

Abstract
Systemic life-threatening fungal infections represent a significant unmet medical need. Cell-based, phenotypic screening can be an effective means of discovering potential novel antifungal compounds, but it does not address target identification, normally required for compound optimization by medicinal chemistry. Here, we demonstrate a combination of screening, genetic, and biochemical approaches to identify and characterize novel antifungal compounds. We isolated a set of novel non-azole antifungal compounds for which no target or mechanism of action is known, using a screen for inhibition of Saccharomyces cerevisiae proliferation. Haploinsufficiency profiling of these compounds in S. cerevisiae suggests that they target Erg11p, a cytochrome P450 family member, which is the target of azoles. Consistent with this, metabolic profiling in S. cerevisiae revealed a buildup of the metabolic intermediates prior to Erg11p activity, following compound treatment. Further, human cytochrome P450 is also inhibited in in vitro assays by these compounds. We modeled the Erg11p protein based on the human CYP51 crystal structure, and in silico docking of these compounds suggests that they interact with the heme center in a manner similar to that of azoles. Consistent with these docking observations, Candida strains carrying azole-resistant alleles of ERG11 are also resistant to the compounds in this study. Thus, we have identified non-azole Erg11p inhibitors, using a systematic approach for ligand and target characterization.
INTRODUCTION
Fungal infections remain a significant health problem, with Candida spp. being the fourth most common cause of nosocomial septicemia in the United States (37). The difficulties in treating fungal infections are multifaceted, including factors such as the difficulty of correct diagnosis, leading to late diagnosis, and the lack of clinically established breakpoints for commonly used drugs (7). These problems are exacerbated because only a small number of antifungal targets have been clinically validated (8). The identification of new targets for antifungal drug discovery is made especially challenging for two reasons: first, there are a large variety of different pathogenic species that can display great diversity in potential target homology (12, 16), and second, the putative antifungal target must be different enough from the human homologue that a suitable therapeutic window is possible (17).
A successful strategy for the identification of antimicrobial agents has been to use simple growth inhibition assays (27). The advantages of such assays is that they monitor multiple factors required for antimicrobial activity, including compound solubility and target accessibility, as well as the ability to sufficiently inhibit the target(s) to inhibit cell growth. However, growth inhibition assays cannot identify the target for a given molecule. Growth inhibition assays targeting specific biochemical pathways provide additional information but still lack the resolution required to identify the molecular target (2).
While target identification is not absolutely essential for drug development, it facilitates the optimization of a compound's inhibitory activity (31). A number of reports have described the use of genetic tools for the identification of compound targets in Saccharomyces (11, 14). This approach has recently been extended to allow such profiling in pathogenic fungal species, i.e., Candida spp. (16, 38). While such genetic methods provide strong evidence for the possible mechanism of a compound, such a hypothesis still needs to be confirmed by alternative methods that use a different assay or detection method.
This report describes the use of genome-wide fitness profiling, resistance mutation analysis, metabolic profiling, biochemical assays, and molecular modeling, using in silico docking to explore the mechanism of action of a group of antifungal compounds identified by high-throughput screening. This set of methods has identified a class of antifungal compounds that target Erg11p. Erg11p is part of the ergosterol biosynthesis pathway, catalyzing the demethylation of lanosterol at C-14 to 4,4-dimethylcholesta-8,14,24-trienol. The ergosterol biosynthesis pathway is the target of a number of antifungal drugs (10, 19). In Saccharomyces cerevisiae, it has been demonstrated that all enzymes required to convert squalene to lanosterol are essential for ergosterol production and cell viability (21). Ergosterol is one of the critical components in fungal membranes and has important biological functions in modulating membrane fluidity and control of the cell cycle (3). Compounds containing an azole moiety, often referred to as azoles, are the best-known examples of Erg11p inhibitors and are static or cytocidal depending on the fungal species (20, 24, 35).
This report describes an integrated drug discovery effort utilizing high-throughput screening to identify compounds inhibiting fungal growth. Fungal genetics suggested Erg11p as the target of this set of compounds. Biochemical assays, focused metabolomic profiling, and molecular modeling were then applied to further support this proposed mechanism of action.
MATERIALS AND METHODS
Fungal strains and biochemicals.
The commercially available fungal strains used in this study are listed in Table 1. Midazolam was purchased from Sequoia Research Products Ltd. (Pangbourne, United Kingdom) and 1′-hydroxymidazolam from Cerilliant Corp. (Round Rock, TX). Drugs and Novartis proprietary compounds were obtained from the Novartis compound store as either 2 or 10 mM stock solutions in dimethyl sulfoxide (DMSO). All other reagents, chemicals, and buffer salts were purchased from Sigma-Aldrich Chemicals (St. Louis, MO) or Fluka (Buchs, Switzerland).
Table 1.
