

Abstract
Viruses employ an alternative translation mechanism to exploit cellular resources at the expense of host mRNAs and to allow preferential translation. Plant RNA viruses often lack both a 5′ cap and a 3′ poly(A) tail in their genomic RNAs. Instead, cap-independent translation enhancer elements (CITEs) located in the 3′ untranslated region (UTR) mediate their translation. Although eukaryotic translation initiation factors (eIFs) or ribosomes have been shown to bind to the 3′CITEs, our knowledge is still limited for the mechanism, especially for cellular factors. Here, we searched for cellular factors that stimulate the 3′CITE-mediated translation of Red clover necrotic mosaic virus (RCNMV) RNA1 using RNA aptamer-based one-step affinity chromatography, followed by mass spectrometry analysis. We identified the poly(A)-binding protein (PABP) as one of the key players in the 3′CITE-mediated translation of RCNMV RNA1. We found that PABP binds to an A-rich sequence (ARS) in the viral 3′ UTR. The ARS is conserved among dianthoviruses. Mutagenesis and a tethering assay revealed that the PABP-ARS interaction stimulates 3′CITE-mediated translation of RCNMV RNA1. We also found that both the ARS and 3′CITE are important for the recruitment of the plant eIF4F and eIFiso4F factors to the 3′ UTR and of the 40S ribosomal subunit to the viral mRNA. Our results suggest that dianthoviruses have evolved the ARS and 3′CITE as substitutes for the 3′ poly(A) tail and the 5′ cap of eukaryotic mRNAs for the efficient recruitment of eIFs, PABP, and ribosomes to the uncapped/nonpolyadenylated viral mRNA.
INTRODUCTION
Initiation is a rate-limiting step in eukaryotic translation, and is tightly regulated. Eukaryotic mRNAs possess an m7GpppN cap structure at the 5′ end and a poly(A) tail at the 3′ end. These two structures cooperate to recruit eukaryotic initiation factors (eIFs) and the 40S ribosome subunit (57) and stimulate translation initiation (19). The m7GpppN cap serves as the binding site for eIF4F, which is composed of eIF4E, eIF4G, and eIF4A. eIF4E is an m7GpppN-cap-binding protein, and eIF4G is a scaffold protein that binds eIF4E, eIF4A, the poly(A)-binding protein (PABP), and mRNA. eIF4A is an RNA helicase that unwinds RNA duplex structures in an ATP-dependent manner (57). In plants, eIF4F is thought to be composed of only eIF4E and eIF4G (7), because eIF4A is purified as a single polypeptide and is not copurified with eIF4F in wheat germ (35). Plants have a second form of eIF4F (eIFiso4F), which is composed of eIFiso4E and eIFiso4G (8). Both eIF4F and eIFiso4F enhance the translation of m7GpppN-capped mRNAs with an unstructured 5′ untranslated region (UTR), whereas only eIF4F can stimulate the translation of capped mRNAs with a highly structured 5′ UTR and uncapped mRNAs, including viral mRNAs (20). PABP binds to a poly(A) tail at the 3′ end of eukaryotic mRNAs via four RNA recognition motifs (RRMs) located in its N-terminal portion and simultaneously interacts with eIF4F via direct binding to eIF4G. This ternary interaction circularizes mRNA (30). The interaction between PABP and eIF4G stabilizes the association of eIF4F with the 5′ cap structure (30) and enhances the recruitment of the 43S ribosomal preinitiation complex (PIC), which is composed of the eIF2–GTP–Met-tRNAi Met ternary complex, eIF5, eIF1, eIF1A, the 40S subunit, and eIF3 (25), to the 5′ capped mRNA through an interaction between eIF4G and eIF3 (7, 25). The PABP-eIF4G interaction also enhances the recruitment of the 60S ribosomal subunit joining (31).
Viruses are obligate intracellular parasites that depend on host cells for their replication. To exploit cellular resources at the expense of host mRNAs and to allow preferential translation or proper translational regulation, positive-strand RNA viruses have developed diverse strategies, which, in many viruses, include cap- and poly(A)-independent translation mechanisms. For example, herpes-, polyoma-, nima-, picorna-, poty-, flavi-, dicistro-, and retroviruses recruit ribosomes at the internal ribosomal entry site (IRES) located in their 5′ UTR or intergenic region, which enhances the 5′-end-independent translation (3). All IRES elements, with the exception of dicistroviruses, bind a subset of eIFs and certain RNA-binding proteins to facilitate translation (18).
Many plant RNA viruses lacking both a 5′ cap and a 3′ poly(A) tail have cap-independent translation elements (CITEs) in the 3′ UTR of their genomic RNAs. To date, at least six distinct classes of 3′CITEs have been identified (46). These different classes of 3′CITEs exhibit no similarity to each other regarding primary or secondary structure (46). The 3′CITEs do not act as an IRES, although some cooperate with an IRES (39). The 3′CITEs interact with eIF4F and eIFiso4F (21, 47, 61, 67, 68) or the 60S ribosomal subunit (58). The binding of the eIF4F and eIFiso4F factors to the 3′CITE are believed to facilitate ribosome recruitment to the 5′ end, either via 5′-3′ interaction that are mediated by a protein factor (21), or via an RNA-RNA interaction (16, 17, 24, 45, 47, 69). However, the nature of the factors, other than eIF4F/eIFiso4F, that are required for the 3′CITE-mediated translation remain unclear (46).
To study the viral 3′CITE-mediated cap-independent translation mechanism, we used Red clover necrotic mosaic virus (RCNMV) as a model. RCNMV is a member of the Dianthovirus genus in the Tombusviridae family, and its genome consists of two RNA molecules, RNA1 and RNA2. These two RNAs lack both the 5′ m7GpppN cap and the 3′ poly(A) tail. Replication proteins and a capsid protein encoded in RNA1 are translated via a 3′CITE-mediated translation mechanism (26, 43, 53). The movement protein that is required for cell-to-cell movement is encoded in RNA2, and translated via an unknown cap-independent translation mechanism (42). The 3′CITE of RCNMV RNA1 is composed of five stem-loop structures (3′SL1 to 3′SL5) and is categorized as a Barley yellow dwarf virus (BYDV)-like CITE (BTE) (46). BTE contains a 17-nucleotide (nt) sequence (17-nt CS) conserved among dianthoviruses, necroviruses, umbraviruses, and luteoviruses. The 17-nt CS is essential for cap-independent translation of these viruses (32, 43, 66). The 17-nt CS contributes to form the 5′-proximal 3′SL1in the 3′ CITE of RCNMV RNA1 (43, 66). Previously, we showed that 5-nt substitutions (termed “Lm1” mutation) in the loop of the 3′SL1 completely abolished cap-independent translation of RCNMV RNA1 (43, 53). The BYDV BTE interacts with eIF4F through eIF4G and enhances BTE-mediated cap-independent translation (61). The BYDV BTE can also bind to eIFiso4F, albeit with lower affinity than to eIF4F (61). BYDV BTE-mediated translation requires a long-distance RNA–RNA interaction between the 5′ and 3′ UTRs of the BYDV RNA (24). In contrast to BYDV, our systematic mutagenesis studies suggest no or an insignificant role for such a long-distance RNA-RNA interaction in RCNMV 3′CITE-mediated cap-independent translation (53). This is supported by the fact that nonviral sequences at the 5′ end are sufficient to enhance RCNMV 3′CITE-mediated cap-independent translation efficiently in the protoplasts of cowpea, a natural host for this virus, although the 5′ UTR is required for the stability of RNA1 and for the enhancement of its translational activity in the protoplasts of tobacco BY-2 cells (53).
In the present study, we searched for host proteins that are associated with the 3′ UTR of RNA1 using RNA aptamer-based affinity chromatography and tandem mass spectrometry analysis in a cell-free viral translation/replication system. We identified PABP as a key player in the 3′CITE-mediated translation of RCNMV RNA1. We showed that PABP interacts directly with an A-rich sequence (ARS) in the 3′ UTR of RNA1, and enhances 3′CITE-mediated translation. We also found that the ARS is needed for the recruitment of eIF4F/eIFiso4F factors to the 3′ UTR and of the 40S ribosome to the viral mRNA.
MATERIALS AND METHODS
Plasmid constructions.
The constructs described previously that were used in the present study include the followings: pUCR1 (43), pUCR1-3′ UTR-S (27), pUCR2-3′ UTR-S (27), pSR1f (28), pR1-Luc-R1 (53), pR1-Luc-Lm1 (53), pSP64-RLUC (43), pColdGST (40), and pBYL2 (41).
pBYLNtPABP.
An ∼72-kDa protein that was copurified with the 3′ UTR of RCNMV RNA1 was excised and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Seventeen peptide matches were obtained for the poly(A)-binding protein (PABP; gi|7673359). PABP cDNA fragments were amplified by reverse transcription-PCR (RT-PCR) from total RNA extracted from tobacco BY-2 cells using the primers oligonucleotide 1 (oligo 1; GCTCAAGGTGCCATAGATAAGTTAAATGGTATG) and oligo 2 (GCAGTCACTTGGACCACCTTGACTCACTCA), which were designed based on the Nicotiana tabacum PABP mRNA partial coding sequence (gi|7673358). Rapid amplification of cDNA ends (RACE) was used to obtain information about the 5′- and 3′-proximal regions of the mRNA. The full-length cDNA of PABP (AB673187) was amplified by RT-PCR from total RNA extracted from tobacco BY-2 cells by using the primers, oligo 3 (CTGGCGCGCCATGGCGCAGATTCAGGTTCAGCACCAG) and oligo 4 (CTGGCGCGCCTCAAGAAACAAGGTTGTCATTG), digested with AscI, and used to replace the corresponding region of pBYL2.
pBYLNtPABP-F.
A DNA fragment was amplified by PCR from pBYLNtPABP using the primers oligo 3 and oligo 5 (CTGGCGCGCCTCACTTGTCATCGTCGTCCTTGTAGTCAGAAACAAGGTTGTCATTGAG), digested with AscI, and used to replace the corresponding region of pBYL2.
pUCSLAB-S, pUCSLDEF-S, and pUCSLCDEF-S.
