

Abstract
Tulathromycin represents the first member of a novel subclass of macrolides, known as triamilides, approved to treat bovine and swine respiratory disease. The objectives of the present study were to assess the concentration-versus-time profile of tulathromycin in the plasma and lung tissue of healthy and neutropenic mice challenged intranasally with lipopolysaccharide (LPS) from Escherichia coli O111:B4. BALB/c mice were randomly allocated into four groups of 40 mice each: groups T-28 (tulathromycin at 28 mg/kg of body weight), T-7, T7-LPS, and T7-LPS-CP (cyclophosphamide). Mice in group T-28 were treated with tulathromycin at 28 mg/kg subcutaneously (s.c.) (time 0 h). The rest of the mice were treated with tulathromycin at 7 mg/kg s.c. (time 0 h). Animals in dose groups T-7-LPS and T7-LPS-CP received a single dose of E. coli LPS intranasally at −7 h. Mice in group T7-LPS-CP were also rendered neutropenic with cyclophosphamide (150 mg/kg intraperitoneally) prior to the administration of tulathromycin. Blood and lung tissue samples were obtained from 5 mice from each dose group at each sampling time over 144 h after the administration of tulathromycin. There were not statistical differences in lung tissue concentrations among groups T-7, T-7-LPS, and T7-LPS-CP. For all dose groups, the distribution of tulathromycin in the lungs was rapid and persisted at relatively high levels during 6 days postadministration. The concentration-versus-time profile of tulathromycin in lung tissue was not influenced by the intranasal administration of E. coli LPS. The results suggest that in mice, neutrophils may not have a positive influence on tulathromycin accumulation in lung tissue when the drug is administered during either a neutrophilic or a neutropenic state.
INTRODUCTION
Tulathromycin is a semisynthetic, 15-membered-ring macrolide derivative of erythromycin. It represents the first member of a novel subclass of macrolides known as triamilides (15). This macrolide shows metaphylactic and therapeutic efficacy in bovine and swine respiratory disease after a single administration (20, 23). Tulathromycin has also been approved by the U.S. Food and Drug Administration (FDA) to treat infectious keratoconjunctivitis caused by Moraxella bovis and bovine interdigital necrobacillosis associated with Fusobacterium necrophorum and Porphyromonas levii (22).
Tulathromycin is characterized by a high rate of absorption and a large volume of distribution (volume of distribution at steady state [Vss] values of 13.2 liters/kg of body weight in pig and 11 liters/kg in cattle) (3, 19). Pharmacokinetic (PK) studies in cattle, swine, and foals revealed the capacity of tulathromycin to rapidly accumulate in lung tissue and persist for a long period of time (the apparent elimination half-life is about 6 days in pigs and cattle) (3, 19). In one study, the reported ratios of the area under the concentration-time curve (AUC) in lung tissue homogenates to the area under the plasma concentration-time curve from 0 to 360 h (AUC0-360) were 94 and 181 for bovines and swine, respectively (3, 8), and the pulmonary epithelial lining fluid apparent half-life was >100 h in Holstein calves (8). These features would explain to some extent the clinical outcomes of tulathromycin treatment when it is used within the label frame.
The mechanism by which tulathromycin accumulates in lung tissue remains unknown. As reported previously for other macrolides, tulathromycin is taken up by resident inflammatory cells (alveolar macrophages) (8). Tulathromycin also accumulates in blood macrophages and neutrophils (27). Increased amounts of azithromycin and in infected lungs have been reported (2, 29). It was suggested previously that the accumulation of azithromycin in tissues appears to be related to its rapid uptake and transport to the infection site by cells such as polymorphonuclear neutrophils, monocytes, alveolar macrophages, and fibroblasts (12, 13). Also, a reduced tissue AUC of azithromycin was reported previously for leucopenic mice (2, 29, 30). These results have been used to link neutrophil migration and the pharmacokinetics of macrolides.
Neutropenia is a common cellular blood shift that occurs during an acute inflammatory process. Tulathromycin is indicated for both metaphylaxis and therapy of bacterial infections. Consequently, drug administration may take place during a neutropenic state. In this scenario, our hypothesis is that the concentration-versus-time profile of tulathromycin in lung tissue is influenced by neutropenia and the acute pulmonary inflammatory response.
The present study has two objectives: to assess the concentration-versus-time profile of tulathromycin at two different dose levels in mice and to assess the concentration-time profile of tulathromycin in the plasma and lung tissues of healthy and neutropenic mice challenged intranasally with lipopolysaccharide (LPS) from Escherichia coli O111:B4.
MATERIALS AND METHODS
Animals and housing.
Female BALB/c mice (n = 205) (17.4 to 22.0 g) were purchased from a local commercial source (Charles River, Wilmington, MA). Drinking water was available ad libitum throughout the study. Animals were acclimatized for a minimum of 7 days prior to the administration of the treatments. Dose calculations were based on body weights determined on the day of the drug administration previous to dosing. Before the initiation of the study, procedures involving the care or use of mice were reviewed and approved by the Pfizer Animal Health Institutional Animal Care and Use Committee (IACUC).
Study design.
Mice were randomly allocated into four dose groups of 40 mice each: groups T-7 (tulathromycin at 7 mg/kg), T-28, T7-LPS, and T7-LPS-CP (cyclophosphamide). Within each dose group, animals were allocated randomly into eight sampling times (from 30 min to 144 h) with five animals each. All animals within a treatment group (n = 5) were kept in a single cage. In addition, 45 mice were allocated into a control group. Animals within the control group were assigned to one of three control groups: (i) CT-NT (no treatment), (ii) CT-LPS (intranasal administration of LPS), and (iii) CT-CP (cyclophosphamide).
Treatment dose groups T-7, T7-LPS, and T7-LPS-CP were administered one subcutaneous (s.c.) injection of tulathromycin at 7 mg/kg in the interscapular space at time zero. The final concentration of tulathromycin was adjusted by using the commercial formulation without tulathromycin, provided by Pfizer Animal Health. Animals in dose group T-28 were administered one s.c. injection of tulathromycin (Draxxin injectable solution) at 28 mg/kg in the interscapular space at time zero. Animals in dose groups T-7-LPS and T7-LPS-CP received a single LPS (E. coli O111:B4) dose (30 μg of LPS diluted in 30 μl of sterile saline solution) at −7 h by intranasal administration under general anesthesia, as described below. Additionally, mice from group T7-LPS-CP were treated with cyclophosphamide (Cytoxan cyclophosphamide for injection, USP; Bristol-Myers Squibb Company, Princeton, NJ) at 150 mg/kg by the intraperitoneal route at −4, −1, and +2 days (when applicable).
Animals within the control group (CT-NT) (not treated with tulathromycin) received no treatment. Mice in the CT-LPS group received an intranasal dose of LPS as described above. Animals in the CT-LPS-CP group were treated with two doses of cyclophosphamide at −4 and −1 days before the sampling time for these animals (day 0). Additionally, these animals were treated with LPS at −7 h as described above.
Preparation of inoculum.
Lipopolysaccharide from E. coli O111:B4 purified by phenol extraction was purchased from Sigma-Aldrich (St. Louis, MO) (lot number 118K4052). The E. coli LPS powder was resuspended in a saline solution (0.9% NaCl) to obtain a final concentration of 1 μg/ml. The same inoculum size of the LPS solution (30 μl) was used for all the E. coli LPS-treated animals.
Administration of inoculum.
