

Abstract
JTK-853 is a novel piperazine derivative nonnucleoside inhibitor of hepatitis C virus (HCV) RNA-dependent RNA polymerase. JTK-853 showed potent inhibitory activity against genotype 1 HCV polymerase, with a 50% inhibitory concentration in the nanomolar range, and showed potent antiviral activity against the genotype 1b replicon, with a 50% effective concentration of 0.035 μM. The presence of human serum at up to 40% had little effect on the antiviral activity of JTK-853. Structure analysis of HCV polymerase with JTK-853 revealed that JTK-853 associates with the palm site and β-hairpin region of HCV polymerase, and JTK-853 showed decreased antiviral activity against HCV replicons bearing the resistance mutations C316Y, M414T, Y452H, and L466V in the palm site region of HCV polymerase. JTK-853 showed an additive combination effect with other DAAs (direct antiviral agents), such as nucleoside polymerase inhibitor, thumb pocket-binding nonnucleoside polymerase inhibitor, NS5A inhibitor, and protease inhibitor. Collectively, these data demonstrate that JTK-853 is a potent and novel nonnucleoside palm site-binding HCV polymerase inhibitor, suggesting JTK-853 as a potentially useful agent in combination with other DAAs for treatment of HCV infections.
INTRODUCTION
Hepatitis C virus (HCV) is the major cause of posttransfusion non-A non-B hepatitis. Approximately 130 million individuals worldwide are estimated to be infected with HCV (3). It has been suggested that the development of liver cirrhosis and hepatocellular carcinoma are consequences of chronic infection with HCV (38). HCV, a member of the Flaviviridae family, has a single-stranded positive-sense linear RNA genome of approximately 9.5 kb (11, 20, 34). The RNA encodes a single precursor polyprotein of approximately 3,010 amino acids (7, 28) that is co- and posttranslationally cleaved by host and viral proteases to produce individual structural (core, E1, E2, and p7) and nonstructural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins (12, 13). Most of the HCV NS proteins are required for viral RNA replication.
The NS5B protein encoding the viral RNA-dependent RNA polymerase (RdRp) is responsible for the replication of HCV (5, 24). Because of its apparent sequence and structural differences from human DNA and RNA polymerases, the HCV RNA polymerase is an attractive target for antiviral drugs. To date, the effects of a variety of nucleoside and nonnucleoside polymerase inhibitors have been reported. Nonnucleoside polymerase inhibitors (NNIs) interact with four distinct allosteric sites on HCV polymerase (4). We previously reported the discovery of several HCV polymerase inhibitors with a benzimidazole and indole core based on high-throughput screening and structure-based drug design (14, 15, 18, 19).
Here, we report the in vitro antiviral activity of a novel and potent nonnucleoside HCV polymerase inhibitor, JTK-853, (2R)-4-(5-cyclopropylthiazolo[4,5-d]pyrimidin-2-yl)-N-[3-fluoro-4-(trifluoromethoxy)benzyl]-1-[4-(trifluoromethyl)phenylsulfonyl]piperazine-2-carboxamide. JTK-853 showed strong inhibitory activity on the replication of the HCV replicon, and the activity remained unchanged even in the presence of human serum (HS) and HS proteins. An in vitro selection study of JTK-853 showed that C316Y, M414T, Y452H, and L466V were the predominant mutations conferring resistance to JTK-853. Structural analysis demonstrated that JTK-853 associates with the palm I site of HCV polymerase differently from other HCV polymerase palm site inhibitors. In patients infected with genotype 1 HCV, JTK-853 effectively reduced the viral load (30), supporting its use as an effective oral antiviral agent in HCV-infected patients.
MATERIALS AND METHODS
Compounds.
JTK-853 (patent WO 2007119889) was synthesized at Japan Tobacco, Inc., Central Pharmaceutical Research Institute (Osaka, Japan) (Fig. 1). PSI-6130, PF-868554, BMS-790052, and TMC435 (2, 10, 22, 33) were also synthesized at Japan Tobacco. Ribavirin was purchased from Sigma-Aldrich (St. Louis, MO). Alpha interferon (IFN-α; Sumiferon 300) was purchased from Dainippon Sumitomo Pharma (Osaka, Japan).
Fig 1.
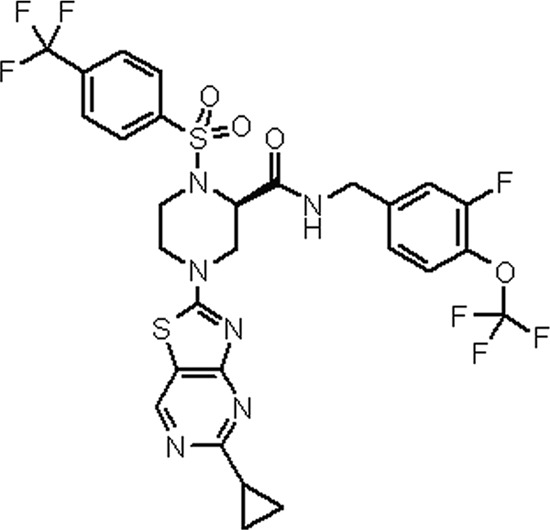
Chemical structure of JTK-853.
