and neither of the lipid amphotericin B preparations was superior to conventional AMBd. Although all regimens showed significant efficacy in one or both organs, no single treatment regimen proved superior in reducing fungal burden from both organs. However, as this is a model of CNS disease, with only secondary spread to the kidney, AmBi at 3 mg/kg could be considered the most efficacious, based on the superior reduction of brain CFU.
Amphotericin B in tissues and serum.
Determination of amphotericin B concentration in various tissues and serum was done on infected and uninfected mice for comparison. As shown in Fig. 2, liver and serum had detectable concentrations 24 h after the last treatment with ABLC, AmBi, or AMBd. ABLC at 10 mg/kg also resulted in substantial concentrations of amphotericin B in kidneys and lungs and in brain in one animal each, infected and uninfected. In contrast, AmBi- and AMBd-treated mice had undetectable or very minimal amphotericin B concentrations in brain, kidneys, or lungs. No animal had detectable concentrations of amphotericin B in the heart, regardless of dose or amphotericin B formulation. AmBi at 10 mg/kg resulted in higher concentrations of amphotericin B in liver and serum than the lower dose and higher serum levels than the ABLC at the 10 mg/kg dose.
Fig 2.

Amphotericin B concentrations in the tissues or serum of mice infected (i) (surviving mice of 5 designated for serum collection or 10 designated for tissue collection) with A. fumigatus and in uninfected mice (u) (n = 5). Data are presented by tissue or serum, with results for infected mice plotted beside those of uninfected mice for comparative purposes and changes. Amphotericin B was extracted from the whole organ for brain, kidney, lung, and heart and from a sample of the liver, which accounts for the differing baselines indicated; the baseline represents the lowest concentration (μg/g) of tissue detectable using the bioassay; serum was also extracted, and amphotericin B concentrations were determined in μg/ml of serum. Abbreviations for drugs are as described in the legend to Fig. 1.
Comparisons between infected and uninfected mice showed for uninfected mice that ABLC at 10 mg/kg had significantly higher levels in the liver (P = 0.02). The amphotericin B levels in other tissues or for other drug doses were not significantly different for infected or uninfected mice. AmBi at 10 mg/kg resulted in higher serum concentrations of amphotericin B than did those given ABLC at 10 mg/kg in infected and uninfected mice (P = 0.016 and 0.008, respectively). Uninfected mice given ABLC at 10 mg/kg had higher concentrations of amphotericin B in kidney and lung than mice given AmBi at 10 mg/kg (P = 0.008), and amphotericin B concentrations trended higher in kidneys of infected mice given ABLC at 10 mg/kg (P = 0.08).
Of note, ABLC produced the highest amphotericin B concentrations in the kidney and yet was not superior to AmBi or AMBd in reducing kidney infection. AmBi at 10 mg/kg produced the highest serum levels and was not superior to AMBd or ABLC in reducing infection in brain or kidney.
The concentrations of amphotericin B in the serum were also determined on unextracted sera (data not shown), and none of the conclusions in the section would have been altered if those values had been used for comparisons.
Histopathology assessment and correlation with CFU.
A. fumigatus infection in the brain and kidney was characterized by extensive necrosis, hemorrhage, histiocytic and neutrophilic inflammation, thrombosis, and fungal vascular invasion. In the brain, microscopic findings consistent with fungal meningoencephalitis were identified in all treatment groups (Fig. 3, Table 1). Assignment of semiquantitative scores for CNS lesion severity showed a decrease in mean severity score in the treatment groups of AmBi at 10 mg/kg and AMBd at 1 mg/kg compared to that of the D5W controls (Table 2); however, the decrease was not statistically significant. CFU findings did not correlate with lesion severity scores. Interestingly, treatment groups with the lowest fungal burden by CFU (AmBi at 3 mg/kg and ABLC at 10 mg/kg had significantly reduced burdens compared to controls) had elevated mean lesion severity scores. Thus, these results demonstrate that conclusions regarding therapeutic benefit should not be based solely on histopathology.
Fig 3.

Brain, cerebrum at the level of the hippocampus. (A) Marked fungal meningoencephalitis characterized by variably sized, random areas of necrosis, inflammation, hemorrhage, and periventricular edema (arrows). (B) Higher magnification of area inside the box, H&E stain. Areas of necrosis were often surrounded by mixed neutrophilic and histiocytic inflammation (i). Intralesional fungal hyphae were abundant, located in the neuropil, blood vessels, and ventricles. (C) Gridley stain demonstrating abundant intralesional fungal hyphae (magenta); the inset shows a higher magnification. V3, third ventricle; VL, lateral ventricle; AQ, cerebral aqueduct.
Table 1.
Incidence and severity of histopathology findings associated with A. fumigatus infection in cyclophosphamide-immunosuppressed CD-1 mice
| Organ and histopathology finding observed | No. of animals positive/total no. of animalsa in antifungal treatment group: |
||||
|---|---|---|---|---|---|
| D5W control | AMBd at 1 mg/kg | AmBi at 3 mg/kg | AmBi at 10 mg/kg | ABLC at 10 mg/kg | |
| Brain | |||||
| Meningoencephalitis, necrotizing | (3/3) | (5/5) | (4/4) | (6/6) | (7/7) |
| Minimal to mild | 1/3 | 4/5 | 1/4 | 4/6 | 4/7 |
| Moderate to marked | 2/3 | 1/5 | 3/4 | 2/6 | 3/7 |
| Intralesional fungi, vascular invasion | 2/3 | 3/4 | 3/6 | 4/7 | |
| Kidney | |||||
| Nephritis, necrotizing, minimal to marked | 2/3 | 2/4 | |||
| Intralesional fungi, parenchyma and/or vascular | 2/3 | 2/4 | |||
Numbers in parentheses represent the number of animals with this finding over the total number of animals examined microscopically.
