

Abstract
Emergence of drug-resistant bacteria represents a high, unmet medical need, and discovery of new antibacterials acting on new bacterial targets is strongly needed. ATP synthase has been validated as an antibacterial target in Mycobacterium tuberculosis, where its activity can be specifically blocked by the diarylquinoline TMC207. However, potency of TMC207 is restricted to mycobacteria with little or no effect on the growth of other Gram-positive or Gram-negative bacteria. Here, we identify diarylquinolines with activity against key Gram-positive pathogens, significantly extending the antibacterial spectrum of the diarylquinoline class of drugs. These compounds inhibited growth of Staphylococcus aureus in planktonic state as well as in metabolically resting bacteria grown in a biofilm culture. Furthermore, time-kill experiments showed that the selected hits are rapidly bactericidal. Drug-resistant mutations were mapped to the ATP synthase enzyme, and biochemical analysis as well as drug-target interaction studies reveal ATP synthase as a target for these compounds. Moreover, knockdown of the ATP synthase expression strongly suppressed growth of S. aureus, revealing a crucial role of this target in bacterial growth and metabolism. Our data represent a proof of principle for using the diarylquinoline class of antibacterials in key Gram-positive pathogens. Our results suggest that broadening the antibacterial spectrum for this chemical class is possible without drifting off from the target. Development of the diarylquinolines class may represent a promising strategy for combating Gram-positive pathogens.
INTRODUCTION
Drug-resistant bacterial infections are a leading cause of mortality worldwide, and their increasing prevalence represents a serious threat to human health (6, 34). As stated in recent public policy documents issued by the Infectious Diseases Society of America (IDSA), the key bacterial pathogens are drug-resistant staphylococci, enterococci, and several Gram-negative pathogens (ESKAPE pathogens) (5, 37). Among the Gram-positive-resistant isolates, it was estimated that multidrug-resistant Staphylococcus aureus infections led to 19,000 deaths per year in the United States, with an associated 3 to 4 billion U.S. dollars in annual health care costs (12). Despite this high mortality rate, there is a paucity of new antibacterial agents within the pharmaceutical pipeline (6). The genomics-driven strategies have been largely ineffective in delivering new antibacterials, and most of the antibiotics developed in the last decade are molecules reengineered from existing antibiotic classes. These reengineered entities represent incremental progress and may retain some of the drawbacks of the parent class. Exploring new compound classes and validating novel target pathways can significantly contribute to addressing the unmet medical need of the increasing antibacterial resistance and its associated burden on human health.
Bacterial energy metabolic pathways are largely unexplored as drug targets for the resistant Gram-positive and Gram-negative pathogens (3, 19). Bacteria can produce ATP by substrate-level phosphorylation of fermentable carbon sources or by oxidative phosphorylation using the respiratory chain and ATP synthase. It has recently been demonstrated that blocking the ATP synthase enzyme by TMC207 (Fig. 1), a new clinical diarylquinoline drug candidate, is a highly efficient strategy for the treatment of drug-resistant Mycobacterium tuberculosis (1, 24). TMC207 is currently in phase IIb clinical trials in patients with multidrug-resistant tuberculosis. However, although TMC207 shows strong potency on a large number of mycobacterial species, it does not display significant activity on the growth of nonmycobacterial pathogenic bacteria (1). Moreover, mycobacteria are quite distinct from other Gram-positive or Gram-negative bacteria in terms of susceptibility for currently used antibacterials (23, 33). As such, it is an open question to what extent small-molecule inhibitors of the diarylquinoline class, acting on energy metabolic pathways, can be used as broad-spectrum antibiotic agents. In this regard, we embarked on novel medicinal chemistry and on chemical-biology approaches to identify new inhibitors with a diarylquinoline chemical scaffold for key Gram-positive bacteria, including S. aureus and Streptococcus pneumoniae. We test the potency of the new compounds on replicating bacteria as well as on bacteria in biofilms and characterize their mode of action. We furthermore demonstrate the promise of these compounds as novel starting points for discovering new drugs to combat bacterial infections.
Fig 1.

Chemical structures of TMC207 and compounds 1 to 5.
MATERIALS AND METHODS
Chemistry of diarylquinolines.
Based on structure-activity relationships of TMC207 analogs, the medicinal chemistry optimization efforts focused on new diarylquinoline chemotypes in order to improve potency against Gram-positive pathogens. The core structures of compounds remain similar to that of TMC207, but the new derivatives are characterized by novel specific lateral chains. More than 500 compounds with a modulation of the chain lengthening between the basic moiety and the carbon bearing the hydroxyl group have been synthesized and tested against Gram-positive bacteria. Another chemical library of 200 compounds has been constructed with increased flexibility around the two stereogenic centers borne by the lateral chain. The addition of spacers between aryl groups and chiral centers reduced significantly the steric hindrance on the lateral chain.
Determination of antibacterial activity.
Determination of MICs were conducted according to the guidelines of the Clinical and Laboratory Standards Institute (CLSI) (8). Briefly, bacteria were grown to logarithmic phase, diluted to ≈105 CFU/ml, and grown in the presence of serial dilutions of the tested compounds for 20 h. MIC was determined as the lowest concentration with no visual growth.
Evaluation of cytotoxicity in HelaM cells.
Cytotoxicity of the compounds was evaluated using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. Human HelaM cells were exposed to serial dilutions of the tested compounds and incubated for 72 h (37°C; 5% CO2). MTT was added, and cell viability was determined by measuring the absorbance of the reduced formazan at 540 nm and 690 nm. The absorbance measured at 690 nm was automatically subtracted from the absorbance at 540 nm to eliminate the effects of nonspecific absorption. The percent cytotoxicity achieved by the compounds was calculated according to standard methods. Cytotoxicity is reported as CC50, the concentration that causes a 50% reduction in cell viability.
Biofilm susceptibility testing.