Enzymes.
Recombinant HCV NS5B RdRp of genotype 1a strain H77 (6), 1b strain BK (1, 34), 1b strain Con1 (25), and 2a strain JFH1 (39) were expressed in Escherichia coli and purified as previously described (1). All these enzymes for RdRp assays have a truncation of 47 amino acids at the C terminus and the addition of a 6His tag (GSHHHHH) at the C terminus and are designated NS5B544. The production of soluble full-length NS5B enzyme (591 amino acids) is very difficult. Alternatively, the enzyme with a truncation of C-terminal amino acid residues (NS5B544) was used (1, 17, 32, 33, 37). The NS5B gene of genotypes 3a and 4a was amplified from commercially available HCV-infected patient serum (ProMedDx, Norton, MA) and then expressed in Escherichia coli and purified as described above. Human DNA polymerases α, β, and γ were purchased from CHIMERx (Milwaukee, WI).
Cells.
Huh-9-13 and Huh-5-2 genotype 1b strain Con1 HCV replicon cells (21, 23, 25) were obtained from ReBLikon GmbH (Heidelberg, Germany). H/SG Neo (L+I) genotype 1a strain H77 HCV replicon cells (6), were obtained from Apath LLC (St. Louis, MO). Huh-9-13 and H/SG Neo (L+I) cells harbor the HCV subgenome (NS3 to the 3′-untranslated region of the HCV RNA genome), and Huh-5-2 cells harbor a luciferase gene as a reporter in addition to the HCV subgenome. All these replicon cells harbor a selectable marker, neomycin phosphotransferase, that confers Geneticin resistance. These replicon cells were propagated in high-glucose Dulbecco's modified Eagle's medium (Nikken BioMedical Laboratory, Kyoto, Japan) containing 10% fetal bovine serum (Moregate Biotech, Bulimba, Australia), 0.1 mM nonessential amino acids (Invitrogen, Carlsbad, CA), 100 units/ml of penicillin (Invitrogen), 100 μg/ml of streptomycin sulfate (Invitrogen), 2 mM l-glutamine (Invitrogen), and 0.5 to 1 mg/ml Geneticin (Invitrogen).
Biochemical RdRp assay.
RdRp assays were carried out using 5 μg/ml of the 3′-terminal HCV RNA (615 nucleotides [Con1]) as a template primer. The reaction mixture contained 0.5 to 1.5 μg/ml HCV polymerase (NS5B544), 0.4 μM [5,6-3H]UTP (1.63 to 1.85 TBq/mmol), 50 μM ATP, 50 μM GTP, 50 μM CTP, 0.3 μM UTP, 20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 50 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 0.01% bovine serum albumin, and dimethyl sulfoxide at a final concentration of 5%, as previously described (1, 9, 15). After incubation for 60 min at 25°C, the RdRp reactions were terminated by the addition of 10% trichloroacetic acid–1% sodium diphosphate solution. RdRp activity was measured by the glass filter trapping method.
DNA-dependent DNA polymerase assays were performed following the manufacturer's instructions (CHIMERx) and previous reports (27, 40).
HCV replicon assay.
Antiviral activity was evaluated in Huh-9-13 cells, H/SG Neo (L+I) cells, and Huh-5-2 cells. These cells were seeded in a 96-well plate at 0.5 × 104 to 1 × 104 cells and cultured at 37°C in 5% CO2. On the following day, the replicon cells were treated with JTK-853 as a 0.3% final concentration of dimethyl sulfoxide and further cultured for 2 days. The HCV replication was determined based on the amount of HCV RNA in the total RNA or by the reporter gene luciferase activity. HCV RNA was quantified by real-time reverse transcription-PCR (RT-PCR) with a TaqMan probe (15, 35). Luciferase activity was measured using the Steady-Glo luciferase assay system (Promega, Fitchburg, WI). The 50% effective concentration (EC50) and EC90 values of JTK-853 were calculated from the inhibitory activity against HCV replication. Cytotoxicity was measured in an assay with [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS; CellTiter 96; Promega), and the 50% cytotoxic concentration (CC50 value) was also determined.
To evaluate the effect of protein binding on the antiviral activity of JTK-853, either 10 to 40% HS or 4.4% human serum albumin (HSA) plus 0.08% α1-acid glycoprotein (AAG) were added to the culture medium. These supplementation levels are thought to be almost equivalent to the physiological concentrations in human blood. HS was purchased from the Interstate Blood Bank (Memphis, TN) or Gemini Bio-Products (West Sacramento, CA); HSA and AAG were purchased from Sigma-Aldrich.
