

Abstract
Major histocompatibility complex class I (MHC-I) molecules are critically important in the host defense against various pathogens through presentation of viral peptides to cytotoxic T lymphocytes (CTLs), a process resulting in the destruction of virus-infected cells. Herpesviruses interfere with CTL-mediated elimination of infected cells by various mechanisms, including inhibition of peptide transport and loading, perturbation of MHC-I trafficking, and rerouting and proteolysis of cell surface MHC-I. In this study, we show that equine herpesvirus type 4 (EHV-4) modulates MHC-I cell surface expression through two different mechanisms. First, EHV-4 can lead to a significant downregulation of MHC-I expression at the cell surface through the product of ORF1, a protein expressed with early kinetics from a gene that is homologous to herpes simplex virus 1 UL56. The EHV-4 UL56 protein reduces cell surface MHC-I as early as 4 h after infection. Second, EHV-4 can interfere with MHC-I antigen presentation, starting at 6 h after infection, by inhibition of the transporter associated with antigen processing (TAP) through its UL49.5 protein. Although pUL49.5 has no immediate effect on overall surface MHC-I levels in infected cells, it blocks the supply of antigenic peptides to the endoplasmic reticulum (ER) and transport of peptide-loaded MHC-I to the cell surface. Taken together, our results show that EHV-4 encodes at least two viral immune evasion proteins: pUL56 reduces MHC-I molecules on the cell surface at early times after infection, and pUL49.5 interferes with MHC-I antigen presentation by blocking peptide transport in the ER.
INTRODUCTION
Equine herpesvirus type 4 (EHV-4) is a member of the subfamily of the Alphaherpesvirinae and the genus Varicellovirus (5, 22). EHV-4 causes respiratory disease predominantly in young horses with symptoms that vary in severity from subclinical to severe and include fever, lethargy, anorexia, nasal discharge, and cough (20, 69, 72). The immune response to EHV-4 infections remains poorly understood, which is partly caused by the lack of immunologically and/or virologically naïve natural hosts (35, 64). As with other respiratory infections, mucosal antibodies and/or cell-mediated immunity may play a role in controlling EHV-4 infections (14). However, it is known that protective immune responses established after either infection or vaccination are short-lived and incomplete (60, 74).
Cytotoxic T lymphocytes (CTLs) represent an essential part of the immune defense against many virus infections, but their role in EHV-4 infections is not well-defined (8). Major histocompatibility complex class I (MHC-I) molecules are present on virtually all somatic cells and essential for the establishment of a sustained immune response against various pathogens (51, 80). The molecules are generated through a complex antigen presentation pathway in which antigenic peptides are first generated by degradation of cytosolic proteins (including virus-encoded proteins) by the proteasome. The peptides are then transported from the cytosol into the lumen of the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP). Upon exit from the ER, MHC-I molecules can rapidly traffic to the cell surface through the default secretory pathway without requirements for specific signals (bulk flow). However, recent evidence of sorting of MHC-I molecules in the trans-Golgi network (TGN) suggests that the regulated expression of MHC-I molecules at the cell surface can be achieved through post-Golgi traffic control (21, 44, 45, 52, 65, 88). CTLs recognize viral peptides presented by MHC-I molecules on the surface of infected cells and induce their lysis (24, 84, 88, 89). The reduction or lack of MHC-I surface expression can render virus-infected cells more resistant to CTL-mediated killing.
Although the complexity of the pathway provides an efficient way for infected cells to eliminate viral pathogens, it also gives viruses numerous opportunities to interfere with the individual steps of the pathway, thereby thwarting the CTL response of the host (36, 66). Thus, many viruses have evolved mechanisms to interfere with MHC-I cell surface expression as a means of evading the host immune response (56, 67).