Three DNA fragments were amplified by PCR from pUCR1 using primer pairs oligo 6 (GCGAGCTCTAATACGACTCACTATAGTGTAGCCTCCACCCGAG) plus oligo 7 (ATCCATGGCACTCTATTTTTTGCAATTTTAC), oligo 8 (GCGAGCTCTAATACGACTCACTATAGGGTTTTCTTTTAGGTAGGAGCAC) plus oligo 9 (ATCCATGGGGTACCTAGCCGTTATACGAC), and oligo 10 (GCGAGCTCTAATACGACTCACTATAGTAGGAGTAGTTCCCGTACCAG) plus oligo 9, respectively, digested with SacI and NcoI, and used to replace the corresponding region of pUCR1-3′ UTR-S, respectively.
pUCR1-3′ UTRΔSLC-S.
A DNA fragment was amplified by PCR from pUCR1-d3′SLC (26) using the primers oligo 6 and oligo 11 (ATCCCGGGATCCGACCGTGGTGCCCTTGCGGGCAGAAGTCCAAATGCGATCCATGGGGTACCTAGCCGTTATACGAC), digested with SacI and SmaI, and used to replace the corresponding region of pUCR1.
pColdAscI.
To create AscI site in pCold I vector (TaKaRa), an oligo-DNA carrying an AscI site (CTGGCGCGCCAGAGCT; the AscI site is in italics) was denatured and self-annealed, and the resulting double-stranded DNA was treated with T4 polynucleotide kinase (TaKaRa). Finally, the DNA fragment was cloned into pCold I vector that was digested with XbaI.
pCold H6-NtPABP-FLAG.
To construct pCold H6-NtPABP-FLAG, pBYLNtPABP-F was digested with AscI, the resulting 2-kb DNA fragment containing C-terminal FLAG epitope-tagged PABP was purified by using 1% agarose gel and cloned into the AscI site of pColdAscI.
pR1-Luc-ΔAms2bs.
A DNA fragment was amplified by PCR from pR1-Luc-R1 using the primer pair oligo 12 (ATTCTAGATTGGTTCTTTTAAGTGTAGCC) plus oligo 13 (CCTCATGTCTGGGATCCGAGACTGCGTGTTCCCCTCTTG). Another fragment was synthesized using the primer pair oligo 14 (TCGGATCCCAGACATGAGGATCACCCATGTCTGCAGCATATGAGTACT) and oligo 15 (ACGGATCCACTAACATGGGTGATCCTCATGTTAGTACTCATATGCTGCAG). A PCR fragment was amplified from the mixture of these two DNA fragments using the primer pair oligo 12 plus oligo 16 (TCCCGCGGGTACGGGAACTACTCCTAGCACGGATCCACTAACATGGGTGATCC). The PCR product was digested with XbaI and SacI and used to replace the corresponding region of pR1-Luc-R1.
pBYLMS2CP-NtPABP.
A DNA fragment containing MS2 bacteriophage coat protein (MS2/CP) was amplified by PCR from pGNC (70), which was a generous gift from Anne E. Simon (University of Maryland), using the primer pair oligo 17 (GCGCCATGGCTTCTAACTTTACTCAGTTCGTTCTC) plus oligo 18 (CCTGAATCTGCGCCTCGAGGTAGATGCCGGAGTTTGCTG). Another fragment containing PABP was amplified from pBYLNtPABP-F using the primer pair oligo 19 (CATCTACCTCGAGGCGCAGATTCAGGTTCAGCACCAGAG) plus oligo 20 (GTAGTTAACGTAACCATAACCAAGGGATCTCCTAG). A PCR fragment was amplified from the mixture of these two DNA fragments using the primer pair oligo 17 plus oligo 20. The PCR product was digested with NcoI and HpaI, and used to replace the corresponding region of pBYLNtPABP-F.
pBYLMS2CP-GFP.
A DNA fragment containing MS2CP was amplified by PCR from pBYLMS2CP-NtPABP using the primer pair oligo 21 (GTTTTCCCAGTCACGAC) plus oligo 22 (CTCCTTTACTCTCGAGGTAGATGCCGGAGTTTGCTGCG). Another fragment containing green fluorescent protein (GFP) was amplified from pBICGFP (59) using the primer pair oligo 23 (CTACCTCGAGAGTAAAGGAGAAGAACTTTTCAC) plus oligo 24 (CTGGCGCGCCTCACTTGTCATCGTCGTCCTTGTAGTCCGCGATCGCTTTGTATAGTTCATCCATGCCATGTG). A PCR fragment was amplified from the mixture of these two DNA fragments using the primer pair oligo 21 plus oligo 24. The PCR product was digested with AscI and used to replace the corresponding region of pBYL2.
pBYLMS2CP-MS2CP.
A DNA fragment containing MS2CP was amplified by PCR from pBYLMS2CP-NtPABP using the primer pair oligo 25 (ACCTCGAGGCTTCTAACTTTACTCAGTTCG) plus oligo 26 (TCCGCGATCGCGTAGATGCCGGAGTTTGCTGCGATTG). The DNA fragment was digested with XhoI and SgfI and used to replace the corresponding region of pBYLMS2CP-GFP.
pUCSLABΔ1-S, pUCSLABΔ2-S, pUCSLABΔ3-S, pUCSLABΔ4-S, pUCSLAB-AC1-S, pUCSLAB-AC2-S, pUCSLAB-AC3-S, pUCSLAB-AC4-S, pUCSLAB-PA25-S, pR1-Luc-SLABΔ1, pR1-Luc-SLABΔ2, pR1-Luc-SLABΔ3, pR1-Luc-SLABΔ4, pR1-Luc-AC1, pR1-Luc-AC2, pR1-Luc-AC3, pR1-Luc-AC4, pR1-Luc-PA25, pRNA1-AC1, pRNA1-AC2, pRNA1-AC3, pRNA1-AC4, and pRNA1-PA25.
For pUCSLAB-S mutants shown in Fig. 2D, pR1-Luc-R1 mutants and pUCR1 mutants used in Fig. 4, which possess mutations in the SLAB region, their constructs were generated using PCR based mutagenesis and standard cloning techniques. Each construct was sequenced across its entire PCR-derived region to ensure that only the desired mutation was present. Details of the modified RNA sequence and/or structure are presented in Fig. 2D.
Fig 2.

NtPABP binds specifically to an internal A-rich sequence in the 3′ UTR of RNA1. (A) Schematic representation of STagT-fused viral RNA fragments. Bold lines indicate virus-derived sequences with the nucleotide numbers of RNA1 at the 5′ and 3′ ends. Dashed lines indicate deleted sequence. (B) STagT pulldown assay using truncated mutants of the 3′ UTR of RNA1 in BYLS20 expressing FLAG-tagged NtPABP. (C) EMSA for NtPABP with SLAB region. The specificity of the NtPABP-SLAB interaction was determined by adding RNA competitors [poly(A), poly(G), and poly(C); Sigma-Aldrich]. (D) At the top is a schematic representation of STagT-fused viral RNA fragments. Bold lines indicate virus-derived sequences. Dashed lines indicate deleted sequences. Substituted sequences are shown in boxes. At the bottom, alignments of the SLAB region of RNA1 in dianthoviruses including RCNMV, Sweet clover necrotic mosaic virus (SCNMV), and Carnation ring spot virus (CRSV) are shown. Conserved nucleotides are highlighted with black boxes and white letters. Adenine residues are highlighted with red letters. (E and F) EMSA for NtPABP using SLAB and its mutants. (G and H) STagT pulldown assay using SLAB and its mutants in BYLS20 expressing FLAG-tagged NtPABP.
Fig 4.

The ARS enhances both cap/poly(A)-independent translation and replication of RNA1. (A) Uncapped R1-Luc-R1 and its derivatives carrying firefly luciferase (FL) were cotransfected with Renilla luciferase (RL) mRNA (internal control) into BY-2 protoplasts via electroporation. The FL/RL luminescence was normalized to the value of the wild-type reporter RNA1 (R1-Luc-R1). Means ± the SD (n = 3) are shown. (B) Uncapped R1-Luc-R1 and its derivatives carrying firefly luciferase (FL) were incubated into BYL. The FL luminescence was normalized to the value of R1-Luc-R1. Means ± the SD (n = 3) are shown. (C) Uncapped RCNMV RNA1 and its derivatives were incubated in BYL. Samples were subjected to SDS-PAGE, which was followed by Western blotting with anti-p27 antiserum. EtBr-stained signals represent the amount of transcripts used in this experiment. Coomassie brilliant blue (CBB)-stained host proteins were used as loading controls. (D) RNA1 and its derivatives were transfected into BY-2 protoplasts via electroporation. Total RNA was extracted and used for Northern blotting with a positive-strand RNA1 detection probe (26). EtBr-stained 18S/26S rRNAs were used as loading controls for Northern blotting. SR1f is a stable degradation product derived from the 3′ UTR of RNA1 (28).
pR1-Luc-AC4-Lm1, pΔ5′-Luc-Lm1, pΔ5′-Luc-AC4, and pΔ5′-Luc-AC4-Lm1.
pR1-Luc-R1 mutants in Fig. 7 were generated using PCR based mutagenesis and standard cloning techniques from the parental plasmid, pR1-Luc-R1. Each construct was sequenced across its entire PCR-derived region to ensure that only the desired mutation was present. Details of the modified RNA sequence and/or structure are presented in Fig. 7A.
Fig 7.

Both the ARS and 3′CITE are required for the efficient cap- and poly(A)-independent translation in BY-2 and cowpea protoplasts. (A) Schematic representation of introduced mutations in the 5′ UTR, ARS, and 3′CITE of reporter RNA1. (B and C) Nonfunctional-capped (A-capped) R1-Luc-R1 and its derivatives, and functional-capped (m7G-capped) and nonfunctional-capped luciferase mRNA with or without poly(A) tail were cotransfected with Renilla luciferase mRNA (internal control) into BY-2 and cowpea protoplasts using PEG transfection regent and electroporation, respectively. The FL/RL luminescence was normalized to the value of Δ5′-Luc-AC4-Lm1. Means ± the SD (n = 3) are shown.
p12-mini-R1, p12-mini-Lm1, and p12-mini-AC4.