Structural analysis.
The cocrystallization analysis of JTK-853 with HCV polymerase genotype 1b BK strain was performed as previously described (1, 18). HCV polymerase with a truncation of 21 amino acids at the C terminus, designated NS5B570, was used in this study because the production of soluble full-length NS5B protein was very difficult (1, 17, 32, 33). The complex of NS5B570 and JTK-853 was prepared by cocrystallization. NS5B570 was concentrated to up to 20 mg/ml and was mixed with 0.5 mM JTK-853 in 5 mM 2-mercaptoethanol containing 1% dimethyl sulfoxide. Cocrystallization of JTK-853 and NS5B570 was performed by the hanging drop vapor diffusion method at 10°C with mixing of 1:1 volumes of the protein with inhibitor solution and a reservoir solution. As the reservoir solution, 13 to 15% (wt/vol) polyethylene glycol 4000, 0.1 M sodium acetate (pH 6.0), 0.3 M sodium chloride, 2-mercaptoethanol, and 2.4 mM n-octanoylsucrose were employed. The diffraction data were collected on the BL17A apparatus in the Photon Factory, KEK (Tsukuba, Japan). The molecular images of the cocrystallization structure of NS5B570 with JTK-853 were generated with the PyMOL Molecular Graphics system (DeLano Scientific, San Carlos, CA).
In vitro selection experiments.
To select JTK-853 resistance mutations in the HCV replicon of genotype 1b, Huh-9-13 HCV replicon cells were cultured in the presence of 1 μM JTK-853 (approximately 25 times higher than the EC50) and 1 mg/ml Geneticin for 24 days. Cells able to grow under these conditions were further cultured in the presence of 3 μM JTK-853 for 18 days. The medium containing JTK-853 was changed twice a week.
Total cellular RNA was extracted from JTK-853-resistant cells by using an RNeasy minikit (Qiagen, Hilden, Germany). The NS5B region in the JTK-853-resistant HCV replicon was amplified by RT-PCR using the primers 5′-GTGAGGACGTCGTCTGCTGC-3′ (forward) and 5′-TGGAGTGTTTAGCTCCCCGT-3′ (reverse). The amplified products were ligated to the pCR Blunt II TOPO vector using the Zero Blunt TOPO PCR cloning kit (Invitrogen) and transformed into E. coli DH5α (Toyobo, Osaka, Japan). Plasmids were purified using a QIAfilter plasmid midikit (Qiagen), and then the nucleoside sequence was determined using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) and ABI Prism 3100 genetic analyzer (Applied Biosystems).
Amino acid substitutions identified in the NS5B region of the JTK-853-resistant replicons were introduced into the wild-type replicon by PCR-based site-directed mutagenesis. The EcoRI site in NS5A and the SpeI site just outside the 3′-untranslated region were used for cloning.
In vitro combination study.
The combination inhibitory effect of JTK-853 on replication of the HCV replicon was analyzed using the Prichard and Shipman MacSynergy II three-dimensional model (29). This model uses the Bliss independence null reference model. Briefly, HCV replicon cells were treated with various concentrations of JTK-853 and other DAAs in a checkerboard fashion, and the antiviral activity was determined. In this model, the antiviral activity was defined as the difference between the percent theoretical additive inhibition of each compound alone and the percent experimental inhibition produced by combinations of two compounds, and the results were plotted on the z axis in the three-dimensional model as the percent inhibition. The combination effect was defined as the extent of synergy or antagonism, which was determined by subtracting the theoretical additive surface (Z = 0) from the actual experimental volume in the three-dimensional graph. Synergism, antagonism, and additive effects were defined as follows: synergism, volume difference of >50; additive, −50 ≤ volume ≤ 50; antagonism, volume difference of below −50. The volume (difference) was calculated with a 95% confidence interval.
Protein structure accession number.
The cocrystallization structure coordinate of JTK-853 bound to HCV polymerase has been deposited in the Protein Data Bank with the PDB code 3VQS.
RESULTS
Biochemical activity of JTK-853 in vitro.
The lead compound of JTK-853 was selected from Japan Tobacco chemical libraries by high-throughput screening. The novel and highly potent HCV polymerase inhibitor JTK-853 (Fig. 1) was generated by optimization of the lead compound.
JTK-853 inhibited the RdRp activity of the genotype 1b Con1 and BK and genotype 1a H77 strains with 50% inhibitory concentration (IC50) values of 0.00832 μM, 0.0178 μM, and 0.0173 μM, respectively. On the other hand, the IC50s against HCV polymerases of genotypes 2, 3, and 4 were >10 μM, 0.277 μM, and 0.214 μM, respectively (Table 1). JTK-853 did not inhibit human DNA polymerases at the highest concentration tested of 100 μM, indicating that JTK-853 is >5,600-fold more selective against HCV polymerase than to human DNA polymerases (Table 1).