Herpesviruses are intracellular pathogens that have acquired diverse mechanisms to prevent clearance by the immune system (32, 34, 41, 53). The immune evasion strategies targeting different stages of the MHC-I antigen processing and presentation pathway allow herpesviruses to establish lifelong latency. Human cytomegalovirus (HCMV) and mouse cytomegalovirus (MCMV) increase MHC-I degradation by the proteasome (27, 28, 57, 58, 73, 85, 87). However, HCMV can also prevent ATP binding to TAP, thereby limiting the energy supply that is necessary for peptide transport (33, 37). Kaposi's sarcoma-associated herpesvirus (KSHV) accelerates endocytosis of cell surface MHC-I molecules (16, 17, 19). Several other herpesviruses have evolved mechanisms to interfere with TAP function through different strategies exerted by unique gene products. Herpes simplex virus 1 (HSV-1) and HSV-2 encode the ICP47 protein that acts as a high-affinity competitor for peptide binding to TAP and thus prevents peptide translocation (3, 4, 25, 29, 38, 63, 79). The bovine herpesvirus 1 (BHV-1) UL49.5 protein induces a conformational arrest of the peptide transporter and degradation of TAP by the proteasome (47, 82). The pUL49.5 homologs encoded by pseudorabies (PrV), EHV-1, and EHV-4 can also inhibit TAP in vitro (49). EHV-1 pUL49.5 and EHV-4 pUL49.5 have been shown to interfere with the binding of ATP to TAP in transfected cells (49). Previous data also indicate that EHV-1 infection results in enhanced endocytosis of cell surface MHC-I, resulting in a dramatic downregulation of MHC-I expression (68). A recent study conducted in our laboratory showed that a newly identified immunomodulatory protein encoded by the UL56 homologue of EHV-1 is involved in MHC-I downregulation. The protein, however, is incapable of performing its function outside the context of viral infection (59). At present, no data on the ability of EHV-4, a close relative of EHV-1, to affect the expression of MHC-I on the surface of infected cells are available.
In the present study, we show that EHV-4 reduces MHC-I cell surface expression through two different mechanisms. First, EHV-4 leads to a significant downregulation of MHC-I expression on the cell surface early after infection through the product of ORF1, a protein expressed with early kinetics from the UL56 homologue and therefore dubbed pUL56 (77). Second, EHV-4 can interfere with MHC-I antigen presentation by inhibition of TAP through the ORF10 gene product a protein which is conserved in all known members of the herpesviruses and which is encoded by the UL49.5 homologue; it is referred to here as pUL49.5 (77). Although pUL49.5 has no readily detectable role in downregulation of cell surface MHC-I, it is able to block MHC-I maturation by blocking TAP and peptide transport into the ER.
MATERIALS AND METHODS
Virus and cells.
All viruses used here are based on EHV-4 strain WA79, which is derived from an infectious bacterial artificial chromosome (BAC) clone (pYO03) that was used for all manipulations and for generation of recombinant viruses as described before (9). Equine dermal (NBL-6) cells were grown in Iscove's modified Dulbecco's medium (IMDM; Invitrogen) supplemented with 20% fetal bovine serum (FBS; Biochrom), 100 U/ml penicillin, and 100 μg/ml streptomycin (1% penicillin-streptomycin). Fetal horse kidney (FHK) cells (generously provided by V. Swanson, University of Iceland) were maintained in Dulbecco's modified Eagle's medium (DMEM; Biochrom) supplemented with 20% FBS. Human embryonic kidney (293), Vero, Madin-Darby bovine kidney (MDBK), murine melanoma B78H1, and B78HC2 cells (with B78HC2 cells expressing equine MHC-I) were kindly provided by Arthur Frampton, University of North Carolina at Wilmington, and propagated in DMEM supplemented with 10% FBS.
Antibodies.
The anti-MHC-I monoclonal antibody (MAb) H58A (isotype immunoglobulin G2a [IgG2a]) was purchased from VMRD. The anti-equine MHC-I MAb CZ3 was provided by Douglas Antczak, Cornell University, Ithaca, NY. Polyclonal anti-pUL56 and anti-pUL49.5 antibodies were generated by Genscript (Table 1). The anti-CD44 MAb was purchased from BioLegend. Polyclonal antibodies specific for gD were kindly provided by Ken Maeda, Yamaguchi University, Japan. MAb P4C2, an α4β1 integrin antagonist, was obtained from Abcam. An unrelated mouse IgG isotype control and rabbit anti-β-actin were purchased from Cell Signaling Technologies. Alexa Fluor 647-labeled goat antimouse secondary antibodies were purchased from Invitrogen. Horseradish peroxidase-conjugated goat antirabbit and goat antimouse antibodies were obtained from Southern Biotech.
Table 1.
Synthetic peptides derived from EHV-4 used to raise polyclonal antibodies
| Peptide | Amino acid sequence | Amino acid residues |
|---|---|---|
| pUL49.5 | EAKQRLDVAREEERRDFWHAAC | 30–51 |
| pUL56 | CSLGSVTYLSEQQLPSRPPS | 75–93 |
Plasmids.
