

Abstract
Hepatitis B virus (HBV) covalently closed circular DNA (cccDNA) plays a central role in viral infection and persistence and is the basis for viral rebound after the cessation of therapy, as well as the elusiveness of a cure even after extended treatment. Therefore, there is an urgent need for the development of novel therapeutic agents that directly target cccDNA formation and maintenance. By employing an innovative cell-based cccDNA assay in which secreted HBV e antigen is a cccDNA-dependent surrogate, we screened an in-house small-molecule library consisting of 85,000 drug-like compounds. Two structurally related disubstituted sulfonamides (DSS), termed CCC-0975 and CCC-0346, emerged and were confirmed as inhibitors of cccDNA production, with low micromolar 50% effective concentrations (EC50s) in cell culture. Further mechanistic studies demonstrated that DSS compound treatment neither directly inhibited HBV DNA replication in cell culture nor reduced viral polymerase activity in the in vitro endogenous polymerase assay but synchronously reduced the levels of HBV cccDNA and its putative precursor, deproteinized relaxed circular DNA (DP-rcDNA). However, DSS compounds did not promote the intracellular decay of HBV DP-rcDNA and cccDNA, suggesting that the compounds interfere primarily with rcDNA conversion into cccDNA. In addition, we demonstrated that CCC-0975 was able to reduce cccDNA biosynthesis in duck HBV-infected primary duck hepatocytes. This is the first attempt, to our knowledge, to identify small molecules that target cccDNA formation, and DSS compounds thus potentially serve as proof-of-concept drug candidates for development into therapeutics to eliminate cccDNA from chronic HBV infection.
INTRODUCTION
It is estimated that 2 billion people worldwide have been infected with hepatitis B virus (HBV). Although most adulthood infections are transient, approximately 5 to 10% of infected adults and over 90% of infected neonates fail to mount a sufficient immune response to clear the virus and develop a life-long chronic infection (23, 27). Chronic hepatitis B is currently a substantial public health burden, affecting approximately 350 million individuals worldwide. These patients have an elevated risk of liver cirrhosis, hepatocellular carcinoma, and other severe clinical sequelae (1, 23). It is therefore a global health priority to cure chronic HBV infection and prevent its dire consequences.
HBV is a noncytopathic, liver-tropic DNA virus belonging to the Hepadnaviridae family. Upon infection, the viral genomic relaxed circular DNA (rcDNA) is transported into the cell nucleus and converted into episomal covalently closed circular DNA (cccDNA), which serves as the transcription template for the viral mRNAs. After transcription and nuclear export, cytoplasmic viral pregenomic RNA (pgRNA) is assembled with HBV polymerase and capsid proteins to form the nucleocapsid, inside which polymerase-catalyzed reverse transcription yields minus-strand DNA, which is subsequently copied into plus-strand DNA to form the progeny rcDNA genome. The mature nucleocapsids are then either packaged with viral envelope proteins to egress as virion particles or shuttled to the nucleus to amplify the cccDNA reservoir through the intracellular cccDNA amplification pathway (reviewed in references 1, 29, and 37).
cccDNA is an essential component of the HBV replication cycle and is responsible for the establishment of infection and viral persistence. The details of the molecular mechanism by which rcDNA is converted into cccDNA remain poorly understood. Considering the subcellular location and unique structures of these two viral DNA molecules, virus trafficking and a number of specific biochemical reactions can be predicted to occur during cccDNA formation. To begin with, the cytoplasmic rcDNA present in nucleocapsids needs to be transported into the nucleus via karyopherin-dependent recognition of nuclear localization signals (NLS) on the capsid protein (19, 34). On the other hand, several enzymatic reactions must occur because of the unique terminal features of rcDNA, including (i) completion of viral plus-strand DNA synthesis, (ii) removal of the 5′-capped RNA primer at the 5′ terminus of plus-strand DNA, (iii) removal of the viral polymerase covalently attached to the 5′ end of minus-strand DNA, (iv) removal of one copy of the terminal redundancies on minus-strand DNA (40), and (v) ligation of both strands to generate cccDNA. Recently, a protein-free rcDNA form without covalently bound viral polymerase has been identified, which was designated deproteinized rcDNA (DP-rcDNA). This viral DNA species is found in infected cells but not in virions. DP-rcDNA was demonstrated as one, if not the only, functional precursor intermediate for cccDNA formation (6, 11, 12). DP-rcDNA thus provides a potential antiviral target for cccDNA intervention.
To date, there is no definitive cure for chronic hepatitis B. Drugs currently approved for HBV treatment include alpha interferon (IFN-α) and five nucleos(t)ide analogues (lamivudine [3TC], adefovir, entecavir, telbivudine, and tenofovir). IFN-α therapy yields sustained virological responses in less than 40% of patients after 48 weeks of treatment, with significant on-treatment side effects. The five nucleos(t)ide analogues all act as potent inhibitors of viral polymerase but rarely cure HBV infection (7), and emergence of drug-resistant virus dramatically limits their long-term efficacy (31). The major limitation of current treatment is the failure to eliminate the preexisting cccDNA pool and/or prevent cccDNA formation from trace levels of wild-type or drug-resistant virus. Thus, there is an urgent need for the development of novel therapeutic agents that directly target cccDNA formation and maintenance.
However, screenings for anti-cccDNA agents have not been conducted because of the lack of efficient in vitro HBV infection models, and a practical approach for measuring cccDNA in a high- to mid-throughput format was unavailable. Although there are primary human hepatocytes and the recently established HepaRG cell culture system available to support cccDNA-dependent HBV replication, the efficiency of HBV infection in both systems is extremely low (8, 36). There is a high degree of variability in primary hepatocytes from donor to donor, with some batches proving to be uninfectable. The HepaRG system requires much manipulation for experimental setup (8). These characteristics restrict the utility of these two HBV infection systems for the screening of compound libraries for cccDNA inhibitors. Alternatively, cccDNA formation can be achieved through the intracellular amplification pathway in stably transfected HBV cell cultures that constitutively or conditionally replicate the HBV genome (2, 11, 22, 39), as represented by the HepG2.2.15 line (35, 38). However, direct cccDNA detection from HBV cell lines by either Southern hybridization or real-time PCR assay would not be amenable to screening because of low sensitivity and high cost, respectively. On the other hand, there is no suitable surrogate marker for cccDNA in HepG2.2.15 cells since most of the viral products are derived from an integrated viral transgene and are indistinguishable from cccDNA contributions. To surmount this problem, we have previously reported that the production of secreted HBV e antigen (HBeAg) was predominantly cccDNA dependent in HepAD38 cells and might serve as a surrogate marker for cccDNA (22, 45). In the present study, we used an upgraded version of a solely cccDNA-dependent HBeAg-producing cell line, termed HepDE19 (11), in a 96-well format assay for the screening of agents that inhibit cccDNA formation and/or maintenance. As developed, the assay is adaptable to high-throughput screening formats and full automation.
This report presents the results of that screening campaign. Our screening effort led to the identification of two disubstituted sulfonamides (DSS) as inhibitors of cccDNA production. Both compounds exhibit low micromolar 50% effective concentrations (EC50s) in cell culture. We further demonstrated that the DSS compounds synchronously reduced the levels of viral cccDNA and DP-rcDNA without directly affecting viral DNA replication in both HBV and duck HBV (DHBV) models. In addition, our work suggested that the DSS compounds primarily interfered with rcDNA conversion into cccDNA. This is the first attempt, to the best of our knowledge, to identify small molecules that directly target cccDNA formation and has resulted in two novel leads for the development of new hepatitis B therapeutics.
