

Abstract
BMS-599793 is a small molecule entry inhibitor that binds to human immunodeficiency virus type 1 (HIV-1) gp120, resulting in the inhibition of CD4-dependent entry into cells. Since BMS-599793 is currently considered a candidate microbicide drug, we evaluated its efficacy against a number of primary patient HIV isolates from different subtypes and circulating recombinant forms (CRFs) and showed that activity varied between ∼3 ρM and 7 μM at 50% effective concentrations (EC50s). Interestingly, CRF01_AE HIV-1 isolates consistently demonstrated natural resistance against this compound. Genotypic analysis of >1,600 sequences (Los Alamos HIV sequence database) indicated that a single amino acid polymorphism in Env, H375, may account for the observed BMS-599793 resistance in CRF01_AE HIV-1. Results of site-directed mutagenesis experiments confirmed this hypothesis, and in silico drug docking simulations identified a drug resistance mechanism at the molecular level. In addition, CRF01_AE viruses were shown to be resistant to multiple broadly neutralizing monoclonal antibodies. Thus, our results not only provide insight into how Env polymorphisms may contribute to entry inhibitor resistance but also may help to elucidate how HIV can evade some broadly neutralizing antibodies. Furthermore, the high frequency of H375 in CRF01_AE HIV-1, and its apparent nonoccurrence in other subtypes, could serve as a means for rapid identification of CRF01_AE infections.
INTRODUCTION
High genetic diversity is characteristic of human immunodeficiency virus type 1 (HIV-1) both within and among infected individuals. Recombination among HIV-1 subtypes occurs frequently, resulting in genetic mosaics termed circulating recombinant forms (CRFs), whereby the two globally predominant CRFs are CRF01_AE and CRF02_AG (23). Interestingly, individuals infected with CRF01_AE HIV-1 can progress to disease faster than those infected with other HIV-1 subtypes or CRFs (52), yet the mechanism(s) responsible for this phenomenon remains unknown. Drug efficacy may also vary according to subtype classification (20, 27).
In addition to HIV-1 genetic diversity among hosts, HIV-1 diversifies within hosts. This diversification is ongoing and tailored by concurrent host selective pressure(s). Hence, each host harbors a unique progeny of HIV-1 genetic variants, commonly termed quasispecies. The development of a quasispecies may be largely attributed to reverse transcriptase (RT) infidelity (32, 42, 65) and genetic recombination (80). A high mutation rate, coupled with the production of up to 1010 to 1012 virions per day (24, 59), permits the founder HIV-1 to rapidly adapt to in vivo selective pressures, such as immune and/or drug pressure (21). HIV-1 genetic variability, both within and among hosts, contributes to the complexity of both vaccine and drug development (4, 10, 28, 34, 43, 46, 48, 50, 51, 54, 57, 61, 63, 69, 81).
The development of entry inhibitors and/or an effective Env-targeted vaccine is particularly challenging since Env is the most mutable of all gene products. Env is the only viral protein exposed to the extracellular environment and is therefore the protein against which selective pressures are easily exerted. Mechanisms of immune or drug escape of Env include (i) incomplete gp160 processing, resulting in decoy Env antigens or drug targets (3), (ii) incorporation of high-mannose glycans, which can shield potential neutralizing antibody (NAb) epitopes or impede access to drug binding sites (66), and (iii) Env structural flexibility/flux, which can impede interactions with NAbs and/or drugs (40). Underpinning the latter two mechanisms is the exceptionally high capacity of env to mutate without unduly compromising the structural and/or functional integrity of the protein.
HIV-1 Env is comprised of two glycoproteins (37), gp120 and gp41, that form the specialized viral membrane fusion complex that mediates HIV-1 entry. In its native state, gp120 contains two distinct regions: a gp41-interacting inner domain that forms the functional Env structure (gp120/gp41 trimer or spike) and a heavily glycosylated outer domain that constitutes most of the exposed surface of the spike (26, 35). HIV-1 gp41 mediates viral-to-target cell membrane fusion only after gp120 binds to cellular CD4 and coreceptor. The current model describes a flexible unbound gp120/41 trimer relative to the more rigid CD4-bound state (30). CD4 binding to gp120 induces a marked conformational change, resulting in the formation of a third domain, termed the “bridging sheet,” that links the inner and outer domains and facilitates Env gp120 interaction with HIV-1 coreceptors, typically CXCR4 or CCR5 (33, 65). Coreceptor binding triggers further conformational change in Env, resulting in the exposure of gp41, and ultimately mediates virion-to-cell membrane fusion. Due to the variety and range of Env conformational changes during the entry process, it is conceivable that many epitopes are fleeting and, therefore, poorly antigenic. Likewise, the poor immunogenicity of cell-free HIV-1 may be due to the inherent flexibility of the unbound Env complex; pre-CD4-bound Env epitopes in a constant state of structural flux are likely weak immunogens.
Although eliciting an effective Ab response is challenging due to the exceptional structural flexibility of Env, small molecules designed to limit this flexibility may serve as potent anti-HIV-1 inhibitors. By locking Env into a more rigid structure, these molecules may not only serve to directly inhibit HIV-1 infection but could help to present Env as a more robust immunogen. The entry inhibitor BMS-378806 (BMS-806) (38) and related compounds (Fig. 1) are highly potent HIV entry inhibitors. Previous work has suggested that these inhibitors compete with CD4 for the gp120-CD4 binding pocket (22, 25), and recent work provides in silico evidence that these compounds may also inhibit HIV-1 entry by affecting bridging sheet formation induced by gp120-CD4 binding (14). Due to the failure of BMS-806 to achieve targeted plasma exposure in humans (78), the clinical pursuit of BMS-806 was abandoned in favor of BMS-488, a newer version of the drug that demonstrated improved in vitro and pharmokinetic properties (78). Eventually, BMS-488 was replaced with BMS-663068 due to the latter's improved potency, a seemingly higher barrier for resistance, and a good safety profile in humans (53). Currently, BMS-663068 is a lead compound in therapeutic clinical trials, while BMS-599793, a structurally related compound, is being developed as an anti-HIV-1 microbicide by the International Partnership for Microbicides. The use of BMS-599793 may be advantageous over other current microbicide candidate antiretrovirals since the drug targets viral protein in lieu of host proteins.
Fig 1.

The two-dimensional (2D) molecular structures of BMS-599793 and related compounds. The 2D structures of BMS-378806, -488043, -599793, -626529, and -63068 are shown.
The high rate of Env mutation makes the preclinical evaluation of entry inhibitors salient, lest the efficacy of an entry inhibitor be compromised by specific Env variations. Thus, we evaluated the efficacy of BMS-599793 and other HIV-1 entry inhibitors against a panel of primary patient isolates and clones of different subtypes in vitro.
MATERIALS AND METHODS
Cells and cell culture.
TZM-bl cells (62) were obtained through the NIH AIDS Research & Reference Reagent Program (http://www.aidsreagent.org/) (catalog no. 8129). Cells were routinely subcultured every 3 to 4 days in 90% Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% (vol/vol) penicillin-streptomycin (P-S), and 1% (vol/vol) glutamine and maintained at 37°C under 5% CO2. Cryopreserved CD4+-enriched peripheral blood mononuclear cells (PBMCs) from a single healthy HIV negative donor were purchased from STEMCELL Technologies, Vancouver, BC, Canada. PBMCs (from cryopreservation) were cultured with RPMI 1640 (1×) (GIBCO, Invitrogen), supplemented with 10% FBS, 1% (vol/vol) P-S, 1% (vol/vol) glutamine, 5% (vol/vol) human interleukin 2 (hIL-2) (ABI Inc.), and 10 μg/ml phytohemagglutinin (30) to facilitate T-cell stimulation. Stimulation medium was removed from PBMCs at 72 h postreanimation and replaced with fresh culture medium (RPMI 1640 [1×; GIBCO, Invitrogen] supplemented with 10% FBS, 1% [vol/vol] P-S, 1% [vol/vol] glutamine, and 5% [vol/vol] hIL-2) prior to infection with primary HIV-1 isolates.
Primary HIV isolates.