The entire EHV-4 UL56 and UL49.5 genes were amplified by PCR using the primers listed in Table 2. The PCR products were digested with the appropriate restriction enzymes and inserted into vector pcDNA3 (Invitrogen), resulting in recombinant plasmids pcDNA_56 and pcDNA_49.5, respectively. To construct pcDNA_56Kan, the kanamycin resistance kanr gene was amplified from plasmid pEPkan-S by PCR using the primers listed in Table 2. The PCR product was digested with the appropriate restriction enzymes and inserted into pcDNA_56. Correct amplification and insertion were confirmed by nucleotide sequencing (Starseq).
Table 2.
Oligonucleotides used in this study
| Primer use and primer | Product | Sequence (5′–3′)a |
|---|---|---|
| Mutagenesis | ||
| UL56FW | UL56 deletion | cagcaaaggctcctcgatgacagcagcgccacgtcgtgctatggctggctttagtgatgcaggatgacgacgataagtaggg |
| UL56RV | gtgaaaataaacataatacaactgtgttgaaccacttgttgacgagcagaaaagtgtaaacaaccaattaaccaattctgattag | |
| UL49.5FW | UL49.53A | ctcgcgcttcccatccattactacattcaaccaccacgatAttgtcagcgagattagtgacaggatgacgacgataagtaggg |
| UL49.5RV | ggcaggttaaaatggccagcgtcactaatctcgctgacaaTatcgtggtggttgaatgtagcaaccaattaaccaattctgattag | |
| Revertant | ||
| UL56FW1 | UL56-Kan | atttagccttccgctcctgtctgcttacactttacacttttctgctcgtcatgaggcccacggaagtttc |
| Ul56RV1 | aggggtgtttgtgaaaataaacataatacaactgtgttgaaccacttgttttaatttttggattttgtga | |
| UL49.5FW1 | UL49.53 M | ctcgcgcttcccatccattactacattcaaccaccacgatGttgtcagcgagattagtgacaggatgacgacgataagtaggg |
| UL49.5RV1 | ggcaggttaaaatggccagcgtcactaatctcgctgacaaCatcgtggtggttgaatgtacaaccaattaaccaattctgattag | |
| Plasmids | ||
| UL56FW2 | UL56 | attgaattcatgaggcccacggaagtttc |
| UL56RV2 | cacggatccttaatttttggattttgtga | |
| UL56FW Kan | Kan | gcggcatgctgtgcacctccgcgtttggaaggatgacgacgataagtaggg |
| UL56RV Kan | acagcatgccgcgtggctgtgttgtggagcaaccaattaaccaattctgattag | |
| UL49.5FW2 | UL49.5 | tatgaattcgcatgcagcctgctccggac |
| UL49.5RV2 | tatggatcctcagtgtaggtgtcgcaaca | |
| Sequencing | ||
| UL56FW3 | gctcctccctaaatgccttg | |
| UL56RV3 | atagtgtaccgtggatttgg | |
| UL49FW3 | tgctgcggcttcccatccat | |
| UL50RV | ttctagtaggtttagcgcta |
Restriction enzyme sites are given in lowercase bold letters; sequences in italics indicate additional bases which are not present in the EHV-1 or -4 sequence. Underlined sequences indicate the template-binding region of the primers for PCR amplification with pEPkan-S. Uppercase bold letters indicate the nucleotides that were mutated.
For generation of NBL-6 cells, which constitutively express EHV-4 UL56 or UL49.5 (NBL/56 or NBL/49.5), NBL-6 cells were transfected with the recombinant pcDNA_56, pcDNA_49.5, or empty pcDNA3 (control) plasmid using electroporation (260 V, 1,050 μF, and 335 Ω) as described before (75). Transfected cells were grown in the presence of 1 mg/ml G418 (Invitrogen) for selection. Resistant cells were selected and analyzed by Western blotting for expression of UL56 and UL49.5. In other experiments, NBL-6 cells were transiently transfected with the pcDNA_49.5 expression plasmid without applying antibiotic selection.
BAC cloning and mutagenesis.