A single LPS dose was administered intranasally under general anesthesia. Mice were placed onto their backs on a solid surface with an inclination of 35°. The LPS solution was gently dropped into one of the nostrils. After the administration of LPS, mice were kept on the same inclined board until they recovered from anesthesia. After recovery, mice were placed back into their corresponding cages.
Sampling.
Five mice from each dose group per sampling time were anesthetized to obtain blood and lung tissue samples at 30 min and at 1, 24, 48, 72, 96, 120, and 144 h after the administration of tulathromycin.
Mice in the control group were euthanized, and lungs were excised from CT-NT (n = 3) and CT-LPS mice at 7 h (n = 3) and 24 h (n = 3) post-LPS administration. In group CT-LPS-CP, lung tissue samples were taken at time zero (7 h post-LPS administration and after two doses of CP). Lungs were filled with 10% formalin (0.7 ml) and placed individually into a container with 10% formalin until a histology evaluation was performed. Also, blood samples were withdrawn from mice of the CT-NT (n = 10), CT-LPS (n = 7), and CT-LPS-CP (n = 7) groups at time zero for white cell counts. White cell counts were performed by using a cell counter (120 hematology system; Advia, Deerfield, IL). The rest of the animals and tissues were used for analytical purposes.
Plasma sampling.
Blood samples were obtained by terminal cardiac puncture under general anesthesia. Briefly, once mice were under an appropriate anesthetic level, animals were placed onto their backs on a solid surface. The xiphoid process was palpated at the caudal aspect of the animal's sternum. A 22-gauge needle attached to a 1-ml syringe was inserted toward the heart, as determined by palpating for the heartbeat. Immediately after blood samples were withdrawn, animals were euthanized by using an overdose of pentobarbital.
Blood samples were placed into tubes with EDTA as an anticoagulant and kept on ice until centrifugation. Blood samples were centrifuged for 15 min at 1,000 rpm at 5°C. Plasma samples were placed into cryotubes and kept at −20°C until analysis.
Lung tissue sampling.
Following euthanasia, lungs from each mouse were removed from the thorax cavity. Lung tissue samples were placed into cryotubes and kept at −20°C until analysis.
Determination of the wet-to-dry ratio.
An approximate 0.5-cm by 0.5-cm subsection of lung tissue sampled for the lung tissue homogenate was taken for determinations of the wet-to-dry ratio. Samples were placed into an aluminum container and weighed. The wet weight was recorded. The samples were then dried in an oven at 60°C for 15 days. Samples were then weighed again for the determination of the wet-to-dry ratio by using the following equation: wet-to-dry ratio = wet weight (g)/dry weight (g).
Anesthesia and euthanasia.
Both LPS administration and sampling under general anesthesia were performed by the intraperitoneal administration of ketamine (100 mg/kg) combined with xylazine (10 mg/kg). All experimental animals were euthanized after blood sampling and before recovery from anesthesia by using an overdose of pentobarbital.
Analytical methods.
All the matrix samples were analyzed for tulathromycin content with a Waters Acquity ultraperformance liquid chromatography (UPLC) system with tandem mass spectrometry (MS/MS) detection (Waters Corp., Milford, MA) according to procedures described previously (8, 11). Roxithromycin was used as an internal standard (IS) (25). We did not evaluate the stability of the analyte in the matrices. We considered previously evaluated stability data generated by a full validation study using lung samples from cattle and pig (11) to be valid. We used the same analytical method validated previously by Galer et al. (11) for the analysis of our study samples. In this validation study, stability was demonstrated under different conditions. In this study, samples were evaluated within a month after collection. Samples were thawed only once for the analysis without any thawing and freezing cycles. All the samples were evaluated in one batch for a period no longer than 36 h after thawing.
Plasma.
Study plasma samples (100 μl) were spiked with 100 μl of an internal standard (25 ng/ml roxithromycin) and then extracted by using solid-phase extraction (SPE) cartridges (Oasis MCX, 1 ml; Waters). The SPE cartridges were preconditioned with 1.0 ml of acetonitrile followed by 1.0 ml of 50 mM K2HPO4 (pH 6.8). A weak vacuum was applied to the SPE manifold. Cartridges were loaded with quality control, blank plasma, and test samples (200 μl). The SPE tubes were then rinsed sequentially with 1.0 ml of 50 mM K2HPO4 (pH 6.8) and 1.0 ml of H2O. Finally, analytes were eluted with 2× 0.5 ml of freshly prepared 5% NH4OH–95% acetonitrile into labeled 13- by 100-mm polypropylene tubes. The collected extracts were evaporated to dryness under a stream of nitrogen at 50°C to 55°C. The extract residues were reconstituted with 200 μl of 20 mM ammonium acetate (pH 4.0) by vortex mixing and transferred into autosampler vials.
Chromatography was accomplished with a BDS Hypersil C8 30- by 2.1-mm column (Thermo Fisher Scientific Inc., Waltham, MA) at a flow rate of 0.300 ml/min with linear-gradient chromatography. The mobile phase initially consisted of 80% solution A (20 mM ammonium acetate, pH 4) and 20% solution B (acetonitrile) and was changed at 0.5 min to 20% solution A and 80% solution B. The retention times were approximately 1 min for tulathromycin and 1.2 min for roxithromycin. The mass spectrometer (Linear Ion Trap 4000 Qtrap equipped with a turbo spray ion source; AB Sciex, Foster City, CA) was used in the positive-ion mode using an electrospray ionization source, and positive ions were monitored with precursor→product ion pairs of 806→577 m/z for tulathromycin and 837→679 m/z for roxithromycin. Calibration curves were generated automatically by using Analyst 1.0 software (Perkin-Elmer, Waltham, MA).
The calibration curve (R2 > 0.99) occurred over the range of 1.95 to 500 ng/ml using nine calibration standards (by duplicating each concentration) and five quality control samples (15.63, 62.5, 125, 250, and 500 ng/ml) (by quadruplicating each concentration level). Standard curves and quality controls were run at the beginning and at the end of the analytical run. The lower limit of quantification (LLOQ) was defined as the standard concentration where the analysis of 3 replicate samples did not exceed a coefficient of variation of 20%. The LLOQ was 3.90 ng/ml. For the quality control samples, the intrarun coefficients of variation (CVs) ranged from 12.9 to 19.0%, and the intrarun biases ranged from −14.0% to 10.0%. For the calibration curve samples, the intrarun coefficients of variation ranged from 2.0 to 19.7%, and the intrarun biases for these samples ranged from −10.6% to 8.38%. All the study samples were analyzed in a single analytical run. The recovery ranges for tulathromycin and the IS in spiked plasma were 60.1 to 66.2% and 51.1 to 62.4%, respectively.
Lung tissue homogenate samples.