Compound 1, 2, 3, and 4 and voriconazole antifungal activities
| Species and strain | MIC (μg/ml) (concn [μM])a |
||||
|---|---|---|---|---|---|
| Compound |
Voriconazole | ||||
| 1 | 2 | 3 | 4 | ||
| S. cerevisiae ATCC 9763 | 4 (12.3) | £0.25 (0.7) | ≤0.25 (0.7) | 16 (49.3) | 0.5 (1.4) |
| C. albicansb ATCC 24433 | 4 (12.3) | 2 (6.1) | ≤0.25 (0.7) | >128 (394.7) | 0.5 (1.4) |
| A. fumigatus ATCC MYA-3627 | >128 (395.9) | >128 (393.5) | 32 (98.0) | >128 (394.7) | 0.5 (1.4) |
| R. oryzae ATCC 4621 | >128 (395.9) | 4 (12.2) | 1 (3.0) | >128 (394.7) | 32 (91.6) |
| Candida tropicalisb ATCC 750 | >128 (395.9) | ND | ND | ND | 0.125 (0.3) |
| C. kruseib ATCC 6258 | 16 (49.4) | ND | ND | ND | 0.5 (1.4) |
| C. albicansb ATCC 64124 (azole resistant, mutations: F126L, E266D, S405F, V437I) | >128 (395.9) | ND | ND | ND | 4.0 (11.1) |
| C. albicansb in-house (azole resistant, mutations: D116E, Y132H, F499L) | 32 (98.9) | ND | ND | ND | 1.0 (2.8) |
ND, not done.
The MIC endpoint determination for azoles is less well defined. A less stringent endpoint of MIC-2 (prominent decrease in turbidity) was used to define the MIC, as described previously (6).
High-throughput screening.
The compounds described in the study were initially identified from a screen of the Novartis compound archive. This compound collection was screened for growth-inhibitory effects using S. cerevisiae (strain BY4743 pdr5Δ/pdr5Δ; purchased from Invitrogen) grown in yeast nitrogen base (Difco) medium supplemented with uracil (Sigma), leucine (Bio101), and histidine (Sigma). A starter culture from a single colony from a minimal agar plate was incubated for 20 h on an orbital shaker (30°C). The cells were then diluted to an optical density at 600 nm (OD600) of 0.01 (i.e., ∼5 × 103 CFU/ml), after which 4 μl was plated into 1,536-well black polystyrene plates (Greiner number 789176-A). The compounds were then transferred to the assay plates from a 2 mM stock solution using a 40-nl, 1,536-slot pin tool. The plates were then incubated (ca. 95% relative humidity) in a Liconic cell culture incubator at 30°C, without added CO2, overnight. Cell growth was monitored by adding 2 μl of a stock solution of resazurin (Sigma; 22.5 μM in 500 mM potassium phosphate buffer, pH 6.4). The detection solution was then incubated for a further 2 h before being read using a Viewlux 1430 multilabel plate imager (Perkin Elmer), using excitation at 525 nm and emission at 598 nm. On each plate, negative controls (i.e., DMSO) and active controls (cycloheximide) were used to normalize the relative growth to 0 and −100%.
Antifungal suspectibility testing.
Assays for antifungal susceptibility testing were performed in RPMI 1640 (HyClone; SH30011.03) with 2.05 mM glutamine and phenol red, without bicarbonate, and buffered with 0.165 mol/liter MOPS [3-(N-morpholino)propanesulfonic acid] (AppliChem; A1076,0250). The medium was adjusted to pH 7.0 and filter sterilized. Antifungal susceptibility testing was performed according to the Clinical and Laboratory Standards Institute (CLSI) guidelines for broth microdilution M27-A3 (yeast) and M38-A2 (mold) (5, 6).
For azoles, MIC endpoint determination is typically more difficult for Saccharomyces. Error in scoring antifungal activity was eliminated by repeating some experiments, supplementing media with 10% resazurin solution (R&D Systems; AR002). The plates were then incubated at 30°C overnight, and fungal growth inhibition was determined by measuring fluorescence (excitation, 544 nm; emission, 590 nm) every 30 min (Molecular Devices SpectraMax M3 and Soft Max Pro 5.4).
Assay for mammalian toxicity.
A WST-1 cytotoxicity/proliferation assay was performed according to the manufacturer's instructions (Roche catalog number 11 644 807 001), testing for growth inhibition after 72 h in three mammalian cell lines: K562 (CRL-1573), HEK293 (CCL-243), and HEPG2 (HB-8065).
Genome-wide analysis.
The potencies of test substances were determined using wild-type S. cerevisiae BY4743. The OD600 values of exponentially growing S. cerevisiae cultures in rich medium were recorded with a robotic system. Twelve-point serial dilutions were assayed in 96-well plates with a reaction volume of 150 μl. Solutions containing DMSO were normalized to 2%. The 30% inhibitory concentration (IC30) values were calculated using logistic regression curve fits generated by Tibco Spotfire v3.2.1 (Tibco Software Inc.).