EHV-4 BAC clone pYO03, generated previously by inserting mini-F-plasmid sequences flanked by loxP sites into the intergenic region between genes 58 and 59 (9), was introduced into Escherichia coli strain GS1783 cells (a kind gift from Greg Smith, Northwestern University, Chicago, IL). Bacteria were maintained in Luria-Bertani (LB) medium containing 30 μg/ml chloramphenicol. Viruses reconstituted from pYO03 were used in this study to make use of enhanced green fluorescent protein (EGFP) expression for rapid identification of infected cells. Deletion of UL56 was done by two-step Red recombination as described before (78). PCR primers (Table 2) were designed to have recombination arms of 60 nucleotides that enabled the replacement of the UL56 gene by the kanr gene amplified out of plasmid pEPKan-S. PCR products were digested with DpnI in order to remove residual template DNA, and the transfer fragments were electroporated into GS1783 containing pYO03. Kanamycin-resistant colonies were purified and screened by PCR and restriction fragment length polymorphism (RFLP) to detect E. coli cells harboring mutant clones. Positive clones were subjected to a second round of Red recombination to obtain the final construct, pYOΔUL56, after removal of the kanr gene. To reintroduce the authentic UL56 sequence in pYOΔ56, we first amplified the UL56Kan fragment from the pcDNA_56Kan plasmid and inserted UL56 back into pYOΔUL56, resulting in pYOUL56R (Table 2).
A point mutation targeting the AUG start codon of the EHV-4 UL49.5 gene (pYO49.53A) was engineered by converting nucleotide 11673 of UL49.5 from a guanine to an adenine, changing the methionine codon into an isoleucine (UL49.53A), using two-step Red-mediated recombination. The change resulted in the inhibition of UL49.5 gene expression without affecting expression of the overlapping gene (UL50). Primers used for generation of both the mutant (UL49.53A) and the revertant (UL49.5R) are listed in Table 2.
Finally, a double mutant that lacked expression of both UL49.5 and UL56 (pYOΔ56_49.53A) was created by targeting the start codon of the UL49.5 gene in the pYOΔUL56 genome and introducing the point mutation as described above. The appropriate revertants were also created to repair both UL56 and UL49.5 as described above. The genotypes of all mutants and revertants were confirmed by PCR, RFLP, and nucleotide sequencing using primers listed in Table 2.
Virus growth assays.
Reconstitution of mutant (pYOΔ56, pYO49.53A, or pYOΔ56_49.53A) and revertant viruses was achieved by transfection of purified BAC DNA into 293 cells using Lipofectamine 2000 (Invitrogen) as described earlier (9, 11). After 3 days, cells and supernatants were collected and used to infect confluent NBL-6 cells.
To compare the in vitro growth properties of EHV-4Δ56, EHV-4_49.53A, and EHV-4Δ56_49.53A with parental and revertant viruses, plaque areas and growth kinetics were determined as described before (12, 83). For plaque size measurements, confluent monolayers of NBL-6 cells in 12-well plates were infected at a multiplicity of infection (MOI) of 0.001 and overlaid at 2 h postinfection (p.i.) with DMEM containing 1.5% methylcellulose (Sigma). For each virus, 50 plaques were photographed at 5 days p.i., and mean plaque sizes were analyzed by using ImageJ software (http://rsb.info.nih.gov/ij/). Values were calculated and compared to plaque areas induced by parental virus, which were set to 100%. The average percentage of plaque areas and standard deviations were determined from three independent experiments. For determining growth kinetics, NBL-6 cells were infected at an MOI of 0.5. Viruses were allowed to attach for 1.5 h at 4°C and then to penetrate for 2 h at 37°C. Infected cultures were harvested at the indicated times p.i. (0, 6, 12, 24, 48, and 72 h), and viral titers were determined by plating onto NBL-6 cells. At day 4 p.i., cells were stained with 0.3% crystal violet, and plaques were counted. Growth curves were determined in three independent experiments.
Western blotting.
For Western blot analysis, FHK cells were seeded in 6-well plates and infected with parental, mutant, or revertant viruses. Pellets of infected cells or stably transfected cell lines (NBL/56 or NBL/49.5) were resuspended in radioimmunoprecipitation assay (RIPA) buffer (1 mM Tris, pH 7.4, 1% Triton X-100, 0.25% sodium deoxycholate, 5 M sodium chloride, 0.5 mM EDTA, 0.1% sodium dodecyl sulfate [SDS]) and protease inhibitor cocktail (Roche). Samples were mixed with sample loading buffer (1 M Tris-HCl, pH 6.8, 0.8% SDS. 0.4% glycerol, 0.15% β-mercaptoethanol, 0.004% bromophenol blue), heated at 65°C for 10 min, and subjected to SDS–10% polyacrylamide gel electrophoresis (PAGE) exactly as described before (83). Expression of EHV-4 UL56, UL49.5, and gD or cellular β-actin was detected with the specific antibodies and peroxidase conjugates listed earlier. Reactive bands were visualized by enhanced chemiluminescence (ECL plus; Amersham).