Primary specimens of HIV-16050, HIV-15512, HIV-16030, HIV-16240, and HIV-16343 were obtained through the Laboratoire de Santé Publique du Quebec (LSPQ) with informed consent. The primary isolates HIV-1MOLE-1, HIV-1MOLE-3, and HIV-1MOLE-13 were obtained courtesy of Max Essex, Harvard AIDS Institute and the Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston, MA. The primary isolates HIV-192UG037, HIV-194UG114, HIV-1CMU06, HIV-1NI1052, HIV-1NP1525, and HIV-192TH001 were obtained from the NIH AIDS Research & Reference Reagent Program, catalog number 11412 (http://www.aidsreagent.org/). The isolate HIV-2MVP was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH; H9/HIV-2MVP was obtained from Lutz Gürtler and Friedrich Deinhardt. HIV-2CBL-23/H9 was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH; HIV-2 CBL-23/H9 was obtained from Robin Weiss. Each sample was amplified using stimulated CD4+-enriched PBMCs by means of a conventional protocol (36, 64). Amplified virus was then sequenced to determine or confirm subtype classification and the presence of any drug resistance (DR) mutations.
Subtype-specific HIV-1 molecular clones.
The subtype C HIV-1 molecular clone, pINDIE-C1, was kindly provided by Michiyuki Matsuda, International Medical Center of Japan, Research Institute, Department of Pathology, Toyama, Shinjuku-ku, Tokyo, Japan (45). The subtype B HIV-1 molecular clones, pNL43 and pBa-L, were obtained from the NIH AIDS Research & Reference Reagent Program (http://www.aidsreagent.org/). The CRF01_AE molecular clone was kindly provided by Nelson Michael, Division of Retrovirology, Walter Reed Army Institute of Research, Rockville, MD (70). To generate enough DNA for production of replication-competent HIV-1, molecular clones were individually transformed into DH5-α cells and cultured overnight at 37°C, after which single colonies were selected for further bacterial culture in LB medium overnight at 37°C. Mini- and maxi-preparations of all molecular clones were accomplished using kits (Qiagen) according to the manufacturer's instructions. Wild-type (WT) sequences and subtype classification were confirmed by sequencing of entire proviruses (REGA HIV-1 subtyping tool version 2.0; www.hivdb.stanford.edu). To introduce the S375H Env mutation into subtype B HIV-1, site-directed mutagenesis (SDM) was performed with pNL4-3 using XL-Gold 2 SDM kits (Stratagene), according to the manufacturer's protocol. Sequencing of Env constant region 3 (C3) confirmed the presence of the mutation in the clones as well as the absence of any spurious mutations. The resulting molecular clone encoding S375H was designated pNL-43-S375H.
Generation of genetically homogeneous HIV-1.
To generate replication-competent HIV-1 that was genetically homogenous, 16 μg of each of the molecular clones was separately transfected into 293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells were subsequently incubated in antibiotic-free DMEM (2% FBS) for 6 h prior to being washed with phosphate-buffered saline (PBS) (GIBCO) and replacement with fresh medium. At 24 h postwash, cell supernatants (containing replication-competent HIV-1) were collected and divided into 1-ml aliquots and stored at −150°C for future use. The replication-competent subtype C HIV-1 strains produced were designated HIV-1INDIE-C1. Replication-competent subtype B HIV-1 viruses generated from pNL-43 and pBa-L were designated HIV-1NL-43 ans HIV-1Ba-L, respectively. The S375H virus derived from pNL-43 was designated HIV-1NL-43-S375H.
Antiviral compounds.
BMS-599793 was kindly provided by the International Partnership for Microbicides (IPM) (Silver Spring, MD). Maraviroc, AMD3100, and enfuvirtide were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. Concentrated stock solutions were prepared from powders using dimethyl sulfoxide (DMSO) as the solvent for each of BMS-599793, maraviroc, and AMD3100. Control experiments ensured that DMSO was not toxic to cells at any of the concentrations that we employed. H2O was used as the solvent for enfuvirtide. Further dilutions of compound stocks were completed immediately prior to experimentation using DMEM. The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH; HIV-1 gp120 monoclonal antibody (MAb) (IgG1 b12) was obtained from Dennis Burton and Carlos Barbas (1, 8), and HIV-1 gp120 MAb (VRC03) was obtained from John Mascola (74).
Inhibition of HIV-1 infection and HIV-1 neutralization assay.
Compounds were tested against clonal or primary HIV-1 in an infectivity assay. Briefly, TZM-bl cells were seeded at a concentration of 10,000 cells per well in a 96-well plate (CellBind; Corning) and permitted to adhere in DMEM (2.5% FBS, 1% [vol/vol] P-S, 1% [vol/vol] glutamine) overnight prior to drug addition. Candidate microbicide compounds were individually diluted from concentrated stock solutions to 4× the predetermined working concentration of half the maximal inhibitory concentration (EC50) of each drug alone (sufficient to inhibit 100% infection). Drugs were then serially diluted to achieve between six and eight total concentrations (in order to generate a dose response curve) and then immediately added to TZM-bl cells followed by infection. Cells were permitted to incubate with compounds (37°C, 5% CO2) throughout the duration of the experiments. No later than 48 h postinfection, cells were rinsed with 100 μl phosphate-buffered saline (PBS) and lysed with 50 μl/well of 1× cell lysis reagent (Promega, Fisher). Cell lysates (30 μl/well) were then transferred to a white, opaque 96-well plate (Corning, Fisher). To determine the extent of HIV-1 infection, 80 μl of luciferase assay reagent (Promega, Fisher) was added to each well, and the number of relative luminescence units (RLU)/well was measured using a luminescence counter (Microbeta2; PerkinElmer). Standardization of virus infections was accomplished by adding virus to result in 200 50% tissue culture infective dose (TCID50) as calculated in TZM-bl cells (∼180,000 RLU [±30,000] by luminescence). The inhibitory/neutralization effect of each drug was calculated as percent inhibition of infection relative to infections in the absence of drug. The 50%, 75%, and 90% inhibitory concentration of each anti-HIV-1 compound was determined by curve fit analysis (four-parameter dose-response curve/variable slope model) with GraphPad Prism 5.0 software. Levels of resistance to BMS-599793 were determined relative to the expected potency of the drug. Unless otherwise specifically stated, the expected potency of BMS-599793 was taken to be the average of the EC50 (0.73 nM), EC75 (1.4 nM), and EC90 (2.7 nM) for the laboratory-adapted subtype B molecular HIV-1 clones NL4-3, NL(AD8), and BAL (given that the original BMS entry inhibitors were designed against similar subtype B laboratory-adapted strains). Moderate, strong, and very strong drug resistance was considered to be 5- to 100-fold, 100- to 1,000-fold, and >1,000-fold, respectively.
Genotyping and analysis of gp120 sequence region C3.
For primary patient HIV-1 and HIV-2 isolates, viral RNA was extracted from stock solutions using a Qiagen QIAmp viral extraction kit (Missassauga, Ontario, Canada). Viral RNA was amplified via RT-PCR, and nested PCR was employed to amplify the HIV-1 env gene (44). For HIV-1 and HIV-2 molecular clones, primers were used to amplify env DNA segments and amplified using PCR. The resulting DNA was purified using a QIAquick PCR purification kit (Qiagen), as specified by the manufacturer. The presence of the expected PCR product was confirmed by running 5 μl of each product on a 1% agarose gel. The samples were directly sequenced with subtype-specific env primers using the BigDye Terminator cycle sequencing kit (version 1.1; Applied Biosystems). The sequences were run on an ABI Prism 3130xl genetic analyzer (Applied Biosystems). The data were analyzed using SeqScape software (version 2.5), and PCR sequences were aligned using Bioedit (version 7.0) and CLC sequence viewer 6 software.
Selection and analysis of the HIV-1 gp120 C3 env domain from the Los Alamos HIV-1 database.