The S. cerevisiae haploinsufficiency profiling (HIP), homozygous profiling (HOP), and microarray analysis were performed as described previously (29). The basic concept behind this assay is that HIP identifies genes in which one functional copy, compared to two, confers hypersensitivity to inhibition by the compound. This indicates pathways directly affected by the compound. HOP (with both gene copies deleted) indicates synthetic lethality and identifies compensating pathways for those directly affected by the compound. Genome-wide hetero- and homozygous deletion libraries of S. cerevisiae strains were purchased (OpenBiosystems; catalog numbers YSC1056 and YSC1055), and pools were constructed as described previously (29). Each HIP strain is haploid, and each HOP strain is completely null for one gene (each strain was identified by a unique DNA sequence, called a “barcode” or “tag,” inserted into the deleted gene). Each test substance was assayed in duplicate at its IC30, and the relative abundances of all strains in the pools were compared to eight no-drug control samples. Specific HIP HOP assay adaptations concerning the starting culture density, reaction culture volume, dilution scheme, and experimental controls will be described elsewhere (D. Hoepfner et al., submitted for publication). For the experimental analysis, we used the same computation of normalized tag intensities, outlier masking, and saturation correction described previously (29). Sensitivity was computed as the median absolute deviation logarithmic (MADL) score for each compound-concentration combination, as described in the supplemental material. The similarities of the obtained HOP profiles were compared by calculating the Pearson correlation coefficients of a compound HOP profile with all other compound profiles. In order to assess the significance of the correlation coefficients, we transformed them by the Fisher transformation and fitted a normal distribution to the transformed values. This empirical normal distribution allowed us to assign a P value to each correlation coefficient. The (negative) logarithm of the false-discovery rate (FDR) corrected P value was then displayed.
Targeted metabolomics profiling.
Targeted metabolic-profiling studies were carried out by extraction of the cellular lipids with methanol-chloroform extraction following compound treatment. The extracted lipids were then subjected to analytical liquid chromatography coupled to a 4000 QTRAP mass spectrometry (LC-MS) analysis, with comparison of ergosterol and lanosterol control samples. A detailed description of the method used is given in the supplemental material.
Human CYP inhibition.
CYP3A4 inhibition was investigated using the formation rate of 1-hydroxy-midazolam from midazolam in human liver microsomes supplemented with NADPH and monitored by LC-MS detection of the substrate. More details are given in the supplemental material.
Molecular modeling and in silico docking.
Human CYP51 (Protein Data Bank [PDB] code 3JUV) was selected as a template based on the results of HHpred (32) for Erg11p modeling. Primer application in Maestro version 9.2.109 (18) was used to generate the homology model of S. cerevisiae Erg11p based on the human CYP51 template. The model was then checked by QMEAN (http://swissmodel.expasy.org/qmean/cgi/index.cgi) (4). Superimposition of the S. cerevisiae Erg11p model with the crystal structure of human CYP51 and A. fumigatus CYP51 (9) was performed with DaliLite (http://www.ebi.ac.uk/Tools/dalilite/) (15).
For in silico docking of compounds using GOLD (Genetic Optimization for Ligand Docking) (36), the structure of human CYP51 was selected as a template. Default GOLD parameters were used to generate solutions, but docking was centered near the heme center of CYP51. All the figures were generated in PyMOL (version 1.3; Schrödinger, LLC).
RESULTS
Screening.
To identify novel compounds with antifungal activity, high-throughput screening was performed using the Novartis compound archive and S. cerevisiae cells with a miniaturized 1,536-well plate assay. The assay monitored proliferation using reduction of resazurin as a surrogate marker for metabolic activity (25). Cycloheximide (100 μM) was used as a positive control, and a final concentration of 1% DMSO was used as a negative control. Typically, robust screening results with Z′ values of >0.7 were obtained (39). Overall, 1,101,408 compounds were tested in the primary screen, and approximately 0.9% of the tested compounds showed >90% inhibition of cell proliferation.
Initially, compounds showing >90% inhibition of cell proliferation in the 16-h assay were designated hits (Fig. 1). These hits were further prioritized based on their effects on mammalian cell viability and their predicted physicochemical properties. Hits were initially validated by dose-response characterization, and compound identity and purity were confirmed by LC-MS. The screening campaign identified a number of known antifungal compounds (which were removed from further characterization), suggesting that novel compounds identified in this screen might serve as useful antifungal compounds for further development. This eventually led to a set of 5,000 high-priority compounds that were then characterized further.
Fig 1.
