

Abstract
Interference with stress granule (SG) accumulation is gaining increased appreciation as a common strategy used by diverse viruses to facilitate their replication and to cope with translational arrest. Here, we examined the impact of infection by herpes simplex virus 2 (HSV-2) on SG accumulation by monitoring the localization of the SG components T cell internal antigen 1 (TIA-1), Ras-GTPase-activating SH3-domain-binding protein (G3BP), and poly(A)-binding protein (PABP). Our results indicate that SGs do not accumulate in HSV-2-infected cells and that HSV-2 can interfere with arsenite-induced SG accumulation early after infection. Surprisingly, SG accumulation was inhibited despite increased phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), implying that HSV-2 encodes previously unrecognized activities designed to maintain translation initiation downstream of eIF2α. SG accumulation was not inhibited in HSV-2-infected cells treated with pateamine A, an inducer that works independently of eIF2α phosphorylation. The SGs that accumulated following pateamine A treatment of infected cells contained G3BP and PABP but were largely devoid of TIA-1. We also identified novel nuclear structures containing TIA-1 that form late in infection. These structures contain the RNA binding protein 68-kDa Src-associated in mitosis (Sam68) and were noticeably absent in infected cells treated with inhibitors of viral DNA replication, suggesting that they arise as a result of late events in the virus replicative cycle.
INTRODUCTION
Cellular responses to stresses, including heat shock, starvation, oxidation, and viral infection, can culminate in the arrest of cap-dependent translation. Translational arrest is problematic for viruses as all viruses depend on the host cell protein synthetic machinery for production of their proteins. Consequently, viruses have evolved many sophisticated strategies to ensure that viral mRNA translation can continue, often at the expense of cellular mRNA translation (59, 61, 63). Oftentimes, these strategies are aimed at blunting innate antiviral responses initiated by stress-activated kinases. Activation of stress-activated kinases by viral infection, principally activation of the double-stranded RNA-dependent protein kinase, PKR, results in increased phosphorylation of the alpha subunit of eukaryotic initiation factor 2α (eIF2α), leading to failed translational initiation and shutdown of protein synthesis (64). There are many examples of viral gene products that act as antagonists of PKR (37), as well as at least one example of a viral gene product that acts as an antagonist of the related stress-activated kinase, the PKR-like endoplasmic reticulum kinase, PERK (53). Viruses can also directly modulate the phosphorylation status of eIF2α (27, 66).
Stress-induced translational arrest results in the accumulation of messenger ribonucleoproteins (mRNPs) consisting of mRNAs that remain associated with stalled translational initiation complexes and the 40S ribosomal subunit. Cellular RNA-binding proteins such as T cell internal antigen 1 (TIA-1) and Ras-GTPase-activating SH3-domain-binding protein (G3BP) bind to mRNPs and to one another and subsequently other cellular proteins are recruited into the complex (3). The result is the formation of cytoplasmic assemblages known as stress granules (SGs). SGs act as centers for sorting and triaging mRNAs and also provide a cache of ready-to-use mRNPs, allowing cells to rapidly resume translation once stress is alleviated (3, 5). Several viruses, including dengue virus, hepatitis C virus, human immunodeficiency virus type 1, poliovirus, and West Nile virus, interfere with SG formation by usurping or degrading specific SG components (1, 2, 4, 20, 39, 65). Other viruses including human T cell leukemia virus type 1, mammalian orthoreovirus, and Semliki Forest virus, have the ability to manipulate SG formation to benefit their replicative cycle and to regulate the shutdown of host protein synthesis (38, 50, 57). In addition, the growth of herpes simplex virus 1 (HSV-1), Sindbis virus, and vesicular stomatitis virus is considerably enhanced in TIA-1 knockout cells compared to parental cells (39). These observations implicate interference with SG formation as another strategy used by diverse viruses to cope with translational arrest (52).
Infection of cells by HSV can activate at least two stress-activated kinases, and yet accumulation of SGs is not observed after infection by HSV-1 strains F and KOS (17, 21). This may be reflective of the combined abilities of the HSV-1 gene products Us11, ICP34.5, and gB to counteract the activities of PKR and PERK (11, 12, 14–16, 53, 54). In addition, the HSV virion host shut off (vhs) protein is capable of degrading most mRNAs (60), resulting in reduced production of cellular proteins that contribute to stress responses. Since mRNA is a key starting ingredient for SG assembly, lower levels of mRNA seen in the presence of vhs may be a contributing factor to the observed lack of SGs in HSV-1-infected cells (5). Indeed, SGs do accumulate in cells infected with vhs-deficient HSV-1 mutants (17, 21).
The importance of TIA-1 and possibly SG formation in suppressing HSV-1 infection is suggested by the enhanced growth of HSV-1 in TIA-1 knockout cells (39). Since the role of TIA-1 and/or SG formation in suppressing HSV infection has not been directly addressed with HSV-2, we examined the impact of HSV-2 infection on SG accumulation. We found that HSV-2 infection does not lead to the accumulation of SGs and analyzed SG accumulation in HSV-2-infected cells following treatment with mechanistically distinct inducers of SG formation. We probe the mechanism of inhibition of SG accumulation by HSV-2 and describe unique nuclear structures containing TIA-1 that form late in infection. Furthermore, we demonstrate that TIA-1 and G3BP, both widely used markers of SGs, report differently on SG accumulation in HSV-2-infected cells.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney cells (Vero), cervical carcinoma cells (HeLa), and T12 cells, life-extended human diploid foreskin fibroblasts, were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS) in a 5% CO2 environment. The T12 cells were generously provided by W. A. Bresnahan, University of Minnesota (8). The HG52 strain of HSV-2 was propagated and titered on Vero cells. Infection of HeLa cells was carried out using a multiplicity of infection (MOI) of 2 to 10. The times postinfection, reported as hours postinfection (hpi), refer to the time elapsed following medium replacement after a 1-h inoculation period. For phosphonoacetic acid (PAA)-treated infections, 200 μg of PAA (Sigma, St. Louis, MO)/ml was applied 1 h prior to inoculation, and the cells were maintained in the continuous presence of 200 μg of PAA/ml for the duration of the experiment. For acyclovir-treated infections, 5 μM acyclovir (Santa Cruz Biotechnology, Santa Cruz, CA) was applied 1 h prior to inoculation, and the cells were maintained in the continuous presence of 5 μM acyclovir for the duration of the experiment. To produce virus-free supernatants for use in control experiments, virus stocks were subjected to centrifugation at 100,000 × g for 20 min in a MLA 130 rotor in an Optima MAX-XP benchtop ultracentrifuge (Beckman Coulter, Mississauga, Ontario, Canada) to pellet the virus, and the resulting supernatants were used as the inoculum.
Plasmids and transfections.
Green fluorescent protein (GFP)-tagged TIA-1 was provided by V. Kruys, Université Libre de Bruxelles (58). GFP-tagged versions of Sam68 were provided by C. Sette, University of Rome Tor Vergata (10). Transfection of HeLa cells for the purpose of subsequent infection, and microscopic analyses of live or fixed specimens was carried out using FuGene HD (Roche, Laval, Quebec, Canada) according to the manufacturer's instructions.
Induction of SGs.
To induce SG formation, HeLa cells were treated with 0.5 mM arsenite (Sigma) for 30 min or 300 nM pateamine A (generously provided by A. Mouland, Lady Davis Institute for Medical Research) for 30 min. In addition, the overexpression of GFP-tagged TIA-1 and GFP-tagged Sam68-GSG for 24 h or longer consistently resulted in SG formation.
Immunological reagents.
Goat polyclonal antiserum against human TIA-1 (Santa Cruz Biotechnology) was used for indirect immunofluorescence microscopy at a dilution of 1:1,000 and for Western blotting at a dilution of 1:500. Mouse monoclonal antibody against human G3BP (BD Biosciences, Mississauga, Ontario, Canada) was used for indirect immunofluorescence microscopy at a dilution of 1:1,000. Goat polyclonal antiserum against human PABP (Santa Cruz Biotechnology) was used for indirect immunofluorescence microscopy at a dilution of 1:100. Mouse monoclonal antibody against HSV-2 ICP8 (Virusys, Taneytown, MD) was used for indirect immunofluorescence microscopy at a dilution of 1:1,000 and for Western blotting at a dilution of 1:4,000. Mouse monoclonal antibody against HSV ICP27 (Virusys) was used for indirect immunofluorescence microscopy at a dilution of 1:1,000 and for Western blotting at a dilution of 1:500. Mouse monoclonal antibody against HSV ICP5 (Virusys) was used for Western blotting at a dilution of 1:3,000. Rabbit monoclonal antibody against human phospho-eIF2α (Epitomics, Burlingame, CA) was used for Western blotting at a dilution of 1:1,000. Rabbit polyclonal antiserum against human eIF2α (Santa Cruz Biotechnology) was used for Western blotting at a dilution of 1:500. Mouse monoclonal antibody against actin (Sigma) was used for Western blotting at a dilution of 1:2,000. Alexa Fluor 488 (Alexa 488)-conjugated donkey anti-goat and Alexa Fluor 568 (Alexa 568)-conjugated donkey anti-mouse (Molecular Probes, Eugene, OR) were used for indirect immunofluorescence at a dilution of 1:500. Finally, horseradish peroxidase (HRP)-conjugated goat anti-mouse and HRP-conjugated goat anti-rabbit (Sigma) were used for Western blotting at a dilution of 1:10,000.