Samples were processed as described previously by Galer et al. (11). Briefly, a lung tissue samples (0.100 g) was mixed with 0.050 ml of 50 mM phosphate buffer (pH 6) and 2.5 ml of 0.04 M phosphoric acid in polypropylene tubes. Samples were homogenized by using a Polytron probe. The homogenate was centrifuged during 10 min at ∼4,500 × g, 0.25 ml of the supernatant was then mixed with 0.25 ml of 50 mM phosphate buffer (pH 6) and 0.25 ml of an internal standard solution, and the entire sample was loaded onto an preconditioned Oasis MCX 1-ml (30-mg) SPE cartridge. The SPE cartridges were then washed with 1 ml of 50 mM phosphate buffer (pH 6), 1 ml of distilled deionized water, and 1 ml of acetonitrile. The drug was then eluted from the cartridge with two 0.5-ml volumes of 5% ammonium hydroxide–95% acetonitrile. After evaporation with nitrogen gas at 50°C, the sample was reconstituted with 0.95 ml of 20 mM ammonium acetate and acetonitrile (pH 4) (80:20, vol/vol). A 10-μl aliquot was then injected into the chromatographic system. The calibration curve (R2 >0.99) occurred over the range of 3.90 to 1,000 ng/g lung tissue using 10 calibration standards, and three quality control samples of lung tissue (7.80, 125, and 1,000 ng/g) were included in each analytical run. The LLOQ was defined as the standard concentration where an analysis of 3 replicate samples did not exceeded a coefficient of variation of 20%. The LLOQ was 3.90 ng/g. For the quality control samples, the intrarun coefficients of variation ranged from 4.9 to 8.1%, and the intrarun biases ranged from −4.0% to 9.4%. For the calibration curve samples, the intrarun coefficients of variation ranged from 1.85 to 8.1%, and the intrarun biases ranged from −7.5% to 13.6%. All the study samples were analyzed in a single analytical run. The recovery ranges for tulathromycin and the IS in incurred lung tissue samples were 87.1 to 93.2% and 71.1 to 76.4%, respectively.
Pharmacokinetic and biometrical analyses.
Plasma and lung tissue homogenate drug concentrations were analyzed by using an analysis of variance (ANOVA) linear mixed model as implemented in the Statistical Analysis System (version 9.2; SAS, Cary, NC). A natural log transformation was applied to the concentration data for both matrices prior to analysis. Least-squared (LS) means (geometric means) and 95% confidence bounds were calculated with natural logarithm-transformed data, and the final results were back-transformed, representing geometric means (8). Standard errors (SE) of LS means were estimated, and a 95% confidence interval (CI) was constructed. Back-transformed LS means and CIs for each treatment and dose group were reported. The back-transformed LS mean concentration was used as an estimate of the treatment means. The dose groups were tested for significant differences for fixed effects at a level of an α value of 0.05, indicating that P values of ≤0.05 showed a significant difference for the effect. The wet-to-dry ratios were summarized as arithmetic means, standard deviations (SDs), and CVs.
Wet-to-dry ratios were compared statistically by the same linear mixed model. Linear regression analysis was performed to test the linear association between the wet-to-dry ratio and raw drug concentration data.
Pharmacokinetic parameters were estimated based on the back-transformed LS means (8). Pharmacokinetic parameters were estimated by using WinNonlin software (standard edition, version 5.1; Pharsight Corporation, Mountain View, CA). The WinNonlin 200 model was used for the noncompartmental analysis. The AUC through 144 h postdose (AUC0–144), the area under the concentration–first-moment time curve through 144 h postdose (AUMC0–144), the mean residence time (MRT), and the terminal elimination half-life (t1/2) were estimated. The AUC0–144 was calculated by the log-linear trapezoidal rule, and λz was estimated with uniform weighting. The observed peak LS mean concentration (Cmax) and time of its occurrence (Tmax) were obtained from an inspection of the back-transformed LS mean concentration.
RESULTS
All experimental animals remained in good health throughout the acclimatization period. One animal (animal 149) of dose group T-7-LPS-CP (72 h) was euthanized due to animal welfare concerns. Animal 145 (48 h), from the same dose group, was found dead. These animals were not replaced. The rest of the experimental animals remained in good health throughout the study period. No adverse reactions or detrimental health effects were observed following the administration of tulathromycin. Animals in dose groups T-7, T-7-LPS, T-7-LPS-CP, and T-28 received actual averaged doses of 6.95, 6.90, 6.99, and 27.57 mg tulathromycin/kg, respectively. For one animal from dose group T-7 (animal 4), there was a leakage of the treatment solution from the administration site. Therefore, this animal was considered underdosed and was excluded from the study.
Plasma.
Tulathromycin was rapidly absorbed in both dose groups. The maximal LS mean concentration of tulathromycin was observed 30 min after the administration of tulathromycin for dose groups T-7, T-28, and T-7-LPS and 1 h after administration for T-7-LPS-CP. The LS mean Cmax values (±SE) of tulathromycin in plasma were 6,790 ± 2,450 (Tmax, 30 min) and 6,920 ± 2,520 ng/ml (Tmax, 1 h) for dose groups T-7-LPS and T-LPS-CP, respectively. Results for plasma concentrations of tulathromycin are included in Table 1 and are shown in Fig. 1 and 2. The pharmacokinetic parameter results are included in Table 3.
Table 1.
LS mean concentrations of tulathromycin in plasma from mice treated with a single s.c. injection of tulathromycin at 7 and 28 mg/kg body weight
| Time point | Group | Tulathromycin (ng/ml) |
|
|---|---|---|---|
| LS meana | 95% CIa | ||
| 30 min | T-28 | 11,700 | 490–18,500 |
| T-7-LPS | 6,790 | 184–11,700 | |
| T-7-LPS-CP | 6,700 | 1,230–12,200 | |
| T-7 | 3,280 | 1,500–5,070 | |
| 1 h | T-28 | 792 | 214–1,370 |
| T-7-LPS | 3,870 | 713–7,030 | |
| T-7-LPS-CP | 6,920 | 1,870–12,000 | |
| T-7 | 2,060 | 378–3,700 | |
| 24 h | T-28 | 145 | 26.7–260 |
| T-7-LPS | 50.8 | 13.7–87.9 | |
| T-7-LPS-CP | 59.1 | 11.0–107 | |
| T-7 | 103 | 28.1–180 | |
| 48 h | T-28 | 100 | 27.2–174 |
| T-7-LPS | 51.1 | 13.8–88.4 | |
| T-7-LPS-CP | 57.0 | 15.4–98.6 | |
| T-7 | 60.0 | 16.2–103 | |
| 72 h | T-28 | 109 | 20.2–199 |
| T-7-LPS | 19.2 | 5.20–33.2 | |
| T-7-LPS-CP | 5.32 | 0.31–10.3 | |
| T-7 | 24.4 | 4.50–44.2 | |
| 96 h | T-28 | 26.2 | 7.10–45.4 |
| T-7-LPS | 16.9 | 4.60–29.3 | |
| T-7-LPS-CP | 15.9 | 2.93–28.9 | |
| T-7 | 9.28 | 2.50–16.0 | |
| 120 h | T-28 | 19.2 | 3.50–34.9 |
| T-o7-LPS | 10.9 | 2.00–19.9 | |
| T-7-LPS-CP | 6.89 | 1.27–12.5 | |
| T-7 | 10.8 | 2.90–18.7 | |
| 144 h | T-28 | 13.6 | 3.70–23.5 |
| T-7-LPS | 7.82 | 0.50–15.2 | |
| T-7-LPS-CP | 6.48 | 1.75–11.2 | |
| T-7 | 15.4 | 4.20–26.7 | |
Back-transformed.
Fig 1.