HIV-1 gp120 env sequences were selected according to the most recent year of submission to the Los Alamos HIV-1 database. For example, all sequences from 2009 were added to those from 2008, which were added to those before 2007, etc., in order to obtain a mutation frequency estimate at gp120 position 375 that would be most reflective of the current pandemic. Variability at gp120 position 375 was expressed as a percentage for each subtype, and entropies at this site were calculated using Shannon's entropy equation.
Building of the BMS-599793 three-dimensional structure.
The three-dimensional representation of BMS-599793 was created manually using the build functionality of PyMol Molecular Graphics System, version 1.3. To generate a more energy-favorable structure, bond lengths, torsions, and local geometry were improved manually. Further geometric optimization was performed with Avogadro version 1.0.3 software, available online (http://avagadro.openmolecules.net), with 500 steps of steepest descent and the MMFF94 force field. The software determined the net charge and the free energy for BMS-599793 at pH 7.4 to be +1 and 1,194 kJ/mol, respectively. The structure was prepared as a flexible docking ligand using Autodock tools (49). Autodock tools automatically calculated and assigned appropriate bond lengths and torsions.
Modeling the binding modes of BMS-599793.
Structures representing HIV-1 subtype B gp120 (PDB:2B4C) (26) and CRF01_AE gp120 (PDB:3SES) (75) were downloaded from the protein database (PDB) (71). Using PDB:2B4C as the template and gp120 sequences pertinent to this study, gp120 homology models were created using the Protein Homology Fold Recognition2 server (PHYRE2) (31). In this manner, homology protein structures were created for subtype B, CRF01_AE, HIV-2, and simian immunodeficiency virus cpz [SIV(cpz)] using sequences from HIV-1HXB2 (GenBank accession number AAK49977), HIV-1CRF01_AE clone, HIV-2MVP, and SIV(cpz) (GenBank accession number X52154). For the gp120 PDB:2B4C sequence, models were built for WT, S375W, S375H, M475I, and S375H/M475I subtype B gp120 proteins. The PDB:3SES gp120 sequence was utilized to generate models for WT, H375S, I475M, and H375S/I475M gp120 proteins. A CD4-unbound structure was solved using PDB:3DNL (39) from the antibody (b12)-bound gp120 trimer cocrystallized structure. All structures created showed excellent global homology to the original template (root main square deviation [RMSD] < 0.5 Å). Additional CD4-bound versions of the homology models were obtained by overlay and extraction of CD4 from the PDB:2B4C structure.
To enable use of crystal structure templates in BMS-599793 docking simulations, the b12 complexes were deleted from PDB:2B4C and PDB:3SES to yield the b12-unbound gp120 structures that we designated 2B4C_4 and 3SES_1. CD4 was then removed from the 2B4C_4 structure to yield 1RZK_1, and CD4 from 1RZK was introduced into the 3SES_1 structure to create 3SES_4. Clashes and improper torsions in all models were reduced by energy minimization using GROMOS96 implementation of SwissPDBViewer version 4.04 software and GROMOS96 43B1 parameters (5). All files were then prepared as docking receptors in Autodock Tools. Docking simulations were carried out by multithreading using Autodock Vina software (73). All Autodock Vina calculations were performed with an exhaustiveness of 100 using a docking box centered on the water-filled channel of gp120 and measuring 50 Å in each of the x,y, and z axes. This box included most of gp120 and included the entire putative CD4 binding interface, the gp120 regions C1 to C4, PS01-2, and the gp120 regions V1 to V5. Three independent dockings were carried out for each structure using Autodock Vina to yield 60 possible poses for the ligand in each structure. The most favorable binding orientation was based on kcal/mol calculated by Autodock Vina. All positions were also visually evaluated to identify previously predicted poses that may not have been deemed energetically favorable using the SwissPDBViewer. Visualization of docking poses, protein models, and all image processing were performed using PyMol Molecular Graphics System software. Minimization of ligand-docked structures was facilitated by use of a UCSF Chimera version 1.5.3 software package (60). To determine if our structures were in agreement with previously published work, the BMS-599793 progenitor drug, BMS-806, was docked to the structures using the same parameters.
Simulation of gp120-CD4+ protein-protein interactions.
Using the processed structures and models of subtype B gp120 (PDB:2B4C, WT, S375W, S375H, M475I, and S375H/M475I) and CRF01_AE gp120 (PDB:3SES, WT, H375S, I475M, H375S/I475M) described above and the CD4 molecule extracted from PDB:2B4C, protein-protein interactions were investigated using the three top-ranked (Critical Assessment of Predicted Interactions [CAPRI] meeting [33]) protein-protein docking servers: ClusPro 2.0 (12), the HADDOCK Web server (15, 16, 18), and the GRAMM-X Protein-Protein Docking Web server version 1.2.0. ClusPro and GRAMM-X were used to perform rigid-ligand/rigid-receptor dockings using global search algorithms, and HADDOCK was used to perform the flexible-ligand/flexible-receptor docking of CD4 to gp120. Initial docking using ClusPro and GRAMM-X was performed with no preconceived bias toward particular residue interactions. In addition, another set of docking simulations was carried out, identifying the CD4-F43 residue as part of the interaction interface. Evaluation of docking orientation was based on respective docking scores and the RMSD of the CD4-gp120 complex less than 1 Å away from the CD4-gp120 complex in the PDB:2B4C structure.
RESULTS
Evaluation of entry inhibitors against HIV-1.
The entry inhibitor BMS-599793 was evaluated against a panel of primary patient HIV-1 isolates. To evaluate the impact of inherent viral variation among patient quasispecies, clonal HIV-1 variants from different subtypes were tested. All HIV-1 subtypes and recombinants demonstrated various degrees of sensitivity to BMS-599793 (Table 1). Both subtype A and the two subtype C HIV-1 isolates, MOLE3 and MOLE13, demonstrated moderate BMS-599793 resistance at EC50s. This was likely due to the already present D185N Env mutation (data not shown) predicted by others to occur naturally in 54.88% (P < 0.0001, n = 82) and 53.46% (P < 0.001, n = 217) of isolates (44) and shown to confer low to moderate resistance to BMS-806 (22). In contrast, we observed that CRF01_AE HIV-1 isolates showed considerable resistance to BMS-599793 (Table 1). For CRF01_AE HIV-1, resistance to BMS-599793 varied between 700- and 20,000-fold in comparison to subtype B HIV-1NL4-3. The magnitude of BMS-599793 sensitivity at average EC50 per subtype group (relative to HIV-1NL4-3) was as follows: CRF01_AE < A < C and D < B and CRF02_AG. All viruses, including CRF01_AE HIV-1, were sensitive to maraviroc (a CCR5 antagonist) and/or AMD3100 (a CXCR4 antagonist) (data not shown).
Table 1.