Indirect immunofluorescence microscopy.
Cells for microscopic analyses were grown either on glass coverslips or on glass-bottom dishes (MatTek, Ashland, MA). Cells to be stained for the presence of PABP were fixed with cold 100% methanol at 4°C for 20 min; otherwise, the cells were fixed and stained as described previously (24). Images were captured using either a Nikon Eclipse TE200 inverted fluorescence microscope and Metamorph 7.1.2.0 software or an Olympus FV1000 laser scanning confocal microscope and Fluoview 1.7.3.0 software. Images from the inverted fluorescence microscope were captured using a 60× (1.40 NA) oil immersion objective lens. Images from the confocal microscope were captured using a 60× (1.42 NA) oil immersion objective lens. The relative fluorescence intensities of TIA-1 and G3BP or PABP and G3BP in individual SGs from six independent fields per specimen were quantified by using Fluoview 1.7.3.0 software. Composites of representative images were prepared using Adobe Photoshop software.
FRAP analyses.
HeLa cells seeded onto glass bottom dishes were transfected with plasmid DNA encoding GFP-tagged TIA-1 or Sam68. At 24 h after transfection, cells were infected with HSV-2 or mock infected. At the indicated times postinfection, the medium was replaced with warm DMEM (lacking phenol red)–10% FCS, and the cells were mounted onto an Olympus FV1000 confocal microscope and maintained at 37°C in a humidified 5% CO2 environment. All image acquisition parameters were controlled using Olympus Fluoview software version 1.7.3.0. GFP was excited using a 488-nm laser line set at 5% power. For fluorescence recovery after photobleaching (FRAP) experiments, 512-pixel-by-512-pixel images were obtained using a 60× (1.42 NA) oil immersion objective lens. Images were collected at a rate of 1.1 frames per s. Five frames were collected before the indicated region was photobleached by repeated scanning of the region of interest with a 405 nm laser set at 100% power for a total of 300 ms. After photobleaching, an additional 45 frames were collected. The fluorescence intensity in bleached and unbleached control regions in each frame were measured using Fluoview 1.7.3.0, and the data were exported into Microsoft Excel for graphical presentation.
Fluorescence loss in photobleaching (FLIP) analyses.
HeLa cells seeded onto glass bottom dishes were transfected with plasmids encoding GFP-tagged TIA-1. At 24 h after transfection, the cells were infected with HSV-2. At various times postinfection, cells were placed in warm DMEM (lacking phenol red)–10% FCS and transferred to an Olympus FV1000 laser scanning confocal microscope and maintained at 37°C in a humidified 5% CO2 environment. All image acquisition parameters were controlled using Fluoview 1.7.3.0 software. Images were collected every 10 s using a 60× (1.42 NA) oil immersion objective lens and a 488-nm laser at 2 to 5% power to excite the specimen. During the interval between capturing each image, a defined area of the cytoplasm of a transfected HeLa cell was repeatedly photobleached through eight consecutive rounds of scanning with a 405-nm laser operating at 100% laser power at scan speed of 8 μs/pixel. The fluorescence intensities in the nucleus and in the cytoplasm of the bleached cells, as well as in the nucleus and the cytoplasm of neighboring transfected cells, were measured over time using Fluoview 1.7.3.0 software, and the data were exported into Microsoft Excel for graphical presentation.
Preparation and analysis of whole-cell extracts.
To prepare whole-cell extracts of HeLa cells for Western blot analyses, the cells were washed once with cold phosphate-buffered saline (PBS) and then scraped into cold PBS containing protease inhibitors (Roche) plus 5 mM sodium fluoride (New England Biolabs, Pickering, Ontario, Canada) and 1 mM sodium orthovanadate (New England Biolabs, Pickering, Onrario, Canada) to inhibit phosphatases. Harvested cells were transferred to a 1.5-ml microfuge tube containing 3× SDS-PAGE loading buffer. The lysate was repeatedly passed through a 28.5-gauge needle to reduce the viscosity and then heated at 100°C for 5 min. For Western blot analysis, 10 to 20 μl of whole-cell extract was electrophoresed through SDS-PAGE gels. Separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA) and probed with appropriate dilutions of primary antibody, followed by appropriate dilutions of HRP-conjugated secondary antibody. The membranes were treated with Pierce ECL Western blotting substrate (Thermo Scientific, Rockford, IL) and exposed to film.
Analysis of protein synthesis.
HeLa cells growing in six-well cluster dishes were infected with HSV-2 strain HG52 at an MOI of 5 or mock infected. At 3.5 hpi, the cells were rinsed three times with warm PBS before methionine-free DMEM containing 5% dialyzed FCS was added to the cells to deplete cellular stores of methionine. At 4 hpi, the medium was removed and methionine-free DMEM–5% dialyzed FCS containing 50 μM l-homopropargylglycine (Invitrogen, Burlington, Ontario, Canada), an alkyne-containing methionine analog, with or without 0.5 mM arsenite, was added to cells. At 5 hpi, the medium was removed, and the cells were rinsed three times with cold PBS. The cells were lysed in 200 μl of lysis buffer (1% SDS in 50 mM Tris-HCl [pH 8.0]) containing 250 U of benzonase (Santa Cruz Biotechnology) and protease inhibitors (Thermo Scientific) for 30 min on ice. Newly synthesized proteins containing l-homopropargylglycine were covalently linked to biotin azide (PEG4 carboxamide-6-azidohexanyl biotin) via a copper-catalyzed click reaction using the Click-iT protein reaction buffer kit (Invitrogen) according to the manufacturer's instructions. Proteins were separated on 10% SDS-PAGE gels and transferred to PVDF membranes (Millipore). Biotinylated proteins were detected using HRP-conjugated streptavidin (Invitrogen). The membranes were treated with Pierce ECL Western blotting substrate (Thermo Scientific) and exposed to film.
RESULTS
HSV-2 infection does not cause accumulation of SGs.
To investigate whether SGs accumulate during HSV-2 infection, HeLa cells were infected at an MOI of 10 and then stained with antisera specific for TIA-1, a marker of SGs (35), at various times postinfection. TIA-1 was diffusely localized throughout the cytoplasm in infected cells at all time points examined and SGs were not readily detected (Fig. 1A). This cytoplasmic staining pattern was indistinguishable from that observed in mock-infected cells. HeLa cells used in our experiments were competent to make SGs as demonstrated by the relocalization of TIA-1 to discrete cytoplasmic puncta when cells were treated for 30 min with 0.5 mM arsenite (see Fig. S1 in the supplemental material; see also Fig. 4 and Fig. 7A). Curiously, at 8 and 20 hpi, distinct nuclear structures that contained TIA-1 were detected in many infected cells (indicated by arrows in Fig. 1A). At 10 hpi, >86% of infected cells contained these nuclear TIA-1 structures. FRAP analyses were performed to assess the kinetics of TIA-1 recruitment into these structures. HeLa cells were transfected with plasmids encoding GFP-tagged TIA-1 and infected with HSV-2, and then the nuclear TIA-1 structures formed at 10 hpi were subjected to FRAP. These analyses demonstrated that TIA-1 was rapidly recruited into these structures, allowing fluorescence intensity to recover within 14 s after photobleaching (Fig. 2A; see Movie S1 in the supplemental material), a rate comparable to the rate of recruitment of TIA-1 into SGs in uninfected cells (Fig. 2B; see Movie S2 in the supplemental material) (34). Importantly, these data also indicated that these nuclear TIA-1 structures are dynamic and do not represent insoluble aggregates of protein. Nuclear TIA-1 structures appeared to be located at the nuclear periphery often in close proximity to Hoechst-stained chromatin (Fig. 1B). These structures did not contain G3BP, another reliable marker of SGs (62) (Fig. 1B) and were not formed when virus-free supernatants were used as inocula (data not shown). In keeping with our observations using TIA-1 as a marker of SGs, no evidence of SG accumulation at 10 hpi was observed using G3BP as a marker (Fig. 1B). In summary, while HSV-2 infection of HeLa cells does not evoke the accumulation of SGs, it does cause a component of SGs, TIA-1, to accumulate in dynamic structures located at the inner periphery of the nucleus. Lack of SG accumulation and the appearance of similar nuclear TIA-1 structures were also observed in HSV-2-infected T12 cells (see Fig. S2 in the supplemental material).
Fig 1.