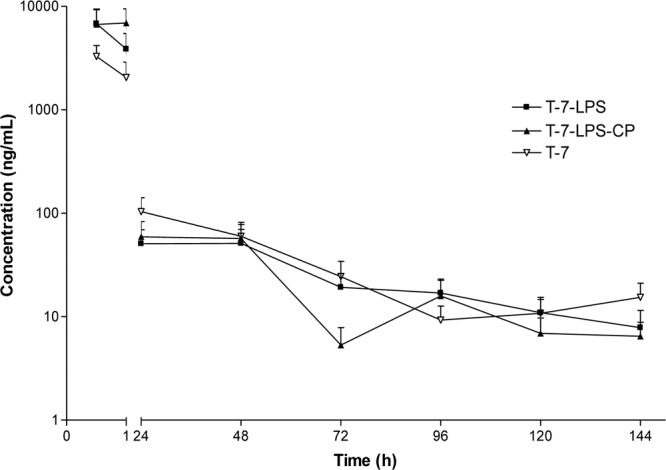
LS mean concentrations (±SE) (ng/ml) of tulathromycin in plasma from mice treated s.c. with tulathromycin at 7 mg/kg (n = 4 for group T-7-LPS at 30 min and 96 h; n = 4 at 30 min, 24 h, and 48 h and n = 3 at 72 h for group T7-LPS-CP; n = 4 for group T-7 at 30 min and 96 h).
Fig 2.
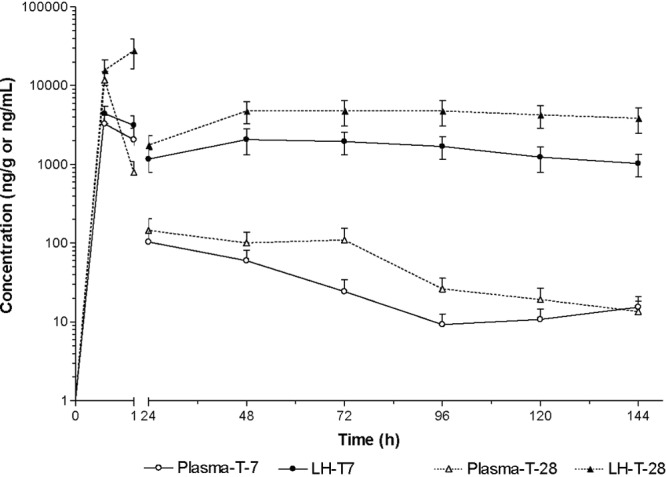
LS mean concentrations (±SE) of tulathromycin in lung tissue homogenates (ng/g) and plasma samples (ng/ml) from mice treated s.c. with tulathromycin at 7 and 28 mg/kg (n = 4 for group T7 at 48 h).
Table 3.
Pharmacokinetic parameters of tulathromycin in lung tissue homogenates and plasma samples of mice treated with a single s.c. injection of tulathromycin at 7 and 28 mg/kg body weight
| Pharmacokinetic parametera | Value for group |
|||||||
|---|---|---|---|---|---|---|---|---|
| T-28 |
T-7 |
T-7-LPS |
T-7-LPS-CP |
|||||
| Plasma | LH | Plasma | LH | Plasma | LH | Plasma | LH | |
| HL_lambda_z (h) | 18.1 | 112 | 13.5 | 82.0 | 16.5 | 213 | 17.6 | 144 |
| Tmax (h) | 0.50 | 1.00 | 0.50 | 0.50 | 0.50 | 0.50 | 1.00 | 1.00 |
| Cmax (ng/ml) | 11,700 | 27,900 | 3,300 | 4,410 | 6,790 | 3,940 | 6,920 | 5,500 |
| Clast (ng/ml) | 13.6 | 3,860 | 15.4 | 1,020 | 7.82 | 1,060 | 6.48 | 750 |
| AUClast (h · ng/ml) | 152,000 | 869,000 | 31,000 | 246,000 | 52,600 | 199,000 | 88,100 | 199,000 |
| AUCinf_obs (h · ng/ml) | 152,000 | 1500,000 | 31,200 | 378,000 | 52,800 | 487,000 | 88,300 | 356,000 |
| AUMClast (h · h · ng/ml) | 599,000 | 45000,000 | 274,000 | 15700,000 | 252,000 | 8670,000 | 259,000 | 9330,000 |
| MRTinf_pred (h) | 4.16 | 148 | 8.91 | 138 | 4.99 | 287 | 3.03 | 177 |
| AUC(0–144) LH/plasma ratio | 5.77 | 7.95 | 3.79 | 2.26 | ||||
Dispersion data of the PK parameters are not presented because PK parameters were estimated based on least-square means derived from the naïve pooling from each sample point. LH, lung homogenate.
Lung tissue homogenate.
Tulathromycin was rapidly and extensively distributed into lung tissue (Table 2 and Fig. 2 and 3). For dose groups T-7 and T-28, the tulathromycin Cmax values were 4,410 and 27,800 ng/g (observed) at 30 min and 1 h after the administration of tulathromycin, respectively (Fig. 2). The Cmax values for dose groups T-7-LPS and T-7-LPS-CP were 3.93 and 5.49 ng/g, occurring at 30 min and 1 h after the administration of tulathromycin, respectively (Fig. 3). There were no statistically significant differences in tissue concentrations between the two dose groups treated with 7 mg/kg of tulathromycin. At all the sample times, the lung tissue LS mean concentrations far exceeded the plasma concentrations in the four treatment groups. In all the dose groups, tulathromycin persisted for a long period of time, with an MRT that ranged between 138 and 287 h. A summary of pharmacokinetic parameters is presented in Table 3. The lung-to-plasma AUC0-144 ratio was 1.3 times higher in dose group T-7 than in group T-28 (AUC0-144 ratios of 7.95 and 5.77, respectively).
Table 2.
LS mean concentrations of tulathromycin in lung tissue homogenates from mice treated with a single s.c. injection of tulathromycin at 7 and 28 mg/kg body weight
| Time point | Group | Tulathromycin (ng/ml) |
|
|---|---|---|---|
| LS meana | 95% CIa | ||
| 30 min | T-28 | 15,700 | 4,550–2,600 |
| T-7-LPS | 3,940 | 1,440–6,430 | |
| T-7-LPS-CP | 5,240 | 1,910–8,560 | |
| T-7 | 4,410 | 2,198–6,621 | |
| 1 h | T-28 | 27,900 | 5,050–50,500 |
| T-7-LPS | 3,250 | 1,192–5,320 | |
| T-7-LPS-CP | 5,500 | 2,010–8,980 | |
| T-7 | 3,150 | 1,150–5,140 | |
| 24 h | T-28 | 1,760 | 643–2,870 |
| T-7-LPS | 1,170 | 213–2,140 | |
| T-7-LPS-CP | 1,680 | 614–2,740 | |
| T-7 | 1,170 | 426–1,910 | |
| 48 h | T-28 | 4,770 | 1,740–7,790 |
| T-7-LPS | 1,050 | 305–1,800 | |
| T-7-LPS-CP | 1,000 | 292–1,720 | |
| T-7 | 2,070 | 603–3,540 | |
| 72 h | T-28 | 4,800 | 1,400–8,210 |
| T-7-LPS | 889 | 325–1,450 | |
| T-7-LPS-CP | 680 | 123–1,240 | |
| T-7 | 1,950 | 715–3,196 | |
| 96 h | T-28 | 4,760 | 1,380–8,130 |
| T-7-LPS | 824 | 301–1,350 | |
| T-7-LPS-CP | 879 | 159–1,600 | |
| T-7 | 1,690 | 619–2,770 | |
| 120 h | T-28 | 4,240 | 1,550–6,940 |
| T-7-LPS | 516 | 189–844 | |
| T-7-LPS-CP | 926 | 338–1,510 | |
| T-7 | 1,230 | 359–2,110 | |
| 144 h | T-28 | 3,860 | 1,120–6,590 |
| T-7-LPS | 1,060 | 308–1,810 | |
| T-7-LPS-CP | 750 | 218–1,280 | |
| T-7 | 1,020 | 375–1,670 | |
Back-transformed.
