

Abstract
Glycoprotein H (gH) is an envelope protein conserved in the Herpesviridae. Together with glycoprotein B (gB), the heterodimeric complex of gH and glycoprotein L (gL) mediates penetration and direct viral cell-to-cell spread. In herpes simplex and pseudorabies virus (PrV), coexpression of gH/gL, gB, and gD induces membrane fusion to form polykaryocytes. The recently determined crystal structure of a core fragment of PrV gH revealed marked structural similarity to other gH proteins (M. Backovic et al., Proc. Natl. Acad. Sci. U. S. A. 107:22635–22640, 2010). Within the membrane-proximal part (domain IV), a conserved negatively charged surface loop (flap) is flanked by intramolecular disulfide bonds. Together with an N-linked carbohydrate moiety, this flap covers an underlying patch of hydrophobic residues. To investigate the functional relevance of these structures, nonconservative amino acid substitutions were introduced by site-directed mutagenesis. The mutated proteins were tested for correct expression, fusion activity, and functional complementation of gH-deleted PrV. Several single amino acid changes within the flap and the hydrophobic patch were tolerated, and deletion of the glycosylation site had only minor effects. However, multiple alanine substitutions within the flap or the hydrophobic patch led to significant defects. gH function was also severely affected by disruption of the disulfide bond at the C terminus of the flap and after introduction of cysteine pairs designed to bridge the central part of the flap with the hydrophobic patch. Interestingly, all mutated gH proteins were able to complement gH-deleted PrV, but fusion-deficient gH mutants resulted in a pronounced delay in virus entry.
INTRODUCTION
Herpesviruses enter their host cells by fusion of the viral envelope with the plasma membrane, although endocytosis of enveloped virus particles has also been described. Three viral envelope glycoproteins, which are conserved in all subfamilies of the Herpesviridae, are required for membrane fusion either at the cell surface or within endosomes (reviewed in references 10 and 58). In herpes simplex virus types 1 and 2 (HSV-1, HSV-2) these glycoproteins have been named gB, gH, and gL (53), and designations have been adopted for most other herpesviruses. Additional, less conserved envelope proteins are required for the initial steps of virus attachment and receptor binding (10, 58).
Like most of their cognates, HSV-1 and the porcine alphaherpesvirus pseudorabies virus (PrV) initially adsorb to heparan sulfate proteoglycans at the cell surface. Although this step enhances the efficiency of infection, it is not required, and the responsible envelope glycoprotein gC is dispensable for productive virus replication (24, 31). Stable binding of PrV and other alphaherpesviruses to specific cellular receptor proteins like nectin 1 or 2 is mediated by the essential glycoprotein D (17). For HSV-1, it has been further shown that a defined domain of gD triggers membrane fusion, presumably via interaction with gB or gH (9). Nevertheless, replication-competent PrV and bovine herpesvirus 1 could be isolated after serial passage of gD-deleted viruses, which had acquired compensatory mutations within gB and gH (55, 56).
In HSV-1, the best conserved herpesvirus glycoprotein, gB, also interacts with heparan sulfate and different cellular surface proteins and, therefore, might contribute to attachment at least to certain cell types (10). However, the major function of gB is membrane fusion after virus attachment. Determination of crystal structures of the HSV-1 and Epstein-Barr virus (EBV) gB proteins revealed that they form homotrimers and exhibit marked structural similarities to fusion proteins found in other virus families, like the G protein of vesicular stomatitis virus and gp64 of baculovirus (4, 23, 29). Structurally conserved “fusion loops” are crucial for the fusogenic activity of gB (3, 22). They might interact with target cell membranes, so that subsequent changes in gB allow juxtaposition of viral and cellular membranes to form a fusion pore (10). However, unlike the single fusion proteins of many RNA viruses, herpesvirus gB homologs require other viral proteins for function.
The conserved heterodimeric gH/gL complex is a second core component of the herpesvirus fusion machinery, although its precise role is still unclear. Like gB, gH is essential for host-cell entry of all hitherto tested members of the Herpesviridae (2, 12). The type I integral membrane protein gH is required for virion incorporation of gL, which lacks a membrane anchor. In most herpesviruses, gL is required for correct processing and virion incorporation of gH (25, 54). In contrast, in PrV, murid herpesvirus 4 (MuHV-4) gH and bovine herpesvirus 4 (BoHV-4) gH are incorporated into virions in the absence of gL (21, 33, 40). However, whereas MuHV-4 and BoHV-4 gL are apparently not essential for fusion, gL-deleted PrV is severely impaired. Infectivity by free virions is blocked, but very limited direct cell-to-cell spread occurs (33). This limited ability to perform direct cell-to-cell spread was used for reversion analysis by serial cell culture passaging of gL-deleted PrV, which resulted in a replication-competent rescuant. In this virus, a spontaneous recombination event led to a hybrid gene encoding a chimeric protein in which the ectodomain of gD is fused to an N-terminally truncated gH. The resulting gDH executed all essential in vitro functions of gD and the gH/gL complex (34), indicating that the expressed parts of gD and gH are fully functional.
Like gB and gD, gH/gL homologs interact with putative cellular virus receptor proteins, in particular integrins, and at least for EBV these interactions appear to be sufficient for attachment to epithelial but not to B cells (6, 47). There is also evidence that gH, together with gB, functions during nuclear egress of nascent HSV-1 virions (13). However, this is not the case in PrV (37). Thus, the conserved and main function of gH appears to be an essential role in fusion during entry.
To elucidate the precise functions of gH, binding sites of gH-specific neutralizing antibodies have been identified, and several mutational studies have been performed (reviewed in reference 10). These studies were facilitated by in vitro fusion assays resulting in formation of polykaryocytes (syncytia) in cells cotransfected with expression plasmids for, e.g., alphaherpesvirus gB, gD, gH, and gL (35, 60). Random linker insertions within either part of the protein, including transmembrane domain and cytoplasmic tail of HSV-1 gH, revealed effects on fusogenic activity and function in virus replication, but many mutations of this type as well as deletions of a conserved N-glycosylation site or of an RGD integrin-binding motif were tolerated (15, 26, 63). Despite the demonstrated relevance of the cytoplasmic tail for gH function, recent studies showed that soluble HSV-1 gH/gL lacking this part, together with soluble gD, induced fusion of cells expressing only gB as a transmembrane protein, although at a very low level. This indicated that gH might not be directly involved in the membrane fusion process but is required for activation of gB after receptor binding of gD (1). In contrast, a similar effect of soluble EBV gH/gL ectodomains on cell fusion was not observed, suggesting that at least the EBV protein has to be membrane anchored (32).