MATERIALS AND METHODS
Cell cultures.
HepG2.2.15 cells (a gift from George Acs, Mt. Sinai Medical College, New York, NY) were maintained in Dulbecco's modified Eagle's medium-F12 (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 400 μg/ml G418. Tetracycline-inducible HBV producer cells, specifically, HepDE19 and HepDES19 cells, were maintained in the same way as HepG2.2.15 cells but with the addition of 1 μg/ml tetracycline (11). To initiate HBV replication and cccDNA formation in HepDE19 and HepDES19 cells, tetracycline was withdrawn from the culture medium and the cells were cultured for the indicated time period.
Compound sources and handling.
The in-house compound library (Institute for Hepatitis and Virus Research [IHVR] small-molecule collection) consists of 85,000 compounds from the complete libraries of ChemDiv, Inc. (San Diego, CA), Asinex Inc. (Winston-Salem, NC), Chembridge Inc. (San Diego, CA), Maybridge Inc. (Cornwall, United Kingdom), LOPAC1280 (Sigma-Aldrich, St. Louis, MO), the MicroSource SPECTRUM Collection (Gaylordsville, CT), and the Johns Hopkins Clinical Compound Library (Baltimore, MD). The compounds were selected from the large libraries of these companies on the basis of their “drug-like” properties and diversity. The library was “cherry-picked” by the vendors as requested using their own cheminformatic tools to generate smaller sets. Web-based tools (especially ADME-Tox) available at http://mobyle.rpbs.univ-paris-diderot.fr were used to characterize the assembled library. While the ∼47,000-compound ChemDiv and Asinex collections are combinatorial, the remainder of the IHVR library (∼38,000) is highly diverse. Overall, the IHVR library has an average molecular mass of ∼350 Da and a maximum cLogP value (the logarithm of a compound's partition coefficient between n-octanol and water) of 5.0. The LOPAC, MicroSource, and John Hopkins collections are annotated compounds that are drugs in clinical use, drug candidates that have reached clinical testing, or experimental small-molecule modulators with known targets. Compounds were purchased as dry powder in 96-well format “mother” plates, resuspended in ultrapure dimethyl sulfoxide (DMSO) to a final concentration of 10 mM, and diluted in DMSO into working stock (“daughter”) plates at 1 mM. Compounds were stored in covered polypropylene plates at −20°C. Resynthesis of CCC-0975 and CCC-0346 was carried out by ChemDiv, Inc. (San Diego, CA), and Pharmabridge Inc. (Doylestown, PA), respectively.
Compound screening with a 96-well format assay.
HepDE19 cells were cultured under tetracycline-containing medium until confluent; the cells were then trypsinized and seeded into 96-well plates at a density of 5.0 × 104 cells/well with tetracycline-free medium to induce HBV replication. Immediately following cell seeding, compounds were added to screening plates by means of automated liquid handling (Beckman Coulter, Brea, CA) at a final concentration of 10 μM in 1.0% DMSO. Each screening plate consisted of 80 compound test wells, 4 wells of cells with 1.0% DMSO only, 4 wells with DMSO and without cells, and 4 wells of cells with DMSO and 50 μM 3TC as a reference inhibitor. Screening plates were incubated at 37°C in a 5.0% CO2 atmosphere for 7 days. Intracellular HBeAg (precore) accumulation was assayed with an in-house-developed indirect enzyme-linked immunosorbent assay (ELISA) with a Z factor of 0.45. Briefly, medium was removed from wells and the cell monolayer was fixed with 100% ice-cold methanol for 20 min, followed by two washes (150 μl each) with phosphate-buffered saline (PBS) containing 0.5% Tween 20 (PBST) with 1 min of incubation at room temperature. Wells were blocked for 12 h at 4°C with 100 μl PBST containing 2% bovine serum albumin (PBST-BSA), followed by incubation with 25 μl of PBST-BSA containing a mouse anti-HBeAg monoclonal antibody (clone M2110146; Fitzgerald International, Acton, MA) diluted 1:3,200 for 1 h at 37°C. Wells were washed three times with PBST as described above, except that the plates were shaken at 600 rpm for 4 min between washes. This was followed by the addition of 25 μl PBST-BSA containing a horseradish peroxidase-conjugated anti-mouse antibody (diluted 1:5,000) and incubation at 37°C for 1 h. This step was followed by three washes with shaking and the addition of 50 μl BM Blue POD substrate (Roche Applied Science, Indianapolis, IN). A signal was allowed to develop for 20 min, and optical absorbance at a wavelength of 650 nm was determined with a reference reading at 490 nm. Wells where the signal was reduced by 50% were defined as containing a hit compound. The primary screening took approximately 5 months at a pace of 4,300 compounds per week.
To confirm the activity of hit compounds, EC50s were determined by incubating cells with compounds in duplicate wells at concentrations of 50 to 0.016 μM (in half-log steps), carrying out ELISAs as described above, and performing best-fit curve analysis of the results with XLfit 4.0 (IDBS; Bridgewater, NJ). Degrees of inhibition were calculated against multiple negative-control samples where only DMSO was incubated with the cells. In addition, each plate had multiple wells containing 50 μM 3TC as a reference inhibitor. Only curves with R2 values of >0.5 were considered to produce valid EC50s. Alternatively, the activity of hit compounds was also tested by the measurement of secreted HBeAg by using 100 μl of medium from each well and a commercial sandwich ELISA for HBeAg and following the manufacturer's directions (International Immuno-Diagnostics, Foster City, CA).
Concurrent with experiments determining EC50s, 50% cytotoxic concentrations (CC50s) were determined by plating cells at 1.0 × 104/well (20% confluence) to detect inhibition of cell growth compared with that which occurred in the absence of compounds. Cell plates were then incubated with compound dilutions and controls for 7 days as described above for the compound screen assay, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma-Aldrich, St. Louis, MO) was added to a final concentration of 0.5 mg/ml (28). Plates were again incubated at 37°C for 4 h, after which 10% SDS–0.01 N HCl was added to each well in a volume equal to the medium (100 μl), followed by overnight incubation to solubilize the reaction product. Colorimetric absorbance was read in a Rainbow spectrophotometer (Tecan US Inc., Durham, NC) at 570 nm (reference wavelength of 630 nm) and analyzed with XLfit 4.0 as described above.
The selectivity index (SI) of each compound was determined as follows: SI = CC50/EC50. Compound hits with SIs of >4 were counterscreened through HepG2.2.15 cells, in which the majority of HBeAg expression is from integrated HBV DNA, not cccDNA. Those compounds that did not significantly reduce the HBeAg level in HepG2.2.15 cells were forwarded to the secondary assay.
Viral nucleic acid analysis.
Total cellular RNA was extracted with TRIzol reagent (Invitrogen, Grand Island, NY) by following the manufacturer's directions. Ten micrograms of total cellular RNA was resolved in a 1.5% agarose gel containing 2.2 M formaldehyde and transferred onto Hybond-XL membrane (GE Healthcare, Piscataway, NJ) in 20× SSC buffer (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). Intracellular hepadnaviral core DNA and cccDNA (Hirt DNA) were extracted as described previously (10–13, 15). Half of the DNA sample recovered from one well of a six-well plate was resolved by electrophoresis into a 1.2% agarose gel and blotted onto Hybond-XL membrane. Membranes were probed with either an [α-32P]UTP (800 Ci/mmol; Perkin-Elmer)-labeled hepadnaviral minus-strand-specific (for detection of DNA) or plus-strand-specific (for detection of RNA) full-length riboprobe and exposed to a phosphorimager screen (Fuji Film, Tokyo, Japan). Hybridization signals were visualized by Typhoon FLA-7000 (GE Healthcare) and quantified with ImageQuant TL software (GE Healthcare).