A DNA fragments was amplified by PCR from psiCHECK2-let-7 8× (29) using the primer pair oligo 27 (ATTCGAGCTCTAATACGACTCACTATAGTCGCCACCACCATGGCTATGTTCATCGAGTCCGACCC) plus oligo 28 (TATAGTTCTAGACGATCGCCTAGAATTACTGC). The DNA fragment was digested with SacI and XbaI and used to replace the corresponding region of pR1-Luc-R1, pR1-Luc-AC4, and pR1-Luc-Lm1.
pR1-3′UTR-Lm1-STagT, pR1-3′UTR-AC4-STagT, and pR1-3′UTR-AC4-Lm1-STagT.
Three PCR fragments were amplified by PCR from pR1-Luc-Lm1, pR1-Luc-AC4, and pR1-Luc-AC4-Lm1, respectively, using the primer pair oligo 6 and oligo 11, digested with SacI and SmaI, and used to replace the corresponding region of pUCR1.
RNA preparation.
RNA transcripts derived from “pBYL” plasmids were synthesized in vitro from NotI-linearized plasmids with T7 RNA polymerase. For m7G-capping of these transcripts, the ScriptCap m7G capping system (Epicentre Biotechnologies) was used. R2-3′ UTR-S was synthesized in vitro from XbaI-linearized pUCR2-3′ UTR-S with T7 RNA polymerase. The nonfunctional-capped (G-capped) 12-mini-R1, 12-mini-Lm1, and 12-mini-AC4 were synthesized in vitro from XmaI-linearized plasmids with T7 RNA polymerase and capped with ScriptCap m7G capping system in the absence of S-adenosyl-l-methionine. Other RNA transcripts were synthesized in vitro from XmaI-linearized plasmids with T7 RNA polymerase in the presence or absence of ApppG cap structure analog (New England Biolabs). The control mRNA, R-Luc mRNA, was transcribed from EcoRI-linearized pSP64-RLUC and capped with ScriptCap m7G capping system. All transcripts were purified with a Sephadex G-50 fine column (Amersham Pharmacia Biotech). The RNA concentration was determined spectrophotometrically and its integrity was verified by agarose gel electrophoresis. All transcripts are named for their parent plasmids minus the “pUC”, “p”, or “pBYL” prefix.
Preparation of plant lysates.
The preparation of BYL and BYLS20 (membrane-depleted supernatant fractions of BYL) was described previously (27, 33). The lysate derived from Arabidopsis suspension cultured cell line MM2d (MM2dL) was prepared in a way similar to that used for BYL preparation (33). Briefly, to remove vacuoles, 1 ml of MM2d protoplasts (packed cell volume) were mixed with 5 ml of 24% (vol/vol) Percoll (GE Healthcare) overlaid on 2 ml of a 70% (vol/vol) and 7 ml of 30% (vol/vol) Percoll gradient and centrifuged at 9,000 rpm for 1.5 h at 25°C using an SW28 rotor (Beckman Coulter). All Percoll solutions contained 0.7 M mannitol, 20 mM MgCl2, and 5 mM PIPES-KOH (pH 7.4). After centrifugation, the evacuolated protoplasts were recovered from the interface between 30 and 70% Percoll solution. The evacuolated protoplasts were suspended in equal volume of TR buffer (without KOAc) (30 mM HEPES-KOH [pH 7.4], Mg(OAc)2, 2 mM dithiothreitol [DTT], and one tablet per 10 ml of Complete Mini protease inhibitor mixture [Roche Diagnostics]), homogenized with a Dounce homogenizer, and centrifuged at 17,000 × g for 10 min at 4°C. The supernatant was recovered and stored at −80°C.
Purification of host proteins that bind to the 3′ UTRs of RCNMV RNA1 and RNA2 in the evacuolated BY-2 extract.
Modified Strepto Tag (STagT) (12)-fused viral RNA fragments (150 pmol) were incubated in 400 μl of the evacuolated BY-2 extract (protein concentration: 15 mg/μl) at 4°C for 20 min. Subsequently, 8 μl of heparin (100 mg/ml) was added to the mixture. After further incubation for 40 min on ice, the sample was applied to a column containing 1.2 ml of streptomycin-conjugated Sepharose that was preequilibrated using column buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, and 3 mM MgCl2). The column was washed with 10 ml of column buffer, and the bait RNA-cellular protein complexes were eluted using 3 ml of column buffer containing 10 μM streptomycin. The elution fractions were concentrated 30-fold via acetone precipitation. The samples were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by silver staining. The protein bands of interest detected on SDS-PAGE were excised and subjected to in-gel digestion with trypsin. The resulting peptides were extracted from the gels and subjected to LC-MS/MS analysis as described previously (60). Peak lists obtained from the MS/MS spectra were used to identify proteins using the Mascot search engine (Matrix Science).
STagT pulldown assay.
RNA–protein interaction experiments using Strepto Tag were performed essentially as described previously (27). Briefly, 200 μl of BYLS20 reaction mixture, in which FLAG-tagged proteins were expressed from 6.25 pmol of mRNAs, was incubated with STagT-fused RNA fragments (75 to 150 pmol) for 20 min on ice. The mixture was further incubated with 4 μl of heparin solution (100 mg/ml) for 40 min on ice. The sample was applied to a column containing 0.6 ml of streptomycin-coupled Sepharose that was preequilibrated with column buffer. The column was washed with 2.5 ml of column buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, and 3 mM MgCl2), and then the protein-STagT-fused RNA complexes were eluted with 1.5 ml of column buffer containing 10 μM streptomycin. The elution fractions were concentrated 50-fold by acetone precipitation. The sample was subjected to SDS-PAGE, followed by ethidium bromide staining and Western blotting with anti-FLAG M2 monoclonal antibody (Sigma-Aldrich).
Luciferase reporter assay and negative-strand RNA synthesis in plant lysates.
Luciferase reporter assays and negative-strand RNA synthesis in BYL and MM2dL were performed essentially as described previously (26). For the tethering assay, MS2/CP-NtPABP or other control proteins were expressed in BYL from capped mRNA (10 ng/μl). After 100 min of incubation, R1-Luc-AC4, R1-Luc-ΔAms2bs, or R1-Luc-R1 (0.5 nM) was added, followed by incubation in the lysate for an additional 2 h. Aliquots of these samples were diluted using the passive lysis buffer (Promega) and assayed using the luciferase reporter assay system (Promega).
Protoplast assay.
Transfection of BY-2 protoplasts via electroporation, subsequent luciferase assay, and Northern blotting were performed essentially as described previously (27, 53). PEG transfection was performed essentially as described previously (44).
Expression and purification of recombinant proteins.
Expression and purification of recombinant proteins were performed essentially as described previously (40). H6-NtPABP-FLAG used in Fig. 3B was purified using Talon CellThru resin (Clontech) and concentrated by VIVA SPIN 500 MWCO 30000 (Sartorius).
Fig 3.

Poly(A) but not poly(C) inhibits the cap- and poly(A)-independent translation and the negative-strand RNA synthesis of RCNMV RNA1 in BYL. (A) R1-Luc-R1 was incubated in BYL in the presence of increasing amounts of poly(A) or poly(C) and was assayed for the expression of firefly luciferase. Means ± the standard deviations (SD; n = 3) are shown. (B) Recombinant NtPABP significantly restored luciferase activity repressed by adding poly(A). R1-Luc-R1 was incubated in BYL in the presence of both 150 ng of inhibitor [poly(A) or poly(C)]/μl and increasing concentrations of recombinant NtPABP (0, 0.5, 1, and 2.5 μM). Recombinant GST (2.5 μM) was used as a control in this assay. The level of expression of luciferase in the presence of poly(C) was defined as 100%. (C, D, and E) RNA1 was incubated in BYL in the presence of increasing amounts of poly(A) or poly(C). Total protein and RNA were used for Western blotting using a p27 antiserum (first panel) and Northern blotting with a negative-strand RNA1 detection probe (third panel) (26), respectively. EtBr-stained signals (second panel) represent the amount of transcripts used in this experiment. Coomassie brilliant blue (CBB)-stained host proteins (second panel) and EtBr-stained 18S/26S rRNAs (fourth panel) were used as loading controls for Western blotting and Northern blotting, respectively. (D and E) Quantification of relative accumulations of p27 and the negative-strand RNA1 shown in panel C by using Image Gauge program (Fujifilm Life Sciences). The level of accumulation of p27 and negative-strand RNA1 in the absence of competitors was defined as 100%. Means ± the SD (n = 3) are shown.
EMSA.
Electrophoretic mobility shift assay (EMSA) was performed essentially as described previously (36). A total of 10 pmol of 32P-body-labeled RNAs (SLAB-S and the mutants) was incubated in 20 μl of binding buffer (25 mM HEPES-KOH [pH 7.5], 1 mM Mg(OAc)2, 0.1 mM EDTA, 100 mM KCl, 10% glycerol, 1 mM DTT, and 0.5 mg of yeast tRNA/ml) for 15 min at 4°C in the presence or absence of RNA competitors [5 μg of poly(A), poly(G), and poly(C); Sigma-Aldrich] and the indicated amount of proteins. After incubation, 2.5 μl of 80% glycerol and 2.5 μl of a 0.25-mg/ml concentration of heparin were added, and the reaction mixture was incubated for an additional 10 min at 4°C. The RNA-protein complexes were resolved on a native 4% polyacrylamide gel, dried, and exposed to an imaging plate. Radioactive signals were detected using FLA-5100 (Fujifilm Life Sciences). The signal intensity of the RNA-protein complexes was quantified using Multi-Gauge software (Fujifilm Life Sciences).
Sucrose density gradient centrifugation.
A 25-μl portion of BYLS20 was incubated with 1 mM cycloheximide (CHX) and 250 μM sinefungin (SIN) in the absence or presence of 1 mM GMP-PNP at 25°C for 20 min. Subsequently, 10 nM 32P-cap-labeled mini reporter RNAs (12-mini-R1, 12-mini-Lm1, and 12-mini-AC4; the cap structures at these 5′ ends were not methylated [Gppp]; thus, canonical translation cannot occur using these transcripts) were added to the mixture, respectively, and incubated further for 20 min at 17°C. Reactions were stopped on ice and then layered over 5 to 25% linear sucrose gradients (10 mM HEPES-KOH [pH 7.5], 10 mM (for the 80S formation) or 5 mM (for the 48S formation) MgCl2, and 100 mM KCl) and sedimented via ultracentrifugation at 36,000 rpm using an SW41 Ti rotor (Beckman Coulter) for 2 h and 50 min at 4°C. Gradients were fractionated and analyzed using the Cerenkov counting technique.