Table 2.
Mean severity score for CNS inflammation in Aspergillus fumigatus-infected cyclophosphamide-immunosuppressed CD-1 mice
| Treatment | Mean lesion severity scorea |
|---|---|
| Control D5W | 2.67 |
| AMBd at 1 mg/kg | 1.80 |
| AmBi at 3 mg/kg | 2.75 |
| AmBi at 10 mg/kg | 1.83 |
| ABLC at 10 mg/kg | 2.57 |
Differences between groups were not statistically significant by a Mann-Whitney U test.
In the kidneys, necrotizing, neutrophilic, histiocytic inflammation consistent with fungal nephritis was noted only in animals treated with D5W or AmBi at 3 mg/kg (Table 1). In contrast, all animals had recoverable CFU in the kidneys regardless of treatment regimen, and no regimen was superior to another (Fig. 1). AmBi at 3 mg/kg was associated with more inflammation in the kidney compared to the other drug regimens. A. fumigatus infection was also identified in the lung (D5W control group only) and liver (D5W control and ABLC at 10 mg/kg groups only); however, the infection was less extensive compared to those in other organs with only random multifocal areas of inflammation and necrosis evident. CFU were not determined in these organs.
Organs from infected and uninfected mice also were evaluated for evidence of drug-related toxicity. Minimal to mild drug-related microscopic findings were identified in the kidney and liver in the drug treatment groups studied (AmBi, ABLC, and AMBd). No such changes were noted in the control group, affirming that the changes described in the treatment groups were drug related. There was also no pattern of difference between infected and uninfected mice. Kidney findings were limited to minimal to mild, focal to multifocal, tubular dilation with intraluminal protein casts, and/or concurrent tubular regeneration (Table 3). The tubular changes were slightly increased in incidence in the treatment groups receiving AmBi or ABLC at 10 mg/kg or AMBd at 1 mg/kg compared to those in the treatment group receiving AmBi at 3 mg/kg (infected and uninfected).
Table 3.
Four-day study: incidence and severity of histological changes in infected and uninfected CD-1 mice given pretreatment with cyclophosphamide
| Histopathology finding in kidney | No. of animals positive/total no. of animalsa by antifungal treatment group, without (−) or with (+) A. fumigatus infection |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No-treatment control |
AMBd at 1.0 mg/kg |
AmBi at 3.0 mg/kg |
AmBi at 10.0 mg/kg |
ABLC at 10 mg/kg |
||||||
| − | + | − | + | − | + | − | + | − | + | |
| Tubular regeneration, minimal to mild | 0/5 | 0/3 | 3/5 | 1/5 | 1/5 | 0/4 | 3/5 | 2/6 | 1/5 | 1/7 |
| Tubular dilation, minimal to mild | 0/5 | 0/3 | 1/5 | 1/5 | 1/5 | 1/4 | 3/5 | 2/6 | 3/5 | 2/7 |
The numerator represents the number of animals with this finding, and the denominator represents the number of animals examined microscopically.
Similarly, minimal liver changes (random mononuclear cell infiltrates and/or individual hepatocyte necrosis) were identified in uninfected and infected animal groups treated with AmBi or ABLC at 10 mg/kg or AMBd at 1 mg/kg but not in the treatment group receiving AmBi at 3 mg/kg (data not shown). These changes were considered to be of limited biological effect. The other organs examined (i.e., brain, heart, lungs) showed no drug-related microscopic changes.
Immunohistochemistry and localization of amphotericin B in the CNS.
Capillaries within and surrounding areas of inflammation, malacia (necrosis), and fungal infection were more prominent, often dilated, and lined by hypertrophic (activated) endothelium. Amphotericin B localized in the activated (hypertrophic) endothelial cells found in these areas of inflammation/infection (Fig. 4, Table 4). Endothelial cell staining for amphotericin B was identified in each amphotericin B formulation treatment group (i.e., AmBi at 3 mg/kg, AmBi at 10 mg/kg, AMBd at 1 mg/kg, and ABLC at 10 mg/kg) of infected animals and was increased in incidence in the ABLC treatment group compared to that in the AmBi and AMBd treatment groups. Positive endothelial cells had 2+ to 3+ diffuse to granular cytoplasmic staining. Fungal hyphae, neutrophils, monocytes/macrophages walling off and surrounding areas of malacia, endothelium lining the blood vessels in unaffected areas of the brain, and the capillary endothelium in the brain of all uninfected drug-treated animals consistently showed no staining for amphotericin B.
Fig 4.

Brain, A. fumigatus infected. Immunohistochemistry staining to localize amphotericin B. Amphotericin B-positive staining identified in reactive (hypertrophic) endothelium lining blood vessels in and surrounding areas of inflammation and intravascular mononuclear cells, presumptive monocytes (arrows). (A) Negative staining of inflammatory infiltrates (i) and capillary endothelium (arrowheads) in the D5W control group. Amphotericin B-positive endothelial cells lining dilated capillaries in 1 mg/kg AMBd (B), 3 mg/kg AmBi (C), and 10 mg/kg ABLC (D) treatment groups. V, blood vessel; i, inflammatory cell infiltrates. Faint nonspecific background staining was present in areas of inflammation in both drug-treated and control groups.
Table 4.