EPR.
The hepadnavirus endogenous DNA polymerase reaction (EPR) was performed as previously described but with modifications (12). Briefly, an EPR mixture was assembled with 10 μl of HBV virion stock (∼5 × 107 genome equivalents) concentrated from the supernatant of HepDE19 cells cultured in tetracycline-free medium (11), or 10 μl of DHBV virion stock (∼2 × 107 genome equivalents) partially purified from DHBV-positive duck serum, plus 15 μl of 2× EPR buffer, which consisted of 0.3 M NaCl; 0.1 M Tris-HCl (pH 8.0); 20 mM MgCl2; 2 mM dithiothreitol; 0.2% (vol/vol) Nonidet P-40; 0.2 mM (each) dATP, dGTP, and dTTP; and 10 μM [α-32P]dCTP. Compounds were added as indicated, and the mixture was supplemented with water to bring the volume to 30 μl. As inhibitory controls, 0.2 mM dGTP in 2× EPR buffer was replaced with 0.2 mM ddGTP in the HBV EPR mixture and 1 mM foscarnet sodium (PFA) was added to the DHBV EPR mixture, respectively. After incubation at 37°C for 1 h, the reaction solution was dotted onto a Whatman 3M filter (Millipore, Billerica, MA), which was rinsed with 10% trichloride acid for 30 min at room temperature and then washed three times with 95% ethanol for 5 min each time. The radioactivity (counts per minute) of acid-insoluble 32P was measured with a liquid scintillation counter (Perkin-Elmer, Waltham, MA).
HBV precore/core immunoblotting.
Cells in one well of a six-well plate were washed once with PBS buffer and lysed in 300 μl of 1× Laemmli buffer. Thirty microliters of the cell lysate was resolved by 12% SDS-PAGE, and proteins were transferred onto an Immobilon-FL polyvinylidene difluoride membrane (Millipore). The membrane was blocked with Western Breeze blocking buffer (Invitrogen) and probed with antibodies against the C-terminal 14 amino acids of HBV precore/core protein (12) and β-actin (Millipore). Bound antibodies were revealed by IRDye secondary antibodies and visualized using the Li-COR Odyssey system (Lincoln, NE).
PDH infection and viral DNA analysis.
One-day-old Pekin ducklings were purchased from Metzer Farms (Gonzales, CA). Primary duck hepatocyte (PDH) cultures were prepared from 1-week-old DHBV-negative ducklings (33). PDHs were plated into 60-mm-diameter tissue culture dishes in L15 medium supplemented with 5% fetal bovine serum and maintained at 37°C. The next day, the cultures were shifted to serum-free L15 medium. The cells were infected on day 2 after seeding with congenitally DHBV-infected duckling serum containing approximately 1.5 × 107 enveloped virus particles. The medium was replaced after 16 h and changed every other day thereafter. Cultures were harvested at the time points indicated. The plates were washed once with chilled PBS, and viral core DNA and cccDNA were extracted and analyzed by Southern blotting as previously described (10). All PDH experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the Fox Chase Cancer Center.
RESULTS
Production of HBeAg is cccDNA dependent in HepDE19 cells.
The cccDNA-dependent assay mechanism of the HepDE19 cell line is illustrated in Fig. 1. Briefly, a more-than-one-genome length of the HBV genome spanning the entire viral pgRNA region under the control of the tetracycline-regulated cytomegalovirus immediate-early (tet-CMV) promoter is integrated into the chromosomal DNA of HepG2 cells. A point mutation of the precore start codon at the 5′ end of the transgene is introduced to prevent the expression of precore/HBeAg from the transgene. Upon tetracycline withdrawal, the transcribed pgRNA will express viral core protein and polymerase and initiate reverse transcription to generate rcDNA, resulting in cccDNA formation via the intracellular amplification pathway. Meanwhile, the start codon of the C-terminally truncated precore open reading frame (ORF) at the 3′ end of the pgRNA is copied into the viral DNA sequence and the precore ORF is restored during rcDNA conversion into cccDNA. Thus, the authentic precore mRNA will be transcribed only from cccDNA, with the translated precore protein product being further processed into HBeAg (42), which is secreted into the culture fluid and serves as a marker for cccDNA in HepDE19 cells.
Fig 1.

HBV nucleic acid metabolism in HepDE19 cells and assay design rationale. (A) The HepDE19 transgene contains a 1.1 overlength HBV genome (genotype D, subtype ayw), starting at nucleotide 1805, under the control of the tet-CMV promoter, in which the precore start codon (AUG) was mutated to UG at the HBV DNA 5′ end, with the second one at the 3′ redundancy (r′) left unchanged. The HBV nucleotide positions are according to the nomenclature of Galibert et al. (5). pC, C, pol, pS1, pS2, S, and X represent ORFs for the precore protein; the core protein; the polymerase; the preS1, preS2, and S domains of the HBV surface antigen; and the X protein, respectively. DR represents identical direct repeat sequences 1 and 2. pA is a polyadenylation site. Upon the removal of tetracycline (tet), pgRNA is transcribed and the viral core protein and polymerase are produced (B), resulting in pgRNA packaging, reverse transcription of pgRNA to minus-strand DNA (C), and sequential plus-strand DNA synthesis and circulation into rcDNA (D). rcDNA is converted to the cccDNA template, in which the precore ORF is restored, giving rise to authentic precore mRNA (E) and pgRNA (F). (G) cccDNA-derived precore mRNA serves as the template to translate the precore, which is further processed into secreted HBeAg through proteolytic cleavage of the N-terminal signal peptide and the C-terminal domain (CTD) (42).
To validate cccDNA-dependent HBeAg production in HepDE19 cells, we compared HBV replication, cccDNA formation, and precore/HBeAg production in parallel in HepG2.2.15 and HepDE19 cells. As shown in Fig. 2, noninducible HepG2.2.15 cells displayed constitutive HBV RNA transcription, DNA replication, and precore/core protein expression when the cells reached confluence (Fig. 2A, left panel); ELISAs showed that the HBeAg level rapidly increased from day 0 to day 4 and continued to climb slowly thereafter in an 8-day incubation period (Fig. 2B). However, cccDNA was undetectable by Southern blot assay at any time point in this experiment (Fig. 2A, lanes 1 to 5), which demonstrated that the HBeAg is expressed predominantly from the HBV transgene and not from cccDNA, if there is any, in HepG2.2.15 cells. In contrast, upon the withdrawal of tetracycline from HepDE19 cells, HBV pgRNA transcription, core protein expression, DNA replication, and cccDNA synthesis gradually increased in a time-dependent manner and precore protein expression was detected only after cccDNA reached detectable levels (Fig. 2A, right panel). Moreover, the levels of secreted HBeAg were proportional to the intracellular cccDNA level (Fig. 2B). Therefore, the results confirmed a quantitative relationship between cccDNA and HBeAg levels and support the use of HepDE19 cells for screening to identify compounds that affect HBV cccDNA formation and/or maintenance.
Fig 2.