Purification of eIF4F and eIFiso4F components from MM2dL.
A 200-μl portion of the MM2dL reaction mixture containing 0.75 mM ATP, 0.1 mM GTP, 25 mM creatine phosphate, 50 mM (each) 20 amino acids mixture, 80 mM spermine, 0.1 U creatine phosphokinase (Calbiochem)/μl, and 100 μl of MM2dL was incubated with 75 pmol of STagT RNA at 17°C for 30 min. The sample was applied to a column containing 0.6 ml of streptomycin-coupled Sepharose that was preequilibrated with column buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, and 3 mM MgCl2). The column was washed with 2.5 ml of column buffer, and subsequently the protein-RNA complexes were eluted with 1.5 ml of column buffer containing 10 μM streptomycin. The elution fractions were concentrated 50-fold by acetone precipitation. The sample was separated on 5 to 20% SDS-PAGE gradient gels, followed by staining with nucleic acid staining reagent GelRed (Biotium) and Western blotting with anti-AteIF4E (15), anti-AteIF4G, anti-AteIFiso4E (15), and anti-AteIFiso4G (37) antiserum (kindly provided by K. S. Browning) and anti-PABP antiserum (14; kindly provided by J.-F. Laliberté).
RESULTS
PABP binds specifically to the 3′ UTR of RCNMV RNA1.
Strepto Tag affinity purification was performed to isolate cellular proteins that bind to cis-acting elements required for cap-independent translation of RCNMV RNA1. Strepto Tag is an RNA aptamer that binds to streptomycin (1, 64). Because the 3′ UTR of RNA1 is essential for 3′CITE-mediated cap-independent translation (43), the 3′ UTR of RNA1 was fused to a modified Strepto Tag (STagT) (12) to obtain R1-3′ UTR-S (Fig. 1A). A similar construct was also obtained for the 3′ UTR of RNA2, which was used as a control. These RNA fragments were incubated with the evacuolated tobacco BY-2 extract. The extract was applied to a streptomycin-coupled column and viral RNA-protein complexes were eluted using a buffer containing streptomycin. Affinity fractions were analyzed using SDS-PAGE and protein bands were visualized using silver staining (Fig. 1B). The band patterns of putative proteins in each purified fraction differed from each other, suggesting that some host proteins were copurified specifically with STagT-fused viral RNA fragments (Fig. 1B). The specific proteins in each affinity fraction were analyzed using MS.
Fig 1.

PABP binds specifically to the 3′ UTR of RCNMV RNA1. (A) Schematic representations of RCNMV genomic RNAs and STagT-fused 3′ UTRs of RNA1 and RNA2 (R1-3′ UTR-S and R2-3′ UTR-S). Bold lines indicate the virus-derived sequence of STagT-fused viral RNA fragments. (B) Silver-stained SDS-PAGE gel showing eluted STagT-RNA fragments and host proteins from a Strepto Tag affinity purification. The 72-kDa band in the left gel was excised, digested with trypsin, and subjected to LC-MS/MS. MM, molecular mass. The protein is NtPABP (AB673187). (C) STagT pulldown assay using R1-3′ UTR-S and R2-3′ UTR-S in BYLS20 expressing FLAG-tagged NtPABP. The rightmost lane represents a mock pulldown sample. The input lane contains 1.6% of the extract used for the STagT pulldown assay. Purified bait RNAs (top) were visualized using ethidium bromide (EtBr) fluorescence. NtPABP-FLAG (bottom) was detected by Western blotting using anti-FLAG M2 monoclonal antibody (Sigma-Aldrich).
Among the proteins identified using the approach described above, we focused on one specific and prominent band, corresponding to a 72-kDa protein that was copurified with the 3′ UTR of RCNMV RNA1 (Fig. 1B). Tryptic digested peptides derived from the 72-kDa band exhibited an MS/MS spectrum similar to that predicted for the PABP of Nicotiana tabacum. We cloned the cDNA of PABP (NtPABP; DDBJ/EMBL/GenBank accession no. AB673187) from tobacco BY-2 cultured cells. To examine the interaction between NtPABP and the 3′ UTR of RNA1, we performed pulldown experiments using STagT-fused viral RNA fragments and the C-terminally FLAG-tagged NtPABP. This FLAG-tagged NtPABP was synthesized by in vitro translation in the 20,000 × g supernatant of an evacuolated tobacco BY-2 lysate reaction mixture (BYLS20) (27, 33). Note that BYLS20 and the membrane containing lysate (BYL) were optimized for the efficient translation and replication of RCNMV RNA1 and RNA2 and they are different from the extract used in Fig. 1B. NtPABP was specifically pulled down by the STagT-fused 3′ UTR of RNA1 (Fig. 1C), thus confirming the specific interaction between PABP and the 3′ UTR of RCNMV RNA1.
NtPABP binds specifically to an internal A-rich sequence in the 5′ side of the 3′ UTR of RNA1.
To define the PABP-binding sequences in the 3′ UTR of RNA1, five STagT-fused RNA fragments (SLAB-S, SLC-S, ΔSLC-S, SLDEF-S, and SLCDEF-S) (Fig. 2A) were tested for their ability to bind FLAG-tagged NtPABP in BYLS20. An STagT pulldown assay showed that NtPABP was pulled down by SLAB-S, ΔSLC-S, but not by SLC-S, SLDEF-S, or SLCDEF-S, indicating that the SLAB region is necessary and sufficient for the interaction with NtPABP (Fig. 2B). To test whether NtPABP binds to SLAB region directly, EMSA was used with bacterially expressed tobacco PABP (H6-NtPABP-FLAG). H6-NtPABP-FLAG bound to SLAB-S, whereas recombinant glutathione S-transferase (GST) did not bind to the RNA fragment. Moreover, the PABP-SLAB interaction was blocked by adding poly(A) and poly(G), but not poly(C) fragments (9, 11, 48), indicating that NtPABP binds to the SLAB region in the 3′ UTR directly and specifically (Fig. 2C).
To define further the nucleotide sequences required for the interaction with NtPABP, four RNA mutants with a series of deletions in SLAB (Fig. 2D) were tested using EMSA. SLABΔ1-S, SLABΔ2-S, and SLABΔ3-S bound to H6-NtPABP-FLAG with an affinity that was similar to that of SLAB-S (Fig. 2E), whereas SLABΔ4-S (lacking the 3′-proximal region of SLAB) did not bind to H6-NtPABP-FLAG (Fig. 2E). Because the region deleted in SLABΔ4-S contains runs of four and six A residues and this A-rich sequence (ARS) is conserved among dianthoviruses (Fig. 2D), we investigated whether these A residues are involved in NtPABP binding. Four SLAB mutants in which each of the A clusters in the ARS were replaced with a C cluster were tested using EMSA. With the exception of SLAB-AC1-S, all mutants failed to bind to H6-NtPABP-FLAG (Fig. 2F), suggesting the importance of the runs of four and six A residues for NtPABP binding. Another type of SLAB-S mutant (SLABPA25-S), in which the ARS was replaced completely by a 25-A cluster, bound to H6-NtPABP-FLAG with high affinity. This result suggests that no specific sequences between the runs of A residues in the ARS are required for the interaction with NtPABP and that the runs of A residues, rather than the structure of the ARS, are important for NtPABP binding. The importance of this ARS in the interaction with NtPABP was confirmed using an STagT pulldown assay. All SLAB mutants except for SLABPA25-S showed no or very low affinity for NtPABP in the pulldown assay (Fig. 2G and H), indicating that the entire SLAB region is required for the binding of PABP in plant lysates. Discrepancies between EMSA and STagT pulldown assay suggest that endogenous RNAs (e.g., tRNA, rRNA, and mRNA) act as strong competitors for the PABP binding to the ARS in plant lysates and that binding of additional host factors and/or modified PABP to the upstream of the ARS is required for the stable interaction between PABP and ARS.
Poly(A) but not poly(C) inhibits the cap- and poly(A)-independent translation and the negative-strand RNA synthesis of RCNMV RNA1 in BYL.
Since PABP is one of the key players in the initiation of translation (57), we hypothesized that PABP is required for cap-independent translation of RCNMV RNA1. To address this, we first tested the effect of poly(A) fragments on 3′CITE-mediated cap-independent translation in BYL using a reporter RNA (R1-Luc-R1) carrying a firefly luciferase open reading frame (ORF) and the 5′ and 3′ UTRs of RCNMV RNA1 (53). The translational activity of R1-Luc-R1 decreased with the addition of poly(A) fragments at concentrations higher than 150 ng/μl, whereas the addition of poly(C) fragments did not inhibit the translation (Fig. 3A). The repressed translational activity of R1-Luc-R1 was significantly restored by the recombinant PABP added back to the lysate (Fig. 3B). These results suggest that the sequestration of PABP by poly(A) fragments inhibits the cap-independent translation of R1-Luc-R1. It should be noted that both poly(C) and poly(A) fragments at low concentrations enhanced the translation of R1-Luc-R1 (Fig. 3B). This translational enhancement may have been caused by stabilization of reporter mRNAs via sequestration of RNases by these polyribonucleotides (55).
Next, we investigated the effects of poly(A) fragments on the translation and negative-strand synthesis of RNA1 in BYL. Poly(A) fragments inhibited the accumulations of p27 and negative-strand RNA1 much more effectively than poly(C) fragments did (Fig. 3C, D, and E), suggesting that the sequestration of PABP by poly(A) fragments compromises the production of replication proteins and negative-strand RNA synthesis and that PABP is important for these processes.
The ARS located in the 3′ UTR of RNA1 enhances 3′CITE-mediated cap-independent translation and replication of RNA1.