Table 1.
Biochemical activity of JTK-853
| Enzyme and strain | IC50 (μM)a |
|---|---|
| HCV NS5B polymerase in viral strain | |
| 1a H77 | 0.0173 ± 0.0011 |
| 1b Con1 | 0.00832 ± 0.00011 |
| 1b BK | 0.0178 ± 0.0008 |
| 2a JFH-1 | >10 |
| 3a | 0.277 ± 0.044 |
| 4a | 0.214 ± 0.024 |
| Human DNA polymerases α, β, and γ | >100 |
Data represent means ± standard errors of three independent experiments.
In vitro antiviral activity of JTK-853.
The antiviral activity of JTK-853 in HCV replicon cells was determined as previously described (15, 35). JTK-853 demonstrated potent antiviral activity against the genotype 1b Con1 strain replicon, with an EC50 of 0.0347 μM and an EC90 of 0.146 μM without any toxicity (Table 2). The EC50 and EC90 values of JTK-853 against the genotype 1a H77 replicon were 0.378 μM and 1.09 μM, respectively.
Table 2.
In vitro antiviral activities of JTK-853
| HCV replicon | EC50 (μM)a | EC90 (μM)a | CC50 (μM) |
|---|---|---|---|
| 1a H77 | 0.378 ± 0.02 | 1.09 ± 0.03 | >10 |
| 1b Con1 | 0.0347 ± 0.0031 | 0.146 ± 0.013 | >10 |
Data represent means ± standard errors of three independent experiments.
Effect of protein binding on the antiviral activity of JTK-853.
To estimate the effect of protein binding on the antiviral activity of JTK-853, HS or HS proteins were added to the culture medium of genotype 1b HCV replicon cells. The EC90 of JTK-853 in 40% HS was 0.276 μM, which was affected little, even in the presence of human serum up to 40% (Table 3). The protein binding percentages of JTK-853 to 100% HS and 40% HS plus 10% fetal bovine serum (FBS) by equilibrium analysis were 99.87% and 99.74%, respectively. Therefore, free JTK-853 from protein binding was 0.13% and 0.26%, suggesting the protein shift of JTK-853 in 100% HS is almost comparable to that of 40% HS plus 10% FBS. In addition, the antiviral activity of JTK-853 was also evaluated in the presence of 4.4% HSA plus 0.08% AAG, suggesting physiological concentrations of these major proteins in human blood. The EC90 of JTK-853 in the presence of 4.4% HSA plus 0.08% AAG was 0.449 μM (Table 3). The antiviral activity of JTK-853 remained potent even in the presence of HS or HS proteins.
Table 3.
In vitro antiviral activities of JTK-853 in the presence of human serum or human serum proteins
| Condition | EC90 (μM)a |
|---|---|
| 0% HS | 0.146 ± 0.013 |
| 10% HS | 0.138 ± 0.010 |
| 20% HS | 0.149 ± 0.010 |
| 40% HS | 0.276 ± 0.017 |
| 4.4% HSA + 0.08% AAG | 0.449 ± 0.074 |
Data represent means ± standard errors of three independent experiments.
Cocrystallization structure of JTK-853 and HCV polymerase.
Next, we investigated the binding mode of JTK-853 to HCV polymerase (BK strain) by structure analysis of the cocrystal. Figure 2 shows, at 1.9-Å resolution, that JTK-853 binds to the palm site of HCV polymerase. The thiazolopyrimidine ring of JTK-853 appeared to participate in a π-stacking interaction with F193, and it formed hydrogen bonds with S288 and water-bridged hydrogen bonds with V179 and Y191 residues of HCV polymerase. The carboxamide of JTK-853 interacted with the Y195 backbone carbonyl by hydrogen bond, and the 3-fluoro-4-trifluoromethoxy benzene moiety appeared to participate in a CH-π interaction with M414. Interestingly, the 4-trifluoromethyl benzene of JTK-853 bound to the hydrophobic pocket of HCV polymerase, which we termed the LWF pocket in the previous report (1). The LWF pocket included residue Y452 of the β-hairpin region. These results indicate that JTK-853 interacts with the β-hairpin region as well as with the palm site I of HCV polymerase. Thus, the binding mode of JTK-853 to HCV polymerase is characteristic and clearly distinct from that of other palm site-binding HCV polymerase inhibitors (16, 32).
Fig 2.