Evaluation of cccDNA-dependent HBeAg production in HepDE19 cells. (A) Kinetics of intracellular virus production in HepG2.2.15 and HepDE19 cells. HepG2.2.15 cells were seeded at a density of 1.2 × 106 cells/well in six-well plates, and day 0 was when cells were completely attached, and then the cells were continuously cultured for 8 days. HepDE19 cells were cultured in tetracycline-containing medium until confluent (day 0), and then tetracycline was removed from the medium and the cells were maintained for another 8 days. HepG2.2.15 and HepDE19 cells and culture fluid were harvested every other day from day 0 to day 8. Total cellular RNA was extracted, and HBV RNA was detected by Northern blotting as described in Materials and Methods; rRNA (28S and 18S) served as a loading control. The positions of HBV pgRNA (3.5 kb) and surface mRNAs (sRNA, including 2.4 kb and 2.1 kb) are indicated. HBV core DNA and cccDNA (Hirt DNA) were extracted and analyzed by Southern hybridization. The positions of rcDNA (RC), single-stranded DNA (SS), DP-rcDNA (DP-rc), and cccDNA are indicated. The expression of the HBV core and precore proteins was detected by Western blotting, with the levels of β-actin serving as a loading control. Since the core antibody recognizes the 14-amino-acid epitope at the C terminus of the core protein (12), the detected precore band represents an HBeAg precursor (p22) with an intact C-terminal domain (18, 42). (B) Correlation between the levels of HBV cccDNA and HBeAg in HepDE19 cells. HBeAg levels in the culture medium harvested from panel A were determined by commercial HBeAg ELISA (International Immuno-Diagnostics, Foster City, CA) and plotted as a function of time (days). The quantified levels of viral cccDNA from the Southern blot assay shown in panel A were superimposed onto the graph. The results are representative of two separate trials.
Identification of DSS compounds as novel cccDNA inhibitors.
Using the HepDE19 cell-based assay described above, we screened an in-house chemical compound library consisting of 85,000 drug-like small molecules at a single concentration, 10 μM. Compounds were added simultaneously with tetracycline withdrawal in order to identify inhibitors of cccDNA establishment and/or maintenance. HBeAg was detected by ELISA as described in Materials and Methods. Nontoxic compounds causing a 50% reduction of the HBeAg level were declared primary hits. Hits were counterscreened in HepG2.2.15 cells, in which HBeAg is produced predominantly in a cccDNA-independent manner. A total of 329 compounds that selectively reduced HBeAg levels in HepDE19 cells, but not HepG2.2.15 cells, were chosen for follow-up assays. Following EC50 and CC50 determinations, eight confirmed hits were selected and advanced to the measurement of HBV cccDNA by Southern blotting, in which HepDE19 cells were incubated with each compound at 10 μM after tetracycline withdrawal for a total incubation time of 14 days to ensure sufficient cccDNA accumulation (data not shown). Two structurally related DSS, termed 2-[benzenesulfonyl-(2-chloro-5-trifluoromethyl-phenyl)-amino]-N-pyridin-4-ylmethyl-acetamide (CCC-0975) and 4-(benzyl-methyl-sulfamoyl)-N-(2-methyl-benzothiazol-5-yl)-benzamide (CCC-0346), which reduced HBeAg with acceptable SIs of >11 and >7 in the screening assays, respectively (Fig. 3), emerged as final confirmed cccDNA inhibitor hits.
Fig 3.

DSS compounds as inhibitors of HBeAg production in HepDE19 cells. The chemical structures and properties of CCC-0975 (A) and CCC-0346 (B) are presented. Graphs represent inhibition of HBeAg production (top) and loss of HepDE19 cell viability (bottom), and each point is the average of duplicate samples. Incubation was for 7 days. EC50 and CC50 values (μM) were calculated with XLfit 4.0 (IDBS, Surrey, United Kingdom). MW, molecular weight.
DSS compounds were deemed interesting in that they had not been identified as hits in other antiviral screenings we had conducted in-house, which suggested that their activity is specific (unpublished data). Moreover, we have found that no close structural relationship with DSS compounds and other sulfonamide-containing antiviral molecules have been described in the literature as inhibitors of viral polymerases, proteases, integrases, transcription, and entry (41); this chemical distinction underscores the novelty of the anti-HBV DSS compounds. Both DSS compounds possess very attractive drug-like properties that meet Lipinski's rule of five (24), including cLogP values of <5.0, molecular weights of <500, <5 H bond donors, <10 H bond acceptors, and acidic pKa values that suggest good oral bioavailability. In addition, the structures are highly chemically tractable for future lead optimization and structure-activity relationship (SAR) investigation. The hit compounds were repurchased from the vendor or prepared by resynthesis and retested with HepDES19 cells, which have a higher level of cccDNA production than HepDE19 cells (11).
DSS compounds inhibit accumulation of HBV cccDNA and DP-rcDNA in HepDES19 cells.
Treatment with CCC-0975 resulted in a dose-dependent reduction of cccDNA in HepDES19 cells with an EC50 of 10 μM, along with a significant reduction of the putative cccDNA precursor DP-rcDNA (Fig. 4A, bottom panels). The reduction of DP-rcDNA and cccDNA was proportional, compared with that in the untreated control. CCC-0346 has a higher level of toxicity than CCC-0975 in cell growth assays (Fig. 3); however, at concentrations that proved nontoxic to confluent cells, CCC-0346 displayed an antiviral effect against DP-rcDNA and cccDNA accumulation similar to that of CCC-0975 but had an EC50 of 3 μM (Fig. 4B, bottom panels).
Fig 4.

DSS compounds reduce the levels of cccDNA and DP-rcDNA in HepDES19 cells. Upon withdrawal of tetracycline, HepDES19 cells were left untreated (UNT) or treated with CCC-0975 (A) or CCC-0346 (B) at the indicated concentrations; tetracycline-free medium was changed every other day with fresh compound supplementation. The concentration of DMSO in all experimental groups was normalized at 0.1%. The cells were harvested at day 12, and viral RNA (top), core DNA (middle), and Hirt DNAs (DP-rcDNA and cccDNA) (bottom) were extracted and analyzed by Northern blotting and Southern blotting, respectively. Shorter exposure of the Hirt DNA blot was used for the quantitative determination of DP-rcDNA signals. The relative intensity of the viral RNA or DNA species in each sample is expressed as a percentage of that from untreated cells and is indicated below each blot.
Although cccDNA formation in HepDES19 cells is driven by transgene-derived pgRNA transcription and DNA replication, the observed slight reduction of viral RNA and DNA replicative intermediates under DSS compound treatment at high concentrations (Fig. 4, top and middle panels) did not quantitatively account for the reduction of cccDNA, and their decrease ought to be a consequence of the reduction of cccDNA, which, once established, contributes approximately 10% of the total viral RNA and DNA production in stably HBV-transfected cell lines (2, 14, 45). We thus speculated that DSS-mediated cccDNA reduction was not, or at least not completely, due to the inhibition of transgene-based viral transcription or DNA replication. This notion was further supported by the observations that DSS compounds did not inhibit HBV polymerase activity in the in vitro endogenous polymerase reaction (Fig. 5), suggesting that CCC-0975 and CCC-0346 do not directly target viral polymerase-catalyzed DNA replication. In addition, DSS compounds did not reduce the steady-state levels of HBV RNA and core DNA in HepDES19 cells at an early time point (day 4) of treatment (Fig. 6, top and middle panels), when the cccDNA was undetectable by Southern blotting (Fig. 6, bottom panel). Strikingly, DSS compounds led to a significant reduction of DP-rcDNA at this early time point, suggesting that HBV rcDNA deproteinization and/or the stability of DP-rcDNA is influenced by DSS compounds. Nevertheless, the possibility that DSS-mediated cccDNA reduction, seen at later time points, is due to DP-rcDNA independent mechanisms is not ruled out.