To investigate the roles of the PABP-binding region in the translation of RCNMV RNA1 in vivo, we first tested reporter R1-Luc-R1 mutants with the same mutations as those of the STagT-fused SLAB fragments (see Fig. 2D) for their translational activity in BY-2 protoplasts. All R1-Luc-R1 mutants, with the exception of R1-Luc-PA25 (in which the ARS was replaced with the sequence of 25 A), exhibited a cap-independent translation activity that was 10 to 40% that of the wild-type R1-Luc-R1 (Fig. 4A). Thus, the cap-independent translational activity of reporter RNA1 mutants correlated well with the PABP-binding capacities of SLAB-S mutants in BYL. Next, we performed a reporter assay in BYL using the same set of mutants. All mutations, with the exception of the replacement of the ARS with the 25-A sequence, decreased 3′CITE-mediated cap-independent translational activity in the reporter mRNAs, although the deleterious effects of the mutations were milder in BYL than in BY-2 protoplasts (Fig. 4A and B). These results suggest a more important role of the ARS in vivo. To validate the importance of the ARS for the cap-independent translation in viral context, we investigated translational activities of full-length RCNMV RNA1 and its derivatives carrying the same mutations as those of the STagT-fused SLAB fragments (see Fig. 2D) in BYL. RNA1-AC4 was dramatically compromised for cap-independent translation (Fig. 4C), suggesting that the ARS is required for the 3′CITE mediated cap-independent translation of full-length RCNMV RNA1.
Next, to investigate the role of the ARS in RNA replication, RNA1 mutants in the ARS were tested for their ability to replicate in BY-2 protoplasts. All RNA1 mutants accumulated only a small amount, if any, of RNA1 (Fig. 4D). These results indicate that both A residues and other nucleotide sequences in the ARS are required for the replication of RCNMV RNA1 in vivo.
Tethered PABP stimulates 3′CITE-mediated translation of reporter RNA1.
To investigate whether the interaction between PABP and the 3′ UTR enhances the 3′CITE-mediated translation of RCNMV RNA1, we used a tethering assay. An MS2 bacteriophage coat protein (MS2CP)-fused NtPABP (MS2CP-NtPABP) was tethered to the 3′ UTR of R1-Luc-R1ΔAms2bs, which is a reporter RNA1 mutant in which the ARS in the 3′ UTR of RNA1 was replaced by the MS2CP binding site (Fig. 5A). The translational activity of R1-Luc-R1ΔAms2bs was similar to that of R1-Luc-AC4 (negative control) in BYL in the absence of MS2CP-NtPABP (Fig. 5B). However, expression of MS2CP-NtPABP increased the translational activity of R1-Luc-R1ΔAms2bs by more than twice that of R1-Luc-AC4 in BYL (Fig. 5C), and the translational activity of R1-Luc-R1ΔAms2bs was comparable to that of R1-Luc-R1 (positive control) (Fig. 5D). Expression of FLAG-tagged NtPABP, MS2CP-fused GFP (MS2CP-GFP), and MS2CP-fused MS2CP (MS2CP-MS2CP) failed to enhance the translational activity of R1-Luc-R1ΔAms2bs (Fig. 5D). These results suggest that MS2CP-NtPABP stimulates the translation of a reporter RNA1 carrying the MS2CP binding site in cis and in a manner that is specific to PABP.
Fig 5.

Tethered PABP stimulated cap-independent translation of a reporter RNA1. (A) Schematic representation of the components used for the assay of tethered PABP function. R1-LucΔAms2bs is a reporter RNA1 variant that lacks the ARS but contains tandem binding sites for the MS2 bacteriophage coat protein (MS2CP) in the 3′ UTR. (B) The translational activity of R1-LucΔAms2bs was similar to that of R1-Luc-AC4 (negative control) in BYL. The level of expression of luciferase from R1-Luc-AC4 was defined as 100%. Means ± the SD (n = 3) are shown. (C) The translational activity of R1-LucΔAms2bs was enhanced in the presence of MS2CP-NtPABP in BYL. The level of expression of luciferase from R1-Luc-AC4 was defined as 100%. Means ± the SD (n = 3) are shown. (D) MS2CP-NtPABP specifically enhanced the translational activity of R1-LucΔAms2bs in cis in BYL. The level of expression of luciferase from R1-Luc-R1 (positive control) in BYL expressing NtPABP was defined as 100%. Means ± the SD (n = 3) are shown.
Both the ARS and 3′CITE are required for the efficient cap- and poly(A)-independent translation of RCNMV RNA1.
PABP has been proposed to stimulate cap- and poly(A)-dependent translation through various mechanisms, including the enhancement of mRNA binding to the 43S PIC and stimulation of joining of the 60S ribosome at the start codon (30, 31). How does PABP stimulate the translation of RCNMV RNA1 lacking both cap and poly(A) tail? First, we examined whether the ARS is involved in the stability of the viral RNA using R1-Luc-R1 and its mutants with deletions in the SLAB region. Mutations in the ARS (R1-Luc-Δ3 and R1-Luc-Δ4) did not enhance degradation of reporter RNAs (Fig. 6A and B), suggesting that PABP binding to the ARS is not involved in the stability of the viral RNAs. Next, we sought to determine whether the 5′ UTR of RNA1 is required for the translational enhancement by PABP. Even though deletion of the viral 5′ UTR decreased the translational activity of R1-Luc-R1 by 70% in BY-2 protoplasts and by 40% in cowpea protoplasts (Fig. 7), the translational activity of this mutant (Δ5′-Luc-R1) was still 100- and 1,500-fold higher than that of a triple mutant (Δ5′-Luc-AC4-Lm1; Fig. 7A) or a reporter mRNA lacking functional cap structure (A-capped LucpA60) in BY-2 and cowpea protoplasts, respectively (Fig. 7B and C). Furthermore, the translational activity of Δ5′-Luc-R1 was 3-fold higher than that of a canonical mRNA (m7G-capped LucpA60) in cowpea protoplasts (Fig. 7C). These results support our previous studies showing that the 5′ UTR functions as a translational enhancer but is not essential for cap-independent translation of RNA1 (53). Consistent with previous studies, the translational activities of the mutants with mutations in 3′CITE were at basal levels (Fig. 7B and C). The mutant lacking the ARS showed 10- to 50-fold decreases in the 3′CITE-dependent translational activity, although the decreased level was lower than that by the mutation in 3′CITE (Fig. 7B and C). These results indicate that the ARS can function in the 3′CITE-dependent translation independently of the 5′ UTR of RNA1, and that both the ARS and 3′CITE are required for the efficient cap- and poly(A)-independent translation of reporter RNA1s in vivo.
Fig 6.
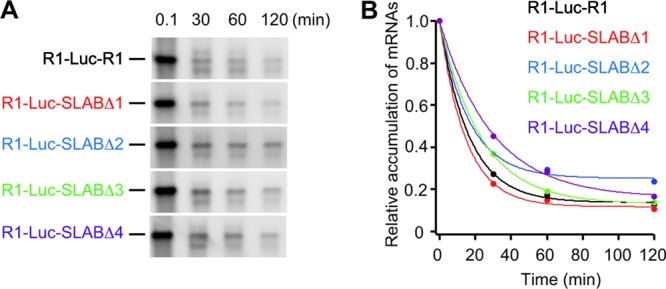
Stability of a reporter RNA1 (R1-Luc-R1) and its derivatives. (A) BYL was incubated with R1-Luc-R1 and its derivatives. Total RNA was extracted at the indicated time points and used for Northern blotting with the positive-strand detection probe (26). (B) The signal intensities were quantified with Image Gauge program (Fujifilm Life Sciences). Transcripts abundance for each reporter mRNA was normalized to 0.1-min time point and fits to decaying exponential with IGOR pro 6 (Wavemetrics).
Both the ARS and 3′CITE are required for the efficient recruitment of the 40S ribosome subunit to the viral genomic RNA.
Next, to investigate which step of translation is associated with the functions of the ARS and 3′CITE, we examined the effects of AC4 and Lm1 mutations on the formation of ribosome complexes using a sucrose density gradient assay. In this assay, we used short reporter mRNAs carrying a nonviral 5′ UTR, a Renilla luciferase-derived short ORF, and the viral 3′ UTR with or without AC4 or Lm1 mutation (12-mini-R1, 12-mini-Lm1, and 12-mini-AC4, respectively; Fig. 8A). We used the short reporter RNAs lacking the viral 5′ UTR because the moderate enhancement of translation by adding the viral 5′ UTR is independent of the ARS and 3′CITE (Fig. 7) and also because shorter mRNAs are preferable for sucrose density gradient assays to provide high-resolution complex profiles (23). These transcripts were cap labeled via a capping enzyme in the absence of S-adenosyl-l-methionine, producing transcripts that possessed a nonfunctional Gp*pp cap (“p*” indicates the position of 32P) at the 5′ ends. The cap-labeled transcripts were stable enough in the lysate to provide complex profiles in the sucrose density gradient assay. These nonfunctional Gp*pp-capped reporter RNAs were incubated in BYLS20 that had been incubated with two different inhibitors, sinefungin (SIN) and cycloheximide (CHX). SIN is a natural S-adenosyl-l-methionine analog that inhibits methyltransferase activity (38). CHX blocks translational elongation, but not initiation. The incubated BYLS20 lysate was subjected to sucrose density gradient centrifugation in order to assess the formation of the 80S and 48S initiation complexes. Both AC4 and Lm1 mutations greatly decreased the formation of the 80S initiation complex (Fig. 8B). The accumulation of the 48S initiation complex was not observed in RNAs with either of the two mutations, or in wild-type 12-mini-R1, suggesting that the ARS and 3′CITE are required for the recruitment of the 40S ribosome subunit to the mRNA, rather than via subsequent scanning and 60S-joining steps. This idea was supported by the fact that the 48S initiation complex accumulated after the addition of GMP-PNP, which blocks 60S joining. As expected, this accumulation of the 48S initiation complex was also inhibited by AC4 and Lm1 mutations (Fig. 8C). Taken together, these results lead us to conclude that both the ARS and 3′CITE located in the 3′ UTR are required for the efficient recruitment of the 40S ribosome subunit to the viral RNA.
Fig 8.

Both the ARS and 3′CITE are required for the efficient recruitment of the 40S ribosome subunit. (A) Schematic representation of a mini reporter RNA1 carrying nonviral 12-nt leader sequence, a Renilla-luciferase-derived short ORF, and the 3′ UTR of RNA1 (12-mini-R1), and of its mutants (12-mini-AC4 and 12-mini-Lm1). (B and C) BYLS20 containing CHX and SIN was incubated with 32P-labeled 12-mini-R1 (black line), 12-mini-AC4 (blue line), and 12-mini-Lm1 (red line) in the absence (B) or presence (C) of GMP-PNP. The complexes were separated by sucrose density gradient centrifugation, and the fractions were analyzed by Cerenkov counting. The radioactivity of each fraction is denoted as the percentage of the total counts recovered.
The ARS and 3′CITE recruit eukaryotic translation initiation factors coordinately to the viral 3′ UTR.