Biofilm cultivation and susceptibility testing were performed using the MBEC assay as previously described (7). Briefly, S. aureus LMG8064 was grown for 24 h (37°C) under orbital agitation in brain heart infusion (BHI) broth to inoculate the minimum biofilm eradication concentration (MBEC) assay and to cultivate statically a biofilm at 37°C. Serial dilutions (100 to 0.2 μg/ml) of the test compound, rifampin, and amoxicillin were prepared in 2-fold dilution steps. Biofilm-containing pegs were transferred into the challenging plate and incubated for 24 h (22°C; 75% relative humidity). The exposed pegs were rinsed in phosphate-buffered saline, placed in freshly prepared BHI broth, and sonicated. After removal of the pegs, MBEC values were determined by measuring the optical density at 600 nm upon a 24-hour incubation (37°C).
Single-step resistance mutation frequencies.
Single-step resistance frequencies were determined for mid-logarithmic-phase S. pneumoniae ATCC 49619 using Todd Hewitt (TH) agar plates containing the drug at 5× and 50× the MIC and using an inoculum size of ±4 × 108 CFU. The colonies on the plates were enumerated following 48 h of incubation at 37°C (5% CO2). The frequency of selection of resistant mutants was calculated as the ratio of the number of resistant colonies arising on plates at 48 h to the number of viable cells plated.
Generation of S. pneumoniae diarylquinoline resistance mutants.
An overnight culture of S. pneumoniae ATCC 49619 (≈109 CFU/ml) grown in TH broth medium (37°C; 5% CO2) was spread on agar plates supplemented with increasing concentrations of compound 5 (5 to 50 μg/ml). The plates were incubated for 48 h (37°C; 5% CO2). The colonies grown in the presence of 50 μg/ml compound 5 were inoculated in Todd Hewitt medium with the same concentration of compound 5 and incubated for 24 h. Genotypic analysis of the complete ATP synthase operon of mutant strains was done by standard methods with amplification and DNA sequencing.
In vitro time-kill assays.
In vitro time-kill studies were performed using 5-ml S. aureus ATCC 29213 cultures (start inoculum, ≈106 CFU/ml) grown with continuous agitation at 37°C over a 24-h period. At selected time intervals, aliquots were taken, and serial dilutions were made in Mueller-Hinton (MH) broth and plated on MH agar plates. CFU were counted after 24 h of incubation.
Determination of minimum bactericidal concentration.
MICs were determined by the broth microdilution method according to CLSI reference methods, and antibacterial susceptibilities were reported using CLSI interpretive criteria. After this primary incubation for 24 h at 37°C, MICs were recorded, and minimum bactericidal concentration (MBC99.9) values were determined by spotting 10 μl from wells at and above the MIC on drug-free MH agar medium. CFU were counted from the sampled test wells and were used to determine the concentration that causes a ≥3-log10 decrease in CFU/ml of the original inoculums (MBC99.9).
Biochemical ATP synthesis inhibition assays in S. aureus.
S. aureus ATP synthesis activity was measured as previously described for Mycobacterium smegmatis (24) with minor modifications. Briefly, mid-logarithmic-phase S. aureus ATCC 29213 cells were lysed in 10 ml lysis buffer (50 mM MOPS [morpholinepropanesulfonic acid], 15 mM MgCl2, 50 μg/ml lysostaphin, protease inhibitors, and DNase I) per 5 g cells (wet weight) and incubated at room temperature for 45 min while shaking. Inverted membrane vesicles were prepared using a precooled French pressure cell (Thermo Electron Corporation) at 14,000 lb/in2. Membrane vesicles were preincubated with the compounds under stirring conditions at room temperature for 10 min. Dicyclohexyl-carbodiimide (DCCD) and levofloxacin were included, respectively, as positive and negative controls. ATP synthesis activity was determined by energizing the membranes with NADH and quantifying the amount of ATP produced using the luciferin/luciferase system (ATP bioluminescence assay kit HSII; Roche Applied Science). Data are presented as averages ± standard deviations (SD).
Inverted membrane vesicles from Escherichia coli were prepared using a French press as described above for S. aureus (12,000 lb/in2). ATP synthesis activity was determined with the glucose-6-phosphate dehydrogenase method as described below for mitochondria.
Isolation of human mitochondria.
The human ovarian cancer cell line OVCAR3 was maintained in 175-cm2 tissue culture flasks (Greiner Bio-One) in Dulbecco's modified Eagle's medium (DMEM) (BioWhittaker, Walkersville, MD), supplemented with 10% fetal bovine serum (PAA Laboratories GmbH) and 100 U/ml penicillin-streptomycin (Lonza BioWhittaker). Cell cultures were maintained at 37°C in 5% CO2, in a humidified atmosphere. Isolation of mitochondria from cells and ATP synthesis was done as described previously (14).
Mitochondrial ATP synthesis inhibition assay.
Human mitochondria (0.25 mg/ml) were incubated in 50 mM morpholino-ethanesulfonic acid (MES; pH 6.5), 5 mM MgCl2, 2 mM ADP, 20 mM KH2PO4, 100 μM P1,P5-di(adenosine-5′) pentaphosphate (Ap5A), 25 mM glucose, 11.8 U/ml hexokinase (Sigma), and protease inhibitors (complete, EDTA free; protease inhibitor cocktail tablets from Roche). Samples (0.25 ml) were incubated at 37°C with vigorous stirring in 18-ml flasks. The reaction was initiated with 5 mM succinate. After 2 h, reactions were stopped with 25 mM EDTA (final concentration) and transferred to ice. Samples were subsequently heated to 100°C for 5 min and centrifuged (10,000 × g, 20 min) to remove denatured protein. In supernatants, the synthesized glucose-6-phosphate was quantified by NADP+ (2.5 mM) reduction in the presence of 3 U/ml of glucose-6-phosphate dehydrogenase (Roche). NADPH formation was monitored spectrophotometrically at 340 nm.
Overexpression and purification of S. aureus ATP synthase subunit c.
S. aureus histidine-tagged ATP synthase subunit c was overexpressed and purified from Escherichia coli strain Lemo21 (39) using a T7 overexpression system. Cells were grown at 30°C in Luria-Bertani medium supplemented with 100 μg/ml chloramphenicol, 100 μg/ml ampicillin, and 0.2% glucose. When an optical density at 600 nm (OD600) of ∼0.4 was reached, cultures were incubated at 25°C and supplemented with 1 mM l-rhamnose and 0.4 mM IPTG (isopropyl-β-d-thiogalactopyranoside; final concentration). Cells were incubated for 14 to 16 h and then harvested by centrifugation.