Recently, the crystal structures of soluble forms of the gH/gL complexes of HSV-2 (8) and EBV (42), as well as of a core fragment of the PrV gH ectodomain in complex with a monoclonal antibody (5), were elucidated. They revealed no homologies of gH to any known fusion protein. Moreover, they demonstrated that previously described lipid-interacting and fusogenic peptide sequences within gH (16, 19) are not exposed at the protein surface. Despite limited sequence homologies, marked structural similarities between the different gH homologs exist. In HSV-2 and EBV, gL is closely associated with the N-terminal part of gH (domain I). This domain is missing in the analyzed gH core fragment of PrV and is also deleted in the above-mentioned PrV mutant which is capable of gL-independent replication by expressing the gDH chimeric protein. The second conserved domain (II) contains an antiparallel β-sheet followed by a bundle of α-helices and includes the integrin binding sites identified in EBV and HSV-2 gH, as well as a proposed interaction site with gB in HSV-2 (5, 8, 42). Domain III also consists in large part of α-helices, whereas the membrane proximal domain IV, which is the best conserved domain, is formed by two opposed four-stranded β-sheets connected by an elongated polypeptide chain designated “flap” (Fig. 1A). Furthermore, domain IV contains a functional N-glycosylation site (Fig. 1B) which is positionally conserved in all hitherto deduced gH proteins. Most gH proteins, excepting, among others, those of HSV-1 and HSV-2, contain a CXXC motif resembling the active site of protein disulfide isomerases (PDI) (7) at the C-terminal side of the flap, which permits formation of an intramolecular disulfide bond (SS4; Fig. 1). A conserved cysteine at the N-terminal side of the flap is involved in another conserved disulfide bond (SS3) as demonstrated in the PrV, EBV, and HSV-2 gH structures (5, 8, 42). Remarkably, all three gH homologs also contain a hydrophobic patch formed by amino acids from different parts of the domain IV sequence, which is covered by the carbohydrate moiety at the conserved glycosylation site and by the flap (Fig. 1B). It had been hypothesized that movement of the flap might unmask the hydrophobic patch as a mechanism involved in fusion promotion by gH (5).
Fig 1.

Structure-based mutagenesis of PrV gH domain IV. (A) Structure of the PrV gH core fragment (5) is shown as space-filled model (left). The amino (N) and carboxy (C) termini of the protein, the three resolved domains (II, III, IV), and the putative position of the membrane (arrow) are indicated. The flap region is highlighted in blue, and the two disulfides clamping its ends (SS3 and SS4) are shown in yellow. A view of domain IV obtained by 90-degree rotation toward the viewer (middle and right) shows that the flap region (displayed as space-filled and wire models, respectively) covers an extensive surface area of the domain. (B) Residues forming the hydrophobic patches of domain IV (shown as space-filled model) are highlighted in cream, the glycosylation site at N627 in light blue, and disulfide bonds SS3 and SS4 in yellow. The residues mutated in this study are underlined. For clarity, the flap region is shown as wire model, with the residues mutated in this study colored in purple. C573 is not visible in the space-filled model due to its buried location. The expected location of L634 (*) is indicated, although it was not resolved in the PrV gH crystal structure. (C) Novel cysteine residues (yellow) were introduced by amino acid substitutions in the flap (C555 and C556, shown as wire model) and in the underlying hydrophobic patch (C630 and C631, shown as surfaces) in order to lock the flap region by additional disulfides (SS5a and SS5b).
Due to these interesting features, we started with structure-based mutational analyses of PrV gH, focusing on the highly conserved domain IV. By site-directed mutagenesis of an expression plasmid, single or multiple codons of the gH-encoding open reading frame (ORF) UL22 of PrV (36) were modified. Besides mutations preventing formation of either of the two conserved disulfide bonds flanking the flap, single and multiple amino acids of the flap and of the underlying hydrophobic patch were substituted by alanine residues. Furthermore, new cysteine pairs were introduced which had been predicted to form additional disulfide bonds between the central parts of the flap and the hydrophobic patch. The conserved N-glycosylation site at amino acid position 627 was also modified by an amino acid exchange. The mutated gH genes were tested for fusogenic activity after coexpression with PrV gB, gD, and gL in transfected eukaryotic cells. Furthermore, all mutated genes were reinserted into the PrV genome, and the in vitro replication properties, including penetration kinetics of the obtained virus recombinants, were investigated.
MATERIALS AND METHODS
Viruses and cells.
Virus mutants were derived from PrV strain Kaplan (PrV-Ka) (30) and propagated in rabbit kidney (RK13) cells or an RK13-derived cell line which constitutively expresses glycoproteins gH and gL of PrV (RK13-gH/gL) (37). Cells were grown at 37°C in minimum essential medium (MEM; Invitrogen) supplemented with 10% fetal calf serum (FCS; Invitrogen). MEM containing only 5% FCS was used after PrV infection or transfection of the cells with plasmids and/or viral DNA. For plaque assays, the virus inoculum was removed 2 h after infection, and cells were overlaid with semisolid medium containing 6 g/liter methylcellulose.
Mutagenesis of the cloned PrV gH gene.
The previously described plasmid pcDNA-gH (35) contains ORF UL22 of PrV-Ka under the control of the human cytomegalovirus (HCMV) immediate-early gene promoter in the eukaryotic expression vector pcDNA3 (Invitrogen). The insert fragment consisting of nucleotides (nt) 60588 to 62712 of the PrV genome sequence (36) (GenBank accession BK001744; Fig. 2A) was cloned via flanking engineered EcoRI and XbaI restriction sites (Fig. 2C). For introduction of point mutations into pcDNA-gH, pairs of complementary oligonucleotide primers (forward strand primers shown in Table 1) exhibiting single base substitutions (lowercase letters) were purchased (Eurofins MWG Operon). Besides mutations resulting in the desired amino acid substitutions, silent mutations leading to creation or deletion of restriction sites were introduced to facilitate identification of recombinant plasmids and viruses. The QuikChange II site-directed mutagenesis kit (Agilent Technologies) was used according to the manufacturer's instructions. The complete DNA sequences of the insert fragments of all mutated plasmids were determined with the BigDye Terminator version 1.1 cycle sequencing kit and a 3130 Genetic Analyzer (Applied Biosystems) using vector-specific T7 and SP6 promoter primers (New England Biolabs) and the gH-specific primer PGH-WH3 (Table 1).
Fig 2.

Plasmids and virus mutants. Open reading frames (pointed rectangles), transcripts (dotted arrows), and relevant restriction sites are indicated. (A) The ca. 140-kbp genome of PrV-Ka consists of unique (UL, US) and inverted repeat sequences (IR, TR). The UL22 (gH) and US4 (gG) genes were targets of mutations. (B) An EGFP expression cassette containing the HCMV immediate-early promoter (PHCMV) and a polyadenylation signal (A+), as well as a mini-F plasmid vector (pMBO131), were inserted at the 5′ end of US4 to permit BAC cloning of the PrV genome. Thereby, expression of the major 3′ part of US4 (crossed) was prevented. (C) For generation of gH-deleted PrV, codons 233 to 637 of UL22 were replaced by a kanamycin resistance gene (KanR). Virus rescuants were generated by homologous recombination with the plasmid-cloned wild-type or mutated gH gene in RK13 cells. Locations of direct repeat sequences (vertical lines) and an origin of replication (OriL) are indicated. (D) Amino acid sequence of the C-terminal part of PrV gH. Residues forming the flap (bold), hydrophobic patches, and the transmembrame domain (bold italics), as well as an N-glycosylation site (underlined), are highlighted. Native and novel disulfide bonds (SS) and amino acid substitutions introduced by site-directed mutagenesis are also indicated.
Table 1.