Immunohistochemical localization of anti-amphotericin B staining in the central nervous systems of infected and uninfected mice
| Treatment group | No. of animals with amphotericin B staining/total no. of animals examined |
||
|---|---|---|---|
| Endotheliuma | Intravascular monocytes | Evidence of infection/inflammation in tissue section | |
| Infected | |||
| D5W control | 0/3 | 0/3 | 2/3 |
| AmBi at 3 mg/kg | 3/4 | 1/4 | 3/4 |
| AmBi at 10 mg/kg | 4/6 | 0/6 | 6/6 |
| AMBd at 1 mg/kg | 2/5 | 0/5 | 4/5 |
| ABLC at 10 mg/kg | 7/7 | 7/7 | 7/7 |
| Uninfected | |||
| D5W control | 0/2 | 0/2 | 0/2 |
| AmBi 3 at mg/kg | 0/2 | 0/2 | 0/2 |
| AmBi at 10 mg/kg | 0/2 | 0/2 | 0/2 |
| AMBd at 1 mg/kg | 0/2 | 0/2 | 0/2 |
| ABLC at 10 mg/kg | 0/2 | 2/2 | 0/2 |
Endothelium staining was observed only in the infected areas.
Additionally, anti-amphotericin B staining was identified in intravascular mononuclear cells (presumptive monocytes) in the brains of all infected and uninfected mice treated with ABLC and in one infected mouse treated with AmBi at 3 mg/kg (Fig. 4, Table 4). These intravascular mononuclear cells had intense amphotericin B (4+) cytoplasmic staining.
Expression of ICAM-1 and P-selectin in activated endothelium.
Capillary endothelium in areas of inflammation and immediately adjacent to areas of inflammation had increased intensity and frequency of ICAM-1 staining in all infected groups, drug treated and control (Fig. 5, Table 5). In uninfected brain or areas distant to the site of infection, only rare capillaries had faint- to mild (1 to 2+)-intensity ICAM-1 staining that is likely due to low-level constitutive expression, whereas ICAM-1 staining was increased in intensity to 3+ to 4+ in capillaries within and adjacent to areas of inflammation. In areas of necrosis, ICAM-1 staining was decreased or absent.
Fig 5.

Brain, A. fumigatus infected. Immunohistochemistry staining for ICAM-1. Increased intensity and frequency of ICAM-1 endothelial cell staining in areas of inflammation identified in all animals, drug treated and control. In areas of necrosis, ICAM staining was decreased or absent. Marked ICAM-1 staining of capillary endothelium in 10 mg/kg ABLC (A), 10 mg/kg AmBi (B), and D5W control (C) treatment groups. Inset, higher magnification of ICAM-1-positive capillary endothelium, D5W control. N, necrosis; i, inflammatory cell infiltrates.
Table 5.
Increased ICAM-1 and P-selectin staining of capillary endothelium associated with inflammation in the CNS (infected animals only)
| Infected treatment group | Marker | Incidence of capillary endothelium staining in areas of inflammation (no. positive/total no.) |
|---|---|---|
| D5W control | ICAM-1+ | 2/2 |
| P-selectin+ | 1/2 | |
| AmBi at 3 mg/kg | ICAM-1+ | 3/3 |
| P-selectin+ | 2/3 | |
| AmBi at 10 mg/kg | ICAM-1+ | 5/5 |
| P-selectin+ | 0/5 | |
| AMBd at 1 mg/kg | ICAM-1+ | 3/3 |
| P-selectin+ | 1/3 | |
| ABLC at 10 mg/kg | ICAM-1+ | 7/7 |
| P-selectin+ | 5/7 |
P-selectin is not constitutively expressed in the mouse cerebral vasculature, which differs from ICAM-1 expression. Therefore, P-selectin was included for study as potentially a more sensitive indicator of endothelial cell activity. P-selectin staining was identified rarely in capillaries and only in areas of inflammation in some infected animals, both in control mice and in all treatment groups, except for the group receiving AmBi at 10 mg/kg (Fig. 6, Table 5). P-selectin-positive capillaries had very faint 1+ granular cytoplasmic staining. The reason for the lack of P-selectin staining in the treatment group receiving AmBi at 10 mg/kg is unknown and warrants further investigation.
Fig 6.
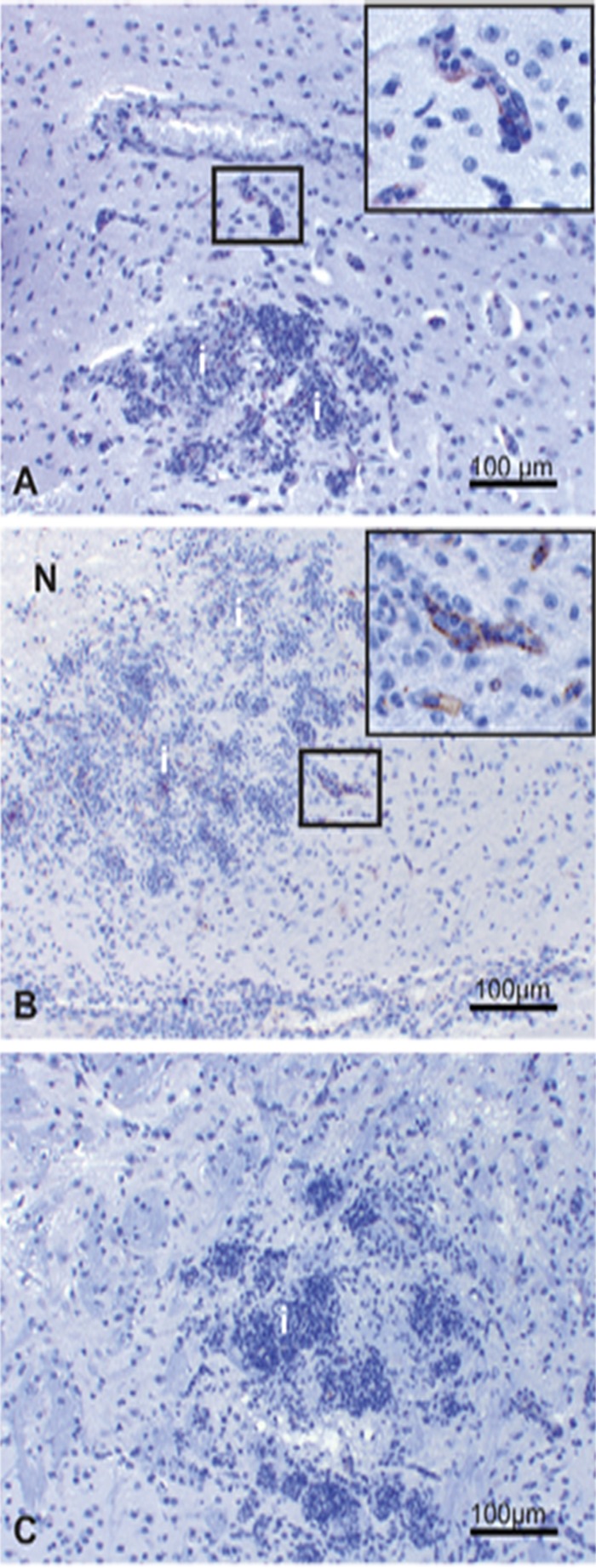