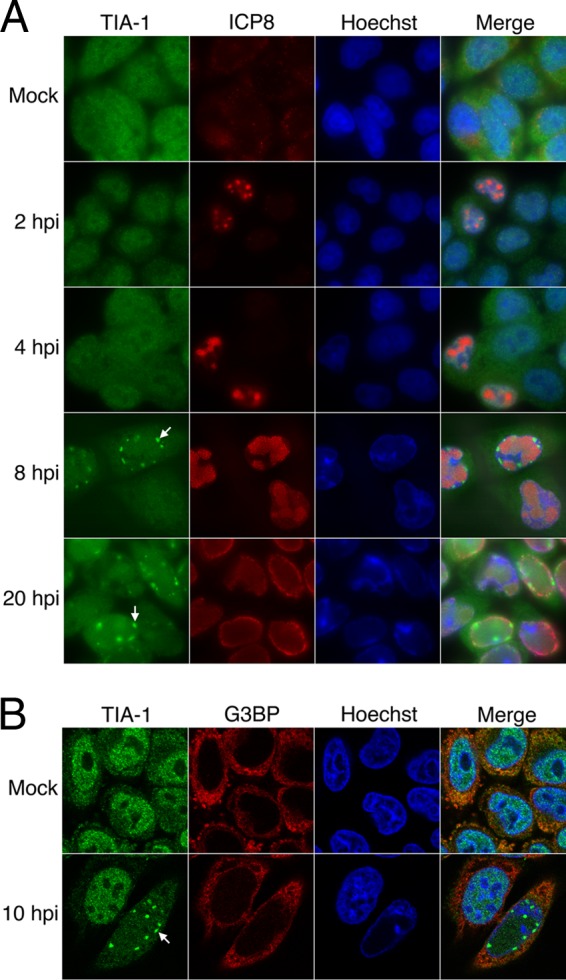
TIA-1 localization and SG accumulation in HSV-2-infected cells. (A) HeLa cells were mock infected or infected with HSV-2 strain HG52 at an MOI of 10. Infected cells were fixed at the indicated times postinfection and stained with goat polyclonal antiserum specific for TIA-1 and mouse monoclonal antibody specific for HSV-2 ICP8, followed by staining with Alexa 488-conjugated donkey anti-goat IgG and Alexa 568-conjugated donkey anti-mouse IgG secondary antibodies. Nuclei were stained with Hoechst 33342. Stained cells were examined by fluorescence microscopy, and representative images are shown. Arrows indicate nuclear TIA-1 structures that accumulate at late times postinfection. (B) HeLa cells were mock infected or infected with HSV-2 strain HG52 at an MOI of 10. Infected cells were fixed at 10 hpi and stained with goat polyclonal antiserum specific for TIA-1 and mouse monoclonal antibody specific for G3BP, followed by staining with Alexa 488-conjugated donkey anti-goat IgG and Alexa 568-conjugated donkey anti-mouse IgG secondary antibodies. Nuclei were stained with Hoechst 33342. Stained cells were examined by confocal microscopy, and representative images of sections through the midpoint of the nuclei are shown. The arrow indicates a nuclear TIA-1 structure.
Fig 4.
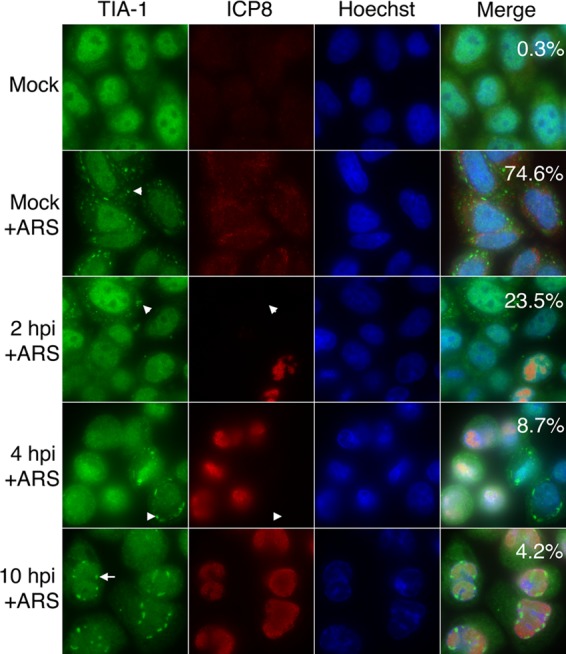
SG accumulation in HSV-2-infected cells following arsenite treatment. HeLa cells were mock infected or infected with HSV-2 strain HG52 at an MOI of 10. At the indicated times postinfection, cells were treated with 0.5 mM arsenite (+ARS) for 30 min. The cells were then fixed and stained with goat polyclonal antiserum specific for TIA-1 and mouse monoclonal antibody specific for HSV-2 ICP8, followed by staining with Alexa 488-conjugated donkey anti-goat IgG and Alexa 568-conjugated donkey anti-mouse IgG secondary antibodies. Nuclei were stained with Hoechst 33342. Stained cells were examined by fluorescence microscopy, and representative images are shown. Numeric values in the merged image panels indicate the fraction of cells containing SGs from 30 independent fields of view; for infected cells, only ICP8-positive cells were scored. Arrowheads indicate SGs in the cytoplasm. Note that in the images shown in the 2- and 4-hpi panels, SGs can be detected in cells that lack detectable ICP8 signal. The arrow indicates a nuclear TIA-1 structure.
Fig 7.

SG accumulation after pateamine A treatment. (A) SG accumulation in HeLa cells treated with arsenite or pateamine A. HeLa cells were treated with 0.5 mM arsenite (+ARS) or 300 nM pateamine A (+PATA) or left untreated for 30 min. The cells were then fixed and stained with goat polyclonal antiserum specific for TIA-1 and mouse monoclonal antibody specific for G3BP, followed by staining with Alexa 488-conjugated donkey anti-goat IgG and Alexa 568-conjugated donkey anti-mouse IgG secondary antibodies. Nuclei were stained with Hoechst 33342. Stained cells were examined by fluorescence microscopy, and representative images are shown. Numeric values in the merged image panels indicate the fraction of cells containing SGs from 30 independent fields of view. Arrowheads indicate SGs in the cytoplasm. Note that SGs formed following either arsenite or pateamine A treatment contain robust and roughly equivalent amounts of TIA-1 and G3BP. (B) SG accumulation in HSV-2-infected cells after arsenite or pateamine A treatment. HeLa cells were infected with HSV-2 strain HG52 at an MOI of 5. At the indicated times postinfection, the cells were treated with 0.5 mM arsenite (+ARS) or 300 nM pateamine A (+PATA) for 30 min. The cells were then fixed and stained with goat polyclonal antiserum specific for TIA-1 and mouse monoclonal antibody specific for HSV ICP27, followed by staining with Alexa 488-conjugated donkey anti-goat IgG and Alexa 568-conjugated donkey anti-mouse IgG secondary antibodies. Nuclei were stained with Hoechst 33342. Stained cells were examined by fluorescence microscopy, and representative images are shown. Numeric values in the merged image panels indicate the fraction of cells containing SGs from 30 independent fields of view; for infected cells, only ICP27-positive cells were scored. Arrowheads indicate SGs in the cytoplasm. Note that in the images shown in the 2 hpi panels, SGs can be detected in cells that lack detectable ICP27 signal. Arrows indicate nuclear TIA-1 structures.
Fig 2.