Cells were harvested and resuspended in 50 mM phosphate buffer (pH 7.5) containing 400 mM NaCl, 50 μg/ml DNase, protease inhibitors (Complete; Roche), 2 mM MgCl2, and 10 mM 2-mercaptoethanol. Subsequently, cells were lysed with a One Shot pressure cell disruptor (ConstantSystems Ltd.). The total membrane fraction was suspended in the same buffer and treated with a final concentration of 1% N-dodecyl β-d-maltoside for 60 min at room temperature to solubilize the lipid membranes and release the overexpressed subunit c. Solubilized samples were subjected to Ni-nitrilotriacetic acid (NTA) metal-affinity chromatography (Qiagen).
Purification of ATP synthase from Bacillus PS3.
ATP synthase from the thermophilic Bacillus PS3 was overexpressed in E. coli strain DK8 and purified as described previously (36). Briefly, cells were disrupted using a French pressure cell (Thermo Scientific), and membrane proteins were extracted using 2% Triton X-100 and 0.5% sodium cholate. The membrane extract was applied to an Ni-NTA affinity column, and ATP synthase was eluted from the column with 300 mM imidazole. The α3β3γ subcomplex of ATP synthase from Bacillus PS3 (comprising the major subunits of the cytoplasmic F1 part) was overexpressed in E. coli strain JM103ΔuncB-D and purified as described previously (4). Briefly, cells were disrupted using a French pressure cell, and the cytosolic fraction was subjected to a heat treatment (30 min, 60°C). Subsequently, the sample was applied to an Ni-NTA affinity column and finally to a gel filtration column (Superdex 200; Amersham).
Surface plasmon resonance studies.
Surface plasmon resonance (SPR) sensing was carried out with a BIAcore 2000 instrument and a carboxymethyl (CM-5) analytical chip (GE Healthcare) at 25°C (17). A total of 70 μl of an equimolar mixture of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) was used to activate the carboxy-methyl surface of the chip. Subsequently, 50 μl of compound 4 (200 μM) in HEPES buffer [10 mM N-(2-hydroxyethyl)-piperazine-N′-ethanesulfonic acid (HEPES)-KOH (pH 7.5), 2 mM MgCl2, 150 mM NaCl] was injected over the activated chip surface (flow rate of 2 μl min−1). Nonreacted EDC/NHS on the chip surface was blocked by injection of 70 μl of 1 M ethanolamine. The chip was then reprimed and rinsed extensively with HEPES/Triton buffer (10 mM HEPES-KOH [pH 7.5], 2 mM MgCl2, 150 mM NaCl, 0.5% Triton X-100). Purified S. aureus ATP synthase subunit c, Bacillus PS3 ATP synthase holoenzyme, Bacillus PS3 a3b3g subcomplex, or mock sample was diluted in HEPES-Triton buffer to a final concentration of 2.5 μg/ml. For each injection, the chip was primed and rinsed for 10 min (flow rate of 15 μl min−1) with HEPES-Triton buffer, after which 325 μl of protein solution was injected at a flow rate of 15 μl/min. Binding to the chip was followed for 20 min, and dissociation was monitored for 10 min.
Construction of S. aureus atpE-inducible antisense strain.
The oligonucleotide primers atpE-for (5′-GTC TAG GGT ACC GTC AAC CAG AAG CAC GTG GTC-3′) and atpE-rev (5′-TAG GCA GGT ACC GTC ATG AAT GCA ATT ACT ACA C-3′) were used to amplify an atpE fragment from S. aureus RN4220(pSTE2) genomic DNA. The bold-faced sequences in the oligonucleotide primers represent the KpnI recognition site. The resulting DNA fragment was cloned into the tetracycline (Tc)-inducible pAJ96 S. aureus/E. coli shuttle vector (kindly provided by Alex O'Neill, University of Leeds, United Kingdom) (28) in the antisense orientation. This recombinant plasmid was electroporated into S. aureus RN4220 (pSTE2) (kindly provided by Tomasz Hauschild, University of Bialystok, Poland) (18) as described previously (2). An empty-vector S. aureus strain harboring the pAJ96 plasmid was also generated and serves as a negative control.
In vitro growth of S. aureus atpE antisense strain.
S. aureus strains were incubated at 37°C overnight in tryptic soy broth (TSB) medium supplemented with chloramphenicol (10 μg/ml) as a selection marker. The cultures were diluted to 5 × 105 CFU/ml in TSB containing chloramphenicol and Tc at concentrations from 0 up to 300 ng/ml. Bacterial growth was monitored at 37°C by measuring the optical density at 600 nm. In addition, overnight cultures were plated onto TSB agar plates containing chloramphenicol and various concentrations of Tc and subsequently incubated at 37°C. After 24 h, the plates were photographed.
RESULTS
Diarylquinolines show antibacterial activity against Gram-positive pathogens.
To identify small-molecule inhibitors with activity against Gram-positive pathogens, libraries of new compounds with diarylquinoline scaffolds were tested in bacterial growth inhibition assays. More than 700 molecules with novel specific lateral chains were synthesized and investigated. Compounds 1 to 5 discussed in this study (Fig. 1) are representative diarylquinoline chemotypes that show antibacterial activity against key Gram-positive human pathogens. The MICs observed against Gram-positive bacteria for compounds 1 to 4 were comparable to reference antibiotics levofloxacin and linezolid (Table 1). Notably, molecules 1 to 3 exhibited potent MICs (0.25 μg/ml) against S. pneumoniae, the causative agent of many respiratory tract infections (Table 1). Compound 5 showed a selective inhibition of S. pneumoniae (MIC 1 μg/ml) without activity against the other Gram-positive pathogens tested. The compounds showed no major activity at the tested concentrations against a selection of key Gram-negative bacteria, except for some minimal activity against Haemophilus influenzae for compounds 1 to 4. Compounds 1 to 3 did not show any activity against a Pseudomonas aeruginosa efflux mutant strain deficient for four important efflux systems, suggesting a lack of potency toward the target in this pathogen. Compounds 1 to 5 have limited activity against M. tuberculosis H37Rv (MIC 8 to 64 μg/ml), in contrast to TMC207, which shows a MIC of 0.03 μg/ml (1). In the presence of 2% human serum, the activity against S. aureus for compounds 1 to 4 significantly decreased (Table 1), suggesting a considerable plasma protein binding for these selected hits. Indeed, higher concentrations of human serum (10 to 50%) resulted in MICs of ≥64 μg/ml for S. aureus and S. pneumoniae. We also assessed the cytotoxicity of compounds 1 to 5 in a mammalian HelaM cell line. As listed in Table 1, target compounds 1 to 5 show CC50 values ranging from 4.5 to >56.9 μg/ml.