Brain, A. fumigatus infected. Immunohistochemistry staining for P-selectin. Rare, faint positive P-selectin endothelial cell staining in capillaries adjacent to the area of inflammation in the D5W control (A) and 10 mg/kg ABLC (B) treatment groups. Boxes indicate areas of P-selectin staining. Insets show high magnification of positive capillary staining in areas demarcated by a box (A, B). (C) Negative P-selectin staining in the 10 mg/kg AmBi treatment group. N, necrosis; i, inflammatory cell infiltrates.
Subacute toxicity: 10-day study.
To better understand the potential for drug-induced toxicity, we compared drug treatment in cyclophosphamide-treated or untreated uninfected mice. Drugs were administered for 10 consecutive days, and blood and tissue samples were collected 4 days after the last treatment. In the course of treatment, deaths occurred only in groups given an amphotericin B preparation and cyclophosphamide; 3 of 5 given ABLC at 10 mg/kg and none given ABLC at 5 mg/kg died, whereas 2 given AmBi at 5 mg/kg died and other treatments had 1 mouse die (Table 6). The liver and kidney histopathologic studies suggested a toxicologic explanation for these deaths.
Table 6.
Ten-day study: incidence and severity of histological changes observed in uninfected CD-1 mice given no cyclophosphamide or pretreated with cyclophosphamide
| Organ and histopathology | No. of surviving animals, with finding indicated, by antifungal treatment group (mg/kg/day), without (−) or with (+) cyclophosphamide treatment |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No tx control |
AMBd 1.0 |
AmBi 1.0 |
AmBi 5.0 |
AmBi 10.0 |
ABLC 1.0 |
ABLC 5.0 |
ABLC 10.0 |
|||||||||
| − | + | − | + | − | + | − | + | − | + | − | + | − | + | − | + | |
| Surviving animals (of 5 per group) | 5 | 5 | 5 | 4 | 5 | 4 | 5 | 3 | 5 | 4 | 5 | 4 | 5 | 5 | 5 | 2 |
| Kidney | ||||||||||||||||
| Any abnormality | 0 | 0 | 3 | 3 | 0 | 1 | 1 | 1 | 0 | 3 | 0 | 1 | 2 | 1 | 2 | 0 |
| Tubular regeneration, minimal to mild | 0 | 0 | 3 | 2 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 2 | 0 |
| Tubular dilation, minimal | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 |
| Nephrosis, moderate | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Liver | ||||||||||||||||
| Any abnormality | 0 | 0 | 1 | 1 | 4 | 0 | 5 | 3 | 5 | 4 | 3 | 2 | 5 | 3 | 5 | 2 |
| Hepatocellular degeneration, minimal | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 3 | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| Hepatocellular necrosis, mild | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 4 | 1 | 0 | 1 | 2 | 0 | 2 | 0 |
| Inflammation, mixed, minimal | 0 | 0 | 1 | 1 | 4 | 0 | 5 | 1 | 5 | 2 | 3 | 1 | 3 | 1 | 3 | 2 |
| Kupffer cell hyperplasia and vacuolizationa | (0) | (0) | (0) | (0) | (1) | (0) | (4) | (3) | (5) | (4) | (0) | (1) | (0) | (3) | (2) | (1) |
| Minimal | 0 | 0 | 0 | 0 | 1 | 0 | 4 | 3 | 2 | 0 | 0 | 1 | 0 | 1 | 2 | 0 |
| Mild | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 2 | 0 | 1 |
| Moderate | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Marked | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
Minimal, mild, moderate, and marked severity modifiers were included: minimal, <25% of tissue affected; mild, 25 to 50%; moderate, >50 to 75%; marked, >75%. The numbers in parentheses represent the total number of animals with the histopathology finding (all severity scores).
AMBd, AmBi, or ABLC treatment with or without cyclophosphamide treatment was associated with renal tubular changes (Table 6). In general, changes were minimal in survivors and included tubular regeneration, dilatation (degenerative change), and protein cast formation. However, nephrosis, defined as extensive tubular damage with accompanying interstitial fibrosis, was identified in 1 of 2 surviving animals treated with ABLC in combination with cyclophosphamide and in none of the 5 survivors treated with ABLC at the same dose without cyclophosphamide, suggesting that cyclophosphamide may accelerate the development of nephrotoxicity.