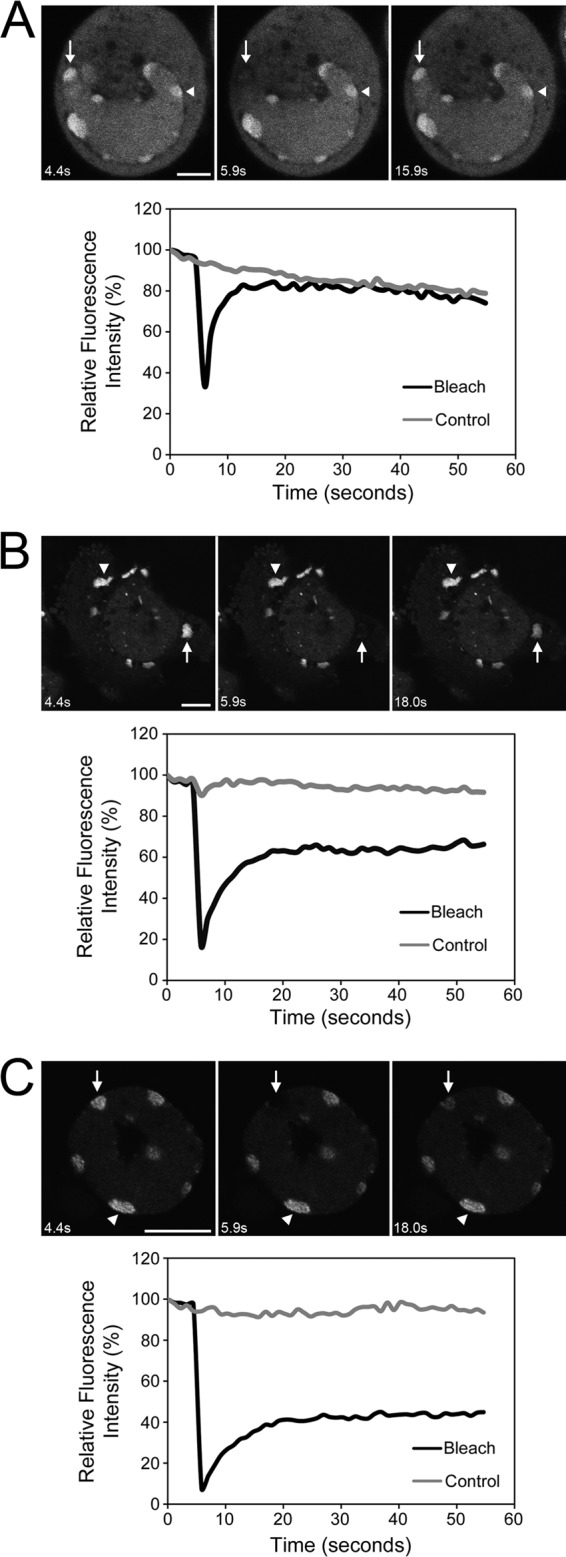
Dynamics of structures formed by TIA-1 and Sam68. HeLa cells were transfected with plasmids encoding GFP-tagged TIA-1 (A and B) or GFP-tagged Sam68 (C). At 24 h after transfection, cells were infected with HSV-2 strain HG52 at an MOI of 5 (A and C) or mock infected (B), and live cells were imaged at 10 hpi (A and C) or 24 h after mock infection (B). Shown in each panel are selected images of cells immediately before, immediately after, or at a later time after the region indicated by the arrow was photobleached. The region indicated by the arrowhead on each image was not photobleached and serves as a control. The scale bar in panel A is 5 μm and 10 μm in panels B and C. For full sets of images corresponding to panels A, B, and C, see Movies S1, S2, and S3, respectively, in the supplemental material. The corresponding quantitation of fluorescence intensity of the photobleached region, as well as the control region over time, is shown below each set of images.
Sam68 colocalizes with nuclear TIA-1 structures formed during HSV-2 infection.
During the course of infection, DNA viruses such as HSV-2 cause profound modifications in the nuclei of cells, including the margination of chromatin. Since the nuclear TIA-1 structures resulting from HSV-2 infection are found late in infection in cells displaying margination of chromatin and are often found in close proximity to chromatin, we speculated that they may arise as a result of cellular responses to DNA damage. One protein recently implicated to respond to DNA damage is the 68-kDa Src-associated in mitosis protein, Sam68 (9). Sam68 is a RNA-binding protein (44) that can be bound and recruited to SGs by TIA-1 in response to oxidative stress (28) and can also be recruited to SGs during poliovirus infection (56). DNA-damaging agents cause Sam68 and other RNA-binding proteins, including TIA-1, to relocalize into distinct nuclear foci (9). Consequently, we wanted to investigate whether Sam68 localized to the nuclear TIA-1 structures formed during HSV-2 infection. HeLa cells were transfected with plasmids encoding GFP-tagged Sam68 or a GFP-tagged derivative of Sam68, Sam68-GSG, which lacks the region implicated in TIA-1 binding. Transfected cells were infected with HSV-2 and then stained for TIA-1 at 4 and 10 hpi. The localization of Sam68 changed dramatically 10 h after HSV-2 infection (Fig. 3). In mock-infected cells and in cells infected for 4 h, Sam68 was evenly distributed throughout the nucleus or found in a few distinct puncta reminiscent of perinucleolar Sam68 nuclear bodies (13, 31), indicated by asterisks in Fig. 3. In contrast, Sam68 localized primarily to nuclear TIA-1 structures in infected cells at 10 hpi (compare the top three panels of Fig. 3). Furthermore, relocalization to nuclear TIA-1 structures was not as dramatic when GFP-Sam68-GSG was used in these assays (bottom two panels of Fig. 3). FRAP analysis of GFP-Sam68 nuclear structures in HSV-2-infected cells at 10 hpi indicated rapid recruitment of Sam68 into these structures at a rate indistinguishable from that observed for TIA-1 (Fig. 2C; see Movie S3 in the supplemental material). These results raise the possibility that nuclear TIA-1 structures form as a result of DNA damage evoked by HSV-2 infection.
Fig 3.
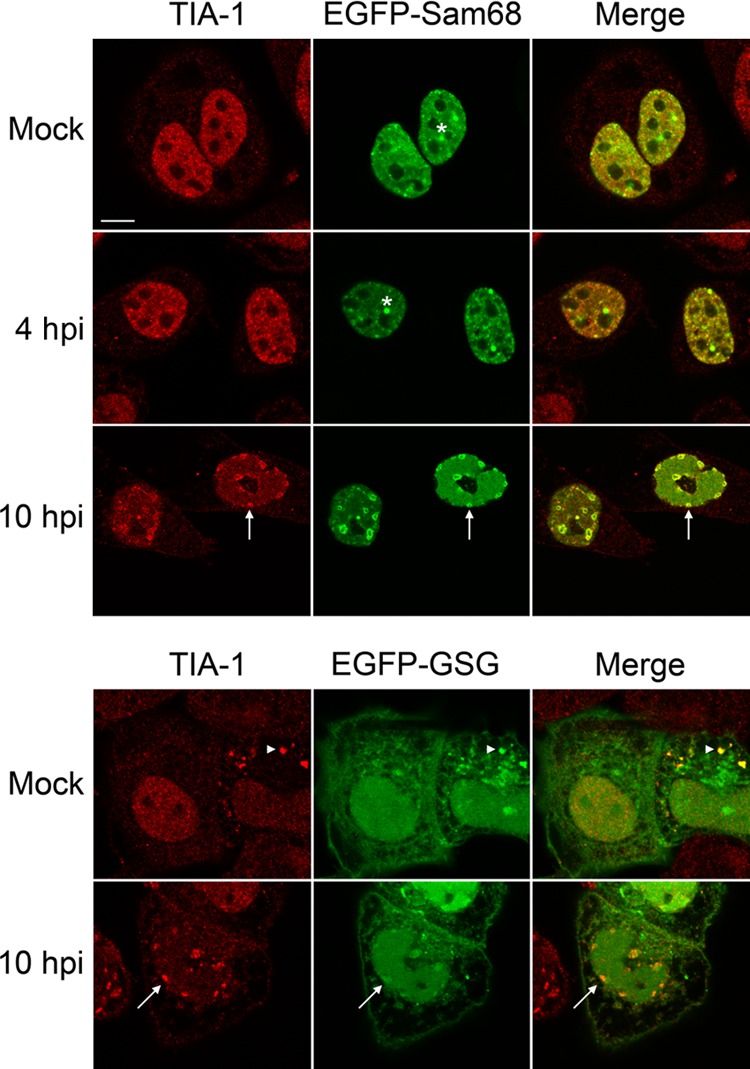
Sam68 localization in HSV-2-infected cells. HeLa cells were transfected with a plasmid encoding GFP-tagged Sam68 or GFP-tagged Sam68-GSG. At 24 h after transfection, the cells were mock infected or infected with HSV-2 strain HG52 at an MOI of 5. Infected cells were fixed at 4 or 10 hpi and stained with goat polyclonal antiserum specific for TIA-1 and Alexa 568-conjugated donkey anti-goat IgG secondary antibody. Stained cells were examined by confocal microscopy, and representative images of sections through the midpoint of the nuclei are shown. Asterisks are placed above examples of perinucleolar Sam68 nuclear bodies; arrows indicate nuclear TIA-1 structures. The arrowheads indicate a SG in the cytoplasm of a cell overexpressing GFP-Sam68-GSG. Scale bar, 10 μm.
HSV-2 interferes with arsenite-induced SG accumulation.
Our observation that HSV-2-infected cells failed to accumulate SGs led us to question whether infected cells would be able to accumulate SGs in response to additional stress. To pursue this, infected HeLa cells were treated with 0.5 mM arsenite for 30 min and probed for the presence of SGs by staining for TIA-1. Arsenite treatment promotes the phosphorylation of eIF2α (19) in a heme-regulated inhibitor kinase (HRI)-dependent manner (43, 49), leading to translational arrest and the subsequent formation of SGs (35, 49). The arsenite treatment conditions used induced the formation of readily detectable SGs in the majority of mock-infected cells (compare the top two panels of Fig. 4). Comparable accumulation of SGs in response to arsenite was not observed in HSV-2-infected cells at 2, 4, and 10 hpi (bottom three panels of Fig. 4) and the fraction of infected cells accumulating SGs in response to arsenite decreased over the course of the infection. Lack of SG accumulation in infected cells following arsenite treatment was observed as early as 30 min postinfection; however, the exposure of cells to virus-free supernatants did not prevent arsenite-induced SG formation (data not shown), indicating that the presence of virus was required for this effect. These results indicate that HSV-2 is able to interfere with SG accumulation caused by arsenite treatment early after infection. Nuclear TIA-1 structures were still observed in many cells following arsenite treatment at late times postinfection (Fig. 4). Interference with arsenite-induced SG accumulation was also observed in HSV-2-infected T12 cells (see Fig. S2 in the supplemental material).