Fig 5.

DSS compounds do not inhibit HBV polymerase activity. HBV virion particles purified from HepDE19 culture fluid were subjected to endogenous polymerase reaction with [32P]dCTP plus CCC-0975 or CCC-0346 at the indicated concentrations. DMSO at 0.1% was used as a solvent control, and ddGTP was a reference chain terminator in the reaction. Incorporated [32P]dCTP radioactivity was measured with a liquid scintillation counter. The relative polymerase activity was plotted as a percentage of the readout of counts per minute in the control reaction (y axis). Values are averages of triplicate samples, and error bars represent standard deviations.
Fig 6.
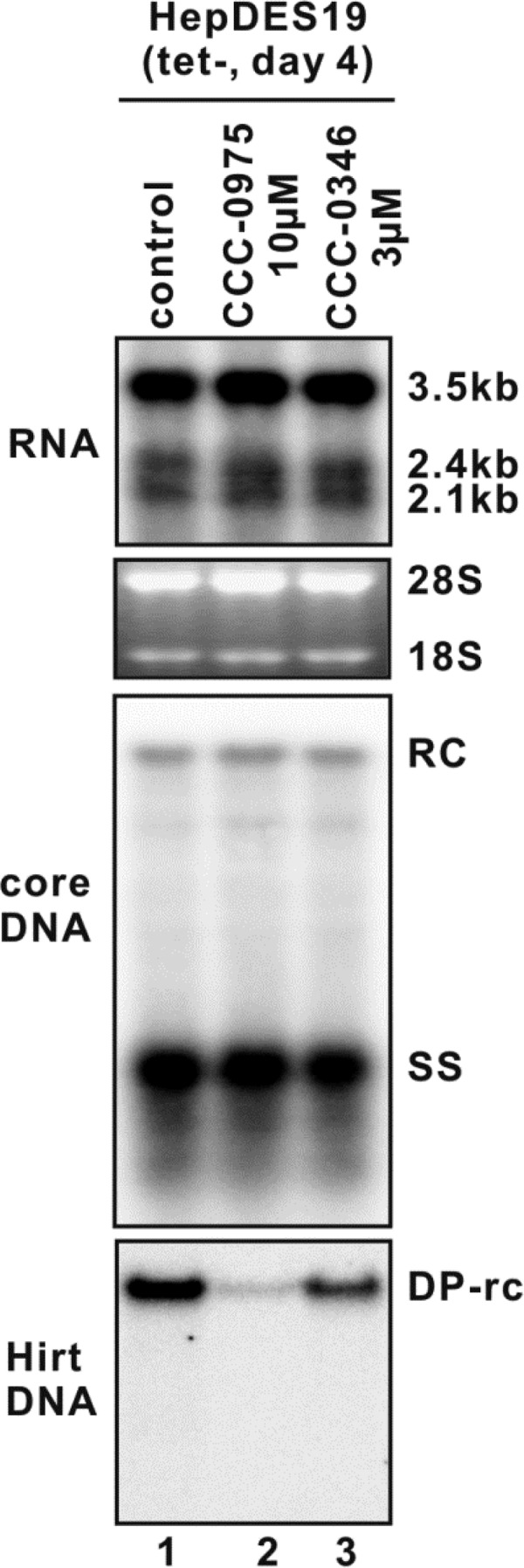
DSS compounds reduce the level of HBV DP-rcDNA, but not viral RNA and core DNA, in HepDES19 cells with short-term treatment. Upon the removal of tetracycline, HepDES19 cells were left untreated or treated with CCC-0975 or CCC-0346 at the indicated concentration; tetracycline-free medium was changed every other day with fresh compound supplementation. The DMSO concentration in the whole experiment was normalized at 0.1%. The cells were harvested at day 4, and viral RNA (top), core DNA (middle), and Hirt DNA (DP-rcDNA and cccDNA) (bottom) were extracted and analyzed by Northern blotting and Southern blotting, respectively.
DSS compounds do not alter the decay kinetics of HBV DP-rcDNA and cccDNA.
To further determine whether DSS compounds reduce HBV DP-rcDNA and cccDNA through direct inhibition of the biosynthesis of these two DNA molecules or by promoting their degradation in cell culture, a HepDES19 cell-based experimental system was used to study the stability of cccDNA upon compound treatment. As shown in Fig. 7, removal of tetracycline from HepDES19 cells led to the accumulation of HBV RNA, core DNA, DP-rcDNA, and cccDNA at day 12 (Fig. 7B, lane 1), at which point the culture fluid was supplemented with tetracycline and 3TC to instantly shut down transgene-based pgRNA transcription and viral DNA replication, respectively, thereby preventing the de novo replenishment of cccDNA formation. After another 4-day period, the levels of viral RNA and core DNA dramatically decreased (Fig. 7B, lane 2, top and middle panels); however, HBV DP-rcDNA and cccDNA remained at high levels (Fig. 7B, lane 2, bottom panel), which was consistent with previous reports demonstrating that hepadnaviral cccDNA per se has a longer half-life than other viral nucleic acids (14, 46). In this scenario, the remaining low level of viral pgRNA was derived predominantly from cccDNA-based transcription and maintained thereafter (Fig. 7B, top panel). The observed slight increase in cccDNA from day 12 to day 16 might have been due to the continued conversion of DP-rcDNA to cccDNA (Fig. 7B [compare lanes 2 and 1], C, and D), which, for unknown reasons, is extremely inefficient in HBV-replicating cell cultures (6, 11, 12, 20).
Fig 7.

DSS compounds do not alter the decay kinetics of HBV DP-rcDNA and cccDNA in HepDES19 cells. (A) HepDES19 cells were cultured in a six-well plate until confluent. Tetracycline was then removed from the culture medium to induce HBV replication and cccDNA formation. After 12 days, tetracycline and 3TC (10 μM) were added back to the culture medium to shut off viral pgRNA transcription from the integrated HBV genome and prevent viral DNA replication. Four days later, one set of cells (lane 3) was cultured further with medium containing tetracycline and 3TC and another two sets of cells were treated with CCC-0975 (10 μM, lanes 4, 7, 10, and 13) or CCC-0346 (3 μM, lanes 5, 8, 11, and 14) in the presence of tetracycline and 3TC for the indicated period of time. The DMSO concentration in all experimental groups was 0.1%. (B) Cells were harvested at the indicated time points, and the levels of viral RNA, core DNA, and Hirt DNA were determined as described in Materials and Methods. The relative intensities of viral DP-rcDNA and cccDNA signals in each sample are expressed as percentages of the signals from the sample at day 12 (lane 2) and are plotted in graphs C and D, respectively. The results are representative of two separate trials.
The decay kinetics of the preexisting DP-rcDNA and cccDNA were then determined without or with DSS compound treatment in the presence of tetracycline and 3TC. As shown in Fig. 7B by Southern blot assay and in Fig. 7C and D by quantitative measurement, both DP-rcDNA and cccDNA gradually degraded over time in the cells following continuous culture with similar half-lives of around 9 days; however, DSS compound treatment apparently did not alter the decay kinetics of DP-rcDNA and cccDNA. Those observations, along with the fact that DSS compounds inhibited the accumulation of DP-rcDNA and cccDNA in the context of HBV DNA replication (Fig. 4 and 6), thus suggested that the antiviral mechanism(s) of DSS compounds is to block the biosynthesis of cccDNA, perhaps through reducing the amount of DP-rcDNA, which is a functional precursor of cccDNA formation (11).