Table 1.
MIC and cytotoxicity (CC50) of compounds 1 to 4 and reference antibiotics
| Strain | MIC (μg/ml) |
|||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | Levofloxacin | Linezolid | |
| Staphylococcus aureus ATCC 29213 | 1 | 1 | 8 | 2 | 0.25 | 2 |
| Staphylococcus aureus ATCC 29213 + 2% human serum | 16 | 16 | 32 | 16 | 0.25 | 4 |
| Staphylococcus aureus MRSA ATCC 700788 | 4 | 4 | 8 | 2 | 16 | 4 |
| Staphylococcus epidermidis CICHEV 8283a | 2 | 1 | 2 | 2 | 0.125 | 1 |
| Streptococcus pneumoniae ATCC 49619 | 0.25 | 0.25 | 0.25 | 2 | 1 | 1 |
| Enterococcus faecalis ATCC29212 | 4 | 4 | 4 | 8 | 1 | 2 |
| Bacillus subtilis ATCC 43639 | 4 | 4 | 8 | 2 | 0.125 | 2 |
| Escherichia coli ATCC 35218 | > 64 | > 64 | > 64 | >64 | 0.125 | 16 |
| Pseudomonas aeruginosa K2732b | > 64 | > 64 | > 64 | >64 | 1 | 32 |
| Pseudomonas aeruginosa K2733c | >64 | >64 | >64 | NDd | 0.125 | 32 |
| Haemophilus influenza ATCC49247 | 32 | 16 | 8 | 8 | 0.125 | 8 |
| Klebsiella pneumoniae ATCC700603 | > 64 | > 64 | > 64 | >64 | 1 | > 64 |
| Cytotoxocity HelaM cells CC50 (μg/ml) | 12.3 | >56.9 | 4.5 | 3.5 | >9.3 | >8.5 |
Clinical isolate obtained from A. Aubry (Hopital Pitie Salepetriere, Paris, France).
PAO1 wild type obtained from K. Poole (Queen's University, Kingston, Canada).
K2733 efflux mutant (K2732 ΔmexB ΔmexX ΔmexCD-oprJΔmexEF-oprN; MexAB-OprM− MexCD-OprJ− MexXY− MexEF-OprN−) obtained from K. Poole (Queen's University, Kingston, Canada).
ND, not determined.
The newly developed diarylquinolines are rapidly bactericidal.
To assess the potential utility of this compound class, we determined the kinetics of bacterial killing in vitro. Time-kill assays demonstrated that compounds 1 and 2 rapidly kill S. aureus ATCC 29213 at a concentration of, respectively, 10× and 50× MIC, with a significant decrease in cell viability of ≥4-log10 CFU/ml from the starting inoculum over 24 h (Fig. 2). At lower concentrations (5× MIC for compound 1; 5× and 10× MIC for compound 2), both compounds exhibited an initial decline in CFU but showed a rebound in growth after 24 h. Compound 1 had no effect on S. aureus cell viability at 1× MIC. Linezolid, a known bacteriostatic molecule, at 10 μg/ml and levofloxacin, a rapid bactericidal compound, at 1 μg/ml were included in this study as control antibiotics. The potent bactericidal activities of compounds 1 and 2 were confirmed by their MBC99.9 values of, respectively, 4 and 2 μg/ml against S. aureus ATCC 29213. This observed in vitro bactericidal effect indicates potential clinical utility of this new class of small-molecule inhibitors.
Fig 2.
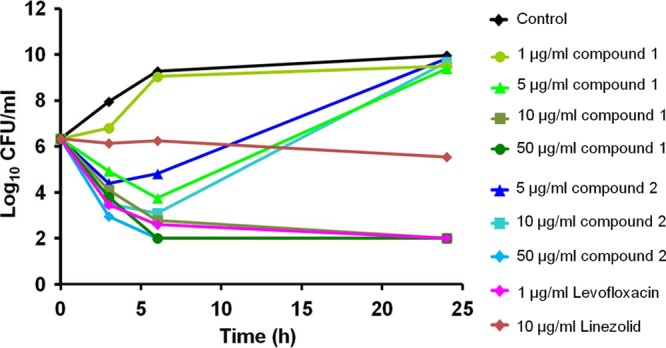
In vitro time-kill studies in S. aureus. The bactericidal activity of selected diarylquinolines against S. aureus ATCC 29213 was determined. Bacterial killing was monitored by measuring CFU/ml up to 24 h. Two independent time-kill studies were performed, and one representative experiment is shown.
In vitro activity against a bacterial metabolic resting state.
Most antibacterials targeting biosynthetic processes in a growing bacterium lack clinical efficacy on slow-growing or nongrowing bacteria, e.g., in biofilms. These bacterial metabolic resting states are a hallmark of chronic infections; consequently, antibacterial activity on biofilms is strongly warranted.
The efficacy of a representative diarylquinoline (compound 2) on S. aureus biofilms was determined (Fig. 3). This compound showed significant potency, with eradication of 1-day-old S. aureus LMG8064 biofilms within 24 h and an MBEC in the range of 15 μg/ml. Compound 2 has a MIC value against planktonic S. aureus LMG8064 of 1 μg/ml. Rifampin and amoxicillin were included, respectively, as a positive (MIC of 0.125 μg/ml; MBEC of ±3 μg/ml) and a negative (MIC of 0.125 μg/ml; MBEC of >100 μg/ml) control.
Fig 3.

In vitro biofilm studies in S. aureus. Activity of compound 2 (squares) and reference antibiotics rifampin (triangles) and amoxicillin (circles) against 1-day-old biofilms of S. aureus LMG8064 treated for 24 h (average from 3 independent experiments). S. aureus LMG8064 biofilms formed on pegs were exposed to increasing compound concentrations for 24 h, followed by optical density measurements at 600 nm (OD600) to monitor biofilm eradication. Three independent biofilm studies were performed, and one representative experiment is shown.