If prevention of arsenite-induced SG accumulation by HSV-2 were an early event, then inhibition of viral DNA replication would not be expected to interfere with this process. Cells were treated with PAA to prevent viral DNA replication and the transcription of true late genes (30, 45). Lack of SG accumulation after infection and arsenite treatment was observed in the presence of PAA at all time points examined (Fig. 5A). The effectiveness of the PAA treatment at blocking late events in HSV-2 infection is evidenced by the differential appearance of ICP8 staining in PAA-treated cells (Fig. 5A), by the lack of chromatin margination in PAA-treated cells (Fig. 5A), and by the specific decrease in a leaky late gene product, ICP5 (40), in whole-cell extracts prepared from PAA-treated cells (Fig. 5B). Control analyses on mock-infected cells demonstrated that treatment with PAA alone did not induce SG accumulation and that the ability of arsenite treatment to induce SG accumulation was not adversely affected by the presence of PAA (see Fig. S1 in the supplemental material). Interestingly, nuclear TIA-1 structures were not observed at 10 hpi in the presence of PAA (compare lower 2 panels of Fig. 5A). Inhibition of viral replication with 5 μM acyclovir also resulted in the absence of nuclear TIA-1 structures (see Fig. S3 in the supplemental material). These results are consistent with the prevention of SG accumulation occurring early in HSV-2 infection and also imply that late events in HSV-2 infection are required for the formation of nuclear TIA-1 structures.
Fig 5.
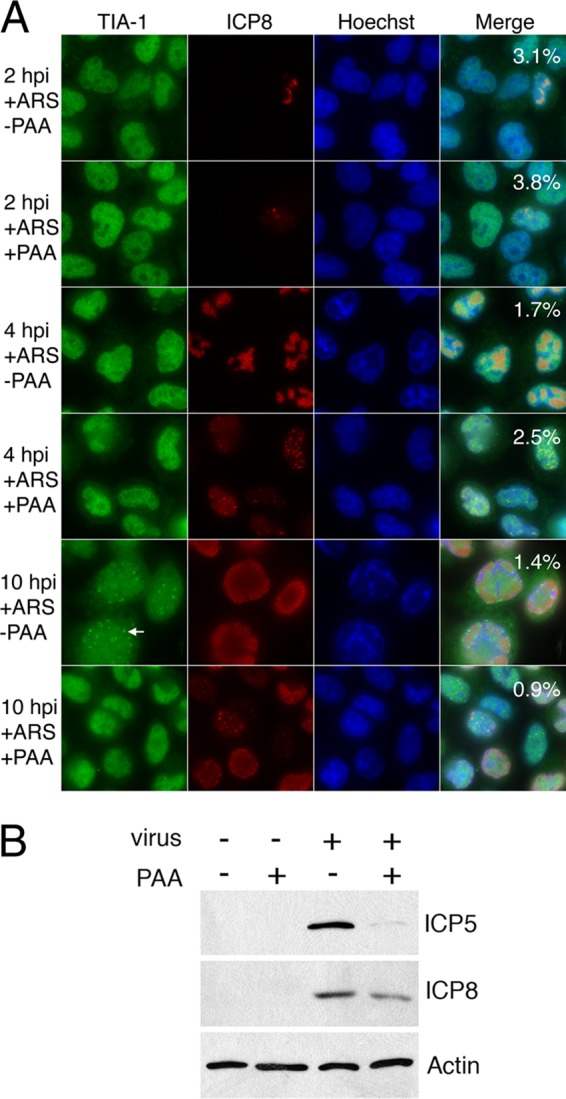
SG accumulation in HSV-2-infected cells in the presence of PAA. (A) HeLa cells were infected with HSV-2 strain HG52 at an MOI of 5 in the presence or absence of 200 μg of phosphonoacetic acid/ml (+PAA or −PAA, respectively). At the indicated times postinfection, the cells were treated with 0.5 mM arsenite (+ARS) for 30 min. The cells were then fixed and stained with goat polyclonal antiserum specific for TIA-1 and mouse monoclonal antibody specific for HSV-2 ICP8, followed by staining with Alexa 488-conjugated donkey anti-goat IgG and Alexa 568-conjugated donkey anti-mouse IgG secondary antibodies. Nuclei were stained with Hoechst 33342. Stained cells were examined by fluorescence microscopy, and representative images are shown. Numeric values in the merged image panels indicate the fraction of cells containing SGs from 10 independent fields of view; for infected cells, only ICP8-positive cells were scored. The arrow indicates a nuclear TIA-1 structure. Note that nuclear TIA-1 structures do not accumulate at late times postinfection in the presence of PAA. (B) HeLa cells were mock infected or infected with HSV-2 strain HG52 at an MOI of 5 in the presence or absence of 200 μg of phosphonoacetic acid/ml, and whole-cell extracts were prepared at 6 hpi. Equal volumes of whole-cell extracts were electrophoresed through 8% polyacrylamide gels and transferred to PVDF membranes. Membranes were probed with antisera indicated on the right.
HSV-2-infected cells phosphorylate eIF2α in response to arsenite treatment.
Since our studies indicate that SGs do not accumulate in HSV-2-infected cells, even following arsenite-induced stress, we wanted to determine whether infected cells could still respond to arsenite in terms of phosphorylating eIF2α. Infected HeLa cells were mock treated or treated with 0.5 mM arsenite for 30 min. Whole-cell extracts were prepared and analyzed by Western blotting with antisera specific for phospho-eIF2α or total eIF2α (Fig. 6). Arsenite treatment consistently resulted in increased levels of phospho-eIF2α in mock-infected cells while the level of total eIF2α remained constant. This same pattern was observed in HSV-2-infected cells at 2 and 4 hpi. However, by 10 hpi the level of phospho-eIF2α was elevated in HSV-2-infected cells in the absence of arsenite relative to earlier times after infection and did not increase appreciably upon arsenite treatment. These data demonstrate that the ability to phosphorylate eIF2α is not compromised in HSV-2-infected HeLa cells. Since lack of SG accumulation does not correspond with lack of eIF2α phosphorylation in infected cells, we conclude that HSV-2 interference with SG accumulation is downstream of eIF2α phosphorylation.
Fig 6.
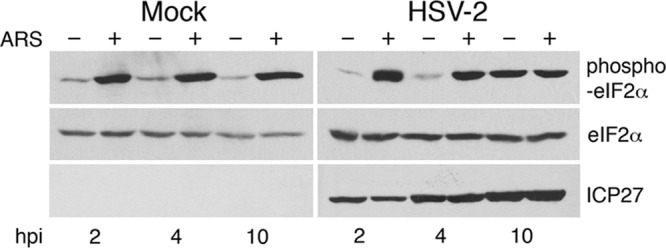
Phosphorylation status of eIF2α in HSV-2-infected cells. HeLa cells were mock infected or infected with HSV-2 strain HG52 at an MOI of 2. At the indicated times postinfection, cells were treated with 0.5 mM arsenite (ARS) for 30 min or mock treated (+ and −, respectively). After treatment, whole-cell extracts were prepared, and equal volumes of whole-cell extracts were electrophoresed through 10% polyacrylamide gels and transferred to PVDF membranes. Membranes were probed with antisera indicated on the right side of each panel.
HSV-2 prevents SG accumulation by eIF2α-independent inducers of SGs.
The data thus far show a disconnect between eIF2α phosphorylation and SG accumulation in HSV-2-infected cells. Therefore, we tested whether HSV-2 would interfere with SG accumulation caused by inducers that operate independently of eIF2α phosphorylation. Overexpression of either TIA-1 or Sam68-GSG is known to result in the formation of SGs (26, 29). In keeping with these studies, HeLa cells transfected with plasmids encoding either GFP–TIA-1 or GFP-Sam68-GSG displayed SGs in 203 of 454 cells examined or in 164 of 374 cells examined, respectively. When HeLa cells transfected with plasmids encoding either protein were subsequently infected with HSV-2, the number of infected cells with SGs was considerably diminished: in the case of GFP–TIA-1, 9 of 290 infected cells examined and 3 of 209 infected cells examined at 4 and 10 hpi, respectively, and in the case of GFP-Sam68-GSG, 1 of 279 infected cells examined and 1 of 118 infected cells examined at 4 and 10 hpi, respectively. These data are consistent with the notion that HSV-2 interference with SG accumulation occurs irrespective of eIF2α phosphorylation.