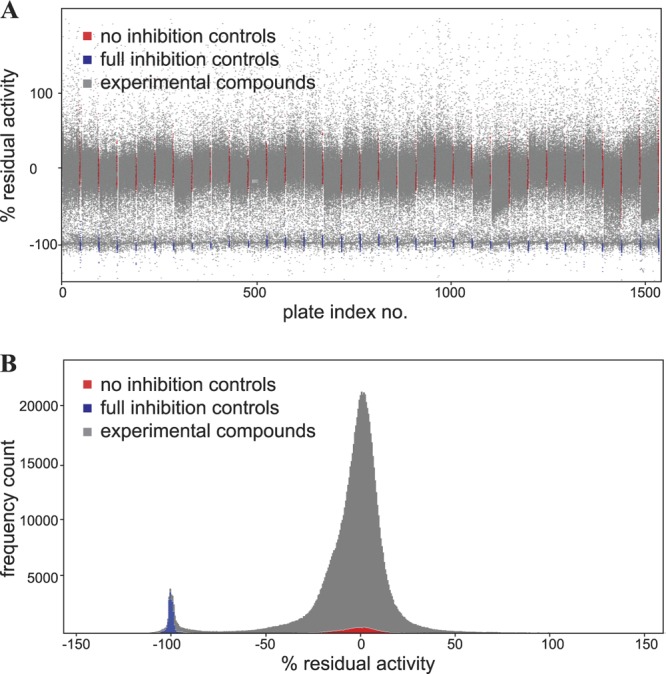
High-throughput screening for inhibition of S. cerevisiae proliferation. (A) Scatter plot, with each point representing the inhibitory activity of one individual compound tested during the screening campaign. Each plate contained 24 no-inhibition controls and 24 full-inhibition controls. (B) Histogram of the activity frequency of the results presented in panel A. Shown is a Gaussian activity distribution with an added peak of activity centered around the active controls, representing the population of compounds with antifungal activity.
Hit characterization and expansion.
Compound 1 (Fig. 2A) was identified from the primary screen and characterized further by determining the MIC against a panel of pathogenic fungi, using the CLSI antifungal susceptibility testing protocol (Table 1). Compound 1 was shown to inhibit the proliferation of S. cerevisiae and pathogenic Candida spp., including Candida albicans and Candida krusei, after incubation for 24 h at 30°C. No MIC was observed for Aspergillus fumigatus or Rhizopus oryzae when tested up to concentrations of 128 μg/ml (395.9 μM) for 48 h. The inhibitory activity of compound 1 is static but not cytocidal, as shown in Fig. S1 in the supplemental material. Thus, compound 1 does have antifungal activity against certain fungal species, demonstrating the utility of the original screen using S. cerevisiae. Further closely related chemical structures (which were not present in the original screening collection of compounds) were selected from the complete Novartis compound collection and tested for antifungal activity (Fig. 2A and Table 1). Compounds 2 and 3 showed slightly more potent antifungal activity; compound 4 did not show activity against any of the species tested, despite its similarity to the other compounds. This clear structure-activity relationship suggests a specific binding target for the active substances.
Fig 2.

HIP HOP profiling results. (A) Structures and S. cerevisiae (S.c.) IC30s of substances tested by HIP HOP profiling. (B to D) HIP HOP profiling of compound 1 and derivatives at the indicated concentrations. The gray squares represent strains with deletions in essential genes and the black dots strains with deletions in nonessential genes. Shared sensitive strains are labeled. In each HIP profile, SET6, a previously published indicator for ergosterol inhibition, is the most sensitive and relevant (as measured by the Z score) hit identified. The second-most-relevant hit is ERG11, the lanosterol 14-alpha-demethylase, the target of azole antifungals. In the HOP profile, the Erg11p regulator DAP1 and a common set of deletion strains that have published genetic interactions with ERG11 or are hypersensitive to azoles scored reproducibly. (E) Compound 4, the inactive derivative of compounds 2 and 3, did not score any relevant sensitive (less than −5/−5) hits at 200 μM. (F) HIP HOP profiles of voriconazole, an established Erg11p inhibitor, show the same hits observed with compound 1 and its active derivatives. (G) Alignment of the HIP HOP Z score of compound 1 with that of voriconazole shows a high degree of correlation and conservation of hits.
To determine the selectivity of these compounds against fungi, the compounds were tested in mammalian cells using the WST-1 cytotoxicity assay against the K562 (CRL-1573), HEK293 (CCL-243), and HEPG2 (HB-8065) cell lines (Table 2). Compounds 1, 2, and 3 showed antiproliferative activity with the K562 and HEK293 cell lines (but at concentrations greater than their activity against fungal cells), while compound 4, which lacks antifungal activity, had only weak cytotoxic activity with the K562 and HEK293 cell lines.
Table 2.
Concentrations (IC50) of compounds tested in the WST-1 cytotoxic assay against the K562, HEK293, and HEPG2 cell lines
| Cell line | IC50 (μM)a |
||||||
|---|---|---|---|---|---|---|---|
| Compound |
AMB | CIP | VOR | ||||
| 1 | 2 | 3 | 4 | ||||
| K562 | 10.3 | 3.3 | 3.6 | 21.1 | 21.5 | 202.0 | >366.4 |
| HEK293 | 25.9 | 27.7 | 62.5 | 145.4 | 6.7 | >205.6 | >366.4 |
| HEPG2 | 60.4 | 43.1 | 53.5 | 55.7 | <1.607 | >205.6 | >366.4 |
Amphotericin B (AMB) and ciprofloxacin (CIP) were used as the positive and negative control for cytotoxicity study. Voriconazole (VOR) was the azole control.
Target hypothesis.