Because both the ARS and 3′CITE are required for the enhancement of the recruitment of the 40S ribosome subunit to RNA1, we hypothesized that these two cis-acting elements function coordinately in recruiting eIFs, together with PABP, to the 3′ UTR of RNA1. To test this hypothesis, we performed STagT pulldown using R1-3′UTR-S and three RNA variants with AC4, Lm1, or both mutations in the 3′ UTR (R1-3′UTR-Lm1-S, R1-3′UTR-AC4-S, and R1-3′UTR-AC4-Lm1-S, respectively) (Fig. 9A). To use antibodies that detect eIFs, we developed an in vitro translation system (MM2dL) using the Arabidopsis MM2d cultured cell line. MM2dL recapitulated cap- and poly(A)-dependent canonical translation and cap-independent translation of RCNMV RNA1 (data not shown). Note that RCNMV translates and replicates efficiently in MM2d cultured cells (H. Iwakawa and T. Okuno, unpublished results).
Fig 9.

ARS and 3′CITE recruit eukaryotic translation initiation factors coordinately to the viral 3′ UTR. (A) Schematic representation of the STagT-fused 3′ UTR of RNA1 and of its mutants. (B and C) STagT pulldown assay using the 3′ UTR of RNA1 and its mutants in an Arabidopsis MM2d cultured cell lysate (MM2dL). The input lane contains 1.6% of the extract used for the STagT pulldown assay. The sample was separated on 5 to 20% SDS–PAGE gradient gels, followed by the staining with nucleic acid staining regent GelRed (Biotium) and Western blotting with AteIF4G, AteIF4E, AteIFiso4G, and AteIFiso4E antisera (kindly provided by K. S. Browning) and a AtPABP2 antiserum (kindly provided by J.-F. Laliberté). The antiserum against AtPABP2 detects three different types of Arabidopsis PABP (AtPABP2, AtPABP4, and AtPABP8).
A. thaliana encodes eight PABP genes that fall into three distinct classes (4). The broadly and highly expressed class II is composed of the PAB2, PAB4, and PAB8 genes, which encode AtPABP2 (68.7 kDa), AtPABP4 (71.7 kDa), and AtPABP8 (72.8 kDa), respectively. These proteins are relatively homologous (4). The serum developed against AtPABP2 detects these three proteins equally well, but cannot be used to distinguish AtPABP4 from AtPABP8 because of identical migration on SDS-PAGE (14). R1-3′UTR-STagT pulled down endogenous eIF4E, eIF4G, eIFiso4G, and PABPs (AtPABP2 and AtPABP4/8) effectively (Fig. 9B), whereas it failed to pull down eIFiso4E (data not shown). The Lm1 mutation in the 3′CITE decreased the copurification of eIF4E, eIF4G, and eIFiso4G, but did not affect that of PABPs (Fig. 9B lane 3), indicating that the 3′CITE is required for binding to eIF4E, eIF4G, and eIFiso4G, but not to PABPs. Conversely, the AC4 mutation in the ARS compromised the pull-down efficiency of both PABP (AtPABP4/8) and the eIF4 proteins (Fig. 9B, lane 4), indicating that this region is important for binding to both PABP (AtPABP4/8) and eIF4 factors. AC4 and Lm1 double mutations further decreased the efficiency of eIF4E pulldown (Fig. 9B, compare lane 5 to lanes 3 and 4).
R1-3′UTR-PA25-S, in which the ARS was replaced with a short poly(A) tract (25 nt; a length that is sufficient for the binding of one PABP molecule), pulled down PABPs (AtPABP2/4/8) more efficiently than the wild type did, whereas the amount of eIF4E and eIFiso4G copurified was not affected much (Fig. 9C). eIF4G was below detectable levels in this experiment. These results suggest that the sequence of the ARS itself is not important; rather, the binding of PABP is important for the binding of eIF4 factors to the 3′ UTR via the 3′CITE of RCNMV RNA1.
DISCUSSION
In this study, we identified PABP as a novel factor in the 3′CITE-mediated cap-independent translation of RCNMV RNA1.
PABP recognizes the internal ARS in the 3′ UTR of RNA1.
PABP contains four RRMs (RRM1 to RRM4) in its N-terminal region. The RRMs are involved in the recognition of poly(A) sequences. PABP requires a minimum of 12 A residues to bind RNA (34, 52), but its packing density is 25 A residues in yeast (52), or ∼27 A residues in mammalian cells (2). In the present study, we demonstrated that PABP binds directly to the SLAB region in the 3′ UTR of RCNMV RNA1. The SLAB region contained an ARS with runs of three, four, and six A residues and these A residues were essential for the binding to PABP. PABP binds to the 3′ UTRs of the oskar mRNA (62), the YB-1 mRNA (56), and the genomic RNA of the dengue virus (50). The 3′ UTRs of these mRNAs contain six tracts of 3 to 10 A residues (62), several tracts of 3 to 4 A residues (56), or runs of 3 and 6 A residues interspersed with a C residue (50), respectively. PABP also binds to the 5′ UTR of its own mRNA (51). In the yeast PABP mRNA, the region upstream of the initiator methionine codon contains runs of 4, 5, 6, 8, and 11 A residues interspersed with U and C residues (51). These reports and the present study indicate that a continuous run of 12 or more A residues is not essential for PABP binding. This conclusion is supported by an in vitro selection/amplification experiment that showed that 5 A residues are sufficient for the specific binding of PABP in the context of a longer oligo (22).
The upstream sequence of the ARS may be important for the stable binding of PABP in plant cells, because deletions of the region upstream of the ARS dramatically reduced the interaction between STagT-fused SLAB and NtPABP in BYLS20. Given that the deletion of the upstream region did not affect the binding affinity between SLAB region and PABP in EMSA using recombinant PABP, it is possible that endogenous RNAs could act as strong competitors for the PABP binding to the ARS in the plant lysates, and binding of additional host factors and/or modified PABP to the upstream of the ARS is required to enhance the binding affinity between PABP and the ARS. A purine-rich sequence that contains a run of 10 A/G residues resides in the upstream of the ARS. Because the full-length wheat PABP binds to both poly(A) and poly(G) (11), this purine-rich sequence is a good candidate for the second PABP binding site in the SLAB region. Because it is known that phosphorylated and hypophosphorylated PABP bind to the poly(A) RNA synergistically and cooperatively (36), two molecules of PABP in different phosphorylation states might bind synergistically to the 5′ purine-rich and 3′ ARS in the SLAB region. Alternatively, some other RNA-binding proteins may bind to the upstream of the ARS and increase binding affinity of PABP to the ARS.
Arabidopsis encodes eight PABPs that fall into three distinct classes (4). To date, little is known about the functional differences between these PABPs. In the present study, we observed that AtPABP4 and/or AtPABP8 recognized both the ARS and the artificial 25-nt oligo(A) sequence in the same region, whereas AtPABP2 recognized only the 25-nt oligo(A) sequence. Therefore, the tobacco PABP identified here may be classified as an AtPABP4/8-class PABP, since the tobacco PABP also recognized both the ARS and the 25-nt oligo(A) sequence. These results suggest that each plant PABP has a distinct preference for RNA sequence recognition, thus exerting different regulatory functions on individual mRNAs in plants.
cis-Elements that mimic poly(A) tail in 3′CITE-dependent translation.
The ARS dramatically enhanced 3′CITE-mediated translation in RCNMV RNA1. Other viruses with BTE-type 3′CITE also require additional sequences or elements in the viral 3′ UTR besides the core 3′CITE to achieve efficient cap-independent translation in vivo. In BYDV, an uncapped reporter RNA carrying the 5′ UTR and the core BTE (109 nt) alone in the 3′ UTR failed to translate efficiently in oat protoplasts. In contrast, a reporter RNA with the long viral 3′ UTR (1,162 nt) translated well in a cap-independent manner. Interestingly, addition of poly(A) tail to the 3′ terminus stimulated cap-independent translation in the reporter RNA with the core BTE alone but did not in the reporter RNA with the long 3′ UTR (65). These results suggest that BYDV possesses poly(A)-mimic elements in the long 3′ UTR. Tobacco necrosis virus (TNV) also has an additional cis-element that is required for cap-independent translation. This element is replaced functionally with a 60-nt poly(A) tail (54), suggesting that the element has a function similar to that of the poly(A) tail. Currently, it remains unclear what factor(s) binds to these cis-elements that enhances the BTE-mediated translation in BYDV and TNV. The poly(A)-mimic element in TNV contains neither an ARS nor an AC(A/G/U)AAY(A/C) consensus heptamer sequence, which is a human PABP-binding sequence (22). Therefore, it is unlikely that PABP directly binds to the 3′ RNA element. Host factors other than PABP may recognize the TNV RNA element and stimulate cap-independent translation. Alternatively, it is still possible that PABP interacts with the non-poly(A) RNA element of TNV via other RNA-binding proteins. Protein-mediated interactions between PABP and RNAs have been reported in a subset of cellular mRNAs in mammalian germ cells (6). The enhancement of 3′CITE-mediated cap-independent translation via direct or indirect PABP-binding to the 3′ UTR might widespread in positive-strand plant RNA viruses.
PABP stimulates eIF4F/iso4F binding to the 3′ UTR.
PABP is thought to exert stimulatory effects at multiple stages of translation (57). PABP stimulates mRNA binding to the 43S PIC, at least partly by enhancing binding of the eIF4F complex to the m7G-capped 5′ end of mRNA (5, 31, 63). To enhance cap-dependent translation, PABP should bind to both poly(A)-tail of mRNA and eIF4G that interacts with 5′ m7G-cap via eIF4E (31). In the present study, we found that PABP stimulated mRNA binding to the 43S PIC at least partly by enhancing the binding of the eIF4F/eIFiso4F components to the 3′ UTR via the 3′CITE. This function of PABP as a strong enhancer for the recruitment of eIF4F/iso4F in the 3′CITE-dependent translation is reminiscent of that in cap-dependent translation. Although currently there is no information on whether PABP interacts with eIF4G/eIFiso4G on the 3′ UTR of RNA1, it is possible that the PABP-eIF4G/eIFiso4G interaction increases the binding affinity between the eIF4F/eIFiso4F complex and 3′CITE of RCNMV RNA1. Alternatively, PABP-binding to the ARS may induce structural changes in the 3′ UTR that make the 3′CITE more accessible to eIF4s. Indeed, stable interaction between PABPs and PA25 did not increase the binding of eIF4s to the 3′ UTR. This result suggests that stable binding of PABP to the 3′ UTR is not required and that moderate affinity interaction is enough for the recruitment of eIF4s to the 3′ UTR. It is possible that once PABP binds to the ARS, RNA sequences near the 3′CITE can be folded properly. Further study will be needed to clarify the molecular mechanism underlying how PABP-ARS interaction enhances recruitment of eIF4s and ribosome to the viral RNA.