Primers for mutagenesis and sequencing of PrV gHa
| Name | DNA sequence (5′ → 3′) | Nucleotide position |
|---|---|---|
| PGH-C547S-F | CAACGACAGCGCCGCtaGCGACTACACGGATC | 62232–62263 |
| PGH-T550A-F | GCGTGCGACTACgCcGATCGCATGCCC | 62245–62271 |
| PGH-D551A-F | GTGCGACTACACcGcTCGCATGCCCGAG | 62247–62274 |
| PGH-P554A-F | CACGGATCGCATGgCCGAGTCCCAGCAC | 62256–62283 |
| PGH-E555A-F | GATCGCATGCCCGcGTCCCAGCACCTG | 62260–62286 |
| PGH-550/4A-F | GTGCGACTACgCGGcTCGCATGgCCGcGTCCCAGCAC | 62247–62283 |
| PGH-E555C-F | CGGATCGCATGCCCtgcTCCCAGCACCTGCC | 62258–62288 |
| PGH-S556C-F | CGCATGCCCGAGTgtCAaCACCTGCCGGCG | 62263–62292 |
| PGH-C571S-F | CGTGTGCGTGTACaGCGACTGCGTGTTC | 62307–62334 |
| PGH-C573S-F | CGTGTACTGCGACaGCGTcTTCGTGCGCTAC | 62313–62343 |
| PGH-N627Q-F | CTGATGCTCTTCCCCcAaGGCACCGTGGTC | 62473–62502 |
| PGH-V630C-F | CCCCAACGGCACCtgcGTCGACCTGCTGTCG | 62484–62514 |
| PGH-V631C-F | CCAACGGCACCGTGtgCGACCTGCTGTCG | 62486–62514 |
| PGH-630/4A-F | CCAACGGCACCGcGGcCGACgcGgcGTCGTTCACGTC | 62486–62522 |
| PGH-PSF | TTCACGTCGGAGATGGGG | 60451–60468 |
| PGH-WH3 | TGCACGAGAGCGACGACTACC | 61319–61339 |
| PGH-PSF2 | GGAAGCCCTTCGACCAG | 61715–61731 |
| PGH-PSR | AGTTATGTCATCCAGCAGCC | 62887–62868 (r) |
| PGH-PSR2 | GTCGAGCAGGCTGAAGG | 61911–61895 (r) |
Cloning of the PrV-Ka genome as a bacterial artificial chromosome (BAC) and generation of a gH-deleted mutant.
To generate a fluorochrome-labeled, wild-type-like infectious full-length clone of PrV-Ka, a 196-bp BamHI fragment (nt 119417 to 119608 of BK001744) including the promoter and 5′-terminal part of the coding region of the nonessential viral glycoprotein gG was deleted from plasmid pU6.3 (28) and replaced by a 8,028-bp BamHI fragment of pBl-GFP/MBO/B. The latter construct was identical to pBl-GFP/MBO/BP (39), except that it possesses a HindIII site instead of an engineered PmeI site. Thus, the resulting plasmid, pU-ΔgGGMBO (Fig. 2B), contained an expression cassette for enhanced green fluorescent protein (EGFP), followed by the mini-F plasmid vector pMBO131 (46). After calcium-phosphate-mediated cotransfection of RK13 cells with pU-ΔgGGMBO and virion DNA of PrV-Ka, an EGFP-expressing PrV recombinant was isolated from virus progeny. Circular DNA of this mutant was prepared from infected cells and used for electroshock transformation of Escherichia coli strain DH10B (38) to obtain the infectious BAC pPrV-ΔgGG.
For partial deletion of the gH gene, a 1,214-bp BmgBI fragment (nt 61306 to 62519 of BK001744) of pcDNA-gH was replaced by a 1,258-bp BstBI fragment of pKD13 (11) containing a kanamycin resistance gene. The insert fragment of the resulting pcDNA-ΔgHK (Fig. 2C) was amplified by PCR using Pfx DNA polymerase (Invitrogen) and vector-specific T7 and SP6 promoter primers and used for mutagenesis of pPrV-ΔgGG in bacteria as described previously (38). The resulting BAC pPrV-ΔgGGΔgHK as well as the parental pPrV-ΔgGG were characterized by DNA analyses and replication studies in cell culture.
Complementation of gH-deleted PrV by mutated gH.
RK13 cells were grown to subconfluency in 12-well plates and transfected (FuGene HD reagent; Roche) with 1 μg/well of mutated or authentic pcDNA-gH expression plasmids or with the empty expression vector pcDNA3. After 24 h, the cells were infected with 103 PFU per well of phenotypically complemented pPrV-ΔgGGΔgHK, which had been prepared from the supernatant of infected R13-gH/gL cells. At day 5 postinfection, EGFP autofluorescence of the infected cells was analyzed by microscopy.
Furthermore, the transfected and subsequently infected cells were harvested and tested for infectious virus progeny by plaque assays on RK13 cells. After repeated plaque purification, genomic DNA of the obtained revertants was characterized by Southern blot hybridization with a UL22-specific probe (results not shown), as well as by PCR amplification and subsequent sequencing of two overlapping fragments of the gH gene using primer pairs PGH-PSF/PGH-PSR2 or PGH-PSF2/PGH-PSR (Table 1).
In vitro replication studies.
For comparative analysis of virus spread, RK13 or RK13-gH/gL cells grown in 12-well plates were infected with serial dilutions of pPrV-ΔgGG, pPrV-ΔgGGΔgHK, or gH rescuants and incubated under semisolid medium for 48 h at 37°C. Then the cells were washed with phosphate-buffered saline (PBS), fixed for 30 min with 3% paraformaldehyde in PBS, and washed again. Areas of 30 plaques per virus were measured by fluorescence microscopy (Eclipse Ti-S with software NIS elements, version 4; Nikon), and average sizes as well as standard deviations were calculated as percentages of the mean plaque area induced by pPrV-ΔgGG. Student's t test was used for estimation of the statistical significance of differences.
For investigation of replication kinetics, RK13 or RK13-gH/gL cells grown in 96-well plates were infected at a multiplicity of infection (MOI) of 5 at 4°C to permit virus adsorption and shifted to 37°C after 1 h for penetration. After 2 h at 37°C, nonpenetrated virus was inactivated by treatment with citric acid (43), and incubation at 37°C was continued. At different times after the temperature shift, cells and supernatants were harvested jointly and lysed by freeze-thawing. Progeny virus titers were determined by plaque assays, and mean titers of two experiments per virus were calculated.
For determination of penetration kinetics, RK13 cells grown in 6-well plates were infected at 4°C with 500 PFU per well of PrV recombinants expressing wild-type or mutated gH. After 1 h, prewarmed medium was added. After 0, 5, 10, 20, or 40 min at 37°C, nonpenetrated virus was inactivated by citric acid treatment. Thereafter, the cells were overlaid with semisolid medium, and 2 days later plaques were counted and compared to the numbers in wells which had been not acid treated. Mean penetration rates of two independent experiments were plotted.
Western blot analyses.
Protein expression of pPrV-ΔgGG, pPrV-ΔgGGΔgHK, and gH revertants was analyzed 20 h after infection of RK13 cells at an MOI of 5. Cell lysates were separated by discontinuous sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and incubated with antibodies as described previously (48). A gH-specific rabbit antiserum (34) or a rabbit serum raised against a bacterially expressed protein containing the capsid triplex protein pUL38 of PrV C-terminally fused to glutathione S-transferase was used at dilutions of 1:50,000. Antibody binding was detected by chemiluminescence (VersaDoc 4000MP; Bio-Rad).
In vitro fusion assays.