Saccharomyces HIP and HOP were performed with compounds 1 to 4 of the compound cluster. HIP HOP is a gene dosage-dependent method that assesses the effects of compounds against potential targets encoded by the S. cerevisiae genome (16). We profiled all active compounds at their IC30 in a HIP HOP assay that was performed essentially as described previously (16). The inactive compound 4 was tested at 200 μM, based on its maximal solubility in DMSO.
The HIP results (Fig. 2B to F) suggest that the active substances, compounds 1, 2, and 3, act by targeting Erg11p, because the heterozygote erg11/ERG11 strain is one of the most sensitive strains in the profile, as is the heterozygote set6/SET6 strain, a key indicator of ergosterol modulation. Erg11p is the established target for azoles and encodes lanosterol 14-alpha-demethylase, which catalyzes an essential step in ergosterol biosynthesis (1, 20, 22, 26). SET6 encodes a protein of unknown function, but the heterozygous deletion strain has been previously linked to ergosterol modulation (11). The observation that the HIP profiles of all active compounds are similar and overlap the HIP profile of voriconazole, an established azole-containing Erg11p inhibitor, strongly suggests that it is the target of these compounds (19, 28, 30) (Fig. 2G). A HOP profile (homozygous profile) is based upon a genome-wide set of homozygous deletions of the nonessential genes in S. cerevisiae and often highlights parallel or compensatory pathways. The HOP profile of compound 1 identifies five genes with significant sensitivity linked to Erg11p function: DAP1 encodes an established Erg11p-regulating protein, and GCS1, RCY1, TVP18, and YPT31 encode membrane-trafficking components already associated with Erg11p inhibition by previous HIP HOP profiling experiments and genetic studies (11, 13, 23, 34). Comparison of the HOP profiles for compounds 1, 2, and 3 again revealed significant correlations with the profiles for other established Erg11p inhibitors, such as voriconazole and other azole-containing antifungals (Fig. 2B to G and Table 3). Compound 4 was tested at 200 μM but failed to identify any significant hits, in agreement with the observation that it is inactive in the fungal growth assays (Fig. 2E).
Table 3.
Compound potencies and similarities of the obtained HOP profilesa
| Substance | IC30 (nM) | Pearson correlation | NegLog FDR |
|---|---|---|---|
| Compound 1 | 8,500 | 1 | |
| Compound 2 | 110 | 0.689 | 11.04 |
| Compound 3 | 150 | 0.680 | 5.11 |
| Compound 4 | >200,000 | <0.15 | <0.3 |
| Voriconazole | 1,100 | 0.580 | 6.18 |
| Cyproconazole | 280 | 0.600 | 2.98 |
| Clotrimazole | 480 | 0.517 | 1.20 |
As measured by computing Pearson correlations and the (negative) logarithm (NegLog) of the FDR.
Targeted metabolomics.
Since Erg11p was identified genetically as the possible target for compounds 1, 2, and 3, we decided to determine if we could measure altered concentrations of the substrate and/or product of Erg11p. To test this hypothesis, we quantified the relative concentrations of ergosterol and lanosterol by a targeted metabolomic-profiling approach following compound exposure for 16 h. In DMSO-treated S. cerevisiae control samples, very low levels of lanosterol were detected (Fig. 3). However, upon treatment with the known Erg11p inhibitor voriconazole, a strong increase in the relative concentration of lanosterol was observed and, thus, a buildup of the Erg11p substrate. Compounds 1, 2, 3, and 4 were analyzed using this metabolomic-profiling protocol to test their effects on lanosterol concentrations. Testing of compound 2, containing a pyridyl moiety, and compound 3, containing a pyrimidyl moiety, showed similar effects on the relative lanosterol concentration, as did voriconazole (Fig. 3 and Table 4). In contrast, treating samples with compound 4, which contains a phenyl moiety but is otherwise identical to compounds 2 and 3, did not result in altered lanosterol concentrations compared to DMSO-treated control samples. The lack of an observable effect on the metabolomic profile caused by compound 4 is consistent with all the data shown above, suggesting that the compound is inactive on the tested fungal species despite high structure similarity to compounds 2 and 3. These results showing a buildup of lanosterol by the active compounds supports inhibition of Erg11p, the target proposed by the genetic-profiling methods, as the mechanism of action of these compounds.
Fig 3.

Inhibition of Erg11p in S. cerevisiae cells led to accumulation of lanosterol, the substrate of Erg11p. (A) Chromatograms of ergosterol mass transition 379.3→69.2 (blue line) and lanosterol mass transition 409.4→191.2 (red line) after treatment of yeast cells with different compounds at a concentration of 3 times the IC30. Small variations in retention times were caused by slight differences in the LC setup at the time of analysis. (B) Normalized lanosterol peak areas after treatment of yeast cells with 3 times the IC30 of the compound. The normalized lanosterol peak areas for compound 2, compound 3, and voriconazole differed statistically significantly from that of the control, with P values of <0.01 (Student's t test). The error bars indicate ± 1 standard deviation.
Table 4.