Average BUN and creatinine values were within normal limits in all groups except the group receiving AmBi at 10 mg/kg and cyclophosphamide. However, data for the treatment group receiving ABLC at 10 mg/kg and cyclophosphamide were biased due to early deaths in this group, which left only 2 animals for evaluation and sampling at day 14; one animal that survived to the time of terminal euthanasia had normal BUN and creatinine values, and the other had a mild increase in BUN, compared to the controls, in the absence of an increase in creatinine (i.e., prerenal azotemia). This finding was suggestive of, but not conclusive for, acute renal impairment.
Liver enzyme values in all drug-treated groups were comparable to the controls and within normal limits for this age and strain of mouse. However, localized hepatic toxicity, characterized by hepatocellular necrosis, degeneration (hydropic), and inflammation, was identified primarily in association with AmBi and ABLC treatment at 5 or 10 mg/kg without cyclophosphamide treatment. The incidence of these hepatocellular changes was greater with AmBi treatment than with ABLC treatment; however, the severity was the same. Overall, the hepatic lesions were decreased with cyclophosphamide treatment, suggesting that the liver injury may be associated with inflammatory mediators, with immunosuppression by the cyclophosphamide reducing the drug-associated injury. In both the AmBi and ABLC treatment groups, hepatocellular inflammation tended to increase in incidence in a dose-dependent fashion. Kupffer hyperplasia and vacuolization occurred with both AmBi and ABLC treatment and increased in incidence and severity in a dose-dependent fashion. In higher-dose groups (i.e., 5 and 10 mg/kg of AmBi or ABLC), the incidence and/or severity of Kupffer cell hyperplasia/vacuolization were increased with AmBi treatment compared to those with ABLC treatment. This suggests that AmBi was more readily phagocytized and sequestered in the hepatic reticuloendothelial cells (Kupffer cells) than was ABLC.
DISCUSSION
Our aims in the current study addressed the observed efficacy profiles of lipid-carried amphotericin B preparations and how related toxicity and tissue distribution might affect these profiles. Overall, our results indicate that each of the drugs tested showed some efficacy against CNS aspergillosis. However, efficacy as judged by quantification of fungal burden versus pathological assessment did not necessarily correlate. For example, AmBi at 3 mg/kg was judged most effective by reduction of CFU in the organs, whereas by pathological assessment AmBi at 10 or AMBd at 1 mg/kg appeared to be the most effective. Determining efficacy solely on the basis of histologic assessment should be cautioned, since in this study, e.g., kidneys judged to be free of infection by histology were shown infected by culture.
Interestingly, treatment groups (e.g., AmBi at 3 mg/kg) showing fewer CFU in the tissues (i.e., an indicator of drug efficacy) also tended to show the most severe inflammatory response and a higher mean lesional severity score. The reasons for this are not clear. It may suggest that an accompanying inflammatory response is a necessary component of an effective therapeutic response.
Recovery of A. fumigatus from the brain showed once again that treatment with high doses of AmBi are not necessary for efficacy and may be less efficacious, as AmBi at 3 mg/kg was superior to AmBi at 10 mg/kg. These data are similar to those from previous experimental studies, where high dosages of AmBi were less effective than lower dosages in CNS disease due to A. fumigatus or Coccidioides posadasii (5, 8) or pulmonary aspergillosis (24). Similar results have been demonstrated in clinical trials for the treatment of opportunistic infections, where higher doses offered no demonstrable benefit but did increase deleterious effects of AmBi (28). We have noted similar results in dose escalation studies of ABLC for treatment of CNS aspergillosis (6). In addition, the concentrations of amphotericin B in the tissues or serum in our study did not correlate with the efficacy results. For instance, ABLC treatment resulted in the highest concentration of amphotericin B in the kidneys but was not the most efficacious and was equivalent to the other treatment groups. This finding may reflect availability of drug in an organ for antifungal effect. Thus, dose escalation of lipid-carried amphotericin B formulations may not be necessary, or even beneficial, for optimal treatment of some fungal infections. In studies using different strains of mice, different immunosuppressive regimens, more doses (six), and a longer interval till sampling than were used in our study, AmBi was detected in kidneys and lungs after doses of 5 or 10 mg of AmBi/kg (30), which is in contrast to our current results, where levels were below the limit of detection in those organs.
Lipid formulations of AMB were developed to overcome the dose limiting nephrotoxicity of conventional amphotericin B described in rodents (38) and humans (15). The safety profile has been significantly improved with lipid formulations, but as we push the dosing limits, toxicities become apparent. Other studies have reported dose-dependent renal toxicity with amphotericin B in mice (38), as well as renal and hepatic toxicity of AmBi in rats (2). Previously, we showed the occurrence of renal nephrosis and hepatic cell necrosis and inflammation in mice treated with AmBi or ABLC and that these toxicities were more severe in conjunction with corticosteroid treatment (7). We found similar nephrotoxicity in this study. We noted that at day 4, high ABLC concentrations were present in the kidney, but significant morphological changes were not. Nephrotoxicity was noted after 10 days of treatment in the ABLC and cyclophosphamide treatment group but not in animals given ABLC alone or AmBi with or without cyclophosphamide. This finding suggests that ABLC-related nephrotoxicity may have been accelerated with cyclophosphamide treatment. Thus, renal toxicity may have contributed to the poor survival in the high-dose ABLC groups. Although prominent morphological changes were not evident in the kidney after 4 days of treatment, high concentrations of amphotericin B as ABLC were present and may be a precursor to late nephrotoxicity.
Hepatotoxicity could also have contributed to the inefficacy of high-dose ABLC and AmBi. Interestingly, hepatotoxicity was decreased when AmBi and ABLC were given in combination with cyclophosphamide, and liver enzymes fell within a normal range. These results are suggestive that renal toxicity was the more important factor.