To analyze the cofusogenic activity of PrV gH, RK13 cells grown in 12-well plates were cotransfected (FuGene HD reagent; Roche) with 200 ng of each of the expression plasmids for EGFP (pEGFP-N1; Clontech), a C-terminally truncated gB (pgB-008) (45), gD (gDgI-CMV) (18), gL (pRc/CMV-gL) (35), and native or mutated gH (pcDNA-gH or derivatives) or the empty expression vector pcDNA3. After 3 days at 37°C, the cells were washed with PBS, fixed with 3% paraformaldehyde, and washed and overlaid with fresh PBS. The cells were then analyzed by fluorescence microscopy, and the numbers of syncytia containing more than 2 nuclei in 20 fields of view (ca. 0.5 mm2 each) and the numbers of nuclei in 20 syncytia were counted in two independent experiments for each gH construct tested. Mean values and standard deviations were calculated, and Student's t test was used to estimate statistical significance of differences.
RESULTS
Isolation of a BAC-based gH deletion mutant of PrV.
Several infectious BAC clones of the PrV genome have been described (14, 38, 57). For the present study, a new BAC construct of PrV-Ka designated pPrV-ΔgGG was generated, which constitutively expresses a reporter protein (EGFP) and is fully replication competent in the presence of the heterologous sequences. To this end, a mini-F plasmid vector and an EGFP expression cassette were inserted into the US4 gene of PrV encoding gG (Fig. 2B). gG is completely dispensable for PrV replication in vitro and in vivo (59). The reporter gene cassette contains the bidirectional polyadenylation signals of simian virus 40 (51), which can substitute for the original mRNA polyadenylation signal of the US3 transcript located downstream of US4 (61), and of the large 8-kbp insertion (Fig. 2B). Unlike a previously described gG-negative BAC clone of PrV (14), the new mutant expressed wild-type-like levels of pUS3 and produced only 2-fold-lower virus titers.
To generate a gH-negative mutant of pPrV-ΔgGG, codons 233 to 637 of UL22 (687 codons) were deleted and replaced by a kanamycin resistance gene (KanR) using lambda Red recombinase-mediated BAC mutagenesis in E. coli (Fig. 2C). This mutation altered virus genome size only negligibly (44-bp increase) and should not affect transcription of adjacent genes such as UL23. As expected, the obtained mutant pPrV-ΔgGGΔgHK did not replicate productively in noncomplementing RK13 cells, and gH expression was undetectable by Western blot analyses of infected cell lysates (Fig. 3A). In contrast, pPrV-ΔgGGΔgHK exhibited wild-type-like plaque sizes and virus titers in RK13-gH/gL cells. This gH and gL-coexpressing cell line provided better complementation of gH-deleted PrV mutants than the available cell lines expressing only gH (results not shown).
Fig 3.
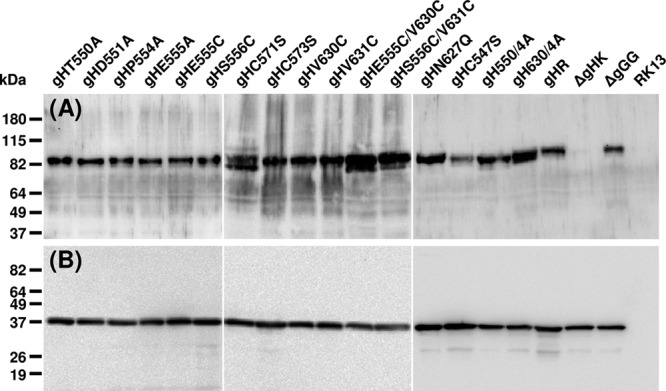
Western blot analyses. RK13 cells were harvested 20 h after infection with pPrV-ΔgGG, pPrV-ΔgGGΔgH K, or virus rescuants expressing the indicated gH mutants at an MOI of 5. Proteins were separated by SDS-PAGE and blots were analyzed with monospecific antisera against gH (A) or capsid protein pUL38 (B). Molecular mass markers are indicated on the left.
Mutations within domain IV of PrV gH have only minor effects on protein expression.
To analyze the functional relevance of the membrane proximal domain IV, which is highly conserved in structure and sequence between herpesvirus gH homologs (5), single or multiple amino acids within the flap region or within the underlying hydrophobic patch were substituted by alanine residues (Fig. 1B, 2D). Furthermore, cysteine residues C547 and C571, which are involved in disulfide bonds 3 and 4 flanking the flap region (Fig. 1A), as well as the unpaired cysteine C573 were substituted by serine residues (Fig. 2D). To allow formation of additional disulfide bonds between the flap and the underlying domain IV region, cysteine pairs were created at the sterically neighboring positions 555 and 630 or 556 and 631 (Fig. 1C, 2D). The residues to be mutated were selected based on their proximity, which should allow formation of disulfide bridges. Corresponding single amino acid substitutions were generated as controls. Finally, the asparagine of the highly conserved N-glycosylation site (N627) was substituted by glutamine (Fig. 2D). All amino acid substitutions (listed in Table 2) were introduced into pcDNA-gH (35) by site-directed mutagenesis. The resulting plasmids were used for in vitro fusion studies and for the generation of PrV rescuants, which were obtained by homologous recombination after transfection of noncomplementing cells, and subsequent infection with phenotypically complemented pPrV-ΔgGGΔgHK (Fig. 2).
Table 2.
Properties of mutated gH proteins and virus mutantsa
| Mutant | Substituted aa (affected element) | In vitro fusion (+gB, gD, gL) | Cell-to-cell spread | Penetration |
|---|---|---|---|---|
| gH (WT) | None | ++++ | ++++ | ++++ |
| gHT550A | T550 → A550 (flap) | ++++ | ++++ | NT |
| gHD551A | D551 → A551 (flap) | ++++ | ++++ | NT |
| gHP554A | P554 → A554 (flap) | ++++ | +++ | NT |
| gHE555A | E 555→ A555 (flap) | ++++ | ++ | ++++ |
| gH550/4A | T550 → A550 (flap) | + | ++ | ++++ |
| D551 → A551 | ||||
| P554 → A554 | ||||
| E555 → A555 | ||||
| gHC547S | C547 → S547 (ΔSS3) | ++ | + | +++ |
| gHC571S | C571 → S571 (ΔSS4) | − | + | + |
| gHC573S | C573 → S573 (flap) | ++++ | +++ | ++++ |
| gHE555S | E555 → S555 (flap) | ++++ | +++ | NT |
| gHV630C | V630 → C630 (HP) | ++++ | ++++ | NT |
| gHE555C/V630C | E555 → C555 (+ SS5a) V630 → C630 | + | + | + |
| gHS556C | S556 → C556 (flap) | ++++ | +++ | NT |
| gHV631C | V631 → C631 (HP) | ++++ | ++++ | NT |
| gHS556C/V631C | S556 → C556 (+ SS5b) V631 → C631 | + | + | ++ |
| gHN627Q | N627 → Q627 (Δ NAG) | +++ | +++ | ++++ |
| gH630/4A | V630 → A630 (HP) | + | ++ | ++ |
| V631 → A631 | ||||
| L633 → A633 | ||||
| L634 → A634 | ||||
| No gH | − | − | NT |
Efficiency of the mutated gH proteins in fusion assays performed in plasmid-transfected RK13 cells, as well as plaque formation and entry kinetics in RK13 cells infected with corresponding PrV mutants, were roughly quantified (++++ to −); gH mutations entailing severe defects with respect to in vitro fusion and/or virus replication are highlighted. Abbreviations: SS, disulfide bridge; HP, hydrophobic patch; NAG, N-glycosylation; NT, not tested; WT, wild type.