BMS-599793 EC50s, EC75s, and EC90s required to inhibit genetically divergent HIV
| Subtype | Coreceptor tropism | HIV-1 virusd | HIV-2 virus | BMS-599793 EC (nM) |
||
|---|---|---|---|---|---|---|
| 50%a | 75% | 90% | ||||
| A | CCR5 | 6050 | 19.50 ± 0.0908 | 64.8 ± 0.107 | 216 ± 0.171 | |
| A | CCR5 | 92UG037 | 3.899 ± 0.1781 | 56.91 ± 0.2605 | 830.6 ± 0.4839 | |
| B | CXCR4 | NL4-3* | 0.354 ± 0.0548 | 0.656 ± 0.0722 | 1.22 ± 0.107 | |
| B | CCR5 | NL(AD8)* | 1.43 ± 0.0211 | 2.22 ± 0.0453 | 3.47 ± 0.0708 | |
| B | CCR5 | Ba-L* | 0.399 ± 0.0467 | 1.19 ± 0.126 | 3.31 ± 0.192 | |
| B | CCR5 | 5512 | 0.01181 ± 0.2599 | 0.3649 ± 0.3812 | 11.28 ± 0.6324 | |
| C | CCR5 | INDIE-C1* | 0.130 ± 0.0603 | 0.265 ± 0.101 | 0.538 ± 0.159 | |
| C | CCR5 | MJ4* | 0.0164 ± 0.0812 | 0.0442 ± 0.0473 | NC (<1)b | |
| C | CCR5 | MOLE1 | 0.300 ± 0.0870 | 1.12 ± 0.123 | 4.18 ± 0.201 | |
| C | CCR5 | MOLE3 | 24.6 ± 0.284 | 144 ± 0.514 | 842 ± 0.790 | |
| C | CCR5 | MOLE13 | 23.2 ± 0.117 | 76.4 ± 0.200 | 251 ± 0.301 | |
| D | CCR5 | 6030 | 1.057 ± 0.2888 | 4.864 ± 0.4372 | 22.37 ± 0.7223 | |
| D | CCR5 | 94UG114 | 0.003119 ± 0.5763 | 0.02506 ± 0.3729 | 0.2013 ± 0.2187 | |
| AE | CXCR4 | 6240 | 363 ± 1.19 | 1,814 ± 0.574 | 9,069 ± 1.255 | |
| AE | CXCR4/CCR5 | 6343 | 273 ± 2.40 | 1,469 ± 1.03 | 7,904 ± 1.71 | |
| AE | CCR5 | 92TH001 | 251 ± 0.615 | 719 ± 0.328 | 2,055 ± 0.366 | |
| AE | CXCR4 | CMU06 | 2,223 ± 0.115 | 4,087 ± 0.190 | 7,515 ± 0.295 | |
| AE | CXCR4 | NI1052 | 7,349 ± 0.02154 | 8,727 ± 0.04071 | 10,364 ± 0.05989 | |
| AE | CXCR4/CCR5 | NP1525 | 5,470 ± 0.02368 | 7,379 ± 0.03707 | 9,953 ± 0.05717 | |
| AG | CCR5 | 97GH-AG1* | 0.101 ± 0.0313 | 0.6931 ± 0.9212 | 4.498 ± 1.433 | |
| A | CXCR4 | MVP | >10,000c | >10,000 | >10,000 | |
| A | CXCR4 | CBL- 20/H9 | >10,000 | >10,000 | >10,000 | |
Mean concentration from 3 independent experiments ± log(SE) at ECs indicated for each HIV-1 tested.
NC denotes “not calculated” since value lies on a nonlinear part of the curve; the value in parentheses indicates maximum nM at the indicated EC.
Values of >10,000 indicate the top DS003 concentration tested at which no inhibition was observed.
*, clonal HIV-1.
A polymorphism unique to CRF01_AE HIV-1 gp120 may account for resistance to BMS-599793.
Amino acid sequences of gp120 of all viruses used in this study were analyzed for known or predicted BMS-806 and/or BMS-488 drug resistance mutations, based on the work of others (11, 41, 47, 55). No previous work has been published on BMS-599793, the inhibitor used in this study. After careful scrutiny and side-by-side comparisons of the gp120 and gp41 amino acid sequences used in this study, we found potential BMS-599793 resistance conferring mutations at CRF01_AE gp120 positions 350 (R350K), 375 (S375H), and 475 (M475I). We considered it unlikely that R350K alone could account for the BMS-599793 resistance characteristic of the CRF01_AE HIV-1 we tested since others have shown that R350K confers 1.7-fold sensitivity to BMS-806 in the absence of M475I (35). Indeed, we observed BMS-599793 sensitivity with a subtype C virus (MJ4), in which R350K was evident and M475I was absent. On the other hand, the presence of R350K and M475I was previously shown to confer BMS-806 resistance, yet only 15-fold (35). Thus, we also thought it unlikely that R350K, in combination with M475I, accounted for the marked BMS-599793 resistance demonstrated by CRF01_AE HIV-1 in our study. Most auspicious, however, was the presence of S375H in all 7 CRF01_AE HIV-1 isolates utilized and its absence in all other non-CRF01_AE HIV-1 isolates tested (Fig. 2). Others have shown that S375W, a mutation that fills in the CD4 binding cavity (of gp120), resulted in significant levels of BMS-806 resistance in vitro (36). Since tryptophan (W) and histidine are residues similar in size, we hypothesized that S375H might contribute to BMS-599793 resistance by filling the gp120 CD4 binding cavity.
Fig 2.

Deduced amino acid sequences aligned to HIV-1HXB2 Env region C3. gp120 C3 amino acids 329 to 393 for HIV-1 and 338 to 407 for HIV-2 are shown using HIV-1HXB2 and HIV-2ROD as the reference sequences, respectively. Colons depict the same amino acid as in the HIV-1 reference sequence. Brackets signify region not present in HIV-1 but present in HIV-2. CRF01_AE HIV-1 H375 and the corresponding HIV-2 W390 are highlighted in red. Sequences represent clones or the dominant viral variant of primary patient HIV isolate quasispecies used in this study. Sensitivity (S) and resistance (R) to BMS-599793, VRC03, and b12 are indicated. ND, not done.
Determining the frequency of mutation at Env amino acid 375 among different subtypes.
To assess the clinical relevance of mutations at Env position 375, 1,668 sequences from HIV-1 subtypes A to D and CRF01_AE (from the Los Alamos HIV sequence database) were analyzed for amino acid variation at the 375 location. The results indicated that subtypes A to D most frequently coded for S375 (84.1% for subtype A, 72.4% for subtype B, 95.8% for subtype C, and 87.4% for subtype D), with various degrees of divergence from S375 with amino acids A, C, F, G, I, K, M, N, P, Q, R, T, and W (Fig. 3A to E). S375T was the most frequent amino acid substitution at this site for subtypes A, B, and D, occurring in 9.5%, 19.8%, and 8.0%, respectively, of the samples analyzed. S375T was not found in the subtype C sample population tested (n = 309), and S375M and S375N were the most frequent substitutions (13%). Remarkably, the CRF01_AE sample population (n = 583) demonstrated 99% invariance at position 375, with histidine present 99% of the time and S or “undetermined” as the only alternatives (each 0.5%). This low frequency of variation was reflected in the low entropy of 0.027331 bits (Shannon's entropy) for the HIV-1 CRF01_AE sample population tested (Fig. 4). In contrast, entropy scores for subtypes A, B, C, and D were relatively high, i.e., 0.250698, 0.372377, 0.105099, and 0.235734 bits, respectively. It is worth noting that S375H was not observed in any of the subtypes or in CRF02_AG HIV-1 sequences sampled; histidine occurred at amino acid position 375 exclusively in the CRF01_AE HIV-1 sequences. Thus, histidine (instead of serine) should be considered the CRF01_AE WT residue at Env position 375 and is apparently unique to this particular group of HIV-1 viruses.
Fig 3.

Frequency of amino acid variability at gp120 position 375 according to HIV-1 subtype designate. Env sequences of subtype A, B, C, D, and CR01_AE (A, B, C, D, and E, respectively) obtained from the Los Alamos database were analyzed in order to determine the frequency of amino acid variability at gp120 position 375. Pie charts depict frequency of the amino acid indicated by color (F). Number of sequences analyzed per subtype designate is indicated at the bottom of each respective pie chart. A total of 1,669 sequences were analyzed.
Fig 4.
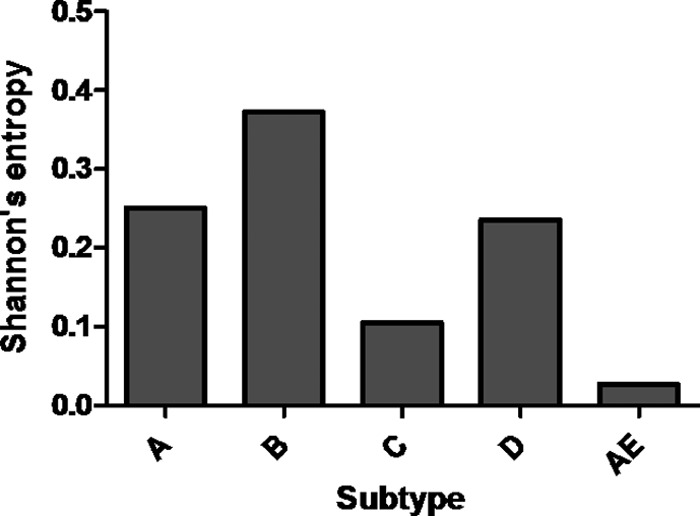
Level of Shannon's entropy for HIV-1 Env amino acid 375 for >1,600 isolates from subtypes A to D and CRF01_AE. An entropy level less than 0.5 bits (bits are depicted on the y axes) may be predicted with >90% accuracy.