Another SG inducer that works independently of eIF2α phosphorylation is pateamine A. Pateamine A modulates the activity of another key translation initiation protein, eIF4A, leading to translational arrest and SG formation (6, 7, 42, 48). To examine whether HSV-2 interfered with SG accumulation induced by pateamine A, infected HeLa cells were treated with either 0.5 mM arsenite or 300 nM pateamine A for 30 min and probed for the presence of SGs by staining for TIA-1. In control analyses on mock-infected cells, treatment with either arsenite or pateamine A resulted in comparable levels of SGs that contained robust and roughly equivalent amounts of both TIA-1 and G3BP (Fig. 7A). The percentage of mock-infected cells with SGs after either treatment was 98% or greater when either TIA-1 or G3BP was used for quantification (Fig. 7A). In contrast, HSV-2-infected cells showed a lack of SG accumulation at all time points examined after treatment with either arsenite or pateamine A when TIA-1 was followed as a SG marker (Fig. 7B). Similar results were observed in infected T12 cells treated with pateamine A (see Fig. S2 in the supplemental material). These data, along with the data described above for HeLa cells over expressing GFP–TIA-1 or GFP-Sam68-GSG, provide independent support for our conclusion that HSV-2 interference with SG accumulation occurs downstream of eIF2α phosphorylation. Nuclear TIA-1 structures were still observed in many cells following pateamine A treatment at late times postinfection.
Assessment of SG accumulation in HSV-2-infected cells using G3BP.
We also wanted to verify whether an independent marker of SGs, G3BP, would report similarly on SG accumulation in cells infected with HSV-2 and subjected to either arsenite- or pateamine A-induced stress. Surprisingly, SGs containing G3BP were detected in >98% of infected cells treated with pateamine A at 4 and 10 hpi (Fig. 8A), a level indistinguishable from that observed in mock-infected cells treated with pateamine A (Fig. 7A). Similar results were also observed at 2 hpi (data not shown). This robust accumulation of SGs containing G3BP was not observed in untreated and arsenite-treated infected cells. Closer inspection of the SGs that accumulate in HSV-2-infected cells following pateamine A treatment confirmed that they were cytoplasmic (Fig. 8B) and contained poly(A)-binding protein (PABP), another common marker of SGs (Fig. 8C), and yet only occasionally contained detectable TIA-1 (indicated by the arrowheads in Fig. 8B). In mock-infected cells treated with pateamine A, all SGs examined contained detectable TIA-1 (n = 132). In contrast, in infected cells treated with pateamine A, only 43% of G3BP containing SGs had detectable TIA-1 at 4 hpi (n = 132). Furthermore, TIA-1 in those SGs was 8 times less abundant than TIA-1 in SGs produced in mock-infected cells. PABP was detectable in 89 and 84% of G3BP containing SGs (n = 150) induced by pateamine A treatment of mock-infected and HSV-2-infected cells, respectively, and the abundance of PABP in these SGs in mock-infected versus HSV-2-infected cells was comparable. Thus, G3BP and TIA-1 report differently on SG accumulation in HSV-2-infected cells. Under conditions of arsenite-induced stress, SG accumulation, as measured by both TIA-1 and G3BP localization, is prevented in HSV-2-infected cells. Conversely, under conditions of pateamine A-induced stress, SG accumulation, as measured by G3BP and PABP localization, is not prevented, and the composition of the SGs produced is distinct.
Fig 8.

SG accumulation in HSV-2-infected cells using different SG markers. (A) HeLa cells were infected with HSV-2 strain HG52 at an MOI of 5. At the indicated times postinfection, cells were treated with 0.5 mM arsenite (+ARS) or 300 nM pateamine A (+PATA) or left untreated for 30 min. The cells were then fixed and stained with goat polyclonal antiserum specific for TIA-1 and mouse monoclonal antibody specific for G3BP, followed by staining with Alexa 488-conjugated donkey anti-goat IgG and Alexa 568-conjugated donkey anti-mouse IgG secondary antibodies. Nuclei were stained with Hoechst 33342. Stained cells were examined by fluorescence microscopy, and representative images are shown. Arrowheads indicate SGs in the cytoplasm. Note that SGs detectable by G3BP accumulate in infected cells after pateamine A treatment. Arrows indicate nuclear TIA-1 structures. (B) HeLa cells were infected with HSV-2 strain HG52 at an MOI of 5. At the indicated times postinfection, cells were treated with 300 nM pateamine A (+PATA) for 30 min. The cells were then fixed and stained with goat polyclonal antiserum specific for TIA-1 and mouse monoclonal antibody specific for G3BP, followed by staining with Alexa 488-conjugated donkey anti-goat IgG and Alexa 568-conjugated donkey anti-mouse IgG secondary antibodies. Nuclei were stained with Hoechst 33342. Stained cells were examined by confocal microscopy and representative images of sections through the midpoint of the nuclei are shown. Arrowheads indicate SGs that contain a detectable amount of TIA-1 in addition to G3BP. (C) HeLa cells were mock infected or infected with HSV-2 strain HG52 at an MOI of 5. At 4 hpi, cells were treated with 300 nM pateamine A (+PATA) or untreated for 30 min. The cells were then fixed and stained with goat polyclonal antiserum specific for PABP and mouse monoclonal antibody specific for G3BP, followed by staining with Alexa 488 conjugated donkey anti-goat IgG and Alexa 568 conjugated donkey anti-mouse IgG secondary antibodies. Nuclei were stained with Hoechst 33342. Stained cells were examined by confocal microscopy, and representative images of sections through the midpoint of the nuclei are shown. Arrowheads indicate SGs in the cytoplasm.
Nuclear export of TIA-1 is not diminished in HSV-2-infected cells.
TIA-1 continuously shuttles between the nucleus and cytoplasm (67). Since TIA-1 accumulates in nuclear structures late in HSV-2 infection and is present in reduced quantities in pateamine A-induced SGs, we considered that nuclear export of TIA-1 might be blocked in infected cells. To address this possibility, we examined nucleocytoplasmic trafficking of TIA-1 in live HeLa cells using fluorescence loss in photobleaching (FLIP) assays (36). HeLa cells were transfected with plasmids encoding GFP-tagged TIA-1 then infected with HSV-2 for various lengths of time prior to FLIP analysis. In live cells expressing GFP–TIA-1, a decrease in fluorescence intensity in the nucleus over time was observed specifically in photobleached cells (solid black lines in Fig. 9B to E), demonstrating that TIA-1 was able to be exported from the nucleus in both mock-infected and HSV-2-infected cells at both early and late times postinfection. The rate of nuclear export, indicated by the slope of the solid black lines in Fig. 9, was observed to be slower for GFP–TIA-1 than for EGFP, a protein small enough to passively diffuse between the nucleus and the cytoplasm. These results are consistent with an active mechanism for nuclear export of TIA-1. Furthermore, the rates of GFP–TIA-1 nuclear export did not vary greatly between mock-infected and HSV-2-infected cells. These analyses demonstrated that nuclear export of TIA-1 is not blocked during HSV-2 infection and therefore is not the reason for the failure of HSV-2-infected cells to accumulate TIA-1 containing SGs. Moreover, the formation of nuclear TIA-1 structures does not occur because of a failure of TIA-1 to be exported from the nucleus in HSV-2-infected cells.
Fig 9.

FLIP analysis of TIA-1 nuclear export. HeLa cells were transfected with plasmids encoding GFP (A) or GFP–TIA-1 (B to E). In panels A and B, the cells were analyzed at 24 h posttransfection; in panel C, the cells were analyzed at 48 h posttransfection; in panels D and E, cells were infected at 24 h posttransfection with HSV-2 strain HG52 at an MOI of 5 and analyzed at 4 and 24 hpi, respectively. Changes in fluorescence in the nucleus and cytoplasm of bleached and control cells over time are shown on the left. For all compartments shown, the total fluorescence was measured in a fixed area using Fluoview 1.7.3.0 software. The total fluorescence measured immediately prior to the first round of bleaching was set to 100%, and subsequent measurements were scored relative to this value. Images corresponding to each graphic representation are shown on the right. Images were captured immediately prior to bleaching and at 420 and 834 s after bleaching. Rectangular boxes indicate the area subjected to repeated bleaching, and the arrows indicate control cells that were not subjected to bleaching.
DISCUSSION
SGs are complex, dynamic cytoplasmic assemblages that form in response to stress induced disruption of translation initiation (3). The impacts of a wide variety of viruses, predominantly RNA viruses, on these structures and their core components have been described in recent years (5, 52). A central theme arising from these studies is that viruses can target SGs for their own benefit: by utilizing SG components to enhance their replication or to avoid the effects of translational arrest associated with SG formation (52). In the present study, we examined the impact of HSV-2 infection on SGs. Our results indicate that SGs do not accumulate in HSV-2-infected cells, that HSV-2 can interfere with arsenite-induced SG accumulation early after infection and, mechanistically, that this interference occurs downstream of eIF2α phosphorylation. These findings imply that HSV-2 encodes previously unrecognized activities designed to maintain translation initiation downstream of eIF2α.
The ability of HSV-2 to block SG accumulation in the presence of arsenite may imply that this is not simply a passive effect and, instead, that HSV-2 actively prevents SG accumulation. While our data support interference with events downstream of eIF2α phosphorylation, we cannot yet distinguish whether SG accumulation in infected cells is prevented by blocking SG assembly and/or by enhancing SG disassembly. However, one observation supports the possibility of enhanced SG disassembly in HSV-2-infected cells. We noted that the number of cells containing SGs as a result of over expression of GFP–TIA-1 or GFP-Sam68-GSG was noticeably diminished when these cells were subsequently infected. These results imply that disassembly of preformed SGs may be enhanced in the presence of HSV-2.