The ability of diarylquinolines to eradicate S. aureus biofilms not only suggests a key role of the target protein in bacteria present in biofilms but also further underscores the clinical potential of these new small-molecule inhibitors.
Chemical genetic studies pinpoint resistance mutations in the atpE gene.
In order to support the expectation that these new small-molecule drug candidates target ATP synthase, we raised drug-resistant mutants in S. pneumoniae, the most susceptible pathogen in our growth inhibition assays (Table 1). These mutants were selected by inoculating S. pneumoniae ATCC 49619 on agar plates containing compound 5 at a concentration of 50× MIC. The single-step mutation frequencies for this compound ranged between <8.1 × 10−7 and 8.3 × 10−7 at 5× MIC and 4.6 × 10−7 and 1.7 × 10−6 at 50× MIC for S. pneumoniae ATCC 49619. Genomic DNA of three resistant mutants was isolated, and the complete ATP synthase operon was sequenced. In all three mutant strains, sequence variations were found in this operon and mapped to the atpE gene, which encodes subunit c of the ATP synthase. ATP synthase is an enzyme that uses the energy of protons flowing across the cytoplasmic membrane in order to synthesize ATP (Fig. 4A; for a review, see reference 20). This subunit c is an oligomeric, membrane-spanning part of ATP synthase that is crucial for the flow of protons across the membrane in order to synthesize ATP. ATP synthase subunit c in mycobacteria had previously been validated as a target of TMC207 (24).
Fig 4.

ATP synthase and the proton-conducting subunit c. (A) Schematic view of ATP synthase subunits. The bacterial enzyme consists of a membrane-bound F0 part (subunits α3β3γδϵ) for conduction of protons, and a hydrophilic F1 part (subunits α3β3γδϵ) responsible for synthesis of ATP. The oligomeric subunit c (AtpE) is shown in gray. (B) Amino acid sequence alignment of ATP synthase subunit c for selected pathogens and human mitochondria. Species used: SPN_S, drug-sensitive strain of Streptococcus pneumoniae D39 (GenBank accession no. ABJ53659); SPN_R, drug-resistant strain of Streptococcus pneumoniae D39 (GenBank accession no. ABJ53659); STA, Staphylococcus aureus MRSA252 (residue 1 to 70; RefSeq accession no. YP_041556); ENT, Enterococcus faecalis V583 (residue 1 to 73; RefSeq accession no. NP_816252); BAC, Bacillus sp. PS3 (Swiss-Prot accession no. P00845); ECO, Escherichia coli (GenBank accession no. AAA24732); MTB, Mycobacterium tuberculosis (Gen Bank accession no. CCE36825); H. sapiens, Homo sapiens (residue 60 to 136; RefSeq accession no. NP_001002027). Single mutations found in drug-resistant S. pneumoniae (V48I and V60A) are indicated by boxes.
Two resistant strains harbored a single point mutation each (V48I and V60A), whereas a third resistant strain contained a double mutation at positions V48I and V60A (Fig. 4B). MIC values of compound 5 against the three mutant strains were >50 μg/ml, resulting in a >100-fold increase in MIC compared to that of wild-type S. pneumoniae.
Both V48 and V60 are completely conserved in subunit c of ATP synthase for S. aureus, S. pneumoniae, Enterococcus faecalis, and Bacillus subtilis but different from that found in E. coli and in human mitochondria (Fig. 4B). In the case of V48I, replacement of the valine residue by the bulkier isoleucine in the resistant strain may cause steric hindrance and interfere with efficient drug binding. Concordantly, subunit c from both human mitochondria and from E. coli displays a bulkier residue at that particular position, respectively, phenylalanine and methionine (Fig. 4B). Taken together, these findings indicate that the proton-conducting subunit c of ATP synthase is likely to be the binding site for diarylquinolines identified in this study.
Biochemical investigation and selectivity of diarylquinolines.
In order to zoom in on the biochemical mechanism of this new compound class, we determined the inhibitory activities of compounds 1 to 3 on ATP synthesis in inverted membrane vesicles derived from S. aureus. As depicted in Table 2, the selected compounds showed significant inhibitory activities, with 50% inhibitory concentrations (IC50s) between 1 and 3 μg/ml. Thus, the concentrations of compound needed to inhibit ATP synthase in the biochemical assay (IC50) correlated well with MIC values for inhibition of S. aureus growth in the whole-cell screening assay. ATP synthesis by membrane vesicles obtained from the Gram-negative E. coli displayed higher IC50s (Table 2). The polymorphisms in the atpE gene on amino acid positions V48 and V60 described above (Fig. 4B) may account for the differences in IC50s determined in the S. aureus and E. coli ATP synthesis assays and might in part explain the lack of activity observed for E. coli in the whole-cell screening assay. However, drug efflux mechanisms, less-pronounced metabolic dependency on ATP synthase, and/or limited uptake of the compound due to the Gram-negative cell membrane cannot be excluded as causes for the lack of whole-cell activity.
Table 2.
Selective inhibition of ATP synthesis by compounds 1 to 3 and reference compounds (average ± SD)
| Compound | ATP synthesis IC50 (μg/ml) |
||
|---|---|---|---|
| S. aureus | E. coli | Mitochondria | |
| 1 | 1.4 ± 0.5 | 7 ± 0.4 | 29 ± 2 |
| 2 | 2.5 ± 0.7 | 8 ± 0.6 | 27 ± 1.6 |
| 3 | 2.7 ± 0.1 | 9 ± 0.6 | 23 ± 2.2 |
| DCCD | 0.9 ± 0.2 | 1 ± 0.1 | 0.2 ± 0.01 |
| Oligomycin | 5.6 | 7 ± 0.7 | 6 ± 0.6 |
As ATP synthase is highly conserved among prokaryotes and eukaryotes, we also determined the effect of the selected compounds on ATP synthesis in mitochondria isolated from the human ovarian cancer cell line OVCAR3. The IC50 of compounds 1 to 3 in the mitochondrial ATP synthase assay was between 23 and 29 μg/ml, with selectivity indices (SI) of >10 (Table 2) compared with S. aureus. In contrast, typical ATP synthase inhibitors, such as oligomycin and DCCD, were nonselective with SI of ≤1.