How does the 3′ UTR of RCNMV RNA1 stimulate the recruitment of the ribosome to the 5′ end?
In the present study, we demonstrated that both the ARS and 3′CITE in the 3′ UTR of RNA1 enhance the cap-independent translation of RCNMV RNA1. Previously, we showed that the cap-independent translation of RNA1 is scanning-dependent (53), suggesting that the ribosome is recruited to the uncapped 5′ end. How does the 3′ UTR of RCNMV RNA1 stimulate the recruitment of the ribosome to the uncapped 5′ end? In a current model for the 3′CITE-mediated cap-independent translation, the RNA-RNA interaction or protein-mediated interaction between the 5′ UTR and the 3′ UTR are thought to be required. The end-to-end communication has been believed to deliver the eIF4F, which is bound to the 3′CITE, to the vicinity of the 5′ end for the recruitment of the ribosomes (24, 61). In fact, in a tombusvirus, the 3′ UTR containing both an eIF4F-bound 3′CITE and a complementary sequence to the 5′ UTR of the virus stimulates cap-independent translation of a reporter RNA in trans (47), supporting the model that the RNA-RNA interaction delivers 3′CITE-bounds to the vicinity of the uncapped 5′ end. However, our present and previous studies (53) clearly demonstrate that the 5′ UTR is not essential for the cap-independent translation of RCNMV RNA1. These results suggest that the long-distant RNA-RNA or protein-mediated interactions between the 5′ and 3′ UTRs of RNA1 are dispensable for the recruitment of the ribosome in RCNMV RNA1. This inexplicable type of cap-independent translation could have several, but not mutually exclusive explanations. First, host factors that bind to the 3′ UTR of RCNMV RNA1 may display an affinity for the highly negatively charged 5′ tri-phosphate terminus of the uncapped mRNA via the positively charged amino acid residues, and this affinity might deliver eIF4F/eIFiso4F factors to the vicinity of the 5′ end to recruit ribosomes. In fact, the N-terminal deleted eIF4G enhances the 5′ end-dependent translation of uncapped mRNA in mammals, suggesting that eIF4G recognizes the uncapped 5′ end of mRNA (13). Second, because the ARS and 3′CITE of RCNMV RNA1 increase the local concentration of eIF4F/eIFiso4F, and probably other translational initiation factors that are required for the recruitment of the ribosome, e.g., eIF4A and eIF4B, the inherent affinity of 43S PIC to the 5′ end of mRNA (49) could be enhanced by these factors. Third, the tertiary structure of the genomic and reporter RNAs may be sufficient to bring the eIF4F/eIFiso4F complex associated with the 3′ UTR of RCNMV RNA1 to the 5′ proximal region where the ribosome small subunit is recruited.
On the other hand, the 5′ UTR of RNA1 enhanced the translation of nonfunctional capped reporter mRNAs ∼3-fold in BY-2 protoplasts (Fig. 7B). These results suggest that the 5′ UTR of RCNMV RNA1 possesses cis-elements that enhance cap-independent translation in addition to the cis elements required for the stability of mRNA (53). This relatively weak translational enhancer element(s) might play a role in the delivery of eIF4F/eIFiso4F factors to the 5′ end. The full-length RNA1 might possess additional RNA elements that function in long-distance interaction. The 3′CITE in Saguaro cactus virus interacts with the 5′ part of genome by RNA-RNA interaction that involves a sequence downstream from the 5′ UTR (10). Such elements could enhance the recruitment of 40S ribosome to the 5′ end of the full-length RNA1.
Role of the ARS in the replication of RCNMV RNA1.
Our results indicate that a 25-nt oligo(A) can replace the original ARS functionally, suggesting that PABP binding property, and not the ARS itself, is important for cap-independent translation of RCNMV RNA1. In contrast, substitution of the ARS with a 25-nt oligo(A) compromised the replication of RNA1. These results suggest that the ARS contains cis-acting elements that are required for viral RNA replication. The cis-elements should be those required for positive-strand RNA synthesis, because our previous study showed that deletion of the ARS has no effect on the negative-strand RNA synthesis of RNA1 (26). Alternatively, the high-affinity binding of PABP to the 25-nt oligo(A) tract may inhibit the negative-strand RNA synthesis of the mutant RNA1 via the blockage of the elongation process by replicase toward the 5′ end. The dengue virus also interacts with PABP with a relatively low affinity via its 3′ UTR (50). The low-affinity binding of PABP to viral genomes may be important for achieving a balance between translation and replication in these viruses.
ACKNOWLEDGMENTS
We thank Karen S. Browning for antibodies against AteIF4E, AteIFiso4E, AteIF4G, and AteIFiso4G; Jean-François Laliberté for the antibody against AtPABP2; Anne E. Simon for plasmids carrying MS2CP (pGNC); James A. H. Murray and Takashi Aoyama for MM2d cell line; and Toshinobu Fujiwara and Akira Fukao for technical advice on sucrose density gradient centrifugation.
This study was supported in part by a Grant-in-Aid for Scientific Research (A) (18208004) and by a Grant-in-Aid for Scientific Research (A) (22248002) from the Japan Society for the Promotion of Science and in part by a Grant-in-Aid for JSPS Fellows.
Footnotes
Published ahead of print 16 May 2012
REFERENCES
- 1. Bachler M, Schroeder R, von Ahsen U. 1999. StreptoTag: a novel method for the isolation of RNA-binding proteins. RNA 5:1509–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baer BW, Kornberg RD. 1983. The protein responsible for the repeating structure of cytoplasmic poly(A)-ribonucleoprotein. J. Cell Biol. 96:717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balvay L, Soto Rifo R, Ricci EP, Decimo D, Ohlmann T. 2009. Structural and functional diversity of viral IRESes. Biochim. Biophys. Acta 1789:542–557 [DOI] [PubMed] [Google Scholar]
- 4. Belostotsky DA. 2003. Unexpected complexity of poly(A)-binding protein gene families in flowering plants: three conserved lineages that are at least 200 million years old and possible auto- and cross-regulation. Genetics 163:311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borman AM, Michel YM, Kean KM. 2000. Biochemical characterisation of cap-poly(A) synergy in rabbit reticulocyte lysates: the eIF4G-PABP interaction increases the functional affinity of eIF4E for the capped mRNA 5′-end. Nucleic Acids Res. 28:4068–4075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brook M, Smith JW, Gray NK. 2009. The DAZL and PABP families: RNA-binding proteins with interrelated roles in translational control in oocytes. Reproduction 137:595–617 [DOI] [PubMed] [Google Scholar]
- 7. Browning KS. 1996. The plant translational apparatus. Plant Mol. Biol. 32:107–144 [DOI] [PubMed] [Google Scholar]
- 8. Browning KS, Webster C, Roberts JK, Ravel JM. 1992. Identification of an isozyme form of protein synthesis initiation factor 4F in plants. J. Biol. Chem. 267:10096–10100 [PubMed] [Google Scholar]
- 9. Burd CG, Matunis EL, Dreyfuss G. 1991. The multiple RNA-binding domains of the mRNA poly(A)-binding protein have different RNA-binding activities. Mol. Cell. Biol. 11:3419–3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chattopadhyay M, Shi K, Yuan X, Simon AE. 2011. Long-distance kissing loop interactions between a 3′ proximal Y-shaped structure and apical loops of 5′ hairpins enhance translation of Saguaro cactus virus. Virology 417:113–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng S, Gallie DR. 2007. eIF4G, eIFiso4G, and eIF4B bind the poly(A)-binding protein through overlapping sites within the RNA recognition motif domains. J. Biol. Chem. 282:25247–25258 [DOI] [PubMed] [Google Scholar]
- 12. Dangerfield JA, Windbichler N, Salmons B, Gunzburg WH, Schroder R. 2006. Enhancement of the StreptoTag method for isolation of endogenously expressed proteins with complex RNA binding targets. Electrophoresis 27:1874–1877 [DOI] [PubMed] [Google Scholar]
- 13. De Gregorio E, Preiss T, Hentze MW. 1998. Translational activation of uncapped mRNAs by the central part of human eIF4G is 5′ end-dependent. RNA 4:828–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dufresne PJ, Ubalijoro E, Fortin MG, Laliberte JF. 2008. Arabidopsis thaliana class II poly(A)-binding proteins are required for efficient multiplication of turnip mosaic virus. J. Gen. Virol. 89:2339–2348 [DOI] [PubMed] [Google Scholar]
- 15. Duprat A, et al. 2002. The Arabidopsis eukaryotic initiation factor (iso)4E is dispensable for plant growth but required for susceptibility to potyviruses. Plant J. 32:927–934 [DOI] [PubMed] [Google Scholar]
- 16. Fabian MR, White KA. 2004. 5′-3′ RNA-RNA interaction facilitates cap- and poly(A) tail-independent translation of tomato bushy stunt virus mRNA: a potential common mechanism for Tombusviridae. J. Biol. Chem. 279:28862–28872 [DOI] [PubMed] [Google Scholar]
- 17. Fabian MR, White KA. 2006. Analysis of a 3′-translation enhancer in a tombusvirus: a dynamic model for RNA-RNA interactions of mRNA termini. RNA 12:1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fitzgerald KD, Semler BL. 2009. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim. Biophys. Acta 1789:518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gallie DR. 1991. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 5:2108–2116 [DOI] [PubMed] [Google Scholar]
- 20. Gallie DR, Browning KS. 2001. eIF4G functionally differs from eIFiso4G in promoting internal initiation, cap-independent translation, and translation of structured mRNAs. J. Biol. Chem. 276:36951–36960 [DOI] [PubMed] [Google Scholar]
- 21. Gazo BM, Murphy P, Gatchel JR, Browning KS. 2004. A novel interaction of Cap-binding protein complexes eukaryotic initiation factor (eIF) 4F and eIF(iso)4F with a region in the 3′-untranslated region of satellite tobacco necrosis virus. J. Biol. Chem. 279:13584–13592 [DOI] [PubMed] [Google Scholar]
- 22. Gorlach M, Burd CG, Dreyfuss G. 1994. The mRNA poly(A)-binding protein: localization, abundance, and RNA-binding specificity. Exp. Cell Res. 211:400–407 [DOI] [PubMed] [Google Scholar]