Indirect immunofluorescence analyses of cells transfected with the different gH expression plasmids or infected with the corresponding PrV recombinants revealed that none of the introduced mutations had apparent effects on the expression or the cytoplasmic distribution of gH (results not shown). Western blot analyses of infected cell lysates confirmed wild-type-like expression levels of all mutated gH proteins (Fig. 3A) compared to those of capsid protein pUL38 (Fig. 3B). All gH proteins exhibited similar apparent molecular masses of >80 kDa, and only in cells infected with mutant pPrV-ΔgGGgHC571S or pPrV-ΔgGGgHE555C/V630C, slightly smaller protein bands were more abundant, which could indicate less efficient protein processing. Interestingly, deletion of an N-glycosylation site in pPrV-ΔgGGgHN627Q had no discernible effect on molecular mass (Fig. 3A).
Effects of gH mutations on virus penetration, productive replication, and direct cell-to-cell spread.
Whereas infection with phenotypically complemented pPrV-ΔgGGΔgHK remained restricted to single RK13 cells (Fig. 4A), preceding transfection of these cells with any of the gH expression plasmids investigated in this study supported at least limited spread of gH-deleted PrV (results not shown). Furthermore, homologous recombination with either of the gH expression plasmids led to replication-competent virus rescuants, demonstrating that none of the introduced mutations completely blocked protein function. PCR amplification and sequencing of the entire gH ORFs of all virus rescuants confirmed that no reversions of the desired mutations, and no mutations elsewhere in the gene, had occurred. One-step growth analyses on noncomplementing RK13 cells after infection at an MOI of 5 revealed no significant effects on maximum titers of most of the obtained virus revertants expressing mutated gH compared to parental or wild-type gH-rescued PrV (Fig. 5A). An approximate 10-fold titer reduction was observed only for pPrV-ΔgGGgHC571S lacking the second cysteine of the CXXC motif involved in formation of disulfide bond 4 (Fig. 2D).
Fig 4.
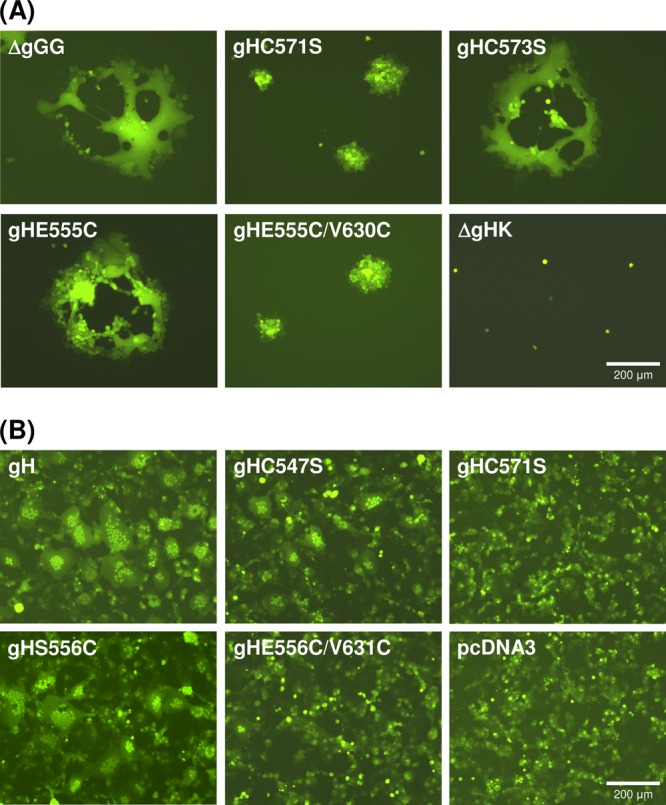
EGFP autofluorescence illustrating gH function. (A) Cell-to-cell spread was analyzed 1 day after infection of RK13 cells with pPrV-ΔgGG, gH deletion mutant pPrV-ΔgGGΔgHK, or virus recombinants possessing the indicated gH mutations (gHC571S, gHC573S, gHE555C, gHE555C/V630C). (B) Induction of syncytium formation was analyzed 3 days after cotransfection of RK13 cells with an EGFP reporter plasmid and expression plasmids for PrV gB, gD, and gL, as well as for wild-type (pcDNA-gH) or mutated gH (gHC547S, gHC571S, gHS556C, gHS556C/V631C) or the empty expression vector (pcDNA3).
Fig 5.

In vitro replication of PrV gH mutants. (A) For analysis of one-step growth kinetics, RK13 cells were infected at an MOI of 5 and harvested together with the supernatant after 3 h, 12 h, 24 h, or 36 h. Mean progeny virus titers of two experiments are shown. (B) For determination of plaque sizes, infected RK13 cells were incubated for 2 days under semisolid medium. Mean areas of 30 plaques per virus were calculated as percentages of wild-type (pPrV-ΔgGG) sizes. Standard deviations (vertical lines) and statistically significant differences from wild-type size are indicated (*, P < 0.05; **, P < 0.01).
In contrast, the effects of the introduced mutations in gH on plaque formation were much more pronounced (Fig. 4A, 5B). Direct viral cell-to-cell spread was impaired in rescuants expressing gH lacking cysteines required for disulfide bond 3 (C547S) or 4 (C571S), whereas removal of the unpaired cysteine 573 had no adverse effect (Fig. 4). Introduction of additional cysteine pairs (E555C/V630C, S556C/V631C) capable of forming new disulfide bonds between the flap and the underlying hydrophobic region was also deleterious, while none of the corresponding single mutations showed similar effects (Fig. 4A, 5B). Considerable reductions in plaque size were also observed after introduction of multiple alanines into the hydrophobic patch (630/4A) or the flap (550/4A), as well as by the single alanine at position 555, whereas the other single alanine substitutions were phenotypically inconspicuous. Deletion of the highly conserved N-glycosylation site (N627Q) led to a moderate but statistically significant reduction of plaque sizes (Fig. 5B). The defects in plaque formation of all affected PrV mutants were corrected on RK13-gH/gL cells (results not shown). Thus, possible accidental mutations elsewhere in the genomes of the PrV mutants were obviously not relevant for the observed defects.
To investigate whether the impaired cell-to-cell spread of several of the PrV gH mutants correlated with defects during virus entry, penetration kinetics in RK13 cells were compared to those of parental and wild-type gH-rescued viruses. After synchronized infection, penetration was permitted for various times prior to acid inactivation of extracellular virus particles. Subsequent incubation under plaque assay conditions revealed that ≥50% of pPrV-ΔgGG or pPrV-ΔgGGgHR virions were internalized within 5 min, and almost complete penetration was achieved after 20 min (Fig. 6). In contrast, only ≤2% of infectious virus particles of pPrV-ΔgGGgHC571S and pPrV-ΔgGGgHE555C/V630C were able to penetrate the cells within 5 min, and after 40 min, less than 30% of the plaques obtained without acid treatment were achieved. pPrV-ΔgGGgHS556C/V631C and pPrV-ΔgGGgH630/4A showed less pronounced but also significant penetration defects, whereas entry of all other virus mutants exhibiting reduced plaque sizes was only marginally affected or not affected at all (Fig. 6).
Fig 6.

Penetration kinetics of PrV gH mutants. After synchronized virus adsorption at 4°C, RK13 cells were incubated for 0, 5, 10, 20, or 40 min at 37°C prior to acid inactivation of nonpenetrated virus. Numbers of plaques after 2 days were compared to those obtained without inactivation. Mean percentages of two independent experiments are shown.