These results demonstrate that for Gram-positive bacteria, inhibition of growth can be achieved via inhibition of ATP synthesis. Further chemical optimization of the diarylquinoline class may potentially further increase the selectivity indices, as has been shown previously for the mycobacterial ATP synthase inhibitor TMC207, for which an SI of >10,000 has been demonstrated (15). Our results support the view that an enzyme should not necessarily be disregarded as a potential target because it has a homologue in eukaryotes.
Diarylquinolines interact with the F0 part of ATP synthase.
To confirm a direct binding between the selected hits and ATP synthase, we performed molecular drug-target interaction studies between a representative diarylquinoline (compound 4) (Fig. 1) and the transmembrane subunit c of ATP synthase from S. aureus using surface plasmon resonance (SPR) sensing. The compound was linked via its secondary amino group onto the carboxymethyl-dextran surface (CM-5) of a BIAcore chip. S. aureus subunit c protein was overexpressed and purified from E. coli and injected onto the compound-linked chip. The experiment demonstrated that subunit c clearly binds to compound 4 (Fig. 5). Control experiments, performed with either a mock purification of subunit c (from E. coli carrying an empty expression vector) or using a chip without immobilized compound, confirmed the specific interaction between subunit c and the immobilized compound (Fig. 5).
Fig 5.
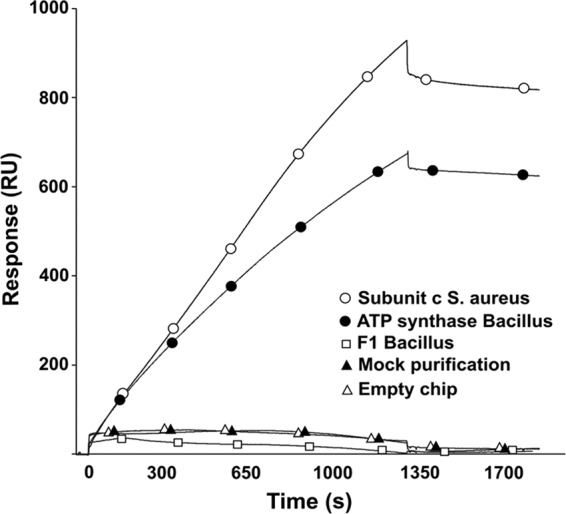
Drug-target interaction studies using Biacore. A selected hit (compound 4) was immobilized on a Biacore CM5 chip. Binding to the compound-linked chip was determined for purified ATP synthase subunit c from S. aureus (empty circles), purified ATP synthase holoenzyme from Bacillus PS3 (filled circles), and the F1 part (subunits α3β3γ) from Bacillus PS3 (empty squares). As controls, a mock purification of subunit c (filled triangles) and binding of purified subunit c from S. aureus to a chip in the absence of immobilized compound (empty triangles) were used. For the mock sample, we performed the purification experiment using cells harboring an empty expression vector. Experiments were done in triplicate, and representative results are shown.
As ATP synthase is a complex, multimeric enzyme with transmembrane subunits (F0) and cytoplasmic subunits (F1) (Fig. 4A), we also evaluated the respective contributions of these two parts to the drug-target interaction. For this, we used either purified whole ATP synthase complex from the Bacillus PS3 strain or the isolated cytoplasmic F1 part (subunits a3b3g). As depicted in Fig. 5, the whole ATP synthase bound to compound 4, whereas no detectable affinity was observed for the cytoplasmic part. These results demonstrate that the transmembrane F0 part of ATP synthase plays a crucial role in interaction with this chemical class.
Downregulating ATP synthase subunit c expression impairs S. aureus growth.
To study the role of ATP synthase in S. aureus, a tetracycline (Tc)-inducible S. aureus antisense strain was generated, in which mRNA levels of subunit c of ATP synthase (atpE) are downregulated. We examined the effect of atpE antisense RNA expression on the growth of S. aureus in liquid cultures and on solid medium, by induction with increasing Tc concentrations. In liquid medium, Tc induction resulted in a strong, dose-dependent decrease in bacterial growth compared to that of the control (empty pAJ96 vector) (Fig. 6B). We also observed that bacterial colony formation on agar plates containing 600 ng/ml Tc was strongly impeded (Fig. 6C), with only a few small colonies appearing on the agar plates. We confirmed downregulation of atpE mRNA levels resulting from Tc induction with qRT-PCR (Fig. 6A).
Fig 6.

Importance of ATP synthase in S. aureus. (A) Downregulation of atpE mRNA transcription in the atpE antisense S. aureus strain after antisense induction with tetracycline (Tc); (B) growth of wild-type (empty pAJ96 vector) and atpE antisense S. aureus strains in liquid MH medium with increasing Tc concentrations (0 to 300 ng/ml); (C) Growth of wild-type (empty pAJ96 vector) and atpE antisense strains on solid MH medium incubated for 24 h at 37°C. atpE antisense is induced by adding 600 ng/ml Tc.
These results reveal an important role of ATP synthase subunit c in the growth of S. aureus and provide a rationale for utilizing small-molecule inhibitors acting on this enzyme as antibacterials.
DISCUSSION
Drug-resistant pathogenic bacteria are a global health care concern and highlight a clear unmet medical need for novel antibacterials with novel mechanisms of action. The diarylquinoline class of antibacterials efficiently kills Mycobacterium tuberculosis by specifically inhibiting ATP synthase; however, the antibacterial spectrum of diarylquinolines reported so far is restricted to mycobacterial strains.
In the current study, we aimed to expand the antibacterial spectrum of diarylquinolines. We identified new diarylquinoline drug candidates with potent inhibitory activity against key Gram-positive pathogens and rapid bactericidal activity on S. aureus. We confirmed that these small-molecule inhibitors interact with subunit c of ATP synthase and act as inhibitors of ATP synthesis. In addition, our studies suggest that subunit c is important for growth and survival of S. aureus. The data described in this paper indicate that diarylquinolines, and potentially other small-molecule inhibitors active on ATP synthase, can have a high potential for combating multidrug-resistant bacterial strains due to a lack of cross-resistance with presently used classes of antibacterials.