Thus far, the only HSV protein with a reported connection to SG accumulation is vhs, the product of the UL41 gene, which promotes the decay of both cellular and viral mRNAs (60). Cytoplasmic structures that contained TIA-1 and the related SG marker protein TIAR were observed to accumulate at late times following infection with vhs-deficient mutants of HSV-1, whereas infection with wild-type viruses did not result in the accumulation of similar cytoplasmic structures (17, 21). These structures were subsequently demonstrated to contain G3BP, PABP, and eIF4A (17). The HSV-2 HG52 strain is deficient in host cell shutoff and contains a frameshift in UL41 resulting in the truncation of the carboxy-terminal 150 amino acids of vhs (22). This truncation encompasses one of the three regions of vhs reported to suppress expression of a reporter gene in cotransfection assays (23, 33). While HG52 has been described as weak in terms of its ability to shut off host cell protein synthesis (22), presumably due to the truncation in its vhs, we did not observe the appearance of cytoplasmic structures similar to those observed in vhs-deficient mutants of HSV-1 at late times postinfection with HG52 (see Fig. 1, 4, 5A, 7B, 8A, and 8B). As expected, HSV-2 strain G, which has been reported to have strong vhs activity (22), performed similarly to HG52 in our SG assays (data not shown).
During the course of these studies we discovered that distinct nuclear structures containing TIA-1 form following HSV-2 infection. These dynamic structures can be detected starting at 8 hpi, are found in close proximity to chromatin, contain Sam68, and do not form in the presence of viral replication inhibitors. Distinct nuclear structures containing both Sam68 and TIA-1 have been described in PC-3 cells treated with the DNA-damaging agent, mitoxantrone (9). However, the nuclear structures described in that study were evenly distributed throughout the nucleus, whereas the nuclear TIA-1 structures described here tended to localize to the periphery of the nucleus. Chromatin becomes displaced to the periphery of the nucleus in HSV-infected cells at late times postinfection due to the expansion of viral replication compartments (51), and so the peripheral localization of nuclear TIA-1 structures may be indicative of their association with chromatin. Our suggestion that nuclear TIA-1 structures form as a result of cellular responses to DNA damage evoked by HSV-2 infection fits our current understanding; however, other mechanisms are plausible. Over 25 years ago, it was reported that small nuclear ribonucleoprotein complex (snRNP) antigens condense and migrate during HSV-1 or HSV-2 infection, giving rise to punctate structures at the periphery of the nucleus (46). The possibility that nuclear TIA-1 structures are coalesced snRNPs fits nicely with the roles of both TIA-1 and Sam68 in alternative mRNA splicing (18, 25, 47). A particularly unique aspect of herpesvirus biology is localized disruptions of the nuclear lamina required for nuclear egress of capsids (32). Perhaps the formation and localization of nuclear TIA-1 structures is related to these focal disruptions of the nuclear lamina. Nuclear TIA-1 structures were not reported in previous studies using TIA-1 to follow SG accumulation in cells infected with HSV-1 at 12 or 16 hpi (17, 21). Since the late events in the virus life cycle that induce these structures would also be expected to occur following HSV-1 infection, perhaps longer infection times are required to visualize similar structures in HSV-1-infected cells.
Our observations that SGs do not accumulate in infected cells following arsenite treatment and yet do accumulate following pateamine A treatment may reflect fundamental differences in the way that HSV-2 copes with stalled translational initiation caused by these mechanistically distinct SG inducers. Arsenite treatment results in increased phosphorylation of eIF2α via HRI (49), which disrupts formation of the 43S preinitiation complex, leading to stalled translational initiation and SG formation (35). Conversely, pateamine A targets eIF4A, an mRNA helicase that is a central component of the eIF4F cap recognition complex (7, 42, 55). In the presence of pateamine A, eIF4A becomes sequestered on RNA thereby lowering free levels of eIF4A, which disrupts formation of eIF4F leading to stalled translational initiation and SG formation (6). Hippuristanol, another eIF4A inhibitor (41, 48), also causes SG accumulation in HSV-1-infected cells (17). Our preliminary analyses indicate that overall viral protein synthesis is repressed the presence of arsenite (see Fig. S4 in the supplemental material). It will be of considerable interest to determine whether this is the case in the presence of a mechanistically distinct SG inducer such as pateamine A and to examine the levels of specific viral proteins in the presence of SG inducers to determine whether the observed differences in SG accumulation correspond to differences in viral protein synthesis.
It is noteworthy that the majority of SGs accumulating in HSV-2-infected cells following pateamine A treatment lack detectable TIA-1. This observation contradicts the general view of TIA-1 as a seminal protein required for initiating SG assembly (3, 35). TIA-1 is typically found in readily detectable levels in SGs induced by a variety of stimuli, including pateamine A treatment (see Fig. 7A) and, furthermore, TIA-1 knockout cells displayed impaired ability to form SGs in response to arsenite and heat shock (26). Our data may suggest that other cellular RNA-binding proteins can functionally substitute for TIA-1 in initiating SG assembly or that only minuscule amounts of TIA-1, below the threshold of detection by immunofluorescence staining, are required to initiate SG assembly. While TIA-1 containing structures accumulated in the nuclei of infected cells, we observed no block in nuclear export of TIA-1 in infected cells that might account for lower levels of TIA-1 in the cytoplasm. In addition, no striking decreases in total cellular levels of TIA-1 were observed in HSV-1-infected cells (21) or in HSV-2-infected cells (see Fig. S5 in the supplemental material). Another possibility is that there is a differential requirement for TIA-1 in SG formation induced by arsenite versus pateamine A, and this difference is exposed upon HSV-2 infection. More detailed analyses of both trafficking and cellular levels of TIA-1 in infected cells following pateamine A treatment should provide insights into why TIA-1 is underrepresented in the SGs formed in HSV-2-infected cells treated with this particular SG inducer.
Interference with SG accumulation by HSV-2 occurs early in infection, whereas the accumulation of nuclear TIA-1 structures occurs much later in infection. Because of this temporal distinction, we currently view interference with arsenite-induced SG accumulation as mechanistically separate from the formation of nuclear TIA-1 structures. The characteristics of TIA-1 localization and SG accumulation in HSV-2 infections, established by the present study, provide the crucial foundation for studies proceeding on two different fronts. Our continued efforts to understand how HSV-2 interferes with SG accumulation will provide fundamental insights into the effects of HSV-2 infection on protein synthesis as well as insights into SG biology. In addition, experiments directed at understanding the formation of novel nuclear TIA-1 structures in infected cells should provide unique insights into nuclear disruptions caused by late events in the HSV-2 infectious cycle.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by CFI award 16389 and NIAID grant AI48626 to B.W.B.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the NIH.
We thank V. Kruys (Université Libre de Bruxelles) for the GFP–TIA-1, C. Sette (University of Rome Tor Vergata) for the GFP-tagged versions of Sam68, W. A. Bresnahan (University of Minnesota) for the T12 cells, and A. Mouland (Lady Davis Institute for Medical Research) for the pateamine A, as well as valuable advice. We acknowledge Sue Johnston for technical expertise and members of the Banfield lab for helpful discussions.