Taken together, our studies strongly indicate that integrities of the hydrophobic patch and of the authentic disulfide bonds at both ends of the flap (Fig. 1A and B) are crucial for proper function of gH during virus entry and direct cell-to-cell spread. The introduction of additional cysteine pairs designed to form disulfide bonds bridging the middle of the flap to the underlying domain IV region (Fig. 1C) also strongly impairs gH function. Although precise sequence motifs within the flap or the hydrophobic patch are obviously not required, alterations of charge by multiple amino acid substitutions in these regions can severely affect protein function. As previously described for HSV-1 (15), glycosylation at the highly conserved asparagine 627 is not required for function of PrV gH but leads only to a reduction in plaque size.
In vitro fusion activity of mutated gH.
To analyze the effects of the point mutations on gH function more directly than in the viral context, in vitro fusion assays were performed. To this end, RK13 cells were cotransfected with expression plasmids for PrV glycoproteins gB, gD, gH, and gL. Although PrV gB, gH, and gL have been previously shown to be sufficient for induction of membrane fusion leading to formation of syncytia (35), gD was included to enhance the efficiency of this process. For the same purpose, native gB was replaced by a truncated protein (gB-008), which lacks the C-terminal 29 amino acids of the cytoplasmic domain, including an internalization signal, and therefore accumulates at the plasma membrane, thus promoting fusion (45). An EGFP expression plasmid was included to control transfection efficiency and to facilitate evaluation of the assays by fluorescence microscopy 3 days after transfection (Fig. 4B, 7). As expected, large syncytia containing numerous nuclei formed after cotransfection with the native gH gene, whereas no fusion was observed in the absence of gH, although the percentages of transfected, EGFP-expressing cells were similar in all experiments (Fig. 4B and data not shown). All mutated gH proteins containing single amino acid substitutions by alanine or cysteine within the flap or the hydrophobic region were still able to mediate cell fusion leading to similar numbers of syncytia and numbers of nuclei per syncytium as wild-type gH (Fig. 7A and B). However, introduction of multiple alanines in either of the two regions (550/4A, 630/4A) severely decreased the size and number of the induced syncytia (Fig. 7). An even more pronounced reduction of the activity of gH occurred after introduction of cysteine pairs designed to form additional disulfide bonds (E555C/V630C, S556C/V631C) (Fig. 4B, 7) between the flap and the hydrophobic patch. An apparent complete loss of fusogenic activity was observed only upon removal of cysteine 571 (Fig. 4B, 7), which is part of the CXCC motif and is required for formation of disulfide bond 4 (Fig. 1, 2D). In contrast, mutation of the highly conserved cysteine 547 required for disulfide bond 3, or of the conserved N-glycosylation site (N627Q), had only moderate effects on syncytium formation (Fig. 4B, 7).
Fig 7.

In vitro fusion activity of mutated gH. RK13 cells were cotransfected with expression plasmids for EGFP, gB, gD, gL, and different gH mutants or the empty expression vector (pcDNA3). After 3 days, the number of polykaryocytes in 20 fields of view (A) and the number of nuclei in 20 syncytia per plasmid set (B) were determined. Mean values and standard deviations are indicated. Statistically significant differences from the numbers obtained with wild-type gH are labeled with asterisks (*, P < 0.05; **, P < 0.01).
Most of these results are in agreement with the in vitro replication studies, with PrV recombinants carrying the corresponding mutations in gH. One exception is the clear effect of the single alanine mutation E555A on cell-to-cell spread (Fig. 3B), which is not reflected by an impairment of fusogenic activity (Fig. 7), indicating that in the viral context the function(s) of gH might be more complex and more dependent on proper protein interactions than in the in vitro fusion assays.
DISCUSSION
Recently, the crystal structures of HSV-2, EBV, and PrV gH have been determined (5, 8, 42). In the present study, the functional relevance of conserved structural features within the C-terminal, membrane-proximal part of the ectodomain of PrV gH (domain IV) (Fig. 1, 2D) was investigated by introduction of 16 different single and multiple nonconservative amino acid substitutions (Fig. 1, Table 2). The fusogenic activity of the mutated gH proteins in the presence of PrV gB, gD, and gL was analyzed in cells cotransfected with expression plasmids, and replication properties of PrV recombinants expressing the mutated gH genes were also investigated. The major findings are the following: (i) substitution of single amino acids within the negatively charged flap by alanine had no or only little effect on protein function, whereas multiple alanine substitutions led to reduced fusion activity; (ii) multiple alanines within the hydrophobic patch, which is masked underneath the flap as revealed by the PrV gH structure, affected fusion as well as penetration and cell-to-cell spread of a corresponding virus mutant; (iii) mutation of the highly conserved N-glycosylation site (N627Q) affected protein function only moderately; (iv) mutation of single cysteines involved in disulfide bonds 3 and 4 flanking the flap (Fig. 2) reduced (C547S) or abrogated (C571S) in vitro fusion activity and impaired virus spread; (v) mutation of cysteine 571 as well as introduction of additional cysteine pairs capable of formation of disulfide bonds between the center of the flap and the hydrophobic patch severely affected virus penetration, cell-to-cell spread, and in vitro fusion activity, whereas corresponding single cysteine insertions had no effects.
Surprisingly, despite the observed defects, none of the gH mutants was completely nonfunctional. Although PrV gH, like its homologs, is essential for productive virus replication (2), PrV recombinants expressing any of the mutated proteins could be isolated and propagated in noncomplementing cells. This finding is in general agreement with previous mutational analyses of HSV-1 or varicella-zoster virus (VZV) gH (15, 26, 62), which also indicated that gH tolerates more sequence alterations than, e.g., gB (41, 52).
After high MOI infection, the maximum virus titers of most of the PrV-expressing mutated gH were very similar to those of parental pPrV-ΔgGG or wild-type gH-rescued virus, and only the recombinant lacking disulfide bond 4 (C571S) exhibited an approximate 10-fold reduction in titer. The absence of significant effects on the amount of infectious progeny might be due to the fact that, after successful penetration, PrV gH is dispensable for subsequent virus replication or virion formation (37). However, our results show that sufficient amounts of all mutated gH proteins were expressed and incorporated to generate infectious virus particles. Western blot analyses of infected cells confirmed that all mutated proteins were present at levels like wild-type gH. Only in PrV mutants carrying gH mutations C571S and E555C/V630C, increased amounts of smaller-than-mature-size gH were detected (Fig. 3A), which may be due to slightly impaired protein processing.
Whereas the introduced gH mutations had little effect on formation of infectious virions, cell-to-cell spread of several of the PrV mutants was significantly impaired. However, while gH-deleted PrV is unable to form plaques in noncomplementing cell cultures (2), all tested gH mutants supported at least limited virus spread in cells transfected with gH expression plasmids or infected with corresponding virus rescuants. The most pronounced defect leading to a plaque size reduction of >80% was again observed after mutating cysteine 571 to serine (C571S), resulting in loss of authentic disulfide bond 4 located C terminal of the flap. Absence of the authentic disulfide bond 3 (C547S) also resulted in a 75% reduction. Mutation of cysteine 571 caused a severe penetration defect of the corresponding virus mutant and abrogated in vitro fusion activity of gH, whereas mutation of cysteine 547 resulted in only moderate effects. Thus, although disulfide bond 4 is not conserved in HSV (8), it seems to be more relevant for stabilization of gH structure in PrV and VZV than the other conserved disulfide bond 3 (62). Since disulfide bond 4 is formed at a CXXC motif, which can function as the active site of protein disulfide isomerases (7), a general role of this sequence in protein processing appears possible. However, there is no experimental evidence for this enzymatic activity in gH, and the absence of the CXXC motif from HSV-1 and HSV-2 gH, as well as the complementation of mutations of this motif by an unrelated amino acid exchange elsewhere in domain IV as described for VZV (62), argues against PDI activity of gH. Interestingly, a nonconserved unpaired cysteine is located at position 573 of PrV gH, and it has been speculated that formation of an alternative disulfide bond (C568-C573 instead of C568-C571) might be involved in the proposed movement of the flap preceding fusion (5). However, mutation of cysteine 573 was phenotypically inconspicuous, arguing against any functional relevance.