Statistical analysis of changes in lanosterol concentration compared to the control
| Compound | Concn | P value | Fold change |
|---|---|---|---|
| 1 | 3 × IC30 | 0.0007 | 47.6 |
| 2 | 3 × IC30 | 0.0020 | 38.8 |
| 3 | 3 × IC30 | 0.0053 | 33.1 |
| 4 | Maximum dose | 0.4174 | 0.8 |
| Voriconazole | 3 × IC30 | 0.0023 | 44.9 |
Human CYP inhibition.
Besides targeting Erg11p, azoles also have an inhibitory effect on the human orthologue sterol 14-demethylase. The crystal structures of cytochrome P450 enzymes, including proteins in complex with azoles, have been determined (PDB codes 3JUS, 3JUV, 3LD6, 2X2N, and 3L4D) (33). The crystal structures reveal that azoles are oriented toward the heme iron, interacting by free electron pairs of the nitrogen of the imidazole moiety.
Since the evidence so far suggests that the compounds in this study interact with Erg11p, we assessed whether they could also inhibit CYP3A4 in vitro despite the absence of a structural azole moiety. Compounds 1, 2, and 3 were indeed potent inhibitors of this enzyme, with IC50s of less than 0.5 μM. Compound 4 exhibited an IC50 of greater than 20 μM. This observation further strengthens the hypothesis that the mode of action of these compounds is similar to those of azole-containing compounds.
Macromolecular modeling supports a conserved azole binding site in humans and S. cerevisiae.
To further explore the experimental observation that compounds 1, 2, and 3 target cytochrome P450 enzymes in a manner similar to azoles, in silico docking was performed with compound 3. Compound 3 was selected because of its structural similarity to compounds 2 and 4 (differing only at the pyridine, pyrimidine, or phenyl moiety) and because it was the most potent compound overall in this study. Using the docking package GOLD, compound 3 was docked into the azole binding site (Fig. 4). The docking experiment provided five solutions with significant docking scores (Table 5). Interestingly, the docking solution with the highest GOLD score oriented one nitrogen in the pyrimidine ring toward the heme iron in a manner similar to that of azoles, allowing conjugation of the free electron pair with the heme iron. As the nitrogen of the pyrimidine ring moves away from the heme iron, disrupting this interaction, the GOLD score decreases. As a control, compound 4, which is structurally similar to compound 3 except that it lacks nitrogen in the phenyl moiety, was docked at the same site. None of the docking poses generated by GOLD oriented the phenyl ring toward the heme iron due to lack of a nitrogen atom. Thus, it appears that heme iron coordination with nitrogen in the phenyl moiety may be important for the activity of these compounds. This is in agreement with the lack of activity of compound 4 in the fungal proliferation assays.
Fig 4.

In silico docking of Erg11p inhibitors into human CYP51 (PDB code 3LD6). Ketoconazole is represented by light yellow, compound 1 by purple, compound 2 by gray, and the heme cofactor by black. (A) Solution of the in silico docking approach with compound 3. (B) Overlay of compounds 1 and 2 with compound 3 in the identified docking solution (compound 2 overlaps perfectly with compound 3, due to high structural conservation). (C) Superimposition of the in silico docking solution of compound 3 with the crystallized ketoconazole. Note that both substances point the nitrogen atom with the free electron pairs toward the heme-bound iron. (D) Binding pocket with labeled residues found to be conserved and mediating cross-resistance to azoles and compound 1 when mutated.
Table 5.
Scoring for the different poses of the compound 3 molecule represented as solutions
| Solution | GOLD score fitness |
|---|---|
| 5 | 48.40 |
| 4 | 47.27 |
| 2 | 46.90 |
| 1 | 45.69 |
We decided to model the 3-dimensional structure of S. cerevisiae Erg11p based on the available crystal structures. Human CYP51 was selected as the template to model S. cerevisiae Erg11p. Based on the sequence alignment, there is 30% identity and 39% similarity between S. cerevisiae Erg11p and human CYP51 (see Fig. S2 in the supplemental material). As shown in Fig. S2 and S3C in the supplemental material, there are a number of conserved residues between CYP51 and the homology model, mostly around the heme-binding pocket, emphasizing the role of heme and its importance for interaction with ligands.
Resistance profiling.
The data presented so far suggest that compounds 1, 2, and 3 target the S. cerevisiae Erg11p enzyme in a manner similar to that of azole-containing compounds. Point mutations in the target, and especially in the binding pocket, can affect binding of a chemical inhibitor and yield resistance. To test the hypothesis that these compounds act on Erg11p in a manner similar to that of azoles in a relevant fungal pathogen, we tested the activities of these compounds against two previously identified azole-resistant C. albicans strains. Each strain carries multiple point mutations in ERG11 (Table 1) and was used to test susceptibility to Erg11p inhibition by elucidating the MICs for voriconazole and compound 1. The two strains with azole resistance-conferring point mutations showed a 30-fold and a 16-fold increase of the MICs against compound 1 and voriconazole, respectively (Table 1). With the exception of E266, all resistance-conferring sites are conserved in S. cerevisiae. Based on our homology model, the corresponding C. albicans azole resistance substitutions (F126L and Y132H) in S. cerevisiae (F134L and Y140H) are located in the azole binding pocket (see Fig. S3C in the supplemental material). In particular, C. albicans Y132 has been shown to be involved in direct interaction with ketoconazole. The observed cross-resistance with compound 1 suggests that in C. albicans, Erg11p is the likely efficacy target of compounds 1, 2, and 3 and that the compounds bind in the same binding pocket supported by the in silico docking approach demonstrated above.