We demonstrated, by immunohistochemistry, that amphotericin B localizes in activated cerebral capillary endothelium in areas of inflammation and infection, as well as in circulating mononuclear cells in the brain. Our study is the first to demonstrate the cellular distribution of amphotericin B in vivo in a model of CNS aspergillosis. Multiple studies have shown relative higher concentrations of AMB in the brain in animals with fungal disease than in uninfected controls (17). For example, Groll et al. (17) noted relatively higher concentrations of amphotericin B given as ABLC or amphotericin B colloidal dispersion (ABCD) in the brain of rabbits with CNS disease due to Candida albicans than in uninfected rabbits; concentrations of AMBd or AmBi were not different between infected and uninfected mice. Those authors also proposed that infection-caused leakiness of the capillaries allowed for diffusion of the amphotericin B, at least when given as ABLC, into the tissues, resulting in concentrations sufficient to be efficacious, whereas the mechanism of penetration by AMBd and AmBi remains unknown (17). Although we found amphotericin B to be taken up by specific cells, we were unable to definitively detect amphotericin B in neuropil by immunohistochemistry. The topographically limited AMB signal identified could explain why drug concentrations of amphotericin B in the brain were below a detectable level, as determined by bioassay of the whole organ. Our novel finding of amphotericin B in activated endothelium clearly demonstrates that AMBd, AmBi, or ABLC could cross the blood-brain barrier and reach the areas of infection, and although efficacious, these concentrations were not curative.
Interestingly, circulating amphotericin B-positive mononuclear cells were identified in infected animals in higher prevalence in animals treated with ABLC than in those treated with AMBi; they also were identified in uninfected animals treated with ABLC but not those treated with AmBi. ABLC and AmBi are known to be recognized by the mononuclear phagocyte system. However, ABLC-formulated amphotericin B distributes differently in tissue than does AmBi-formulated drug. In a recent study in rats, ABLC-derived amphotericin B was found in significantly greater concentrations in the spleen than AmBi-derived amphotericin B, which was found in higher concentration in the liver (16). Others have shown similar differences in biodistribution in the mononuclear phagocytic system (37). Phagocytosis of ABLC and AmBi has typically been thought of as a means of clearance of drug from the bloodstream. However, perhaps amphotericin B within mononuclear cells is delivered to the site of CNS infection via systemic recruitment of mononuclear cells. Additional studies are needed to assess mononuclear cell recruitment and entry of drug-laden mononuclear cells into the CNS in areas of infection and inflammation.
The mechanism by which AMBd and its lipid formulations are taken up by activated endothelial cells is unknown. One possibility is that of amphotericin B binding to serum albumin. Serum albumin can act as a chaperone protein to move hydrophobic molecules, including drugs, across the endothelial cell barrier, which may be an important mechanism of drug transport to specific organs (27). Using an in vitro model of invasive pulmonary aspergillosis, Lestner et al. (25) showed that amphotericin B derived from deoxycholate or lipid formulations had a higher affinity for endothelial cells than for the fluid-phase compartment or the extracellular compartment. Our study is in agreement and demonstrates amphotericin B uptake by cerebral endothelial cells in vivo at the site of infection. In addition, amphotericin B-positive endothelium had no evidence of cell injury by light microscopy. Vascular necrosis was identified only in areas of active fungal infection, and amphotericin B was not detected in these vessels. The lack of endothelial cell staining in areas of necrosis, or in abscesses containing many aspergilli, may be indicative of why there is a lack of cure by these formulations in that the drug is not able to penetrate established fungal abscesses.
Immediately after CNS injury, cell adhesion and selectin molecules on the luminal surface of activated endothelial cells are upregulated. These adhesion molecules make it possible for their ligand counterparts on leukocytes and platelets to tether to the endothelial surface and eventually for transendothelial cell transport of sensitized leukocytes to occur (31). ICAM-1 is a cell adhesion molecule expressed by several cell types, including endothelial cells. This molecule is induced by a variety of factors (e.g., cytokines, steroids, etc.) and plays a role in leukocyte migration in general but notably into the brain through the blood-brain barrier (13, 40). Furthermore, upregulation of ICAM-1 expression on CNS vascular endothelial cells occurs during brain inflammation (13, 20). P-selectin (CD62P) is an adhesion glycoprotein that is stored in Weibel-Palade bodies of endothelial cells and platelets, which are released upon activation. Both adhesion molecules are nonspecific markers of inflammation but have potential utility in monitoring vascular injury or maintenance of the blood-brain barrier in CNS injury due to noninfectious or infectious processes. Although E-selectin is a sensitive, specific marker for endothelial cell activation, it was not assayed in these studies, because it is not expressed (or expressed below detectable levels) by endothelial cells in the CNS of mice. However, E-selectin may have utility as a marker of endothelial cell activation in humans. Our ICAM-1 and P-selectin results suggest the expression of ICAM-1 during experimental CNS aspergillosis plays an important role in recruitment of leukocytes to the area of infection. In contrast to the increased expression of ICAM-1 by activated endothelial cells, upregulation of P-selectin was less prevalent and is likely indicative that it plays a much less significant role in CNS aspergillosis.
Amphotericin B is known to have immunomodulatory activities, including the upregulation of ICAM-1 expression by human monocytes (32, 33). Although we showed localization of amphotericin B in capillary endothelium with upregulation of ICAM-1, it is unclear whether or not amphotericin B played a role in the upregulation. Induction of ICAM-1 by the endothelial cells may promote leukocyte migration to sites of infection; however, amphotericin B also inhibits chemotaxis of neutrophils and possibly depresses the initial immune response. Tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β), which are induced by amphotericin B (10, 26, 32, 35), could aid in the recruitment of mononuclear cells to the CNS through ICAM-1 expression by endothelial cells (9, 29) and induction of MCP-1 (14). These cytokines have also been implicated in amphotericin B infusion-related toxicities (1, 41), probably a result of promoting leakiness of the endothelial vasculature (e.g., kidneys or brain).