Reduction of the hydrophilicity of the flap, or of the hydrophobicity of the patch, by multiple alanine substitutions also significantly affected fusogenic activity of gH and cell-to-cell spread of the respective PrV mutants, whereas most single amino acid exchanges were fully tolerated. This indicated that not precise sequence motifs but the overall hydrophilicity of these regions is relevant for function. The alanine substitutions in the hydrophobic patch, but not in the flap, also led to significantly delayed penetration kinetics. Such differences between the phenotypes of several of the investigated gH mutants in plaque and penetration assays might reflect different protein requirements for direct cell-to-cell spread than for infection by free virus particles. This is highlighted, e.g., by the fact that PrV gD is required for infection by virions but dispensable for direct viral spread at synapses and other cell junctions (44, 50). Thus, since gD has been shown to interact with gH during the fusion process in HSV-1 (20, 49), it is conceivable that different structural features of gH are required during different ways of virus entry.
Interestingly, the exchange of glutamic acid at position 555 by alanine, but not by cysteine, led to significantly reduced plaque sizes of the corresponding PrV rescuant without exhibiting any adverse effect on syncytium formation in in vitro fusion assays. The observed defect was unlikely to be caused by a mutation elsewhere in the PrV genome since, like the defects of all other PrV mutants analyzed in this study, it was complemented on gH-expressing cells. However, a mutation in gL cannot be excluded, since wild-type gL was coexpressed in RK13-gH/gL cells (37). In contrast, unwanted mutations at other than the desired sites in gH were excluded by amplification and sequencing of the gH genes of all generated virus mutants. Thus, the structural requirements for gH function in fusion of two adjacent plasma membranes might also be different from those for fusion of the plasma membrane with the envelopes of attached virions during cell-to-cell spread, which occurs at tight junctions (27). Such mechanistic differences might also explain that removal of cysteine 571-abrogated fusogenic activity of gH but did not block either direct spread or infectivity of released virus particles.
Like the C571S mutation, the introduction of two pairs of cysteines at adjacent positions in the gH structure predicted to form bridges between central parts of the flap and the hydrophobic patch (E555C/V630C, S556C/V631C) caused severe defects in all types of assays performed, whereas neither of the corresponding single cysteine substitutions significantly affected fusogenic activity of gH or in vitro replication properties of the virus mutants. This finding strongly indicated that the additional disulfide bonds were formed (Fig. 1C) (5) and impeded structural changes within domain IV during the fusion process.
As previously demonstrated for HSV-1 (15), removal of the highly conserved, functional N-glycosylation site at position 627 of PrV gH had no effect on replication kinetics and maximum virus titers. Nevertheless, the mutation resulted in a moderate reduction of plaque size and of syncytia in fusion assays, indicating that glycosylation at this site is beneficial, although masking of the hydrophobic part of domain IV by the flap is obviously sufficient to keep gH largely functional.
Based on the conserved structures of HSV-2, EBV, and PrV gH, targeted point mutations have been also introduced into the gH of VZV (62). In these studies, most mutations in the α-helical central part of gH (domains II and III) abolished protein function, whereas mutations introduced into the N-terminal, gL-binding part affected skin pathogenesis in SCID mice only. Disruption of the disulfide bonds flanking the flap in domain IV (in VZV gH designated domain III) by Cys-to-Ala mutations also severely affected fusogenic activity in vitro. Mutation of the first cysteine in the CXXC motif was lethal for VZV replication, but the other cysteine mutants were still replication competent, although they exhibited reduced virulence. These results parallel our findings demonstrating that disulfide bonds 3 and 4 are important for gH function. Interestingly, the defects associated with mutation of either cysteine residue in the CXXC motif of disulfide bond 4 of VZV gH were rescued by a compensatory mutation, S694F, located at an edge of the hydrophobic patch underneath the flap, basically expanding the hydrophobic patch (62), which we did not observe in our PrV studies. Based on the effects of mutations of the flanking disulfide bonds and the compensatory S694F mutation, which was proposed to rigidify the domain by providing a bulkier hydrophobic side chain, the VZV gH data had been interpreted as suggesting that domain IV rigidity is a major requirement for gH function. Our finding that disulfides SS3 and SS4 of PrV gH domain IV are functionally important could be explained by their involvement in the maintenance of the domain IV fold, consistent with the observation in VZV. Our observation that newly introduced cysteine pairs predicted to form bridges between the flap and the hydrophobic patch impair gH function supports the interpretation that displacement of the flap is involved in gH function. The extended polypeptide chain forming the flap, located at the surface of domain IV, could thus move independently of the rest of domain IV, thereby not compromising its rigidity. However, this remains to be tested experimentally.
In summary, our results define a functionally important region in domain IV of PrV gH and demonstrate that structural conservation of herpesvirus gH homologs correlates with functional conservation of individual structural elements within domain IV. In particular, the results confirm the relevance of the hydrophobic region in the membrane proximal part of the gH ectodomain for fusion, which had been indicated by earlier studies on HSV-1 (15, 16). They also demonstrate the importance of hydrophilic residues in the flap region overlaying the hydrophobic patch. Both regions contain residues which are important for in vitro fusion-promoting activity of gH and, although nonessential, also for gH function during viral replication.
ACKNOWLEDGMENTS
This study was supported by DFG grant Me 854/11-1.
We thank S. Böttcher and G. Strebelow for their help with BAC cloning of the PrV genome and DNA sequencing, respectively. The technical assistance of C. Ehrlich and E. Eckroth is greatly appreciated.