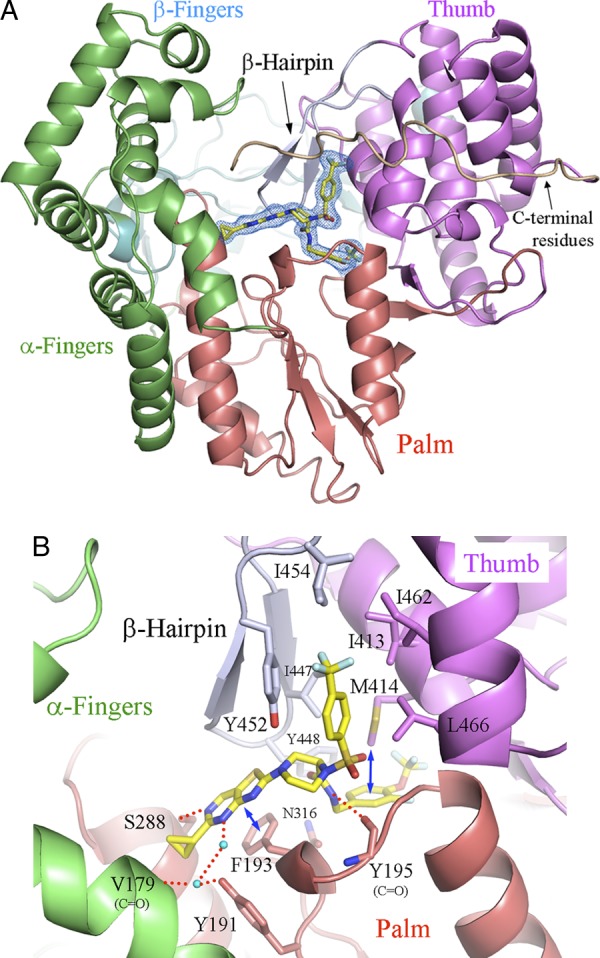
Cocrystallization structure of JTK-853 and the HCV polymerase of strain BK, at 1.9-Å resolution. JTK-853 is indicated as a yellow stick model. Nitrogen, oxygen, sulfur, and fluorine atoms are shown in blue, red, orange and light blue, respectively. The α-fingers, β-fingers, and palm and thumb domains of HCV polymerase are shown in green, cyan, red, and purple, respectively. The β-hairpin and C-terminal residues are shown in gray and wheat, respectively. (A) Overall structure of JTK-853 and HCV polymerase. the Omit Fo − Fc electron density of JTK-853 is shown as a blue mesh, contoured at 3 σ. (B) The JTK-853-binding site of HCV polymerase. The red broken lines indicate hydrogen bonds between JTK853 and HCV polymerase. Some of them are mediated by water molecules (cyan spheres). The blue arrows indicate a hydrophobic interaction.
Phenotypic and genotypic analysis of JTK-853-resistant replicon cells.
JTK-853-resistant mutations were selected using genotype 1b Con1 HCV replicon cells. JTK-853-resistant replicon cells were obtained by culturing the cells with 1 μM JTK-853 for 24 days. JTK-853 showed a 59-fold reduction of antiviral activity in the JTK-853-resistant replicon cells. Then, the JTK-853-resistant replicon cells were cultured with 3 μM JTK-853 for a further 18 days, and the nucleotide sequences of the NS5B region in the resistant cells were obtained by clonal sequencing analysis. C316Y, M414T, Y452H, and L466V mutations were detected in the JTK-853-resistant replicon cells, reflecting the association of JTK-853 with the β-hairpin region and the palm site I of HCV polymerase. In addition to the single mutations, C316Y Y452H and Y452H L466V double mutations were also detected.
Next, these mutations were introduced into wild-type HCV replicon cells, and the antiviral activities of JTK-853 in these mutant replicon cells were evaluated. The EC50s of JTK-853 in the C316Y, M414T, Y452H, and L466V mutant replicon cells were attenuated by fold increases of 58 ± 10 (mean ± standard error), 44, 44 ± 3, and 21 ± 3 compared with that in wild-type replicon cells, respectively. The antiviral activity of JTK-853 was attenuated more in cells with the double mutation C316Y with Y452H or double mutation Y452H with L466V (>198-fold change in the EC50 for both) than in the single mutation cells. On the other hand, no apparent change in susceptibility to IFN-α was found, since the fold changes in the EC50 for IFN-α were between 2 and 4. Several sporadic mutations, such as Q184R and T418A, were also detected, but they were not related to susceptibility to JTK-853 (data not shown). It is likely that the C316Y, M414T, Y452H, and L466V mutations are involved in resistance to JTK-853 in the HCV replicon cells.
Structure analysis showed that the residues C316 (N316 in strain BK), M414, Y452, and L466 are located around JTK-853 of the HCV polymerase–JTK-853 complex (Fig. 2). This suggests that these amino acid substitutions impair the binding affinity of JTK-853 to HCV polymerase.