The diarylquinolines described in this study showed significant human plasma protein binding and a moderate selectivity versus the human ATP synthase. However, our results represent a proof of principle for using the diarylquinoline chemical class as broad-spectrum agents against different Gram-positive bacteria and under different physiological conditions. As such, further chemical optimization of the current inhibitors or, alternatively, screening of pharmaceutical compound libraries for ATP synthesis inhibitors may result in hits that are more amenable for lead optimization in terms of improving potency, addressing the issue of plasma protein binding, and better bacterial selectivity.
Essentiality of ATP synthase may stem from a variety of factors, such as meeting high cellular energy demands, as proposed for mycobacteria (10, 16), or maintenance of the proton motive force, as suggested for Chlorobium limicola (41). Moreover, ATP synthase may play a role in pH homeostasis, as has been reported previously for Listeria monocytogenes and Salmonella enterica serovar Typhimurium (9, 13) or may act as a “relief valve” for protons to facilitate continuous NADH oxidation and to maintain a cellular redox balance (10).
Although ATP synthase appears to be present in all bacteria, the essentiality of the enzyme varies significantly across species. Whereas in some species blocking ATP synthase function dramatically diminishes growth, in other species apparently increased glycolytic flux may in part compensate for deficiencies in respiratory ATP synthesis. For mycobacteria, deletion mutants in the atpD gene demonstrated a central function of ATP synthase for growth (38), and blocking respiratory ATP synthesis by small molecule inhibitors causes depletion of cellular ATP levels and bacterial killing (24, 27). For S. aureus, it has been reported that transposon insertion in the atpD, atpG, and atpA genes did not lead to complete absence of bacterial colonies (22). Our results show a strong, although not a complete, decrease of CFU upon downregulation of atpE gene transcription, encoding the F0 subunit c of ATP synthase, indicating an important function of ATP synthase enzyme in growth and survival of S. aureus. Our work also extends previous mutagenesis studies on streptococci, where insertion of mutations in the atpC gene dramatically affected cell viability (11).
For E. coli and B. subtilis, deletion mutants in ATP synthase are able to grow, although at reduced rates compared to that of the wild type (21, 32). E coli was largely resistant to the selected diarylquinolines, which in addition to the less-pronounced metabolic dependency on ATP synthase and polymorphisms in the atpE gene may be due to drug efflux mechanisms or limited uptake of the compound through the Gram-negative cell membrane. B. subtilis showed sensitivity to selected compounds, although at slightly higher concentrations than S. aureus and S. pneumoniae. The employed growth conditions or other, unexplored factors may influence the relative importance of respiratory ATP synthesis.
Analysis of bacterial energy requirements using a systems biology approach may clarify the physiological basis for ATP synthase essentiality or lack thereof in bacteria. Further unraveling the energy metabolism in Gram-positive bacteria may also reveal additional points of vulnerability, e.g., targeting NADH dehydrogenase or glycolysis/glyconeogenesis (31, 40), which may be exploited for development of new antibiotics.
The reduced antibiotic susceptibility frequently observed in slow-growing or nongrowing bacteria, such as those within biofilms associated with persistent or chronic infections, can be attributed to extensive remodeling of metabolic pathways in persistent bacterial cells (19, 23, 26, 30, 35). Previous experiments, aimed mainly at mycobacterial dormancy, have provided hints that despite absence of growth during a resting metabolic state, a minimal level of energy conversion within the bacterial cell is required for survival (25, 29). It has been hypothesized that more generally targeting energy metabolism in persistent or resting bacteria, such as also found in biofilms, may be an attractive strategy for eradicating these therapeutically recalcitrant bacteria (19). In line with this hypothesis, we demonstrated that diarylquinolines are capable of eradicating S. aureus biofilms as well as planktonic bacteria, a highly desirable property for any new antibiotic. Factors that determine the efficacy of small-molecule drugs on bacteria in biofilms need to be elucidated. Moreover, function and regulation of energy metabolic pathways in bacterial resting states need to be investigated.
Small-molecule inhibitors interfering with cellular ATP homeostasis, in addition to acting on a potential Achilles' heel of persistent bacteria, may also interfere with the function of ATP-driven efflux pumps in drug-resistant S. aureus or S. pneumoniae strains. Our results demonstrate that small-molecule inhibitors with diarylquinoline scaffold are not only active on mycobacteria but may be applicable as broad-spectrum antibacterials against Gram-positive pathogens. Targeting energy metabolic pathways, in spite of the structural and functional homology with eukaryotic ATP synthase, may represent an attractive approach for the discovery of novel antibacterial agents.
ACKNOWLEDGMENTS
P. aeruginosa strains K2732 and K2733 were obtained from Keith Poole (Queen's University, Kingston, Canada). The authors acknowledge Reginald Clayton for his critical review of the manuscript.
This work was supported by an R&D grant (IWT O&O grant 080538) from the Flemish Government Agency for Innovation by Science and Technology (I.W.T.) to Anil Koul.