Footnotes
Published ahead of print 23 May 2012
REFERENCES
- 1. Atanasiu DW, Saw T, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Babic N, et al. 1996. Glycoprotein gH of pseudorabies virus is essential for penetration and propagation in cell culture and in the nervous system of mice. J. Gen. Virol. 77:2277–2285 [DOI] [PubMed] [Google Scholar]
- 3. Backovic M, Jardetzky TS, Longnecker R. 2007. Hydrophobic residues that form putative fusion loops of Epstein-Barr virus glycoprotein B are critical for fusion activity. J. Virol. 81:9596–9600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Backovic M, et al. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc. Natl. Acad. Sci. U. S. A. 107:22635–22640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. U. S. A. 106:20464–20469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chivers PT, Laboissière MC, Raines RT. 1996. The CXXC motif: imperatives for the formation of native disulfide bonds in the cell. EMBO J. 15:2659–2667 [PMC free article] [PubMed] [Google Scholar]
- 8. Chowdary TK, et al. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cocchi F, et al. 2004. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl. Acad. Sci. U. S. A. 101:7445–7450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Desai PJ, Schaffer PA, Minson AC. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1: evidence that gH is essential for virion infectivity. J. Gen. Virol. 69:1147–1156 [DOI] [PubMed] [Google Scholar]
- 13. Farnsworth A, et al. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. U. S. A. 104:10187–10192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76:364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Galdiero M, et al. 1997. Site-directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J. Virol. 71:2163–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Galdiero S, et al. 2007. Evidence for a role of the membrane-proximal region of herpes simplex virus type 1 glycoprotein H in membrane fusion and virus inhibition. Chembiochem 8:885–895 [DOI] [PubMed] [Google Scholar]
- 17. Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620 [DOI] [PubMed] [Google Scholar]
- 18. Gerdts V, Jöns Mettenleiter ATC. 1999. Potency of an experimental DNA vaccine against Aujeszky's disease in pigs. Vet. Microbiol. 66:1–13 [DOI] [PubMed] [Google Scholar]
- 19. Gianni T, Fato R, Bergamini C, Lenaz G, Campadelli-Fiume G. 2006. Hydrophobic a-helices 1 and 2 of herpes simplex virus gH interact with lipids, and their mimetic peptides enhance virus infection and fusion. J. Virol. 80:8190–8198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J. Biol. Chem. 284:17370–17382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gillet L, May JS, Colaco S, Stevenson PG. 2007. Glycoprotein L disruption reveals two functional forms of the murine gammaherpesvirus 68 glycoprotein H. J. Virol. 81:280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hannah BP, et al. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heldwein EE, et al. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 24. Herold BC, WuDunn D, Soltys N, Spear PG. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 65:1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hutchinson L, et al. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 66:2240–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jackson JO, Lin E, Spear PG, Longnecker R. 2010. Insertion mutations in herpes simplex virus 1 glycoprotein H reduce cell surface expression, slow the rate of cell fusion, or abrogate functions in cell fusion and viral entry. J. Virol. 84:2038–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson DC, Webb M, Wisner TW, Brunetti C. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 75:821–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jöns A, Mettenleiter TC. 1997. Green fluorescent protein expressed by recombinant pseudorabies virus as an in vivo marker for viral replication. J. Virol. Methods 66:283–292 [DOI] [PubMed] [Google Scholar]
- 29. Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 15:1024–1030 [DOI] [PubMed] [Google Scholar]
- 30. Kaplan AS, Vatter AE. 1959. A comparison of herpes simplex and pseudorabies viruses. Virology 7:394–407 [DOI] [PubMed] [Google Scholar]
- 31. Karger A, Saalmüller A, Tufaro F, Banfield BW, Mettenleiter TC. 1995. Cell surface proteoglycans are not essential for infection by pseudorabies virus. J. Virol. 69:3482–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kirschner AN, Omerovic J, Popov B, Longnecker R, Jardetzky TS. 2006. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol. 80:9444–9454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klupp BG, Fuchs W, Weiland E, Mettenleiter TC. 1997. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J. Virol. 71:7687–7695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klupp BG, Mettenleiter TC. 1999. Glycoprotein gL-independent infectivity of pseudorabies virus is mediated by a gD-gH fusion protein. J. Virol. 73:3014–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klupp BG, Nixdorf R, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J. Virol. 74:6760–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klupp BG, Hengartner CJ, Mettenleiter TC, Enquist LW. 2004. Complete, annotated sequence of the pseudorabies virus genome. J. Virol. 78:424–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Klupp BG, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J. Virol. 82:6299–6309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kopp M, et al. 2003. The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J. Virol. 77:5339–5351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leege T, et al. 2009. Effects of simultaneous deletion of pUL11 and glycoprotein M on virion maturation of herpes simplex virus type 1. J. Virol. 83:896–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lété C, Machiels B, Stevenson PG, Vanderplasschen A, Gillet L. 2012. Bovine herpesvirus type 4 glycoprotein L is nonessential for infectivity but triggers virion endocytosis during entry. J. Virol. 86:2653–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin E, Spear PG. 2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 104:13140–13145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. U. S. A. 107:22641–22646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mettenleiter TC. 1989. Glycoprotein gIII deletion mutants of pseudorabies virus are impaired in virus entry. Virology 171:623–625 [DOI] [PubMed] [Google Scholar]
- 44. Mulder W, et al. 1996. Glycoprotein D-negative pseudorabies virus can spread transneuronally via direct neuron-to-neuron transmission in its natural host, the pig, but not after additional inactivation of gE or gI. J. Virol. 70:2191–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nixdorf R, Klupp BG, Karger A, Mettenleiter TC. 2000. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J. Virol. 74:7137–7145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. O'Connor M, Peifer M, Bender W. 1989. Construction of large DNA segments in Escherichia coli. Science 244:1307–1312 [DOI] [PubMed] [Google Scholar]
- 47. Parry C, Bell S, Minson T, Browne H. 2005. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J. Gen. Virol. 86:7–10 [DOI] [PubMed] [Google Scholar]
- 48. Pavlova SP, Veits J, Keil GM, Mettenleiter TC, Fuchs W. 2009. Protection of chickens against H5N1 highly pathogenic avian influenza virus infection by live vaccination with infectious laryngotracheitis virus recombinants expressing H5 hemagglutinin and N1 neuraminidase. Vaccine 27:773–785 [DOI] [PubMed] [Google Scholar]
- 49. Perez-Romero P, Perez A, Capul A, Montgomery R, Fuller AO. 2005. Herpes simplex virus entry mediator associates in infected cells in a complex with viral proteins gD and at least gH. J. Virol. 79:4540–4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rauh I, Mettenleiter TC. 1991. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J. Virol. 65:5348–5356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reddy VB, et al. 1978. The genome of simian virus 40. Science 200:494–502 [DOI] [PubMed] [Google Scholar]
- 52. Reimer JJ, Backovic M, Deshpande CG, Jardetzky T, Longnecker R. 2009. Analysis of Epstein-Barr virus glycoprotein B functional domains via linker insertion mutagenesis. J. Virol. 83:734–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roizman B, Knipe DM, Whitney RJ. 2007. Herpes simplex viruses, p 2501–2601 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 54. Roop C, Hutchinson L, Johnson DC. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol. 67:2285–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schmidt J, Gerdts V, Beyer J, Klupp BG, Mettenleiter TC. 2001. Glycoprotein D-independent infectivity of pseudorabies virus results in an alteration of in vivo host range and correlates with mutations in glycoproteins B and H. J. Virol. 75:10054–10064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schröder C, Keil GM. 1999. Bovine herpesvirus 1 requires glycoprotein H for infectivity and direct spreading and glycoproteins gH(W450) and gB for glycoprotein D-independent cell-to-cell spread. J. Gen. Virol. 80:57–61 [DOI] [PubMed] [Google Scholar]
- 57. Smith GA, Enquist LW. 2000. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 97:4873–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Spear PG, Longnecker R. 2003. Herpesvirus entry: an update. J. Virol. 77:10179–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thomsen DR, Marchioli CC, Yancey RJ, Jr, Post LE. 1987. Replication and virulence of pseudorabies virus mutants lacking glycoprotein gX. J. Virol. 61:229–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van Zijl M, van der Gulden H, de Wind N, Gielkens A, Berns A. 1990. Identification of two genes in the unique short region of pseudorabies virus; comparison with herpes simplex virus and varicella-zoster virus. J. Gen. Virol. 71:1747–1755 [DOI] [PubMed] [Google Scholar]
- 62. Vleck SE, et al. 2011. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc. Natl. Acad. Sci. U. S. A. 108:18412–18417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilson DW, Davis-Poynter N, Minson AC. 1994. Mutations in the cytoplasmic tail of herpes simplex virus glycoprotein H suppress cell fusion by a syncytial strain. J. Virol. 68:6985–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]