Evaluation of BMS-599793 against HIV-2 and HIV-1NL4-3 S375H.
Previous work suggested that Env S375 directly interacts with BMS-488 in the CD4 binding cavity (13). Since the structure of BMS-488 is very similar to that of BMS-599793, a histidine instead of a serine may obstruct inhibitor binding given the stark contrast in size and chemical properties of these residues. Further, the mutation S375W was previously shown to decrease the activity of BMS-378806 (22), although the nuances of the molecular mechanism accounting for BMS-378806 resistance were not described. It is conceivable that a larger Van der Waals interaction sphere and increased hydrophobicity might allow S375W to stabilize gp120 into the CD4-bound state by partially occupying the same physical space as the CD4-F43 (necessary for CD4 binding). Since histidine is closer in size to tryptophan than serine, we hypothesized that histidine might have a similar effect as tryptophan in terms of locking gp120 into the CD4-bound state, thereby disrupting the gp120 structure required for BMS-599793 binding. This could account for the BMS-599793 resistance observed.
Given that HIV-2 gp125 is naturally a tryptophan at the site considered equivalent to HIV-1 position 375, we tested the susceptibility of HIV-2 to BMS-599793. We found that both HIV-2 primary isolates tested were resistant to BMS-599793 at the concentrations used (Fig. 5A). This not only confirms observations of others when using BMS-806 and/or BMS-488 but suggests real-world relevance of this amino acid position in the context of HIV gp120 evolution and BMS-599793 susceptibility.
Fig 5.

Resistance to BMS-599793 demonstrated by WT HIV-2 and HIV-1 encoding the gp120 S375H mutation in vitro. Percent inhibition of HIV-2 isolates MVP (circles) and CBL-20/H9 (squares) (A) and HIV-1 WT (circles) and S375H (squares) (B) infection (y axis) in the presence of increasing BMS-599793 concentrations (34) (x axis) in an infectivity assay. Data points depict the means and standard errors from 3 independent experiments.
To investigate if H375 alone could generate strong BMS-599793 resistance, SDM was employed to introduce S375H into HIV-1NL4-3, and the resulting virus was tested against BMS-599793. The results show that the S375H mutation alone conferred strong BMS-599793 resistance (Fig. 5B).
Evaluation of the broadly NAbs, b12 and VRC03, against subtype B and CRF01_AE HIV-1.
To further characterize the H375 mutation in the context of CRF01_AE HIV-1, the broadly NAbs, b12 (9) and VRC03 (74), were utilized in our infectivity assay. Both broadly NAb epitopes overlap the CD4 binding site (58). When tested against VRC03, HIV-16050 and HIV-1Ba-L (subtype A and subtype B, respectively) were neutralized, while subtype C (HIV-1INDIE-C1 and HIV-1MOLE13), subtype D (HIV-16030), and CRF01_AE (HIV-16240 and HIV-192TH001) were not. The NAb b12 did not neutralize either CRF01_AE HIV-1CMU06 or the HIV-2 isolates tested but did neutralize the subtype B primary isolate HIV-15512. Since both VRC03 and b12 neutralize HIV-1 by binding in the CD4 binding pocket and did not neutralize CRF01_AE HIV-1, our results suggest that H375 may alter the CD4 binding pocket in a similar fashion to that described previously for the S375W mutation (76).
Mechanism of BMS-599793 facilitated subtype B HIV-1 inhibition.
The envelope protein gp120 had similar global structures in multiple HIV-1 subtypes as well as in HIV-2 as seen by structural alignments of crystal structures and modeled structures (Fig. 6A). In silico subtype B gp120 docking simulations, performed in the presence and absence of CD4, indicated that the phenolic moiety of BMS-599793 (Fig. 6B) inserts into the CD4-F43 binding cavity of subtype B Env (Fig. 6C) but not CRF01_AE Env (Fig. 6D and E), suggesting that BMS-599793 directly competes with CD4 for the CD4-gp120 binding pocket. Since CD4-F43 insertion into the CD4-F43 cavity (Fig. 6H, I) accounts for 23% of the CD4-gp120 interaction face (6, 32) and is critical for HIV-1 entry into host cells, this competition (Fig. 6F, G, K) could account for the strong subtype B HIV-1 inhibition demonstrated by this drug. Specifically, BMS-599793 interacted with subtype B gp120 residues A281, T283, D368, E370, I371, S375, N425, M426, W427, T455, M475, and D477, and these interactions were identical in the crystallized (Fig. 6C) and modeled (Fig. 6E) forms. Of these, D368, E370, S375, M426, M475, D477, and modeled forms (Fig. 6E) have been previously identified as being essential for subtype B BMS-806-gp120 binding (7, 41). Although BMS-806 docked in the same orientation as BMS-599793, the phenolic moiety of BMS-806 inserted deeper into the CD4-F43 cavity, forming a salt bridge and hydrogen bonds with subtype B gp120 residues D368 and S375, respectively. BMS-806 also interacted with gp120 residues W112, T257, F382, W427, M475, and V430 hydrophobically and through Van der Waals bonds (Fig. 6K). These observed interactions indicate why all of these residues have been reported to contribute to BMS-806 resistance (41). Furthermore, the insertion of BMS-806 deeper into the cleft (relative to BMS-599793) may also allow for the binding of CD4, albeit in a suboptimal manner (57). This is in contrast to BMS-599793, which does not insert as deeply into the CD4-F43 cavity and, consequently, occupies more of the CD4 binding interface. Further, the formation of a salt bridge between the main chain amide of W427 and the BMS-599793 nitrile group was unique to BMS-599793. Given that interactions between CD4 and gp120 rely on electrostatic interplay between the basic residues on CD4 and the acidic residues surrounding the CD4-F43 cavity, the salt bridge may contribute to the superior BMS-599793 efficacy relative to BMS-806. Additionally, BMS-599793 is more basic than BMS-806 and may have more repulsive interactions with CD4 than BMS-806.
Fig 6.

In silico modeling and docking simulations. Alignment of multiple gp120 models and structures showed excellent global structural similarities (A). Three-dimensional structure of BMS-599793 (B). Close-ups of BMS-599793 (white) docked to subtype B gp120 (PDB ID:2B4C) (C), CRF01_AE gp120 (PDB ID: 3SE8) (D), and modeled subtype B HIV-1HXB2 gp120 (E); interaction face on gp120 is covered with mesh and residues involved in interactions are shown as sticks (C, D, and E). The relative positions of residues 375 and 475 to BMS-599793 (white) in subtype B gp120 (F) (orange) and CRF01_AE gp120 (G) (blue); mesh represents internal cavities in gp120 (F and G). The relative positions of residues 375 and 475 to F-43 of superimposed CD4 in subtype B gp120 (H) and CRF01_AE gp120 (I). Close-up of BMS-806 docked to subtype B gp120 (J); key residues in interaction are shown in stick form; buried portion of BMS-806 is shown only as sticks while solvent-exposed portion is shown as sticks within partially transparent space-filling structure. Superimposition of BMS-806 (white), BMS-599793 (black), and CD4 F-43 (green) showing their relative depths of penetration into the F-43 cavity of subtype-B gp120 (K). Hydrophilic and electrostatic interactions that potentially disrupt and prevent insertion of BMS-599793 into the F43 cavity of CRF01_AE gp120 (L). All dotted lines in the figure refer to postulated hydrogen bonding distances ≥ 3Å or postulated salt-bridge interaction distances ≥ 4Å.
Innate CRF01_AE HIV-1 BMS-599793 resistance.