Fig 3.
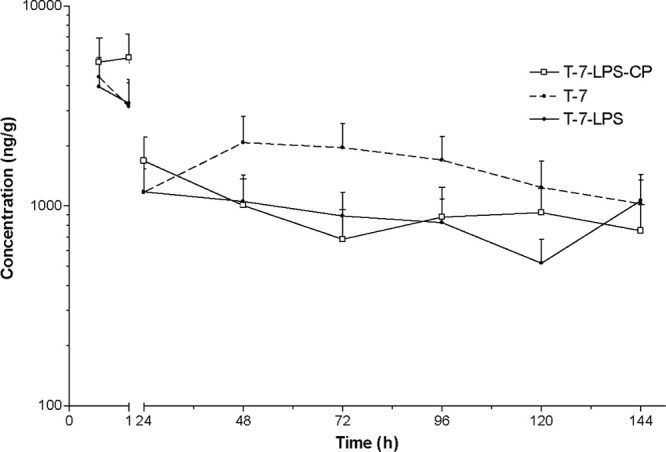
LS mean concentrations (±SE) (ng/ml) of tulathromycin in lung tissue homogenates from mice treated s.c. with tulathromycin at 7 mg/kg (n = 4 for group T7-LPS-CP at 48 h and 72 h; n = 4 for group T7 at 48 h).
Wet-to-dry ratios for lung tissue.
Wet-to-dry-weight ratios were determined for all the dose groups. Summarized data are presented in Table 4. There were statistically significant differences between dose groups T7 and T-7-LPS (P < 0.01) and between groups T7 and T-7-LPS-CP (P < 0.001) between 72 and 144 h. No significant differences were observed between the T-7-LPS and T-7-LPS-CP dose groups (P > 0.05). Likewise, there were no statistically significant differences among the three dose groups during the first 48 h after the administration of tulathromycin. There was a negative linear relationship (P < 0.01) between the raw data concentration and the wet-to-dry ratio for the T-7-LPS and T7-LPS-CP dose groups (Fig. 4 and 5). However, the fraction of the variation of the drug concentration explained by the wet-to-dry ratio (72 to 144 h) was low (R2 <5%) for both dose groups.
Table 4.
Wet-to-dry-weight ratios of lung tissue from BALB/c mice
| Time point | Dose group | Wet-to-dry ratio |
||
|---|---|---|---|---|
| Mean | SD | % CV | ||
| 30 min | T-7 | 3.18 | 0.65 | 20.3 |
| T-7-LPS | 4.22 | 0.38 | 9.07 | |
| T-7-LPS-CP | 4.69 | 0.14 | 3.16 | |
| 1 h | T-7 | 4.37 | 0.11 | 2.53 |
| T-7-LPS | 4.26 | 0.15 | 3.53 | |
| T-7-LPS-CP | 4.75 | 0.17 | 3.65 | |
| 24 h | T-7 | 4.18 | 0.21 | 5.08 |
| T-7-LPS | 4.47 | 0.12 | 2.85 | |
| T-7-LPS-CP | 5.05 | 0.10 | 2.02 | |
| 48 h | T-7 | 4.78 | 1.30 | 27.2 |
| T-7-LPS | 4.64 | 0.15 | 3.24 | |
| T-7-LPS-CP | 4.81 | 0.46 | 9.60 | |
| 72 h | T-7 | 3.95 | 0.28 | 7.05 |
| T-7-LP | 4.87 | 0.22 | 4.70 | |
| T-7-LPS-CP | 5.18 | 0.23 | 4.49 | |
| 96 h | T-7 | 4.32 | 0.25 | 5.73 |
| T-7-LPS | 4.96 | 0.62 | 12.6 | |
| T-7-LPS-CP | 4.95 | 0.28 | 5.67 | |
| 120 h | T-7 | 4.59 | 0.20 | 4.28 |
| T-7-LPS | 4.71 | 0.32 | 6.81 | |
| T-7-LPS-CP | 5.09 | 0.19 | 3.90 | |
| 144 h | T-7 | 4.28 | 0.26 | 6.12 |
| T-7-LPS | 4.97 | 0.77 | 6.45 | |
| T-7-LPS-CP | 5.03 | 0.20 | 3.99 | |
Fig 4.
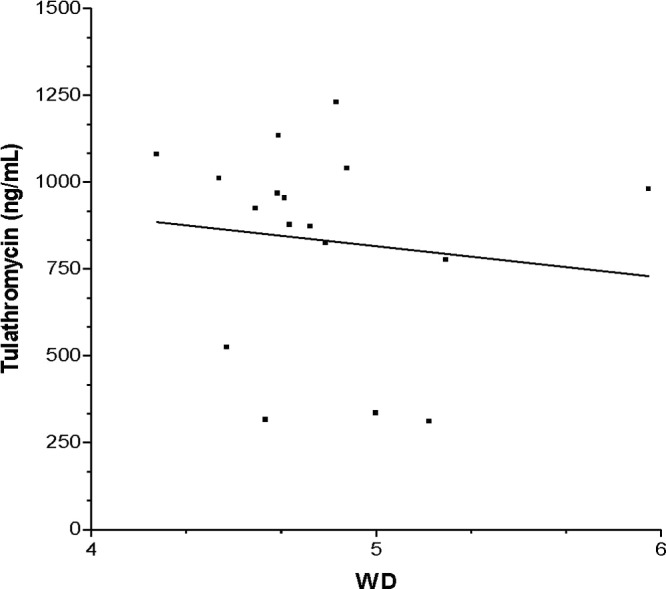
Linear regression analysis of the concentration of tulathromycin in lung tissue versus the wet-to-dry ratio (WD) (from 72 to 144 h) for group T-7-LPS (R2 = 0.01).
Fig 5.
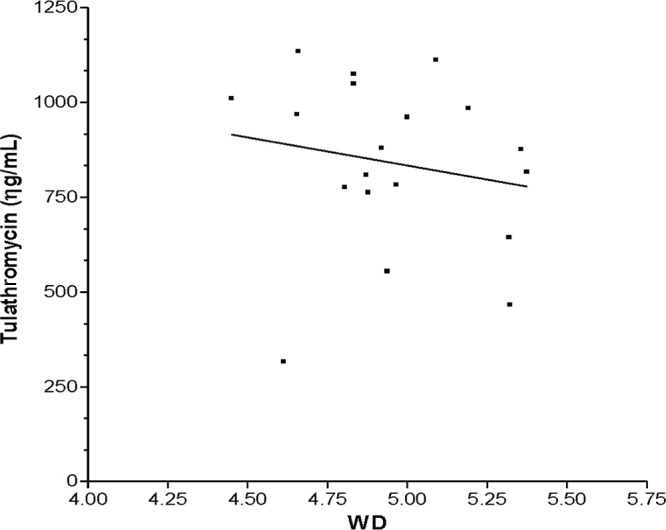
Linear regression analysis of the concentration of tulathromycin in lung tissue versus the wet-to-dry ratio (from 72 to 144 h) for group T7-LPS-CP (R2 = 0.03).
Lung tissue histology.
Neutrophil accumulations were observed in the lumen of bronchioles (minimal to mild), interstitial area around bronchioles (minimal to mild), alveolar spaces (minimal), and alveolar septa (minimal to moderate) of animals treated with LPS alone (LPS for 7 h and LPS for 24 h) but not in nontreated animals (CT-NT) or in animals pretreated with cyclophosphamide and then treated with LPS.
White blood cell count.