We further evaluated the antiviral activity of JTK-853 in other DAA-derived resistant replicons bearing an amino acid substitution: S282T from HCV polymerase nucleotide inhibitors; L419M, M423T, or P495A from HCV polymerase thumb pocket inhibitors (17, 26, 33, 37); R155K, A156V, or D168A from protease inhibitors (22, 31); or L31V or Y93H from NS5A inhibitors (10). The antiviral activity of JTK-853 in these resistant replicons remained as potent as that of the wild-type replicon (data not shown).
Combination effects of JTK-853 with other DAAs on HCV replication.
Finally, we investigated combination effects of JTK-853 with other DAAs on replication of the HCV replicon. HCV replicon cells were treated with various concentrations of JTK-853 and other DAAs in a checkerboard fashion, and the antiviral activities were determined. The combination effects were analyzed by using the Bliss independence model (MacSynergy II) (29). The combinations of JTK-853 with PSI-6130 (nucleoside inhibitor), PF-868554 (thumb pocket nonnucleoside inhibitor), TMC435 (protease inhibitor), BMS-790052 (NS5A inhibitor), or IFN-α with ribavirin resulted in additive inhibitory effects on HCV replication, as shown in Table 4 and Fig. 3. In contrast, 2′-C-methylcytidine with ribavirin showed an antagonistic effect in this model, as described elsewhere (8). The cytotoxicity of each combination was also evaluated at the maximum combination concentration. None of the combinations produced cytotoxicity (data not shown). These results suggested JTK-853 as potentially useful in combination with other DAAs and IFN-α with ribavirin for treatment of HCV infections.
Table 4.
In vitro combination effects of JTK-853
| Drug combinationa | Vol difference for synergy/antagonism (from synergy plot with 95% CI) | Combination effect |
|---|---|---|
| JTK-853 + PSI-6130 | 0.03/−7.50 | Additive |
| JTK-853 + PF-868554 | 0.00/0.00 | Additive |
| JTK-853 + BMS-790052 | 30.94/−15.36 | Additive |
| JTK-853 + TMC435 | 0.00/−18.91 | Additive |
| JTK-853 + IFN-α + ribavirin | 1.38/−1.97 | Additive |
| Ribavirin + 2′-C-methylcytidineb | 0.00/−252.42 | Antagonism |
With the exceptions of the JTK-853 + PF-868554 and JTK-853 + BMS-790052 experiments, the representative results of three independent experiments are shown.
Positive control for antagonism.
Fig 3.

Combination effect on replication of the HCV replicon. The Prichard and Shipman MacSynergy II three-dimensional model was used to evaluate this effect. The percent inhibition on the z axis was defined as the difference between the theoretical additive percent inhibition of each compound alone and that of the experimental percent inhibition for the two compounds in combination. The combination effect was defined as the volume of synergy or antagonism, which was determined based on the difference of the theoretical additive surface (Z = 0) from the actual experimental volume in the three-dimensional graph. Synergism, antagonism, and additive effects were defined as follows: synergism, volume difference of >5; additive, −50 ≤ volume ≤ 50; antagonism, volume difference below −50. The 95% confidence interval was determined. (A to E) Combination effects of JTK-853 with PSI-6130, PF-868554, BMS-790052, TMC435, or of IFN-α plus ribavirin. (F) Combination effect of ribavirin with 2′-C-methylcytidine.
DISCUSSION
There remains a significant unmet need for a targeted, efficacious chemotherapy against HCV genotype 1. HCV genotype 1 is prevalent worldwide, especially in the United States, Europe, and Japan, but the current therapy is poorly tolerated and is not sufficiently effective in HCV genotype 1-infected patients (36, 41). Two protease inhibitors (Telaprevir and Boceprevir) were already launched for HCV treatment in 2011. The treatment outcome was improved by these new drugs but not to a sufficient extent and, furthermore, drug-resistant variant viruses emerged easily in the clinical trials.
We have demonstrated here that JTK-853 produces potent and selective inhibition of the HCV RdRp of genotypes 1a and 1b. Notably, the 316th amino acid residue of HCV polymerase is polymorphic, as seen with Con1 with cysteine (C316) and BK with asparagine (N316). JTK-853 showed inhibitory activities against HCV polymerases of Con1 and BK strains in the same range, suggesting that JTK-853 has inhibitory activity against both HCV polymerases regardless of the amino acid polymorphism at amino acid 316.
The attenuation of the antiviral activity of JTK-853 by the addition of HS to the HCV replicon cells was very small. The protein binding levels of JTK-853 to 100% HS or 40% HS plus 10% FBS by equilibrium analysis were 99.87% and 99.74%, respectively. Therefore, free JTK-853 from protein binding was 0.13% and 0.26%, suggesting the protein shift of JTK-853 in 100% HS is almost comparable to that in 40% HS plus 10% FBS.