Overall, our data are indicative that efficacy of an amphotericin B preparation in the CNS does not correlate with tissue or serum concentrations or histopathology. Although immunohistochemistry staining showed that amphotericin B can be demonstrated locally at the sites of infection in the brain, it was not found in close proximity to fungal hyphae or within more severe lesions. Thus, the lack of curative efficacy is likely a result of failure of the amphotericin B to reach viable organisms present in areas of necrosis or abscess. Various degrees of nephrotoxicity occurred with all three of the amphotericin B preparations tested and thus would remain a concern when treating CNS aspergillosis. Interestingly, the concurrent administration of cyclophosphamide apparently decreased hepatic toxicity, which is in contrast to the increased renal damage noted in those same animals. The contribution of the immunomodulatory effects of amphotericin B to the toxicity profile and to the overall efficacy of the drug should be examined in future studies.
ACKNOWLEDGMENTS
These studies were funded in part by a grant from Gilead Sciences, Inc.
We thank James Raymond, Daniel Pekala, and Marife Martinez for their assistance during the course of these studies.
Footnotes
Published ahead of print 11 June 2012
REFERENCES
- 1. Arning M, Kliche KO, Heer-Sonderhoff AH, Wehmeier A. 1995. Infusion-related toxicity of three different amphotericin B formulations and its relation to cytokine plasma levels. Mycoses 38:459–465 [DOI] [PubMed] [Google Scholar]
- 2. Boswell GW, Bekersky I, Buell D, Hiles R, Walsh TJ. 1998. Toxicological profile and pharmacokinetics of a unilamellar liposomal vesicle formulation of amphotericin B in rats. Antimicrob. Agents Chemother. 42:263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chiller TM, et al. 2002. Development of a murine model of cerebral aspergillosis. J. Infect. Dis. 186:574–577 [DOI] [PubMed] [Google Scholar]
- 4. Chiller TM, Sobel RA, Luque JC, Clemons KV, Stevens DA. 2003. Efficacy of amphotericin B or itraconazole in a murine model of central nervous system Aspergillus infection. Antimicrob. Agents Chemother. 47:813–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clemons KV, Espiritu M, Parmar R, Stevens DA. 2005. Comparative efficacies of conventional amphotericin B, liposomal amphotericin B (AmBisome), caspofungin, micafungin, and voriconazole alone and in combination against experimental murine central nervous system aspergillosis. Antimicrob. Agents Chemother. 49:4867–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clemons KV, Parmar R, Martinez M, Stevens DA. 2006. Efficacy of Abelcet alone, or in combination therapy, against experimental central nervous system aspergillosis. J. Antimicrob. Chemother. 58:466–469 [DOI] [PubMed] [Google Scholar]
- 7. Clemons KV, Schwartz JA, Stevens DA. 2011. Therapeutic and toxicologic studies in a murine model of invasive pulmonary aspergillosis. Med. Mycol. 49:834–847 [DOI] [PubMed] [Google Scholar]
- 8. Clemons KV, Sobel RA, Williams PL, Pappagianis D, Stevens DA. 2002. Efficacy of intravenous liposomal amphotericin B (AmBisome) against coccidioidal meningitis in rabbits. Antimicrob. Agents Chemother. 46:2420–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coisne C, et al. 2006. Differential expression of selectins by mouse brain capillary endothelial cells in vitro in response to distinct inflammatory stimuli. Neurosci. Lett. 392:216–220 [DOI] [PubMed] [Google Scholar]
- 10. Darisipudi MN, Allam R, Rupanagudi KV, Anders HJ. 2011. Polyene macrolide antifungal drugs trigger interleukin-1β secretion by activating the NLRP3 inflammasome. PLoS One 6:e19588 doi:10.1371/journal.pone.0019588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denning DW, Clemons KV, Stevens DA. 1992. Quantitative preservation of viability of Aspergillus fumigatus. J. Med. Vet. Mycol. 30:485–488 [DOI] [PubMed] [Google Scholar]
- 12. Denning DW, Stevens DA. 1990. Antifungal and surgical treatment of invasive aspergillosis: review of 2,121 published cases. Rev. Infect. Dis. 12:1147–1201 [DOI] [PubMed] [Google Scholar]
- 13. Dietrich JB. 2002. The adhesion molecule ICAM-1 and its regulation in relation with the blood-brain barrier. J. Neuroimmunol. 128:58–68 [DOI] [PubMed] [Google Scholar]
- 14. D'Mello C, Le T, Swain MG. 2009. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factor-α signaling during peripheral organ inflammation. J. Neurosci. 29:2089–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gallis HA, Drew RH, Pickard WW. 1990. Amphotericin B: 30 years of clinical experience. Rev. Infect. Dis. 12:308–329 [DOI] [PubMed] [Google Scholar]
- 16. Gershkovich P, et al. 2009. Pharmacokinetics and biodistribution of amphotericin B in rats following oral administration in a novel lipid-based formulation. J. Antimicrob. Chemother. 64:101–108 [DOI] [PubMed] [Google Scholar]
- 17. Groll AH, et al. 2000. Comparative efficacy and distribution of lipid formulations of amphotericin B in experimental Candida albicans infection of the central nervous system. J. Infect. Dis. 182:274–282 [DOI] [PubMed] [Google Scholar]
- 18. Hanson LH, Clemons KV, Denning DW, Stevens DA. 1995. Efficacy of oral saperconazole in systemic murine aspergillosis. J. Med. Vet. Mycol. 33:311–317 [DOI] [PubMed] [Google Scholar]