DSS compounds inhibit DHBV cccDNA formation.
All of the above-described evaluations of DSS compound potency against cccDNA biosynthesis were done with stably HBV-transfected cell lines, in which cccDNA formation relies largely on transgene-derived viral replication. Therefore, a critical issue was whether or not DSS compounds can inhibit hepadnavirus cccDNA formation and subsequent viral replication during an authentic hepadnavirus infection. To test this, we utilized PDHs, which support DHBV infection and cccDNA formation with high efficiency (26). In comparison to the HBV systems, the faster kinetics of DHBV replication and cccDNA formation permit shorter incubation times.
As shown in Fig. 8, pretreatment of PDHs for 2 h, DHBV inoculation, and continued treatment with CCC-0975 for 5 days demonstrated a dose-dependent inhibition of DHBV DP-rcDNA/cccDNA biosynthesis, with an EC50 of 3 μM (Fig. 8A, top panel). Because viral replication totally relies on cccDNA in DHBV-infected PDHs, viral core DNA was reduced proportionally with cccDNA inhibition (Fig. 8A, bottom panel). Interestingly, CCC-0346 did not exhibit significant toxicity in PDHs compared to HepDE19 cells but reduced DHBV cccDNA by only 50% at 30 μM (Fig. 8B). Such a discrepancy might be due to cell type- or virus-specific conditions. In a parallel experiment, treatment of PDH with DSS compounds at a single dose of 10 μM postinoculation with DHBV resulted in similar effects against cccDNA formation and viral DNA replication (Fig. 8C). The primary hepatocytes were monitored daily by microscopy following compound treatment and virus infection, and no cytopathic effects were observed throughout the experiments (data not shown). In addition, neither DSS compound inhibited DHBV polymerase activity in an EPR assay (Fig. 9), thus supporting the idea that DSS compounds, especially CCC-0975, exhibit antiviral activity against cccDNA formation in multiple hepadnavirus systems.
Fig 8.

Antiviral effects of DSS compounds in DHBV-infected PDHs. PDHs were plated in 60-mm dishes and pretreated with control solvent (0.1% DMSO) or with CCC-0975 (A) or CCC-0346 (B) at the indicated concentrations. Two hours later, PDHs were infected in the presence of the test compounds with DHBV-positive duck serum containing approximately 1.5 × 107 virions. After overnight incubation, the PDHs were rinsed twice with regular medium and the treatments were continued with medium and compound replenishment every other day. In a parallel experiment (C), PDHs were infected overnight without compound pretreatment, followed by postinfection treatment with either CCC-0975 or CCC-0346 at 10 μM, with a medium change every other day. In all of the experiments, cells were harvested at 6 days postinfection and DHBV Hirt DNA (top) and core DNA (bottom panels) were analyzed. The relative intensities of viral DNA species in each sample are expressed as percentages of those in the untreated (UNT) cells and are presented below each blot.
Fig 9.
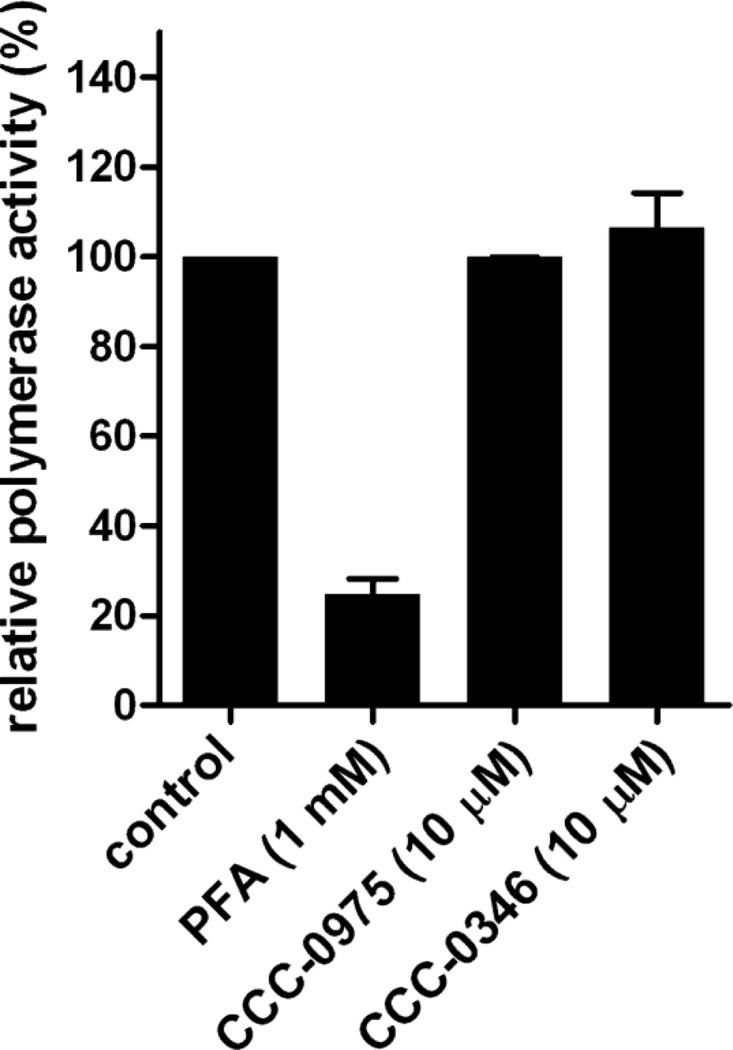
DSS compounds do not inhibit DHBV polymerase activity. DHBV virion particles derived from the sera of congenitally DHBV-infected ducks were subjected to an endogenous polymerase reaction with [32P]dCTP plus CCC-0975 or CCC-0346 at 10 μM. DMSO at 0.1% was used as a solvent control. PFA served as a positive control for polymerase inhibitor. Incorporated [32P]dCTP radioactivity was measured with a liquid scintillation counter. Relative polymerase activity was plotted as a percentage of the counts per minute read in the control reaction mixture (y axis). Values are averages of triplicate samples, and error bars represent standard deviations.
DISCUSSION
Current treatments of chronic hepatitis B are limited to IFN therapy and nucleos(t)ide analogue reverse transcriptase inhibitors, all of which are used for prolonged periods but cure the infection in only a small minority of cases and display no benefit when used in combination (16, 17). The underlying cause of HBV chronicity is the intracellular persistence of a multicopy episomal form of the virus genome, cccDNA, which is inherently stable in the nuclei of infected hepatocytes and is only indirectly affected by current therapies (25). In order to clear the infection, a durable, curative antiviral therapy that directly reduces the level of HBV cccDNA without killing infected hepatocytes is needed.
It is generally acknowledged that cccDNA formation and persistence involve many viral and cellular factors, and there are likely to be many molecular opportunities for intervention, ranging from biosynthesis to maintenance of cccDNA. The putative approaches to cccDNA inhibition include, but are not limited to, the following. The first is inhibition of cccDNA establishment. Such an inhibitor would work by abrogating any of the several steps that lead from mature cytoplasmic rcDNA to the presence of cccDNA in the nucleus (12). The second is inhibition of cccDNA maintenance factors. Although it is still unclear whether a cccDNA-specific maintenance factor(s) exists, a small molecule may specifically recognize and alter the stability of the cccDNA minichromosome. The third is chemical alteration of cccDNA. Compared to the host chromatin, cccDNA should exhibit increased sensitivity to DNA damage due to the very dense coding of the HBV genome (30). The fourth is epigenetic silencing of cccDNA transcriptional activity (32). The fifth is induction of the intracellular innate defense. It has been demonstrated that clearance of cccDNA during acute infection can occur through a noncytolytic mechanism that is largely independent of adaptive immune function (9, 43), indicating that cccDNA can be reduced by innate cellular defenses that may be activated by a biologically active stimulator or a small-molecule compound.