A white blood cell (WBC) count was performed for all control animals in groups CT-NT, CT-LPS, and CT-LPS-CP. The WBC results are summarized in Table 5, and neutrophil count results are presented graphically in Fig. 6.
Table 5.
Summary of white blood cell counts in BALB/c micea
| Dose group and value | No. of WBC (103/μl) | % N | % L | % M | % E | % B | % R | No. of N (103cells/μl) | No. of L (103cells/μl) | No. of M (103cells/μl) |
|---|---|---|---|---|---|---|---|---|---|---|
| CT-LPS-CP (n = 7) | ||||||||||
| Mean | 0.93 | 5.93 | 89.1 | 0.37 | 2.79 | 0.26 | 0.08 | 0.04 | 0.84 | 0.00 |
| SD | 0.27 | 4.79 | 6.88 | 0.44 | 1.77 | 0.34 | 0.02 | 0.01 | 0.26 | 0.00 |
| CV | 0.29 | 0.81 | 0.08 | 1.18 | 0.64 | 1.33 | 0.32 | 0.29 | 0.32 | 2.65 |
| CT-LPS (n = 7) | ||||||||||
| Mean | 7.24 | 43.2 | 52.9 | 0.97 | 1.75 | 0.15 | 4.03 | 3.17 | 3.81 | 0.07 |
| SD | 1.64 | 7.00 | 6.92 | 0.32 | 0.98 | 0.05 | 0.64 | 1.02 | 0.83 | 0.03 |
| CV | 0.23 | 0.16 | 0.13 | 0.34 | 0.56 | 0.33 | 0.16 | 0.32 | 0.22 | 0.38 |
| CT-NT (n = 10) | ||||||||||
| Mean | 6.19 | 10.6 | 83.5 | 1.15 | 1.95 | 0.25 | 3.24 | 0.69 | 5.18 | 0.07 |
| SD | 2.94 | 2.05 | 1.33 | 0.28 | 0.51 | 0.11 | 0.48 | 0.37 | 2.45 | 0.04 |
| CV | 0.47 | 0.19 | 0.02 | 0.24 | 0.26 | 0.44 | 0.15 | 0.54 | 0.47 | 0.54 |
WBC, white blood cells; N, neutrophils; L, lymphocytes; M, macrophages; E, eosinophils; B, basophils; R, reticulocytes.
Fig 6.
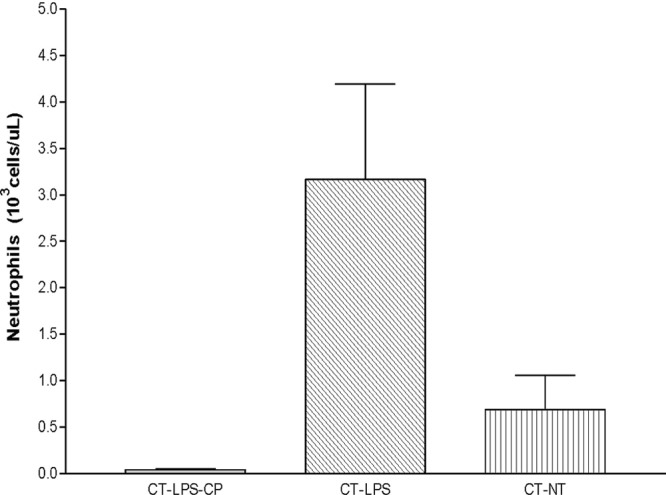
Number of neutrophils (means ± SD) in blood from BALB/c mice.
DISCUSSION
The concentration-versus-time profile of tulathromycin in lung tissue homogenates and plasma was evaluated for all dose groups. Also, pharmacokinetic parameters of tulathromycin were estimated in both matrices.
In dose groups T-7 and T-28, tulathromycin was rapidly absorbed (Fig. 2). Tulathromycin was distributed extensively and rapidly in lung tissue in both of these dose groups, as previously reported for other species (8, 19). Tulathromycin was slowly eliminated from lung tissues of both dose groups T-7 and T-28 with an MRT of about ∼6 days (Table 3).
A similar pharmacokinetic trend in mice was reported previously for other macrolides, including erythromycin, spiramycin, clarithromycin, and azithromycin (30). Furthermore, the results suggested that tulathromycin has a higher level of accumulation and persists in lung tissue longer than other macrolides in mice (30). The high affinity of the drug for lung tissue and the long persistence of tulathromycin was also reported previously for cattle (8, 19), pigs (3, 19), and foals (31). Multiple factors have been hypothesized to explain the accumulation of macrolides in lung tissue, including interactions with phospholipids (17), intracellular accumulation (bronchial and alveolar epithelium and bronchoalveolar cells) due to ion trapping (6), active transport (carriers such as members of the organic anion-transporting protein group) (26), and endocytosis (5). However, the identification and description of the mechanism(s) involved in tulathromycin lung tissue accumulation still remain unknown.
Tulathromycin is a lipid-soluble triamilide (15). Therefore, the overall molecule contains three basic functional groups with pKa values ranging from 8.6 to 9.9. These features may favor the penetration and intracellular accumulation of the drug in an acidic environment (ion trapping). Further research is necessary to elucidate the mechanism(s) and factor(s) involved in the intracellular accumulation of the drug.
We evaluated the pharmacokinetic profile of tulathromycin at two different dose levels in order to predict drug concentrations for future studies. In plasma, the AUC0–144 ratio adjusted by dose between dose groups T-7 and T-28 was 1.22. In lung tissue, the AUC0–144 ratio adjusted by dose between dose groups T-7 and T-28 was 0.89 (mean actual doses of 6.95 and 27.6 mg/kg, respectively). The results obtained indicate that at the two dose levels evaluated, the dose responses in plasma and lung tissue could be different. We cannot confirm dose proportionality or a lack thereof because the data are insufficient. It is important to stress that both parallel design and destructive sampling are associated with an increased imprecision of the data due to interindividual variability. In plasma, the concentration at the 1-h time point for the injection of 28 mg/kg of tulathromycin was much lower than the concentrations achieved at 1 h for lower doses of 7 mg/kg. These data do not agree with the proportional differences in lung concentrations. We could not find a specific reason to explain this discrepancy. However, considering that we used terminal sampling (one data point per animal) and that the apparent discrepancy occurred during the early portion of the curve (1 h), when absorption, distribution, and elimination processes are taking place concurrently, it may have been the case that some of the animals sampled at 1 h in the 28-mg/kg group exhibited a shorter distribution half-life, an increased clearance, and/or a decreased absorption rate that would have resulted in a higher Tmax and lower concentrations in the early fraction of the curve.
The AUClast (AUC measured to the last measured time point) and the AUC to infinity are exactly the same for plasma concentration AUC values in the 28-mg/kg group. The values for the other groups were almost exactly the same when these two values were compared. The reason for the similitude of the values is 2-fold. The area extrapolated for estimations of the AUC to infinity (AUCinf) is very small. The other factor that determines the similitude of the values is that we are reporting the 3 significant figures. Therefore, the numerical differences between the AUClast and AUC to infinity (in particular for the 28-mg/kg group) is lost in the process of rounding up the values.
In this study, two experimental models were implemented: experimentally induced neutropenic mice and animals treated intratracheally with E. coli LPS. Lipopolysaccharides are structural and functional components of the Gram-negative bacterial outer membrane (7). The administration of LPS in the respiratory system has been extensively used to trigger an acute inflammatory response (1). The administration of LPS in the respiratory system prompts an acute infiltration of polymorphonuclear neutrophils and the release of a myriad of inflammatory mediators. The model implemented in the present study was fully described previously (28). The neutropenic mouse model has been extensively used to unravel the pharmacokinetics and pharmacodynamics of antimicrobials (9).