BMS-599793 docked to CRF01_AE gp120 in an opposite orientation to that observed with subtype B gp120 in our in silico simulations. The CRF01_AE gp120 residues observed to specifically interact with BMS-599793 were Q258, R269, P364, G366, G367, D368, M426, W427, P470, G471, N474, and I475 (Fig. 6D). However, the apparent interaction of BMS-599793 to CRF01_AE gp120 with glycines at positions 366, 367, 368, and 471 indicates a high level of disorder in the binding dynamic, suggesting that the binding observed could be transient and may thus not occur in vivo. This is supported by the simulated free energy of binding calculated by Autodock Vina for the CRF01_AE gp120-BMS-599793 complex (5.9 kcal/mol) compared to that of subtype B-BMS-599793 (8.0 kcal/mol). Of the residues identified to be uniquely involved in simulated CRF01_AE gp120 binding, none has been previously reported to be associated with BMS-806 resistance.
Since mutations at position 375 in subtype B gp120 are associated with BMS-806 resistance (4, 7, 22, 41, 72), we performed BMS-599793 docking simulations with HIV-2 and SIV(cpz) gp120s that contain W375 and M375, respectively. In both simulations, BMS-599793 failed to yield an identical binding mode to that observed with subtype B gp120: BMS-599793 docked in a similar mode to CRF01_AE in the SIV model and in a completely different location in HIV-2 (data not shown). Further, the lower free energy of binding observed for CRF01_AE, SIV, and HIV-2, relative to HIV-1 subtype B, implies that BMS-599793 can bind only HIV-1 gp120 in the subtype B-like orientation to exert its AIDS-associated retrovirus (ARV) effect. Thus, gp120 amino acid position 375 appears to be associated with altered BMS-599793 binding.
In stark contrast to dockings performed with subtype B gp120, the phenolic group of BMS-599793 did not insert into the CD4-F43 cavity of CRF01_AE gp120. Thus, key hydrophobic and Van der Waals interactions observed between BMS-599793 and subtype B gp120 may be lost in the context of CRF01-AE gp120. Binding of BMS-806 to the non-B structures also did not result in phenolic moiety insertion into the F43 cavity, likely due to the steric hindrance provided by the presence of H (Fig. 6D, G), W, and M (data not shown) at position 375 for HIV-1 CRF01_AE, HIV-2, and SIV(cpz), respectively. Additionally, there is a propensity for multiple hydrogen bonding and intramolecular electrostatic interactions between H375 and surrounding residues in the case of CRF01_AE HIV-1 (Fig. 6L).
DISCUSSION
The distribution of HIV-1 subtypes and recombinants differs across the world, with the greatest diversity found in central Africa, where all subtypes and CRFs are present. Although the global distribution of HIV-1 subtypes was largely stable from 2000 to 2007, an increase was observed for recombinants (23). Thus, an effective microbicide must be active against a wide array of HIV-1. It cannot be assumed, especially in the context of Env binding compounds, that a drug designed against subtype B HIV-1 Env will be equally as potent against Env classified as non-B. Indeed, the candidate microbicide compound, BMS-599793, demonstrated different inhibitory capacities against both intra- and intersubtype HIV-1 variants. Among all subtypes tested, exceptional BMS-599793 resistance was demonstrated by CRF01_AE HIV-1. This is a concern for BMS-599793-based microbicide use in Southeast Asia, where CRF01_AE HIV-1 is currently responsible for the vast majority of infections (23). CRF01_AE HIV-1 is also the fifth most common subtype worldwide, also being found in eastern and central Africa (23). Given the limitations of epidemiological data and the lapse of time since 2007, it is possible that the current incidence of CRF01_AE HIV-1 infection may be even higher. Thus, naturally occurring BMS-599793 resistance may be more prevalent as well.
The CRF01_AE HIV-1 isolates utilized in this study demonstrated strong resistance to the entry inhibitor BMS-599793. Molecular docking simulations in silico indicated that the bulkiness of Env H375, coupled with other CRF01_AE Env residue variations, may prevent BMS-599793 binding. However, the S375H substitution alone was sufficient to confer CRF01_AE-like resistance to subtype B HIV-1 in vitro. Compared to other subtypes, the most notable and consistent difference in the CRF01_AE HIV-1 amino acid sequences analyzed is H375, located in the constant region 3 (C3) of gp120. In all other subtypes analyzed, S is observed at position 375. In addition to the isolates and clones tested here, we found that the H375 polymorphism is remarkably frequent in CRF01_AE HIV-1, occurring in >99% of the CRF01_AE sequences submitted to the Los Alamos HIV database. Furthermore, CRF01_AE variation at H375 is minimal, even though the C3 region showed the highest rates of amino acid diversity among conserved regions of gp120 and appeared important in CRF01_AE HIV-1 quasispecies evolution (6). Coupled with the finding that all other subtypes and CRFs analyzed have greater entropy and an absolute nonoccurrence of H at gp120 position 375, our observations suggest that H375 may confer an evolutionary advantage/constraint in the context of CRF01_AE HIV-1 infection, distinct from the drug resistance observed in this study.
The C3 region of Env is central to CD4 binding, which is the first step in target cell-virus interaction. It is integral in forming the CD4-F43 cavity, and thus it is thought that the high conservation of amino acids in this region reflects the importance of the structural integrity of this domain for productive HIV-1 infection. Thus, alteration of the CD4-F43 cavity amino acid repertoire would be predicted to lead to either (i) defective virus particles or (ii) CD4-independent infections; the latter possibility would be captivating in terms of pathogenicity and viral evolution. In this context, primordial HIV-1 (SIV) has been hypothesized to use CCR5 as the primary receptor for entry, independent of CD4. In nature, CD4-independent strains of HIV-2 and SIV are encountered, specifically in tissue compartments in which cells expressing CD4 are scarce (2, 19, 67, 68). Despite any potential advantage for augmentation of numbers of target cells as a result of low CD4 dependence for infection, however, HIV-1 strains demonstrating CD4 independence are very rare in the clinic (77, 79), and HIV-2 and SIV are considered to be less pathogenic than HIV-1. Thus, H375 likely does not confer CD4 independence, and CD4 independence does not account for the mechanism of BMS-599793 resistance seen in CRF01_AE HIV-1. In contrast, it is possible that H375 may permit CRF01_AE HIV-1 to bind to CD4 more efficiently than its S375 counterpart or other S375 exhibiting subtypes. This idea stems from observations by others that S375W (and other aromatic amino acid substitutions at position 375) might bind soluble CD4 (sCD4) with greater affinity than WT gp120 (76). As in the case of W, H is also an aromatic amino acid. Thus, H375 may serve to fill the CD4-F43 cavity, much like S375W, conferring greater CD4 binding affinity to CRF01_AE HIV-1. Further, the MAb b12 resistance observed with the CRF01_AE HIV-1 isolates here is reminiscent of that reported to occur with subtype B S375W HIV-1 and supports the idea that CRF01_AE HIV-1 may share characteristics with S375W subtype B mutants (more so than with WT subtype B HIV-1). Given that the S375W mutation stabilizes unbound subtype B HIV-1 gp120 in a CD4-bound-like state (13, 17, 29, 56), we propose that CRF01_AE HIV-1 may already assume the CD4-bound conformation (similar to subtype B S375W Env) prior to CD4 binding. However, other interpretations are possible, and further research is needed in this area. At a molecular level, this polymorphism might contribute to the aggressive pathogenesis and/or transmission rates observed with CRF01_AE HIV-1 infections, and we are hopeful that ongoing work in our laboratory will confirm/refute these notions. It is possible that these data could be further confirmed by studies on the neutralization sensitivity of the NL4-3 S375H virus we have constructed, and such work will be performed in future studies.
Although the resistance to BMS-599793 observed in our study was significant, it was not absolute. Thus, BMS-599793 may still serve as a potent microbicide compound. In geographic areas in which CRF01_AE HIV-1 (and subtype A HIV-1) are common, it is important to recognize the potential for strong resistance to BMS-599793. Thus, it may be sagacious to formulate BMS-599793 in combination with a different ARV. Further, it is worth noting that others have recently observed that CRF01_AE HIV-1 clinical isolates demonstrate marked resistance to BMS-626529, the prodrug to the lead Bristol Myers Squibb therapeutic drug, BMS-663068 (53). Finally, we note that similar patterns of resistance were obtained with each of the BMS-806, BMS-488, and BMS-599793 compounds. Although a fuller examination of the basis for these similarities is warranted, we believe that the molecular mechanism of BMS-626529 resistance is the same as that we have described for BMS-599793, given that these compounds are structurally related.