- 19. Hanson LH, Stevens DA. 1992. Comparison of antifungal activity of amphotericin B deoxycholate suspension with that of amphotericin B cholesteryl sulfate colloidal dispersion. Antimicrob. Agents Chemother. 36:486–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hofer S, et al. 2008. Injury of the blood brain barrier and up-regulation of ICAM-1 in polymicrobial sepsis. J. Surg. Res. 146:276–281 [DOI] [PubMed] [Google Scholar]
- 21. Imai J, Singh G, Fernandez B, Clemons KV, Stevens DA. 2005. Efficacy of Abelcet and caspofungin, alone or in combination, against CNS aspergillosis in a murine model. J. Antimicrob. Chemother. 56:166–171 [DOI] [PubMed] [Google Scholar]
- 22. Imai JK, Singh G, Clemons KV, Stevens DA. 2004. Efficacy of posaconazole in a murine model of central nervous system aspergillosis. Antimicrob. Agents Chemother. 48:4063–4066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lachin JM. 1999. Worst-rank score analysis with informatively missing observations in clinical trials. Control. Clin. Trials 20:408–422 [DOI] [PubMed] [Google Scholar]
- 24. Leenders AC, de Marie S, ten Kate MT, Bakker-Woudenberg IA, Verbrugh HA. 1996. Liposomal amphotericin B (AmBisome) reduces dissemination of infection compared with amphotericin B deoxycholate (Fungizone) in a rat model of pulmonary aspergillosis. J. Antimicrob. Chemother. 38:215–225 [DOI] [PubMed] [Google Scholar]
- 25. Lestner JM, et al. 2010. Pharmacokinetics and pharmacodynamics of amphotericin B deoxycholate, liposomal amphotericin B, and amphotericin B lipid complex in an in vitro model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother. 54:3432–3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Louie A, Baltch AL, Franke MA, Smith RP, Gordon MA. 1994. Comparative capacity of four antifungal agents to stimulate murine macrophages to produce tumour necrosis factor alpha: an effect that is attenuated by pentoxifylline, liposomal vesicles, and dexamethasone. J. Antimicrob. Chemother. 34:975–987 [DOI] [PubMed] [Google Scholar]
- 27. Mehta D, Malik AB. 2006. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86:279–367 [DOI] [PubMed] [Google Scholar]
- 28. Munoz P, Guinea J, Narbona MT, Bouza E. 2008. Treatment of invasive fungal infections in immunocompromised and transplant patients: AmBiLoad trial and other new data. Int. J. Antimicrob. Agents 32(Suppl 2):S125–S131 [DOI] [PubMed] [Google Scholar]
- 29. Murray HW. 2000. Mononuclear cell recruitment, granuloma assembly, and response to treatment in experimental visceral leishmaniasis: intracellular adhesion molecule 1-dependent and -independent regulation. Infect. Immun. 68:6294–6299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olson JA, et al. 2010. Liposomal amphotericin B and echinocandins as monotherapy or sequential or concomitant therapy in murine disseminated and pulmonary Aspergillus fumigatus infections. Antimicrob. Agents Chemother. 54:3884–3894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prendergast CT, Anderton SM. 2009. Immune cell entry to central nervous system—current understanding and prospective therapeutic targets. Endocr. Metab. Immune Disord. Drug Targets 9:315–327 [DOI] [PubMed] [Google Scholar]
- 32. Rogers PD, Pearson MM, Cleary JD, Sullivan DC, Chapman SW. 2002. Differential expression of genes encoding immunomodulatory proteins in response to amphotericin B in human mononuclear cells identified by cDNA microarray analysis. J. Antimicrob. Chemother. 50:811–817 [DOI] [PubMed] [Google Scholar]
- 33. Rogers PD, Stiles JK, Chapman SW, Cleary JD. 2000. Amphotericin B induces expression of genes encoding chemokines and cell adhesion molecules in the human monocytic cell line THP-1. J. Infect. Dis. 182:1280–1283 [DOI] [PubMed] [Google Scholar]
- 34. Shih W. 2002. Problems in dealing with missing data and informative censoring in clinical trials. Curr. Control Trials Cardiovasc. Med. 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simitsopoulou M, et al. 2005. Differential expression of cytokines and chemokines in human monocytes induced by lipid formulations of amphotericin B. Antimicrob. Agents Chemother. 49:1397–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Singh G, Imai J, Clemons KV, Stevens DA. 2005. Efficacy of caspofungin against central nervous system Aspergillus fumigatus infection in mice determined by TaqMan PCR and CFU methods. Antimicrob. Agents Chemother. 49:1369–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith PJ, et al. 2007. Effects of dosing regimen on accumulation, retention and prophylactic efficacy of liposomal amphotericin B. J. Antimicrob. Chemother. 59:941–951 [DOI] [PubMed] [Google Scholar]
- 38. Tonomura Y, et al. 2009. Amphotericin B-induced nephrotoxicity: characterization of blood and urinary biochemistry and renal morphology in mice. Hum. Exp. Toxicol. 28:293–300 [DOI] [PubMed] [Google Scholar]
- 39. Tucker R, Williams P, Arathoon E, Stevens D. 1988. Treatment of mycoses with itraconazole. Ann. N. Y. Acad. Sci. 544:451–470 [DOI] [PubMed] [Google Scholar]
- 40. van de Stolpe A, van der Saag PT. 1996. Intercellular adhesion molecule-1. J. Mol. Med. 74:13–33 [DOI] [PubMed] [Google Scholar]
- 41. Vonk AG, et al. 1998. Modulation of the pro- and anti-inflammatory cytokine balance by amphotericin B. J. Antimicrob. Chemother. 42:469–474 [DOI] [PubMed] [Google Scholar]