Footnotes
Published ahead of print 21 May 2012
REFERENCES
- 1. Andries K, et al. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227 [DOI] [PubMed] [Google Scholar]
- 2. Augustin J, Gotz F. 1990. Transformation of Staphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS Microbiol. Lett. 54:203–207 [DOI] [PubMed] [Google Scholar]
- 3. Bald D, Koul A. 2010. Respiratory ATP synthesis: the new generation of mycobacterial drug targets? FEMS Microbiol. Lett. 308:1–7 [DOI] [PubMed] [Google Scholar]
- 4. Bald D, Noji H, Stumpp MT, Yoshida M, Hisabori T. 2000. ATPase activity of a highly stable alpha(3)beta(3)gamma subcomplex of thermophilic F(1) can be regulated by the introduced regulatory region of gamma subunit of chloroplast F(1). J. Biol. Chem. 275:12757–12762 [DOI] [PubMed] [Google Scholar]
- 5. Boucher HW, et al. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America Clin. Infect. Dis. 48:1–12 [DOI] [PubMed] [Google Scholar]
- 6. Braine T. 2011. Race against time to develop new antibiotics. Bull. World Health Organ. 89:88–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ceri H, et al. 1999. The Calgary Biofilm Device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 37:1771–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. CLSI 2011. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard M07-A8, 8th edition, vol. 29, no. 2 CLSI, Wayne, PA [Google Scholar]
- 9. Cotter PD, Gahan CG, Hill C. 2000. Analysis of the role of the Listeria monocytogenes F0F1-AtPase operon in the acid tolerance response. Int. J. Food Microbiol. 60:137–146 [DOI] [PubMed] [Google Scholar]
- 10. Cox RA, Cook GM. 2007. Growth regulation in the mycobacterial cell. Curr. Mol. Med. 7:231–245 [DOI] [PubMed] [Google Scholar]
- 11. Ferrandiz MJ, de la Campa AG. 2002. The membrane-associated F(0)F(1) ATPase is essential for the viability of Streptococcus pneumoniae. FEMS Microbiol. Lett. 212:133–138 [DOI] [PubMed] [Google Scholar]
- 12. Fischbach MA, Walsh CT. 2009. Antibiotics for emerging pathogens. Science 325:1089–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foster JW, Hall HK. 1991. Inducible pH homeostasis and the acid tolerance response of Salmonella typhimurium. J. Bacteriol. 173:5129–5135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garcia JJ, Ogilvie I, Robinson BH, Capaldi RA. 2000. Structure, functioning, and assembly of the ATP synthase in cells from patients with the T8993G mitochondrial DNA mutation. Comparison with the enzyme in Rho(0) cells completely lacking mtdna. J. Biol. Chem. 275:11075–11081 [DOI] [PubMed] [Google Scholar]
- 15. Haagsma AC, et al. 2009. Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob. Agents Chemother. 53:1290–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haagsma AC, Driessen NN, Hahn MM, Lill H, Bald D. 2010. ATP synthase in slow- and fast-growing mycobacteria is active in ATP synthesis and blocked in ATP hydrolysis direction. FEMS Microbiol. Lett. 313:68–74 [DOI] [PubMed] [Google Scholar]
- 17. Haagsma AC, et al. 2011. Probing the interaction of the diarylquinoline TMC207 with its target mycobacterial ATP synthase. PLoS One 6:e23575 doi:10.1371/journal.pone.0023575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hauschild T, Luthje P, Schwarz S. 2005. Staphylococcal tetracycline-MLSB resistance plasmid pSTE2 is the product of an RSA-mediated in vivo recombination. J. Antimicrob. Chemother. 56:399–402 [DOI] [PubMed] [Google Scholar]
- 19. Hurdle JG, O'Neill AJ, Chopra I, Lee RE. 2011. Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 9:62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Junge W, Sielaff H, Engelbrecht S. 2009. Torque generation and elastic power transmission in the rotary F(O)F(1)-ATPase. Nature 459:364–370 [DOI] [PubMed] [Google Scholar]
- 21. Klionsky DJ, Brusilow WS, Simoni RD. 1984. In vivo evidence for the role of the epsilon subunit as an inhibitor of the proton-translocating ATPase of Escherichia coli. J. Bacteriol. 160:1055–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ko HSY, et al. 2011. Screening of essential genes in Staphylococcus aureus N315 using comparative genomics and allelic replacement mutagenesis. J. Microbiol. Biotechnol. 16:623–632 [Google Scholar]
- 23. Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. 2011. The challenge of new drug discovery for tuberculosis. Nature 469:483–490 [DOI] [PubMed] [Google Scholar]
- 24. Koul A, et al. 2007. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 3:323–324 [DOI] [PubMed] [Google Scholar]
- 25. Koul A, et al. 2008. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 283:25273–25280 [DOI] [PubMed] [Google Scholar]
- 26. Lewis K. 2007. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5:48–56 [DOI] [PubMed] [Google Scholar]
- 27. Lu P, et al. 2011. Pyrazinoic acid decreases the proton motive force, respiratory ATP synthesis activity, and cellular ATP levels. Antimicrob. Agents Chemother. 55:5354–5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Neill AJ, McLaws F, Kahlmeter G, Henriksen AS, Chopra I. 2007. Genetic basis of resistance to fusidic acid in staphylococci. Antimicrob. Agents Chemother. 51:1737–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rao SP, Alonso S, Rand L, Dick T, Pethe K. 2008. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 105:11945–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Resch A, Rosenstein R, Nerz C, Gotz F. 2005. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl. Environ. Microbiol. 71:2663–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rhee KY, et al. 2011. Central carbon metabolism in Mycobacterium tuberculosis: an unexpected frontier. Trends Microbiol. 19:307–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Santana M, et al. 1994. Bacillus subtilis F0F1 ATPase: DNA sequence of the atp operon and characterization of atp mutants. J. Bacteriol. 176:6802–6811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sassetti CM, Rubin EJ. 2007. The open book of infectious diseases. Nat. Med. 13:279–280 [DOI] [PubMed] [Google Scholar]
- 34. Spellberg B, et al. 2011. Combating antimicrobial resistance: policy recommendations to save lives. Clin. Infect. Dis. 52(Suppl 5):S397–S428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stewart PS, Franklin MJ. 2008. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 6:199–210 [DOI] [PubMed] [Google Scholar]
- 36. Suzuki T, Ueno H, Mitome N, Suzuki J, Yoshida M. 2002. F(0) of ATP synthase is a rotary proton channel. Obligatory coupling of proton translocation with rotation of c-subunit ring. J. Biol. Chem. 277:13281–13285 [DOI] [PubMed] [Google Scholar]
- 37. Talbot GH, et al. 2006. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 42:657–668 [DOI] [PubMed] [Google Scholar]
- 38. Tran SL, Cook GM. 2005. The F1Fo-ATP synthase of Mycobacterium smegmatis is essential for growth. J. Bacteriol. 187:5023–5028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wagner S, et al. 2008. Tuning Escherichia coli for membrane protein overexpression. Proc. Natl. Acad. Sci. U. S. A. 105:14371–14376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weinstein EA, et al. 2005. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc. Natl. Acad. Sci. U. S. A. 102:4548–4553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie DL, et al. 1993. The atp2 operon of the green bacterium Chlorobium limicola. Biochim. Biophys. Acta 1172:267–273 [DOI] [PubMed] [Google Scholar]