Footnotes
Published ahead of print 23 May 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Abrahamyan LG, et al. 2010. Novel Staufen1 ribonucleoproteins prevent formation of stress granules but favour encapsidation of HIV-1 genomic RNA. J. Cell Sci. 123:369–383 [DOI] [PubMed] [Google Scholar]
- 2. Alvarez E, Castello A, Menendez-Arias L, Carrasco L. 2006. HIV protease cleaves poly(A)-binding protein. Biochem. J. 396:219–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson P, Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33:141–150 [DOI] [PubMed] [Google Scholar]
- 4. Ariumi Y, et al. 2011. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J. Virol. 85:6882–6892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beckham CJ, Parker R. 2008. P bodies, stress granules, and viral life cycles. Cell Host Microbe 3:206–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bordeleau ME, et al. 2006. RNA-mediated sequestration of the RNA helicase eIF4A by pateamine A inhibits translation initiation. Chem. Biol. 13:1287–1295 [DOI] [PubMed] [Google Scholar]
- 7. Bordeleau ME, et al. 2005. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc. Natl. Acad. Sci. U. S. A. 102:10460–10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bresnahan WA, Hultman GE, Shenk T. 2000. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J. Virol. 74:10816–10818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Busa R, Geremia R, Sette C. 2010. Genotoxic stress causes the accumulation of the splicing regulator Sam68 in nuclear foci of transcriptionally active chromatin. Nucleic Acids Res. 38:3005–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Busa R, et al. 2007. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene 26:4372–4382 [DOI] [PubMed] [Google Scholar]
- 11. Cassady KA, Gross M. 2002. The herpes simplex virus type 1 U(S)11 protein interacts with protein kinase R in infected cells and requires a 30-amino-acid sequence adjacent to a kinase substrate domain. J. Virol. 76:2029–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cassady KA, Gross M, Roizman B. 1998. The herpes simplex virus US11 protein effectively compensates for the γ1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 72:8620–8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen T, Boisvert FM, Bazett-Jones DP, Richard S. 1999. A role for the GSG domain in localizing Sam68 to novel nuclear structures in cancer cell lines. Mol. Biol. Cell 10:3015–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng G, Feng Z, He B. 2005. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2α dephosphorylation by the γ(1)34.5 protein. J. Virol. 79:1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng G, Yang K, He B. 2003. Dephosphorylation of eIF-2α mediated by the γ(1)34.5 protein of herpes simplex virus type 1 is required for viral response to interferon but is not sufficient for efficient viral replication. J. Virol. 77:10154–10161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chou J, Chen JJ, Gross M, Roizman B. 1995. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2α and premature shutoff of protein synthesis after infection with γ(1)34.5− mutants of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 92:10516–10520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dauber B, Pelletier J, Smiley JR. 2011. The herpes simplex virus 1 vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J. Virol. 85:5363–5373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Del Gatto-Konczak F, et al. 2000. The RNA-binding protein TIA-1 is a novel mammalian splicing regulator acting through intron sequences adjacent to a 5′ splice site. Mol. Cell. Biol. 20:6287–6299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duncan RF, Hershey JW. 1987. Translational repression by chemical inducers of the stress response occurs by different pathways. Arch. Biochem. Biophys. 256:651–661 [DOI] [PubMed] [Google Scholar]
- 20. Emara MM, Brinton MA. 2007. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. U. S. A. 104:9041–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Esclatine A, Taddeo B, Roizman B. 2004. Herpes simplex virus 1 induces cytoplasmic accumulation of TIA-1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU-rich RNAs. J. Virol. 78:8582–8592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Everett RD, Fenwick ML. 1990. Comparative DNA sequence analysis of the host shutoff genes of different strains of herpes simplex virus: type 2 strain HG52 encodes a truncated UL41 product. J. Gen. Virol. 71(Pt 6):1387–1390 [DOI] [PubMed] [Google Scholar]
- 23. Everly DN, Jr, Read GS. 1999. Site-directed mutagenesis of the virion host shutoff gene (UL41) of herpes simplex virus (HSV): analysis of functional differences between HSV type 1 (HSV-1) and HSV-2 alleles. J. Virol. 73:9117–9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Finnen RL, Roy BB, Zhang H, Banfield BW. 2010. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 397:23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Forch P, et al. 2000. The apoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol. Cell 6:1089–1098 [DOI] [PubMed] [Google Scholar]
- 26. Gilks N, et al. 2004. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15:5383–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He B, Gross M, Roizman B. 1997. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 94:843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Henao-Mejia J, He JJ. 2009. Sam68 relocalization into stress granules in response to oxidative stress through complexing with TIA-1. Exp. Cell Res. 315:3381–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Henao-Mejia J, et al. 2009. Suppression of HIV-1 Nef translation by Sam68 mutant-induced stress granules and nef mRNA sequestration. Mol. Cell 33:87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Honess RW, Roizman B. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang S. 2000. Review: perinucleolar structures. J. Struct. Biol. 129:233–240 [DOI] [PubMed] [Google Scholar]
- 32. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 33. Jones FE, Smibert CA, Smiley JR. 1995. Mutational analysis of the herpes simplex virus virion host shutoff protein: evidence that vhs functions in the absence of other viral proteins. J. Virol. 69:4863–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kedersha N, et al. 2000. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 151:1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kedersha NL, Gupta M, Li W, Miller I, Anderson P. 1999. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. J. Cell Biol. 147:1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koster M, Frahm T, Hauser H. 2005. Nucleocytoplasmic shuttling revealed by FRAP and FLIP technologies. Curr. Opin. Biotechnol. 16:28–34 [DOI] [PubMed] [Google Scholar]
- 37. Langland JO, Cameron JM, Heck MC, Jancovich JK, Jacobs BL. 2006. Inhibition of PKR by RNA and DNA viruses. Virus Res. 119:100–110 [DOI] [PubMed] [Google Scholar]
- 38. Legros S, et al. 2011. The HTLV-1 Tax protein inhibits formation of stress granules by interacting with histone deacetylase 6. Oncogene 30:4050–4062 [DOI] [PubMed] [Google Scholar]
- 39. Li W, et al. 2002. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 76:11989–12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lieu PT, Wagner EK. 2000. Two leaky-late HSV-1 promoters differ significantly in structural architecture. Virology 272:191–203 [DOI] [PubMed] [Google Scholar]
- 41. Lindqvist L, et al. 2008. Selective pharmacological targeting of a DEAD box RNA helicase. PLoS One 3:e1583 doi:10.1371/journal.pone.0001583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Low WK, et al. 2005. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol. Cell 20:709–722 [DOI] [PubMed] [Google Scholar]
- 43. Lu L, Han AP, Chen JJ. 2001. Translation initiation control by heme-regulated eukaryotic initiation factor 2α kinase in erythroid cells under cytoplasmic stresses. Mol. Cell. Biol. 21:7971–7980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lukong KE, Richard S. 2003. Sam68, the KH domain-containing superSTAR. Biochim. Biophys. Acta 1653:73–86 [DOI] [PubMed] [Google Scholar]
- 45. Mao JC, Robishaw EE. 1975. Mode of inhibition of herpes simplex virus DNA polymerase by phosphonoacetate. Biochemistry 14:5475–5479 [DOI] [PubMed] [Google Scholar]
- 46. Martin TE, Barghusen SC, Leser GP, Spear PG. 1987. Redistribution of nuclear ribonucleoprotein antigens during herpes simplex virus infection. J. Cell Biol. 105:2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matter N, Herrlich P, Konig H. 2002. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420:691–695 [DOI] [PubMed] [Google Scholar]
- 48. Mazroui R, et al. 2006. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2α phosphorylation. Mol. Biol. Cell 17:4212–4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McEwen E, et al. 2005. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 280:16925–16933 [DOI] [PubMed] [Google Scholar]
- 50. McInerney GM, Kedersha NL, Kaufman RJ, Anderson P, Liljestrom P. 2005. Importance of eIF2α phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell 16:3753–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Monier K, Armas JC, Etteldorf S, Ghazal P, Sullivan KF. 2000. Annexation of the interchromosomal space during viral infection. Nat. Cell Biol. 2:661–665 [DOI] [PubMed] [Google Scholar]
- 52. Montero H, Trujillo-Alonso V. 2011. Stress granules in the viral replication cycle. Viruses 3:2328–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mulvey M, Arias C, Mohr I. 2007. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 81:3377–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mulvey M, Arias C, Mohr I. 2006. Resistance of mRNA translation to acute endoplasmic reticulum stress-inducing agents in herpes simplex virus type 1-infected cells requires multiple virus-encoded functions. J. Virol. 80:7354–7363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Parsyan A, et al. 2011. mRNA helicases: the tacticians of translational control. Nat. Rev. Mol. Cell. Biol. 12:235–245 [DOI] [PubMed] [Google Scholar]
- 56. Piotrowska J, et al. 2010. Stable formation of compositionally unique stress granules in virus-infected cells. J. Virol. 84:3654–3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Qin Q, Hastings C, Miller CL. 2009. Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection. J. Virol. 83:11090–11101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rothe F, Gueydan C, Bellefroid E, Huez G, Kruys V. 2006. Identification of FUSE-binding proteins as interacting partners of TIA proteins. Biochem. Biophys. Res. Commun. 343:57–68 [DOI] [PubMed] [Google Scholar]
- 59. Schneider RJ, Mohr I. 2003. Translation initiation and viral tricks. Trends Biochem. Sci. 28:130–136 [DOI] [PubMed] [Google Scholar]
- 60. Smiley JR. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J. Virol. 78:1063–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Smith RW, Graham SV, Gray NK. 2008. Regulation of translation initiation by herpesviruses. Biochem. Soc. Trans. 36:701–707 [DOI] [PubMed] [Google Scholar]
- 62. Tourriere H, et al. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160:823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63. Walsh D, Mohr I. 2011. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 9:860–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wek RC, Jiang HY, Anthony TG. 2006. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34:7–11 [DOI] [PubMed] [Google Scholar]
- 65. White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305 [DOI] [PubMed] [Google Scholar]
- 66. Zhang F, Moon A, Childs K, Goodbourn S, Dixon LK. 2010. The African swine fever virus DP71L protein recruits the protein phosphatase 1 catalytic subunit to dephosphorylate eIF2α and inhibits CHOP induction but is dispensable for these activities during virus infection. J. Virol. 84:10681–10689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang T, Delestienne N, Huez G, Kruys V, Gueydan C. 2005. Identification of the sequence determinants mediating the nucleo-cytoplasmic shuttling of TIAR and TIA-1 RNA-binding proteins. J. Cell Sci. 118:5453–5463 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.