In this study, by employing a cell-based screening strategy that monitors the HBV cccDNA level through proportional expression of HBeAg, we discovered two structurally related sulfonamide compounds that significantly reduced the levels of HBV cccDNA in cell cultures. Further mechanistic studies revealed that the DSS compounds directly block the conversion of HBV rcDNA into cccDNA, rather than inhibiting the production of rcDNA as existing HBV drugs do. Therefore, we have provided the first proof-of-concept evidence that it is feasible to develop small-molecule inhibitors that prevent the formation of cccDNA from the rcDNA precursor, an essential but unexploited step in the HBV replication cycle.
The exact target(s) of DSS compounds is still unclear, considering that there are many molecular details yet to be elucidated in the understanding of cccDNA formation and metabolism. cccDNA is formed from both the incoming viral genome during initiation of infection and newly synthesized mature viral DNA, through conversion of the viral rcDNA genome, most possibly by employing the host DNA repair machinery in the nucleus (1). In one attempt to identify the potential intermediate(s) bridging rcDNA-to-cccDNA conversion, a linear woodchuck hepatitis virus genome that contains a terminal duplication of the cohesive region (between DR1 and DR2) from rcDNA by displacement synthesis through the cohesive overlap was proposed to be a cccDNA precursor by serving as a substrate for DNA repair through legitimate recombination. The authors found that some integrated viral DNA appeared to have this linear DNA as a precursor; however, the extrachromosomal form of such hypothetic DNA species has not been directly detected and thus remains mysterious (44). Our recent studies focusing on a DP-rcDNA molecule which loses the covalently bonded viral polymerase revealed that DP-rcDNA is a bona fide precursor of cccDNA formation, albeit at low efficiency (11, 12). In addition, a model of the molecular pathway of cccDNA formation has been proposed in which the completion of viral plus-strand DNA inside the nucleocapsid triggers the removal of viral polymerase from minus-strand DNA. The deproteinization reaction is tightly associated with a nucleocapsid structural shift, resulting in exposure of the NLS at the C terminus of capsid protein, which in turn initiates the karyopherin-mediated nuclear import of capsid/DP-rcDNA complexes. Finally, DP-rcDNA is released from the capsid in the nucleus and is converted into cccDNA presumably through DNA repair (11, 12). Interestingly, DSS compounds reduce DP-rcDNA, as well as cccDNA, without directly affecting viral DNA replication, thus providing another line of evidence that DP-rcDNA is a precursor of cccDNA formation. Nevertheless, it is still possible that DSS compounds may inhibit the production of another undiscovered cccDNA precursor(s) or DNA repair mechanism(s) involved in cccDNA formation.
The mechanisms underlying the deproteinization of hepadnavirus rcDNA remain elusive. The viral polymerase is covalently linked to the 5′ phosphate group of the rcDNA minus strand through a tyrosine residue (Y63 for HBV, Y96 for DHBV) in the terminal protein (TP) domain, as a consequence of TP-mediated protein priming during the initial reverse transcription of viral pgRNA (29). It is conceivable that the removal of polymerase from rcDNA ought to be an essential step in cccDNA formation. Our previous work ruled out the possibility that the viral polymerase is removed by endonuclease cleavage of the sequences proximal to the linkage between the rcDNA terminus and polymerase, based upon the fact that the 5′ end of the DP-rcDNA minus strand is the same as the end of rcDNA (11). Thus, the anticipated mechanisms of rcDNA deproteinization are narrowed down to the following. (i) The phosphodiester bond is hydrolyzed by a phosphodiesterase, one candidate being a recently discovered human 5′-tyrosyl DNA phosphodiesterase (TDP2) that removes topoisomerase covalently conjugated at the 5′ end of a chromosomal DNA break (3). (ii) Polymerase is removed by proteolytic digestion, which results in a small peptide, or at least the tyrosine residue, being left on the DP-rcDNA. Therefore, it will be important to evaluate the potential phosphodiesterase or protease inhibitor activity of DSS compounds in the future.
The pool size of cccDNA ranges from 1 to 50 copies per cell, and once established, nascent cccDNA converted from progeny rcDNA, coupled with its stability within the infected hepatocyte, helps to maintain viral persistence. The basis of cccDNA longevity is poorly understood. The precise half-life of HBV cccDNA has been reported to range from days to months in different animal and tissue culture models (47), and our results herein showed that the half-life of cccDNA was around 9 days in confluent HepDES19 cells under 3TC treatment (Fig. 7). Because cccDNA plays a central role in HBV persistence, elimination of cccDNA is the ultimate goal of antiviral therapy. Unfortunately, current antiviral treatment with nucleos(t)ide analogues fails to eliminate the preexisting cccDNA pool and/or prevent cccDNA formation from trace level wild-type or drug-resistant virus (48). Although DSS compounds are unable to promote the degradation of the preexisting cccDNA in cell cultures, their mechanism of action is certainly distinct from that of the nucleos(t)ide analogues, and thus, they will likely retain activity in preventing cccDNA formation even when the virus has become resistant to polymerase inhibitors. In addition, DSS compounds may also exhibit synergistic effects in combination therapy with replication inhibitors. It is envisioned that the combination of inhibition of viral replication by nucleos(t)ide analogue with its amelioration in liver function (25) and the inhibition of de novo cccDNA formation by a DSS compound may enhance the eventual clearance of cccDNA. The antiviral effect of DSS compounds in combination with a nucleos(t)ide analogue and the potential inhibitory effect of DSS compounds against drug-resistant virus cccDNA formation will be tested in future studies.
We have confirmed that the DSS compounds, especially CCC-0975, also inhibited DHBV cccDNA formation in virus-infected PDHs. This observation indicates a wide spectrum for these compounds in hepadnavirus cccDNA intervention and also provides an opportunity to evaluate their antiviral activity (alone or in combination with nucleos[t]ide analogues) in the duck model. Other hepadnavirus animal models, including humanized uPA/SCID mice and woodchucks (4, 21), are also available for in vivo testing of DSS compounds. First, however, the potency of the hit compounds needs to be improved. This work is under way through a SAR study. The structural features of these DSS compounds offer multiple modification opportunities for lead optimization. For example, with the common key sulfonamide moiety being maintained, both linkages and substituents will be structurally explored for SAR (e.g., electronic effect, steric effect, etc.) investigation and to improve antiviral profiles.
In summary, we have, for the first time, discovered two structurally related novel inhibitors of HBV cccDNA biosynthesis through an innovative screening approach. Distinct from those of current antiviral agents, the unique antiviral mechanism of DSS compounds is inhibition of the formation of cccDNA from its rcDNA precursor and may involve the inhibition of rcDNA deproteinization, a possible intermediate step during cccDNA formation. Thus, these two inhibitors also provide a promising research tool to identify a viral/host protein(s) involved in cccDNA formation. Clearance of cccDNA is the ultimate goal for the cure of hepatitis B, and it cannot be reliably accomplished by currently available therapeutics alone; the further development of DSS compounds may ultimately lead to drugs that change the landscape of hepatitis B management.