In the present study, we evaluated the LPS response in lung tissues of only a subset of control mice (untreated and treated with LPS and LPS plus CP). At the time of tulathromycin administration, neutrophils were present to a moderate extent in the alveolar septa. Mild congestion in capillaries was noted only for the mice treated with LPS. Neither neutrophils nor any other change was observed for the control and CT-LPS-CP animals. We also assessed the WBC counts for a control group at the time of tulathromycin administration. The administration of LPS induced a remarkable change in the average blood cell count compared with that for control animals (6.19 × 103 ± 2.94 × 103 cells/μl for control mice versus 7.24 × 103 ± 1.64 × 103 cells/μl for LPS-treated mice) and with reference values for anesthetized BALB/c mice (Table 5) (18).
The WBC count for the animals treated with LPS and CP was lower than those of the other groups (0.93 × 103 ± 0.27 × 103 cells/μl). These changes were also reflected in the neutrophil count. Previous studies considered animals to be neutropenic when the cell count was <100 cells/μl (10, 24). In the present study, the mice were severely neutropenic regardless of the administration of LPS (0.04 × 103 ± 0.01 × 103, 3.17 × 103 ± 1.02 × 103, and 0.69 × 103 ± 0.37 × 103 cells/μl for the CT-LPS-CP, CT-LPS, and CT-NT groups, respectively) (Table 5 and Fig. 6) (18). Therefore, at the time of tulathromycin administration, animals treated with LPS were neutrophilic (abnormally higher numbers of neutrophils in blood) and exhibited an accumulation of neutrophils in the lung. In contrast, mice treated with LPS plus CP were neutropenic, without a recruitment of neutrophils to the lung.
The AUC0–144 values of tulathromycin in plasma of neutropenic mice were 1.7 and 2.8 times higher than those in plasma of healthy mice (T-7) for dose groups T-7-LPS and T-LPS-CP, respectively. The reasons for this change in drug exposure are unknown.
For both LPS-treated groups, tulathromycin was distributed extensively and rapidly in lung tissue, as previously observed for group T-7. For the LPS-treated animals, there was a trend toward lower AUC0–144 values than for non-LPS-treated (T-7) animals (Table 3). The reasons for lower AUC0–144 values for the LPS-treated animals are unclear. This trend was not compared statistically because of the lack of dispersion parameters as a consequence of the destructive sampling used in this study.
An increase of the wet-to-dry ratio at 48 h post-LPS administration could be caused by the edema associated with the inflammatory process. This could have a dilution effect on the drug in lung tissue homogenates, explaining the lower LS mean concentrations observed for LPS-treated animals. However, the wet-to-dry ratio explained less than 5% of the change in the lung tissue drug concentration of LPS-treated mice (Fig. 4 and 5). Changes in the pharmacokinetic parameters estimated from plasma and lung tissue have also been reported for other macrolides in infected animals (30). The AUCs of spiramycin, roxithromycin, and azithromycin in lung tissue increased; the AUC of clarithromycin decreased; and the AUC of erythromycin did not change when the drugs were administered at 48 h postinfection. Because the plasma AUC0–144 values of tulathromycin for group T-7 were 41% and 64% lower than those for groups T-7-LPS and T-7-LPS-CP, respectively, the decrease of the AUC0–144 in lung tissue accumulation (AUC0–144) may not be attributable to changes in plasma concentrations (Table 3 and Fig. 1). Even though the plasma AUC was numerically higher in LPS-treated animals, this was not reflected in the lung figures. The lung/plasma ratios of the AUC0–144 were 3.79 and 2.26 for dose groups T-7-LPS and T-7-LPS-CP, respectively, and 7.95 for dose group T-7. Tulathromycin was slowly eliminated from lung tissues of both the T-7-LPS and T-7-LPS-CP dose groups and resided somewhat longer in LPS-treated than in non-LPS-treated animals (Table 3).
Tissue-directed pharmacokinetics have been proposed to be one of the mechanisms of local drug accumulation (2, 12, 29). When azithromycin was administered to infected mice, the AUC in lung tissue was much lower in leucopenic animals (2). Based on those results, those authors suggested that leukocytes may help to transport macrolides to the site of infection. However, the results might be confounded by a lower plasma concentration in leucopenic animals. The results obtained in the present study do not allow a delineation of a relationship between neutrophils and lung tissue drug accumulation in mice. Several scenarios may explain this lack of a relationship. First, neutrophils may be truly irrelevant in the pulmonary accumulation of tulathromycin in mice. Second, tulathromycin may not concentrate in neutrophils from mice, or it may concentrate at a low level at the given dose. Third, the accumulation of cells in lung tissue may not have been enough to generate an evident change in the lung tissue pharmacokinetics of the drug. Last, the relationship between the time of drug administration after the challenge (and concentration-time profile) and kinetics (movement of cells from blood to lung) of neutrophils did not match.
In this study, we administered the drug after the challenge based on the initial manifestation of abnormal clinical signs observed in preliminary studies simulating a field scenario. Azoulay-Dupuis et al. (2) determined previously that the accumulation of azithromycin at the site of infection was influenced by the time of administration and the progression of the disease (2). In the present study, we evaluated drug pharmacokinetics after administration at a fixed time with regard to the challenge administration. The evaluation of multiple scenarios would provide valuable information about the potential relevance of inflammation and drug tissue accumulation or drug kinetics. It is important to highlight that the rate and extent of intracellular accumulation and release depend on multiple factors, including the chemical structure of the drug (5), cell type (4, 12, 16), cell status (21), extracellular conditions (pH) (14), and type of stimulus (12). Those factors should be taken into account in the design of future studies.
In mice, neutrophilia also did not affect drug accumulation in the lung. From a therapeutic standpoint, this may represent an advantage, since the dose regimen may not need to be adjusted based on the cell count in order to reach target therapeutic concentrations. A prospective evaluation of the above-mentioned statement using multiple scenarios would certainly provide valuable information. Ideally, this should be evaluated with target species, such as pig and cattle, under field conditions.
In conclusion, we evaluated the pharmacokinetics of tulathromycin in mouse lung tissue homogenates and plasma in three different dose groups: healthy mice and LPS-challenged neutropenic and neutrophilic animals. For all dose groups, the distribution of the drug in the lungs was rapid and persisted at relatively high levels during 6 days postadministration. The concentration-versus-time profile of tulathromycin in lung tissue was not influenced by the intranasal administration of E. coli LPS. The results suggest that neutrophils may not have a positive influence on tulathromycin accumulation in mouse lung tissue when the drug is administered during either a neutrophilic or a neutropenic state. The potential role of neutrophils in the accumulation of tulathromycin in lung tissue should be evaluated with other species and probably for other macrolides. The microbiological and clinical impacts of these scenarios need to be evaluated.
ACKNOWLEDGMENTS
We are grateful to the Metabolism and Safety Unit staff of Pfizer Animal Health, Kalamazoo, MI, who assisted with this study. We also thank Misty Bailey (University of Tennessee) for her comments on the manuscript.
The present study was funded by Pfizer Animal Health, Kalamazoo, MI.