ACKNOWLEDGMENTS
This work was supported by grants from the International Partnership for Microbicides (IPM), Rockville, MD, and by the Canadian Institutes for Health Research (CIHR). Susan M. Schader was the recipient of a Frederick Banting and Charles Best doctoral research award, awarded by CIHR. P.K.Q. is a recipient of a CAHR/CIHR doctoral scholarship. T.M. is the recipient of the BMS/CTN postdoctoral fellowship.
This work was performed mostly by S.M.S. in partial fulfillment of the requirements of a Ph.D. degree, Faculty of Graduate Studies and Research, McGill University, Montreal, Quebec, Canada. Susan M. Schader designed all experiments, executed most tissue culture experiments involving HIV-1 clones, and analyzed all the data generated. Susan P. Colby-Germinario carried out tissue culture experiments and assisted in data analysis. Peter K. Quashie performed in silico docking simulations and assisted in analysis/interpretation. Maureen Oliveira expanded the primary patient HIV-1 isolates used in this study. Ruxandra-Ilinca Ibanescu and Daniela Moisi sequenced the HIV-1 isolates and clones. Thibault Mespléde performed SDM on proviral DNA. Mark A. Wainberg supervised this research and obtained grant support for its performance.
Footnotes
Published ahead of print 21 May 2012
REFERENCES
- 1. Barbas CF, III, et al. 1992. Recombinant human Fab fragments neutralize human type 1 immunodeficiency virus in vitro. Proc. Natl. Acad. Sci. U. S. A. 89:9339–9343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bhattacharya J, Peters PJ, Clapham PR. 2003. CD4-independent infection of HIV and SIV: implications for envelope conformation and cell tropism in vivo. AIDS 17(Suppl 4):S35–S43 [DOI] [PubMed] [Google Scholar]
- 3. Blay WM, Kasprzyk T, Misher L, Richardson BA, Haigwood NL. 2007. Mutations in envelope gp120 can impact proteolytic processing of the gp160 precursor and thereby affect neutralization sensitivity of human immunodeficiency virus type 1 pseudoviruses. J. Virol. 81:13037–13049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blish CA, Nedellec R, Mandaliya K, Mosier DE, Overbaugh J. 2007. HIV-1 subtype A envelope variants from early in infection have variable sensitivity to neutralization and to inhibitors of viral entry. AIDS 21:693–702 [DOI] [PubMed] [Google Scholar]
- 5. Bonvin A, Mark A, van Gunsteren W. 2000. The GROMOS96 benchmarks for molecular simulation. Comp. Phys. Comm. 128:550–557 [Google Scholar]
- 6. Boonchawalit S, et al. 2011. Molecular evolution of HIV-1 CRF01_AE Env in Thai patients. PLoS One 6:e27098 doi:10.1371/journal.pone.0027098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Briz V, Poveda E, Soriano V. 2006. HIV entry inhibitors: mechanisms of action and resistance pathways. J. Antimicrob. Chemother. 57:619–627 [DOI] [PubMed] [Google Scholar]
- 8. Burton DR, et al. 1991. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc. Natl. Acad. Sci. U. S. A. 88:10134–10137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burton DR, et al. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024–1027 [DOI] [PubMed] [Google Scholar]
- 10. Callahan KM, Fort MM, Obah EA, Reinherz EL, Siliciano RF. 1990. Genetic variability in HIV-1 gp120 affects interactions with HLA molecules and T cell receptor. J. Immunol. 144:3341–3346 [PubMed] [Google Scholar]
- 11. Charpentier C, et al. 2012. Prevalence of subtype-related polymorphisms associated with in vitro resistance to attachment inhibitor BMS-626529 in HIV-1 ‘non-B’-infected patients. J. Antimicrob. Chemother. 67:1459–1461 [DOI] [PubMed] [Google Scholar]
- 12. Comeau S, et al. 2007. ClusPro: performance in CAPRI rounds 6-11 and the new server. Prot. Struct. Funct. Bioinformatics 69:781–785 [DOI] [PubMed] [Google Scholar]
- 13. Da LT, Quan JM, Wu YD. 2009. Understanding of the bridging sheet formation of HIV-1 glycoprotein gp120. J. Phys Chem. B 113:14536–14543 [DOI] [PubMed] [Google Scholar]
- 14. Da LT, Quan JM, Wu YD. 2011. Understanding the binding mode and function of BMS-488043 against HIV-1 viral entry. Proteins 79:1810–1819 [DOI] [PubMed] [Google Scholar]
- 15. de Vries SJ, et al. 2007. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 69:726–733 [DOI] [PubMed] [Google Scholar]
- 16. de Vries SJ, van Dijk M, Bonvin AM. 2010. The HADDOCK Web server for data-driven biomolecular docking. Nat. Protoc. 5:883–897 [DOI] [PubMed] [Google Scholar]
- 17. Dey B, et al. 2007. Characterization of human immunodeficiency virus type 1 monomeric and trimeric gp120 glycoproteins stabilized in the CD4-bound state: antigenicity, biophysics, and immunogenicity. J. Virol. 81:5579–5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dominguez C, Boelens R, Bonvin AM. 2003. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125:1731–1737 [DOI] [PubMed] [Google Scholar]
- 19. Edinger AL, et al. 1997. CD4-independent, CCR5-dependent infection of brain capillary endothelial cells by a neurovirulent simian immunodeficiency virus strain. Proc. Natl. Acad. Sci. U. S. A. 94:14742–14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fleury HJ, et al. 2006. Susceptibility to antiretroviral drugs of CRF01_AE, CRF02_AG, and subtype C viruses from untreated patients of Africa and Asia: comparative genotypic and phenotypic data. AIDS Res. Hum. Retroviruses 22:357–366 [DOI] [PubMed] [Google Scholar]
- 21. Galiano L, et al. 2009. Drug pressure selected mutations in HIV-1 protease alter flap conformations. J. Am. Chem. Soc. 131:430–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo Q, et al. 2003. Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J. Virol. 77:10528–10536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hemelaar J, Gouws E, Ghys PD, Osmanov S. 2011. Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS 25:679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ho DD, et al. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123–126 [DOI] [PubMed] [Google Scholar]
- 25. Ho HT, et al. 2006. Envelope conformational changes induced by human immunodeficiency virus type 1 attachment inhibitors prevent CD4 binding and downstream entry events. J. Virol. 80:4017–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang CC, et al. 2005. Structure of a V3-containing HIV-1 gp120 core. Science 310:1025–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Invernizzi CF, et al. 2009. Signature nucleotide polymorphisms at positions 64 and 65 in reverse transcriptase favor the selection of the K65R resistance mutation in HIV-1 subtype C. J. Infect. Dis. 200:1202–1206 [DOI] [PubMed] [Google Scholar]
- 28. Joyce JG, ter Meulen J. 2010. Pushing the envelope on HIV-1 neutralization. Nat. Biotechnol. 28:929–931 [DOI] [PubMed] [Google Scholar]
- 29. Kassa A, et al. 2009. Identification of a human immunodeficiency virus type 1 envelope glycoprotein variant resistant to cold inactivation. J. Virol. 83:4476–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keele BF, et al. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kelley L, Sternberg M. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 32. Keulen W, Nijhuis M, Schuurman R, Berkhout B, Boucher C. 1997. Reverse transcriptase fidelity and HIV-1 variation. Science 275:229–231 [PubMed] [Google Scholar]
- 33. Kozakov D, et al. 2010. Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SOU, and stability analysis in CAPRI rounds 13–19. Prot. Struct. Funct. Bioinformatics 78:3124–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kulkarni SS, et al. 2009. Highly complex neutralization determinants on a monophyletic lineage of newly transmitted subtype C HIV-1 Env clones from India. Virology 385:505–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kwong PD, et al. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lane J. 1999. Methods in molecular medicine: HIV-1 protocols. Humana Press Inc., Totowa, NJ [Google Scholar]
- 37. Lansdon EB, et al. 2010. Visualizing the molecular interactions of a nucleotide analog, GS-9148, with HIV-1 reverse transcriptase-DNA complex. J. Mol. Biol. 397:967–978 [DOI] [PubMed] [Google Scholar]