ACKNOWLEDGMENTS
This work was supported by the NIH grants R56AI066024, R56AI066024-1A2 (to Andrea Cuconati and Haitao Guo), and R01AI094474 (to Haitao Guo and Andrea Cuconati) and by the Hepatitis B Foundation through an appropriation of the Commonwealth of Pennsylvania. Work in William S. Mason's lab was supported by NIH grants R01AI018641 and CA06927. Haitao Guo is the Bruce Witte fellow of the Hepatitis B Foundation.
Footnotes
Published ahead of print 29 May 2012
REFERENCES
- 1. Block TM, Guo H, Guo JT. 2007. Molecular virology of hepatitis B virus for clinicians. Clin. Liver Dis. 11:685–706, vii [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chou YC, et al. 2005. Evaluation of transcriptional efficiency of hepatitis B virus covalently closed circular DNA by reverse transcription-PCR combined with the restriction enzyme digestion method. J. Virol. 79:1813–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. 2009. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461:674–678 [DOI] [PubMed] [Google Scholar]
- 4. Dandri M, Petersen J. 2012. Chimeric mouse model of hepatitis B virus infection. J. Hepatol. 56:493–495 [DOI] [PubMed] [Google Scholar]
- 5. Galibert F, Mandart E, Fitoussi F, Charnay P. 1979. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 281:646–650 [DOI] [PubMed] [Google Scholar]
- 6. Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J. Virol. 81:6164–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gish RG, et al. 2007. Entecavir therapy for up to 96 weeks in patients with HBeAg-positive chronic hepatitis B. Gastroenterology 133:1437–1444 [DOI] [PubMed] [Google Scholar]
- 8. Gripon P, et al. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U S A. 99:15655–15660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guidotti LG, et al. 1999. Viral clearance without destruction of infected cells during acute HBV infection. Science 284:825–829 [DOI] [PubMed] [Google Scholar]
- 10. Guo H, Aldrich CE, Saputelli J, Xu C, Mason WS. 2006. The insertion domain of the duck hepatitis B virus core protein plays a role in nucleocapsid assembly. Virology 353:443–450 [DOI] [PubMed] [Google Scholar]
- 11. Guo H, et al. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J. Virol. 81:12472–12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J. Virol. 84:387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo H, et al. 2005. Identification and characterization of avihepadnaviruses isolated from exotic anseriformes maintained in captivity. J. Virol. 79:2729–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo H, et al. 2007. Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-akt signal transduction pathway. J. Virol. 81:10072–10080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365–369 [DOI] [PubMed] [Google Scholar]
- 16. Hoofnagle JH, di Bisceglie AM. 1997. The treatment of chronic viral hepatitis. N. Engl. J. Med. 336:347–356 [DOI] [PubMed] [Google Scholar]
- 17. Hoofnagle JH, Doo E, Liang TJ, Fleischer R, Lok AS. 2007. Management of hepatitis B: summary of a clinical research workshop. Hepatology 45:1056–1075 [DOI] [PubMed] [Google Scholar]
- 18. Ito K, Kim KH, Lok AS, Tong S. 2009. Characterization of genotype-specific carboxyl-terminal cleavage sites of hepatitis B virus e antigen precursor and identification of furin as the candidate enzyme. J. Virol. 83:3507–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kann M, Schmitz A, Rabe B. 2007. Intracellular transport of hepatitis B virus. World J. Gastroenterol. 13:39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Köck J, et al. 2010. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 6:e1001082 doi:10.1371/journal.ppat.1001082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kulkarni K, Jacobson IM, Tennant BC. 2007. The role of the woodchuck model in the treatment of hepatitis B virus infection. Clin. Liver Dis. 11:707–725, vii [DOI] [PubMed] [Google Scholar]
- 22. Ladner SK, et al. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 41:1715–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang TJ. 2009. Hepatitis B: the virus and disease. Hepatology 49:S13–S21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46:3–26 [DOI] [PubMed] [Google Scholar]
- 25. Lok AS. 2009. Evolution of nucleoside/tide analogues for hepatitis B: is the ideal drug here yet? J. Hepatol. 51:416–418 [DOI] [PubMed] [Google Scholar]
- 26. Mason WS, Taylor JM. 1989. Experimental systems for the study of hepadnavirus and hepatitis delta virus infections. Hepatology 9:635–645 [DOI] [PubMed] [Google Scholar]
- 27. McMahon BJ. 2005. Epidemiology and natural history of hepatitis B. Semin. Liver Dis. 25(Suppl 1):3–8 [DOI] [PubMed] [Google Scholar]
- 28. Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65:55–63 [DOI] [PubMed] [Google Scholar]
- 29. Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res. 134:235–249 [DOI] [PubMed] [Google Scholar]
- 30. Newbold JE, et al. 1995. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 69:3350–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pawlotsky JM, et al. 2008. Virologic monitoring of hepatitis B virus therapy in clinical trials and practice: recommendations for a standardized approach. Gastroenterology 134:405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pollicino T, et al. 2006. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 130:823–837 [DOI] [PubMed] [Google Scholar]
- 33. Pugh JC, Summers JW. 1989. Infection and uptake of duck hepatitis B virus by duck hepatocytes maintained in the presence of dimethyl sulfoxide. Virology 172:564–572 [DOI] [PubMed] [Google Scholar]
- 34. Rabe B, Vlachou A, Pante N, Helenius A, Kann M. 2003. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. U S A. 100:9849–9854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schmidt K, Korba B. 2000. Hepatitis B virus cell culture assays for antiviral activity. Methods Mol. Med. 24:51–67 [DOI] [PubMed] [Google Scholar]
- 36. Schulze-Bergkamen H, et al. 2003. Primary human hepatocytes—a valuable tool for investigation of apoptosis and hepatitis B virus infection. J. Hepatol. 38:736–744 [DOI] [PubMed] [Google Scholar]
- 37. Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sells MA, Chen M, Acs G. 1987. Production of hepatitis B virus particles in hepG2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA. 84:1005–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sells MA, Zelent AZ, Shvartsman M, Acs G. 1988. Replicative intermediates of hepatitis B virus in HepG2 cells that produce infectious virions. J. Virol. 62:2836–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sohn JA, Litwin S, Seeger C. 2009. Mechanism for CCC DNA synthesis in hepadnaviruses. PLoS One 4:e8093 doi:10.1371/journal.pone.0008093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Supuran CT, Innocenti A, Mastrolorenzo A, Scozzafava A. 2004. Antiviral sulfonamide derivatives. Mini Rev. Med. Chem. 4:189–200 [DOI] [PubMed] [Google Scholar]
- 42. Wang J, Lee AS, Ou JH. 1991. Proteolytic conversion of hepatitis B virus e antigen precursor to end product occurs in a postendoplasmic reticulum compartment. J. Virol. 65:5080–5083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wieland SF, Spangenberg HC, Thimme R, Purcell RH, Chisari FV. 2004. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc. Natl. Acad. Sci. U S A. 101:2129–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang W, Mason WS, Summers J. 1996. Covalently closed circular viral DNA formed from two types of linear DNA in woodchuck hepatitis virus-infected liver. J. Virol. 70:4567–4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou T, et al. 2006. Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antiviral Res. 72:116–124 [DOI] [PubMed] [Google Scholar]
- 46. Zhu Y, et al. 2001. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J. Virol. 75:311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zoulim F. 2005. New insight on hepatitis B virus persistence from the study of intrahepatic viral cccDNA. J. Hepatol. 42:302–308 [DOI] [PubMed] [Google Scholar]
- 48. Zoulim F, Locarnini S. 2009. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 137:1593–1608.e1-2 [DOI] [PubMed] [Google Scholar]