Similarly, the physiological concentrations of major HS proteins (4.4% HSA plus 0.08% AAG) produced just a slight attenuation of the antiviral activity of JTK-853, with an EC90 of 0.449 μM. The results of the protein shift with JTK-853 in 100% HS and the EC90 value of JTK-853 in the physiological concentration of human serum protein strongly demonstrate that the antiviral activity of JTK-853 might be maintained even upon protein binding. This is in clear agreement with the demonstration of the compound's antiviral activity in HCV-infected patients (30).
We previously demonstrated that the HCV polymerase C-terminal residues LWF (Leu547, Trp550, and Phe551) have an autoinhibitory effect on RdRp activity (1). This inhibition is caused by occupation of a putative RNA-binding cleft in HCV polymerase by the LWF residues per se. The LWF residue-binding region has been designated the LWF pocket and is located in the palm and thumb domain. Our structural analysis demonstrated that JTK-853 interacts with the LWF pocket in the palm site as well as with the β-hairpin region, by a distinctly different mode from that of other palm site-binding HCV polymerase inhibitors. Particularly, the association of JTK-853 with the LWF pocket of HCV polymerase is characteristic, because JTK-853 occupies the LWF pocket and displaces the LWF residues of HCV polymerase at the LWF pocket, suggesting the strong inhibitory activity of JTK-853 on HCV polymerase.
Our in vitro resistance selection study of JTK-853 identified the amino acid substitutions C316Y, M414T, Y452H, and L466V at the palm site of the HCV polymerase. Importantly, the antiviral activity of JTK-853 remained unchanged by the resistance mutations from other DAAs. In addition, JTK-853 showed an additive combination effect with other DAAs. These results suggest that JTK-853 could be used in combination with DAAs, such as other class polymerase inhibitors, protease inhibitors, and NS5A inhibitors.
In conclusion, we have presented the results of a preclinical in vitro study of JTK-853 and demonstrated a potent and specific antiviral activity against HCV genotype 1. In fact, JTK-853 administered in a twice-daily dosing regimen has demonstrated potent antiviral activity in genotype 1 HCV-infected patients (30), supporting its beneficial use as an oral antiviral agent for hepatitis C.
ACKNOWLEDGMENTS
We acknowledge all members of the HCV polymerase project team at the Japan Tobacco Central Pharmaceutical Research Institute (Takatsuki, Osaka, Japan), the Pharmaceutical Frontier Research Laboratories (Yokohama, Kanagawa, Japan), the Toxicology Research Laboratories (Hadano, Kanagawa, Japan), and the Clinical Research Planning Department (Tokyo). We thank Tsutomu Shibata, Kunihiro Hirahara, and Mitsuki Kano for coordinating the research and Satoki Doi for crystal structure determination. We also thank Soichi Wakatsuki (High Energy Accelerator Research Organization, Tsukuba, Japan) for data collection at the Photon Factory, KEK.
Footnotes
Published ahead of print 21 May 2012
REFERENCES
- 1. Adachi T, et al. 2002. The essential role of C-terminal residues in regulating the activity of hepatitis C virus RNA-dependent RNA polymerase. Biochim. Biophys. Acta 1601:38–48 [DOI] [PubMed] [Google Scholar]
- 2. Ali S, et al. 2008. Selected replicon variants with low-level in vitro resistance to the hepatitis C virus NS5B polymerase inhibitor PSI-6130 lack cross-resistance with R1479. Antimicrob. Agents Chemother. 52:4356–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alter MJ. 2007. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 13:2436–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beaulieu PL. 2009. Recent advances in the development of NS5B polymerase inhibitors for the treatment of hepatitis C virus infection. Expert Opin. Ther. Pat. 19:145–164 [DOI] [PubMed] [Google Scholar]
- 5. Behrens S-E, Tomei L, De Francesco R. 1996. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 15:12–22 [PMC free article] [PubMed] [Google Scholar]
- 6. Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Choo QI, et al. 1991. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 88:2451–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coelmont L, et al. 2006. Ribavirin antagonizes the in vitro anti-Hepatitis C virus activity of 2′-C-methylcytidine, the active component of valopicitabine. Antimicrob. Agents Chemother. 50:3444–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Francesco R, Behrens SE, Tomei L, Altamura S, Jiricny J. 1996. RNA-dependent RNA polymerase of hepatitis C virus. Methods Enzymol. 275:58–67 [DOI] [PubMed] [Google Scholar]
- 10. Fridell RA, Qiu D, Wang C, Valera L, Gao M. 2010. Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob. Agents Chemother. 54:3641–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hijikata M, Kato N, Ootsuyama Y, Nakagawa M, Shimotohno K. 1991. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. U. S. A. 88:5547–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hijikata M, et al. 1993. Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J. Virol. 67:4665–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hijikata M, et al. 1993. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 90:10773–10777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hirashima S, et al. 2007. Further studies on hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors toward improved replicon cell activities: benzimidazole and structurally related compounds bearing the 2-morpholinophenyl moiety. Bioorg. Med. Chem. Lett. 17:3181–3186 [DOI] [PubMed] [Google Scholar]