DISCUSSION
This report described the characterization of a class of compounds identified in a fungal proliferation inhibition screen. Compound 1 was shown to inhibit the proliferation of S. cerevisiae and pathogenic Candida spp. Subsequent testing of compounds with similar structures revealed a series of compounds with an interesting spectrum of activity and selectivity. The related compounds, 2 and 3, demonstrated antifungal activity consistent with that of compound 1 (Table 1). The genome-wide genetic profiling identified Erg11p as a possible target for these compounds. Resistance mutations were also identified in the ERG11 gene, and fungal strains showing resistance to azole-containing compounds also showed cross-resistance to compound 1 and related compounds. The effect of Erg11p inhibition was measured by metabolic-profiling experiments, which revealed that treatment of S. cerevisiae cells with compound 1 and related molecules had an effect similar to that of azoles, resulting in lanosterol accumulation in the treated cells. Inhibition occurred at a stage before lanosterol in the metabolic pathway, consistent with Erg11p being the target for inhibition.
The fungal Erg11p protein is a cytochrome P450 enzyme. To test the specificity of compound 1 and related molecules for the fungal enzyme, biochemical experiments were performed with human CYP3A4. Compound 1, compound 2, and compound 3 were potent inhibitors of CYP3A4, with IC50s of less than 0.5 μM. Thus, these compounds also inhibited one member of the mammalian cytochrome P450s, as has been observed for many azole class antifungals.
Molecular docking was performed to characterize how these compounds could complex with the heme catalytic center of human CYP51. Consistent with the activity of the compound series and the docking results, nitrogen in the phenyl moiety providing free electrons for conjugation with the heme iron is needed. Further support for the proposed binding mode comes from testing azole-resistant Candida strains against compound 1. The azole-resistant Candida strains showed a 30-fold increase in the MIC against compound 1, indicating that in C. albicians, mutations in Erg11p confer resistance to compound 1. Macromolecular modeling of Erg11p, based on human CYP51, demonstrated that the amino acids involved in azole interaction are conserved and are clustered around the heme center. These conserved residues may interact with this class of compounds. While the spectrum and potency of activity for this class of compounds are of interest, the observed activity against human cytochrome P450s and cross-resistance with existing azole-resistant Candida mutants suggest that substantial medicinal-chemistry efforts might be required to exploit the clinical and commercial potential of these compounds.
In summary, this report describes how modern genetic methods can be used to support the generation of a target hypothesis for compounds identified in whole-cell, phenotypic, high-throughput screens. The availability of such a target hypothesis can then be used to guide further experiments to test the mechanism of action of a class of compounds.
Supplementary Material
ACKNOWLEDGMENTS
We thank Thomas Aust and Ralph Riedl for operation of the HIP HOP robots; Lukas Baeriswyl, Mark Altorfer, Jürg Eichenberger, and Nicole Hartmann for processing of the TAG16k microarrays; and Sven Schuierer and Uwe Plikat for handling of the microarray data and processing of the HIP HOP profiles.
Footnotes
Published ahead of print 21 May 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Aoyama Y, Okikawa T, Yoshida Y. 1981. Evidence for the presence of cytochrome P-450 functional in lanosterol 14 alpha-demethylation in microsomes of aerobically grown respiring yeast. Biochim. Biophys. Acta 665:596–601 [DOI] [PubMed] [Google Scholar]
- 2. Barbosa MDFS, Yang G, Fang J, Kurilla MG, Pompliano DL. 2002. Development of a whole-cell assay for peptidoglycan biosynthesis inhibitors. Antimicrob. Agents Chemother. 46:943–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bard M, et al. 1993. Sterol synthesis and viability of erg11 (cytochrome P450 lanosterol demethylase) mutations in Saccharomyces cerevisiae and Candida albicans. Lipids 28:963–967 [DOI] [PubMed] [Google Scholar]
- 4. Benkert P, Künzli M, Schwede T. 2009. QMEAN server for protein model quality estimation. Nucleic Acids Res. 37:W510–W514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. CLSI 2008. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard, 2nd ed. CLSI document M38-2A. CLSI, Wayne, PA [Google Scholar]
- 6. CLSI 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard, 3rd ed. CLSI document M27-A3. CLSI, Wayne PA [Google Scholar]
- 7. Denning DW, Hope WW. 2010. Therapy for fungal diseases: opportunities and priorities. Trends Microbiol. 18:195–204 [DOI] [PubMed] [Google Scholar]
- 8. Fostel JM, Lartey PA. 2000. Emerging novel antifungal agents. Drug Discov. Today 5:25–32 [DOI] [PubMed] [Google Scholar]