- 38. Lin PF, et al. 2003. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U. S. A. 100:11013–11018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luftig MA, et al. 2006. Structural basis for HIV-1 neutralization by a gp41 fusion intermediate-directed antibody. Nat. Struct. Mol. Biol. 13:740–747 [DOI] [PubMed] [Google Scholar]
- 41. Madani N, et al. 2004. Localized changes in the gp120 envelope glycoprotein confer resistance to human immunodeficiency virus entry inhibitors BMS-806 and #155. J. Virol. 78:3742–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mansky LM, Temin HM. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marconi V, et al. 2008. Viral dynamics and in vivo fitness of HIV-1 in the presence and absence of enfuvirtide. J. Acquir. Immune Defic. Syndr. 48:572–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McGovern RA, Harrigan PR, Swenson LC. 2010. Genotypic inference of HIV-1 tropism using population-based sequencing of V3. J. Vis. Exp. doi:10.3791/2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mochizuki N, et al. 1999. An infectious DNA clone of HIV type 1 subtype C. AIDS Res. Hum. Retroviruses 15:1321–1324 [DOI] [PubMed] [Google Scholar]
- 46. Moore JP, Ho DD. 1995. HIV-1 neutralization: the consequences of viral adaptation to growth on transformed T cells. AIDS 9(Suppl A):S117–S136 [PubMed] [Google Scholar]
- 47. Moore PL, Cilliers T, Morris L. 2004. Predicted genotypic resistance to the novel entry inhibitor, BMS-378806, among HIV-1 isolates of subtypes A to G. AIDS 18:2327–2330 [DOI] [PubMed] [Google Scholar]
- 48. Moore PL, et al. 2009. Limited neutralizing antibody specificities drive neutralization escape in early HIV-1 subtype C infection. PLoS Pathog. 5:e1000598 doi:10.1371/journal.ppat.1000598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morris G, Goodsell D, Huey R, Olson A. 1996. Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J. Comp. Aid. Mol. Des. 10:293–304 [DOI] [PubMed] [Google Scholar]
- 50. Nakowitsch S, et al. 2005. HIV-1 mutants escaping neutralization by the human antibodies 2F5, 2G12, and 4E10: in vitro experiments versus clinical studies. AIDS 19:1957–1966 [DOI] [PubMed] [Google Scholar]
- 51. Nara PL, Lin G. 2005. HIV-1: the confounding variables of virus neutralization. Curr. Drug Targets Infect. Disord. 5:157–170 [DOI] [PubMed] [Google Scholar]
- 52. Ng OT, et al. 2011. Increased rate of CD4+ T-cell decline and faster time to antiretroviral therapy in HIV-1 subtype CRF01_AE infected seroconverters in Singapore. PLoS One 6:e15738 doi:10.1371/journal.pone.0015738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nowicka-Sans B, et al. 30 April 2012. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob. Agents Chemother. [Epub ahead of print.] doi:10.1128/AAC.00426-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nyambi P, et al. 2008. Neutralization patterns and evolution of sequential HIV type 1 envelope sequences in HIV type 1 subtype B-infected drug-naive individuals. AIDS Res. Hum. Retroviruses 24:1507–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Olson WC, Maddon PJ. 2003. Resistance to HIV-1 entry inhibitors. Curr. Drug Targets Infect. Disord. 3:283–294 [DOI] [PubMed] [Google Scholar]
- 56. Pan Y, Ma B, Keskin O, Nussinov R. 2004. Characterization of the conformational state and flexibility of HIV-1 glycoprotein gp120 core domain. J. Biol. Chem. 279:30523–30530 [DOI] [PubMed] [Google Scholar]
- 57. Pantophlet R. 2010. Antibody epitope exposure and neutralization of HIV-1. Curr. Pharm. Des. 16:3729–3743 [DOI] [PubMed] [Google Scholar]
- 58. Pantophlet R, et al. 2003. Fine mapping of the interaction of neutralizing and nonneutralizing monoclonal antibodies with the CD4 binding site of human immunodeficiency virus type 1 gp120. J. Virol. 77:642–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582–1586 [DOI] [PubMed] [Google Scholar]
- 60. Pettersen E, et al. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comp. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 61. Pinter A. 2007. Roles of HIV-1 Env variable regions in viral neutralization and vaccine development. Curr. HIV Res. 5:542–553 [DOI] [PubMed] [Google Scholar]
- 62. Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72:2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Prado I, Fouts TR, Dimitrov AS. 2009. Neutralization of HIV by antibodies. Methods Mol. Biol. 525:517–531 [DOI] [PubMed] [Google Scholar]
- 64. Prasad VRK, Ganjam V. (ed). 2009. HIV protocols, 2nd ed, vol 485 Springer, Humana Press, New York, NY [Google Scholar]
- 65. Preston BD. 1997. Reverse transcriptase fidelity and HIV-1 variation. Science 275:228–231 [DOI] [PubMed] [Google Scholar]
- 66. Quinones-Kochs MI, Buonocore L, Rose JK. 2002. Role of N-linked glycans in a human immunodeficiency virus envelope glycoprotein: effects on protein function and the neutralizing antibody response. J. Virol. 76:4199–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Reeves JD, Doms RW. 2002. Human immunodeficiency virus type 2. J. Gen. Virol. 83:1253–1265 [DOI] [PubMed] [Google Scholar]
- 68. Reeves JD, et al. 1999. Primary human immunodeficiency virus type 2 (HIV-2) isolates infect CD4-negative cells via CCR5 and CXCR4: comparison with HIV-1 and simian immunodeficiency virus and relevance to cell tropism in vivo. J. Virol. 73:7795–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ringe R, Thakar M, Bhattacharya J. 2010. Variations in autologous neutralization and CD4 dependence of b12 resistant HIV-1 clade C env clones obtained at different time points from antiretroviral naive Indian patients with recent infection. Retrovirology 7:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Salminen MO, et al. 2000. Construction and biological characterization of infectious molecular clones of HIV-1 subtypes B and E (CRF01_AE) generated by the polymerase chain reaction. Virology 278:103–110 [DOI] [PubMed] [Google Scholar]
- 71. Sussman J, et al. 1998. Protein data bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. D Biol. Crystallogr. 54:1078–1084 [DOI] [PubMed] [Google Scholar]
- 72. Teixeira C, Serradji N, Maurel F, Barbault F. 2009. Docking and 3D-QSAR studies of BMS-806 analogs as HIV-1 gp120 entry inhibitors. Eur. J. Med. Chem. 44:3524–3532 [DOI] [PubMed] [Google Scholar]
- 73. Trott O, Olson A. 2010. Software news and update AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Computat. Chem. 31:455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu X, et al. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu X, et al. 2011. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 333:1593–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xiang SH, et al. 2002. Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J. Virol. 76:9888–9899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xiao P, et al. 2008. Characterization of a CD4-independent clinical HIV-1 that can efficiently infect human hepatocytes through chemokine (C-X-C motif) receptor 4. AIDS 22:1749–1757 [DOI] [PubMed] [Google Scholar]
- 78. Xue YJ, Yan JH, Arnold M, Grasela D, Unger S. 2007. Quantitative determination of BMS-378806 in human plasma and urine by high-performance liquid chromatography/tandem mass spectrometry. J. Sep. Sci. 30:1267–1275 [DOI] [PubMed] [Google Scholar]
- 79. Zerhouni B, Nelson JA, Saha K. 2004. Isolation of CD4-independent primary human immunodeficiency virus type 1 isolates that are syncytium inducing and acutely cytopathic for CD8+ lymphocytes. J. Virol. 78:1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhuang J, et al. 2002. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J. Virol. 76:11273–11282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zwick MB, Burton DR. 2007. HIV-1 neutralization: mechanisms and relevance to vaccine design. Curr. HIV Res. 5:608–624 [DOI] [PubMed] [Google Scholar]