Footnotes
Published ahead of print 14 May 2012
REFERENCES
- 1. Asti C, et al. 2000. Lipopolysaccharide-induced lung injury in mice. I. Concomitant evaluation of inflammatory cells and haemorrhagic lung damage. Pulm. Pharmacol. Ther. 13:61–69 [DOI] [PubMed] [Google Scholar]
- 2. Azoulay-Dupuis E, Vallée E, Bedos JP, Muffat-Joly M, Pocidalo JJ. 1991. Prophylactic and therapeutic activities of azithromycin in a mouse model of pneumococcal pneumonia. Antimicrob. Agents Chemother. 35:1024–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Benchaoui HA, Nowakowski M, Sherington J, Rowan TG, Sunderland SJ. 2004. Pharmacokinetics and lung tissue concentrations of tulathromycin in swine. J. Vet. Pharmacol. Ther. 27:203–210 [DOI] [PubMed] [Google Scholar]
- 4. Bergogne-Berezin E. 1995. New concepts in the pulmonary disposition of antibiotics. Pulm. Pharmacol. 8:65–81 [DOI] [PubMed] [Google Scholar]
- 5. Bosnar M, Kelnerić Z, Munić V, Eraković V, Parnham MJ. 2005. Cellular uptake and efflux of azithromycin, erythromycin, clarithromycin, telithromycin, and cethromycin. Antimicrob. Agents Chemother. 49:2372–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carlier MB, Zenebergh A, Tulkens PM. 1987. Cellular uptake and subcellular distribution of roxithromycin and erythromycin in phagocytic cells. J. Antimicrob. Chemother. 20(Suppl B):47–56 [DOI] [PubMed] [Google Scholar]
- 7. Caroff M, Karibian D, Cavaillon JM, Haeffner-Cavaillon N. 2002. Structural and functional analyses of bacterial lipopolysaccharides. Microbes Infect. 4:915–926 [DOI] [PubMed] [Google Scholar]
- 8. Cox SR, et al. 2010. Rapid and prolonged distribution of tulathromycin into lung homogenate and pulmonary epithelial lining fluid of Holstein calves following a single subcutaneous administration of 2.5 mg/kg body weight. Int. J. Appl. Res. Vet. Med. 8:129–137 [Google Scholar]
- 9. Craig WA, Andes DR. 2008. In vivo pharmacodynamics of ceftobiprole against multiple bacterial pathogens in murine thigh and lung infection models. Antimicrob. Agents Chemother. 52:3492–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Craig WA, Redington J, Ebert SC. 1991. Pharmacodynamics of amikacin in vitro and in mouse thigh and lung infections. J. Antimicrob. Chemother. 27(Suppl C):29–40 [DOI] [PubMed] [Google Scholar]
- 11. Galer D, et al. 2004. An analytical method for the analysis of tulathromycin, an equilibrating triamilide, in bovine and porcine plasma and lung. J. Agric. Food Chem. 52:2179–2191 [DOI] [PubMed] [Google Scholar]
- 12. Gladue RP, Bright GM, Isaacson RE, Newborg MF. 1989. In vitro and in vivo uptake of azithromycin (CP-62,993) by phagocytic cells: possible mechanism of delivery and release at sites of infection. Antimicrob. Agents Chemother. 33:277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gladue RP, Snider ME. 1990. Intracellular accumulation of azithromycin by cultured human fibroblasts. Antimicrob. Agents Chemother. 34:1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hand WL, Hand DL. 2001. Characteristics and mechanisms of azithromycin accumulation and efflux in human polymorphonuclear leukocytes. Int. J. Antimicrob. Agents 18:419–425 [DOI] [PubMed] [Google Scholar]
- 15. Letavic MA, et al. 2002. Synthesis and activity of a novel class of tribasic macrocyclic antibiotics: the triamilides. Bioorg. Med. Chem. Lett. 12:2771–2774 [DOI] [PubMed] [Google Scholar]
- 16. Mandell GL, Coleman E. 2001. Uptake, transport, and delivery of antimicrobial agents by human polymorphonuclear neutrophils. Antimicrob. Agents Chemother. 45:1794–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Montenez JP, et al. 1996. Interaction of the macrolide azithromycin with phospholipids. II. Biophysical and computer-aided conformational studies. Eur. J. Pharmacol. 314:215–227 [DOI] [PubMed] [Google Scholar]
- 18. Nemzek JA, Bolgos GL, Williams BA, Remick DG. 2001. Differences in normal values for murine white blood cell counts and other hematological parameters based on sampling site. Inflamm. Res. 50:523–527 [DOI] [PubMed] [Google Scholar]
- 19. Nowakowski MA, et al. 2004. Pharmacokinetics and lung tissue concentrations of tulathromycin, a new triamilide antibiotic, in cattle. Vet. Ther. 5:60–74 [PubMed] [Google Scholar]
- 20. Nutsch RG, et al. 2005. Efficacy of tulathromycin injectable solution for the treatment of naturally occurring swine respiratory disease. Vet. Ther. 6:214–224 [PubMed] [Google Scholar]
- 21. Pascual A, Conejo MC, GarcíA I, Perea EJ. 1995. Factors affecting the intracellular accumulation and activity of azithromycin. J. Antimicrob. Chemother. 35:85–93 [DOI] [PubMed] [Google Scholar]
- 22. Pfizer 2005. Tulathromycin injection package insert—Draxxin. Pfizer Inc, New York, NY: https://animalhealth.pfizer.com/sites/pahweb/US/EN/Documents/Prescribing%20Info%20or%20Package%20Inserts/DRAXXIN_Comp_Full_8.5x11_2.17.10.pdf [Google Scholar]
- 23. Rooney KA, et al. 2005. Efficacy of tulathromycin compared with tilmicosin and florfenicol for the control of respiratory disease in cattle at high risk of developing bovine respiratory disease. Vet. Ther. 6:154–166 [PubMed] [Google Scholar]
- 24. Safdar N, Andes D, Craig WA. 2004. In vivo pharmacodynamic activity of daptomycin. Antimicrob. Agents Chemother. 48:63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scheuch E, Spieker J, Venner M, Siegmund W. 2007. Quantitative determination of the macrolide antibiotic tulathromycin in plasma and broncho-alveolar cells of foals using tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 850:464–470 [DOI] [PubMed] [Google Scholar]
- 26. Seithel A, et al. 2007. The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab. Dispos. 35:779–786 [DOI] [PubMed] [Google Scholar]
- 27. Siegel T, et al. 2004. Cellular uptake of the triamilide tulathromycin by bovine and porcine phagocytic cells in vitro. J. Anim. Sci. Suppl. 1:186 [Google Scholar]
- 28. Szarka RJ, Wang N, Gordon L, Nation PN, Smith RH. 1997. A murine model of pulmonary damage induced by lipopolysaccharide via intranasal instillation. J. Immunol. Methods 202:49–57 [DOI] [PubMed] [Google Scholar]
- 29. Vallée E, Azoulay-Dupuis E, Pocidalo JJ, Bergogne-Bérézin E. 1992. Activity and local delivery of azithromycin in a mouse model of Haemophilus influenzae lung infection. Antimicrob. Agents Chemother. 36:1412–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Veber B, Vallée E, Desmonts JM, Pocidalo JJ, Azoulay-Dupuis E. 1993. Correlation between macrolide lung pharmacokinetics and therapeutic efficacy in a mouse model of pneumococcal pneumonia. J. Antimicrob. Chemother. 32:473–482 [DOI] [PubMed] [Google Scholar]
- 31. Venner M, Kerth R, Klug E. 2007. Evaluation of tulathromycin in the treatment of pulmonary abscesses in foals. Vet. J. 174:418–421 [DOI] [PubMed] [Google Scholar]