- 9. Fraczek MG, Bromley M, Bowyer P. 2011. An improved model of the Aspergillus fumigatus CYP51A protein. Antimicrob. Agents Chemother. 55:2483–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghannoum MA, Rice LB. 1999. Antifungal agents: mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin. Microbiol. Rev. 12:501–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Giaever G, et al. 2004. Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. U. S. A. 101:793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hawksworth DL. 2001. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol. Res. 105:1422–1432 [Google Scholar]
- 13. Hillenmeyer ME, et al. 2008. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320:362–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ho CH, et al. 2009. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat. Biotechnol. 27:369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holm L, Park J. 2000. DaliLite workbench for protein structure comparison. Bioinformatics 16:566–567 [DOI] [PubMed] [Google Scholar]
- 16. Hoon S, St Onge RP, Giaever G, Nislow C. 2008. Yeast chemical genomics and drug discovery: an update. Trends Pharmacol. Sci. 29:499–504 [DOI] [PubMed] [Google Scholar]
- 17. Huang Y, Maraia RJ. 2001. Comparison of the RNA polymerase III transcription machinery in Schizosaccharomyces pombe, Saccharomyces cerevisiae and human. Nucleic Acids Res. 29:2675–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacobson MP, et al. 2004. A hierarchical approach to all-atom protein loop prediction. Proteins 55:351–367 [DOI] [PubMed] [Google Scholar]
- 19. Johnson LB, Kauffman CA. 2003. Voriconazole: a new triazole antifungal agent. Clin. Infect. Dis. 36:630–637 [DOI] [PubMed] [Google Scholar]
- 20. Kontoyiannis DP, Sagar N, Hirschi KD. 1999. Overexpression of Erg11p by the regulatable GAL1 promoter confers fluconazole resistance in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 43:2798–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lees N, Skaggs B, Kirsch D, Bard M. 1995. Cloning of the late genes in the ergosterol biosynthetic pathway of Saccharomyces cerevisiae—a review. Lipids 30:221–226 [DOI] [PubMed] [Google Scholar]
- 22. Lees NDD. 1999. Biochemistry and molecular biology of sterol synthesis in Saccharomyces cerevisiae. Crit. Rev. Biochem. Mol. Biol. 34:33–47 [PubMed] [Google Scholar]
- 23. Mallory JC, et al. 2005. Dap1p, a heme-binding protein that regulates the cytochrome P450 protein Erg11p/Cyp51p in Saccharomyces cerevisiae. Mol. Cell Biol. 25:1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Manavathu EK, Cutright JL, Chandrasekar PH. 1998. Organism-dependent fungicidal activities of azoles. Antimicrob. Agents Chemother. 42:3018–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Brien J, Wilson I, Orton T, Pognan F. 2000. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 267:5421–5426 [DOI] [PubMed] [Google Scholar]
- 26. Parks L, Smith S, Crowley J. 1995. Biochemical and physiological effects of sterol alterations in yeast—a review. Lipids 30:227–230 [DOI] [PubMed] [Google Scholar]
- 27. Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6:29–40 [DOI] [PubMed] [Google Scholar]
- 28. Pearson MM, Rogers PD, Cleary JD, Chapman SW. 2003. Voriconazole: a new triazole antifungal agent. Ann. Pharmacother. 37:420–432 [DOI] [PubMed] [Google Scholar]
- 29. Pierce SE, Davis RW, Nislow C, Giaever G. 2007. Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nat. Protoc. 2:2958–2974 [DOI] [PubMed] [Google Scholar]
- 30. Radford SA, Johnson EM, Warnock DW. 1997. In vitro studies of activity of voriconazole (UK-109,496), a new triazole antifungal agent, against emerging and less-common mold pathogens. Antimicrob. Agents Chemother. 41:841–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith C. 2003. Drug target validation: hitting the target. Nature 422:341–347 [DOI] [PubMed] [Google Scholar]
- 32. Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33:W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strushkevich N, Usanov SA, Park H-W. 2010. Structural basis of human CYP51 inhibition by antifungal azoles. J. Mol. Biol. 397:1067–1078 [DOI] [PubMed] [Google Scholar]
- 34. Thevissen K, et al. 2007. Miconazole induces changes in actin cytoskeleton prior to reactive oxygen species induction in yeast. J. Biol. Chem. 282:21592–21597 [DOI] [PubMed] [Google Scholar]
- 35. Venkateswarlu K, Kelly DE, Manning NJ, Kelly SL. 1998. NADPH cytochrome P-450 oxidoreductase and susceptibility to ketoconazole. Antimicrob. Agents Chemother. 42:1756–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. 2003. Improved protein-ligand docking using GOLD. Proteins 52:609–623 [DOI] [PubMed] [Google Scholar]
- 37. Wisplinghoff H, et al. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 39:309–317 [DOI] [PubMed] [Google Scholar]
- 38. Xu D, et al. 2007. Genome-wide fitness test and mechanism-of-action studies of inhibitory compounds in Candida albicans. PLoS Pathog. 3:e92 doi:10.1371/journal.ppat.0030092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang J-H, Chung TDY, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.