- 23. Gray NK, Hentze MW. 1994. Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J. 13:3882–3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo L, Allen EM, Miller WA. 2001. Base-pairing between untranslated regions facilitates translation of uncapped, nonpolyadenylated viral RNA. Mol. Cell 7:1103–1109 [DOI] [PubMed] [Google Scholar]
- 25. Hinnebusch AG. 2006. eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem. Sci. 31:553–562 [DOI] [PubMed] [Google Scholar]
- 26. Iwakawa HO, Kaido M, Mise K, Okuno T. 2007. cis-Acting core RNA elements required for negative-strand RNA synthesis and cap-independent translation are separated in the 3′-untranslated region of Red clover necrotic mosaic virus RNA1. Virology 369:168–181 [DOI] [PubMed] [Google Scholar]
- 27. Iwakawa HO, et al. 2011. Template recognition mechanisms by replicase proteins differ between bipartite positive-strand genomic RNAs of a plant virus. J. Virol. 85:497–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwakawa HO, et al. 2008. A viral noncoding RNA generated by cis-element-mediated protection against 5′→3′ RNA decay represses both cap-independent and cap-dependent translation. J. Virol. 82:10162–10174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iwasaki S, Kawamata T, Tomari Y. 2009. Drosophila argonaute1 and argonaute2 employ distinct mechanisms for translational repression. Mol. Cell 34:58–67 [DOI] [PubMed] [Google Scholar]
- 30. Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell. Biol. 11:113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kahvejian A, Svitkin YV, Sukarieh R, M'Boutchou MN, Sonenberg N. 2005. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 19:104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kneller EL, Rakotondrafara AM, Miller WA. 2006. Cap-independent translation of plant viral RNAs. Virus Res. 119:63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Komoda K, Naito S, Ishikawa M. 2004. Replication of plant RNA virus genomes in a cell-free extract of evacuolated plant protoplasts. Proc. Natl. Acad. Sci. U. S. A. 101:1863–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuhn U, Pieler T. 1996. Xenopus poly(A) binding protein: functional domains in RNA binding and protein-protein interaction. J. Mol. Biol. 256:20–30 [DOI] [PubMed] [Google Scholar]
- 35. Lax SR, Lauer SJ, Browning KS, Ravel JM. 1986. Purification and properties of protein synthesis initiation and elongation factors from wheat germ. Methods Enzymol. 118:109–128 [DOI] [PubMed] [Google Scholar]
- 36. Le H, Browning KS, Gallie DR. 2000. The phosphorylation state of poly(A)-binding protein specifies its binding to poly(A) RNA and its interaction with eukaryotic initiation factor (eIF) 4F, eIFiso4F, and eIF4B. J. Biol. Chem. 275:17452–17462 [DOI] [PubMed] [Google Scholar]
- 37. Lellis AD, et al. 2010. Deletion of the eIFiso4G subunit of the Arabidopsis eIFiso4F translation initiation complex impairs health and viability. Plant Mol. Biol. 74:249–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li J, Chorba JS, Whelan SP. 2007. Vesicular stomatitis viruses resistant to the methylase inhibitor sinefungin upregulate RNA synthesis and reveal mutations that affect mRNA cap methylation. J. Virol. 81:4104–4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miller WA, Wang Z, Treder K. 2007. The amazing diversity of cap-independent translation elements in the 3′-untranslated regions of plant viral RNAs. Biochem. Soc. Trans. 35:1629–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mine A, et al. 2010. Interactions between p27 and p88 replicase proteins of Red clover necrotic mosaic virus play an essential role in viral RNA replication and suppression of RNA silencing via the 480-kDa viral replicase complex assembly. Virology 407:213–224 [DOI] [PubMed] [Google Scholar]
- 41. Mine A, et al. 2010. Identification and characterization of the 480-kilodalton template-specific RNA-dependent RNA polymerase complex of red clover necrotic mosaic virus. J. Virol. 84:6070–6081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mizumoto H, Iwakawa HO, Kaido M, Mise K, Okuno T. 2006. Cap-independent translation mechanism of red clover necrotic mosaic virus RNA2 differs from that of RNA1 and is linked to RNA replication. J. Virol. 80:3781–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mizumoto H, Tatsuta M, Kaido M, Mise K, Okuno T. 2003. Cap-independent translational enhancement by the 3′ untranslated region of red clover necrotic mosaic virus RNA1. J. Virol. 77:12113–12121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Narabayashi T, Iwahashi F, Kaido M, Okuno T, Mise K. 2009. Melandrium yellow fleck bromovirus infects Arabidopsis thaliana and has genomic RNA sequence characteristics that are unique among bromoviruses. Arch. Virol. 154:1381–1389 [DOI] [PubMed] [Google Scholar]
- 45. Nicholson BL, White KA. 2008. Context-influenced cap-independent translation of Tombusvirus mRNAs in vitro. Virology 380:203–212 [DOI] [PubMed] [Google Scholar]
- 46. Nicholson BL, White KA. 2011. 3′ Cap-independent translation enhancers of positive-strand RNA plant viruses. Curr. Opin. Virol. 1:373–380 [DOI] [PubMed] [Google Scholar]
- 47. Nicholson BL, Wu B, Chevtchenko I, White KA. 2010. Tombusvirus recruitment of host translational machinery via the 3′ UTR. RNA 16:1402–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nietfeld W, Mentzel H, Pieler T. 1990. The Xenopus laevis poly(A) binding protein is composed of multiple functionally independent RNA binding domains. EMBO J. 9:3699–3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pestova TV, Kolupaeva VG. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 16:2906–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Polacek C, Friebe P, Harris E. 2009. Poly(A)-binding protein binds to the non-polyadenylated 3′ untranslated region of dengue virus and modulates translation efficiency. J. Gen. Virol. 90:687–692 [DOI] [PubMed] [Google Scholar]
- 51. Sachs AB, Bond MW, Kornberg RD. 1986. A single gene from yeast for both nuclear and cytoplasmic polyadenylate-binding proteins: domain structure and expression. Cell 45:827–835 [DOI] [PubMed] [Google Scholar]
- 52. Sachs AB, Davis RW, Kornberg RD. 1987. A single domain of yeast poly(A)-binding protein is necessary and sufficient for RNA binding and cell viability. Mol. Cell. Biol. 7:3268–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sarawaneeyaruk S, et al. 2009. Host-dependent roles of the viral 5′ untranslated region (UTR) in RNA stabilization and cap-independent translational enhancement mediated by the 3′ UTR of Red clover necrotic mosaic virus RNA1. Virology 391:107–118 [DOI] [PubMed] [Google Scholar]
- 54. Shen R, Miller WA. 2007. Structures required for poly(A) tail-independent translation overlap with, but are distinct from, cap-independent translation and RNA replication signals at the 3′ end of Tobacco necrosis virus RNA. Virology 358:448–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shen X, et al. 1999. Poly [G] improved protein productivity of cell-free translation by inhibiting mRNase in wheat germ extract. J. Biotechnol. 75:221–228 [Google Scholar]
- 56. Skabkina OV, et al. 2003. Poly(A)-binding protein positively affects YB-1 mRNA translation through specific interaction with YB-1 mRNA. J. Biol. Chem. 278:18191–18198 [DOI] [PubMed] [Google Scholar]
- 57. Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stupina VA, et al. 2008. The 3′ proximal translational enhancer of Turnip crinkle virus binds to 60S ribosomal subunits. RNA. 14:2379–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Takeda A, et al. 2005. A plant RNA virus suppresses RNA silencing through viral RNA replication. EMBO J. 24:3147–3157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Taniguchi T, et al. 2010. Induction of endosomal/lysosomal pathways in differentiating osteoblasts as revealed by combined proteomic and transcriptomic analyses. FEBS Lett. 584:3969–3974 [DOI] [PubMed] [Google Scholar]
- 61. Treder K, et al. 2008. The 3′ cap-independent translation element of Barley yellow dwarf virus binds eIF4F via the eIF4G subunit to initiate translation. RNA. 14:134–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vazquez-Pianzola P, Urlaub H, Suter B. 2011. Pabp binds to the osk 3′UTR and specifically contributes to osk mRNA stability and oocyte accumulation. Dev. Biol. 357:404–418 [DOI] [PubMed] [Google Scholar]
- 63. von Der Haar T, Ball PD, McCarthy JE. 2000. Stabilization of eukaryotic initiation factor 4E binding to the mRNA 5′-Cap by domains of eIF4G. J. Biol. Chem. 275:30551–30555 [DOI] [PubMed] [Google Scholar]
- 64. Wallace ST, Schroeder R. 1998. In vitro selection and characterization of streptomycin-binding RNAs: recognition discrimination between antibiotics. RNA 4:112–123 [PMC free article] [PubMed] [Google Scholar]
- 65. Wang S, Browning KS, Miller WA. 1997. A viral sequence in the 3′-untranslated region mimics a 5′ cap in facilitating translation of uncapped mRNA. EMBO J. 16:4107–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang Z, Kraft JJ, Hui AY, Miller WA. 2010. Structural plasticity of Barley yellow dwarf virus-like cap-independent translation elements in four genera of plant viral RNAs. Virology 402:177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang Z, Parisien M, Scheets K, Miller WA. 2011. The cap-binding translation initiation factor, eIF4E, binds a pseudoknot in a viral cap-independent translation element. Structure 19:868–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang Z, Treder K, Miller WA. 2009. Structure of a viral cap-independent translation element that functions via high-affinity binding to the eIF4E subunit of eIF4F. J. Biol. Chem. 284:14189–14202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu W, White KA. 2009. RNA-based regulation of transcription and translation of aureusvirus subgenomic mRNA1. J. Virol. 83:10096–10105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang F, Simon AE. 2003. A novel procedure for the localization of viral RNAs in protoplasts and whole plants. Plant J. 35:665–673 [DOI] [PubMed] [Google Scholar]