- 15. Hirashima S, et al. 2006. Benzimidazole derivatives bearing substituted biphenyls as hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors: structure-activity relationship studies and identification of a potent and highly selective inhibitor JTK-109. J. Med. Chem. 49:4721–4736 [DOI] [PubMed] [Google Scholar]
- 16. Howe AY, et al. 2008. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob. Agents Chemother. 52:3327–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Howe AY, et al. 2006. Molecular mechanism of a thumb domain hepatitis C virus nonnucleoside RNA-dependent RNA polymerase inhibitor. Antimicrob. Agents Chemother. 50:4103–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ikegashira K, et al. 2006. Discovery of conformationally constrained tetracyclic compounds as potent hepatitis C virus NS5B RNA polymerase inhibitors. J. Med. Chem. 49:6950–6953 [DOI] [PubMed] [Google Scholar]
- 19. Ishida T, et al. 2006. Benzimidazole inhibitors of hepatitis C virus NS5B polymerase: identification of 2-[(4-diarylmethoxy)phenyl]-benzimidazole. Bioorg. Med. Chem. Lett. 16:1859–1863 [DOI] [PubMed] [Google Scholar]
- 20. Kato N, et al. 1990. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc. Natl. Acad. Sci. U. S. A. 87:9524–9528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krieger N, Lohmann V, Bartenschlager R. 2001. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75:4614–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lenz O, et al. 2010. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob. Agents Chemother. 54:1878–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lohmann V, Körner F, Dobierzewska A, Bartenschlager R. 2001. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J. Virol. 75:1437–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lohmann V, Körner F, Herian U, Bartenschlager R. 1997. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 71:8416–8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lohmann V, et al. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 26. Migliaccio G, et al. 2003. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 278:49164–49170 [DOI] [PubMed] [Google Scholar]
- 27. Naviaux RK, Markusic D, Barshop BA, Nyhan WL, Haas RH. 1999. Sensitive assay for mitochondrial DNA polymerase γ. Clin. Chem. 45:1725–1733 [PubMed] [Google Scholar]
- 28. Okamoto H, et al. 1991. Nucleotide sequence of the genomic RNA of hepatitis C virus isolated from a human carrier: comparison with reported isolates for conserved and divergent regions. J. Gen. Virol. 72:2697–2704 [DOI] [PubMed] [Google Scholar]
- 29. Prichard MN, Prichard LE, Shipman C., Jr 1993. A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 14:181–206 [DOI] [PubMed] [Google Scholar]
- 30. Rodriguez-Torres M, et al. 2011. Abstr. HEP DART 2011: frontiers in drug development for viral hepatitis, abstr 57. IHL Press, Tucker, GA [Google Scholar]
- 31. Sarrazin C, et al. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777 [DOI] [PubMed] [Google Scholar]
- 32. Shaw AN, et al. 2009. Substituted benzothiadizine inhibitors of hepatitis C virus polymerase. Bioorg. Med. Chem. Lett. 19:4350–4353 [DOI] [PubMed] [Google Scholar]
- 33. Shi ST, et al. 2009. Preclinical characterization of PF-00868554, a potent nonnucleoside inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 53:2544–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takamizawa A, et al. 1991. Structure and organization of the hepatitis C virus genome isolated from human carriers. J. Virol. 65:1105–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takeuchi T, et al. 1999. Real-time detection system for quantification of hepatitis C virus genome. Gastroenterology 16:636–642 [DOI] [PubMed] [Google Scholar]
- 36. Tellinghuisen TL. 2011. An overview of the hepatitis C virus life cycle, p 1–48 In Tan SL, Yupeng H. (ed), Hepatitis C: antiviral drug discovery and development. Caister Academic Press, Hethersett, Norfolk, United Kingdom [Google Scholar]
- 37. Tomei L, et al. 2003. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 77:13225–13231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tong CY, Gilmore IT, Hart CA. 1995. HCV-associated liver cancer. Lancet 345:1058–1059 [DOI] [PubMed] [Google Scholar]
- 39. Wakita T, et al. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang G, et al. 2007. Highly selective action of triphosphate metabolite of 4′-ethynyl D4T: a novel anti-HIV compound against HIV-1 RT. Antiviral Res. 73:185–191 [DOI] [PubMed] [Google Scholar]
- 41. Zein NN. 2000. Clinical significance of hepatitis C virus genotypes. Clin. Microbiol. Rev. 13:223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]