Temporal inhibition of viral protein expression.
To limit viral gene expression to immediate-early (IE) and early (E) genes, NBL-6 cells were infected with EHV-4 at an MOI of 5 in the presence of the viral DNA synthesis inhibitor phosphonoacetic acid (PAA; Sigma) at a concentration of 300 μg/ml, followed by incubation in fresh medium containing PAA for 24 h (40). Cycloheximide (CX; Sigma) at a concentration of 100 μg/ml was used to inhibit viral protein synthesis in EHV-4-infected cells (62). In order to differentiate between involvement of IE and E genes, NBL-6 cells were infected with EHV-4 in the presence of CX to allow accumulation of EHV-4 IE proteins in cells. After 5 h, the cells were extensively washed and incubated for 24 h with either fresh medium or medium containing the transcriptional inhibitor actinomycin D (Act-D; Sigma) at 5 μg/ml. Act-D allows the translation of already accumulated IE mRNA, while preventing further transcription of mRNA. In all experiments, the cells were analyzed by using flow cytometry.
To detect the life span of surface MHC-I molecules, cells were incubated with either CX or brefeldin A (Sigma) at a concentration of 50 μg/ml and 1 μg/ml, respectively. At different time points (24, 48, and 72 h), cells were checked to detect the expression level of surface MHC-I molecules.
Citrate wash experiments.
To analyze the kinetics of MHC-I surface restoration, NBL-6 cells were either mock infected or infected with parental or mutant viruses. The cells were incubated at 37°C for 6 h until the start of UL49.5 expression (time point, 0 h). After removal of the medium, the cells were washed with citrate buffer (0.062 M Na2HPO4, 0.132 M citric acid, 0.5% bovine serum albumin, pH 3) for 1 min as described previously (76). The cells were then washed three times with phosphate-buffered saline (PBS), harvested at different time points (0, 8, 16, and 24 h), and subjected to analysis by flow cytometry.
Flow cytometry.
To evaluate expression of MHC-I molecules on the cell surface, cells (NBL-6, FHK, MDBK, Vero, B78HI, and B78HC2 cells) were either mock or virus infected. The cells were trypsinized and immediately suspended in fluorescent-activated cell sorter (FACS) buffer (PBS containing 2% FBS and 0.01% sodium azide) at the indicated times p.i. Cells were added in duplicate into 96-well round-bottom plates and incubated with 2 μg/ml of MAbs targeting MHC-I (H58A or CZ3), CD44, or α4β1 integrin (P4C2) or an isotype control anti-mouse IgG for 1 h at 4°C. Then, the cells were washed three times in FACS buffer and stained with Alexa Fluor 647-labeled goat anti-mouse IgG (5 μg/ml) for 30 min at 4°C. After three washing steps, 10,000 cells were analyzed with a FACSCalibur flow cytometer (BD Biosciences), and the intensity of fluorescence was analyzed using FlowJo software (TreeStar).
In vitro peptide transport assay.
NBL-6 cells were either mock infected or infected with parental, mutant, or revertant viruses. The cells were trypsinized and washed twice in growth medium and twice in ice-cold transport buffer (130 mM KCl, 10 mM NaCl, 1 mM CaCl2, 2 mM EGTA, 2 mM MgCl2, and 5 mM HEPES [pH 7.3] with KOH). Cells were permeabilized with streptolysin O (SLO; Sigma) at a concentration of 2 U/ml for 10 min at 37°C (13, 47). Subsequently, cells were washed and incubated with 4 μM of the fluorescence-conjugated synthetic peptide CVNKTERAY (Genescript) in the presence of ATP (200 mM, Sigma) for 10 min at 37°C. Peptide translocation was terminated by adding ice-cold stopping buffer (transport buffer containing 10 mM EDTA and 0.01% NaN3) at 4°C. The samples were centrifuged at 16,000 × g for 20 min, and the supernatant was removed. The cell pellets were then incubated with ice-cold lysis buffer (1% Triton X-100, 500 mM NaCl, 2 mM MgCl2, 50 mM Tris HCl, pH 8.0) for 30 min at 4°C. After lysis, cell debris was removed by centrifugation and cell lysates were incubated with concanavalin A (ConA)-Sepharose beads (Sigma) for 2 h at 4°C with gentle agitation. Finally, the glycosylated peptides were eluted with buffer (500 mM mannopyranoside, 10 mM EDTA, 50 mM Tris HCl, pH 8.0) during a 2-h incubation step at room temperature with vigorous shaking and separated from ConA-Sepharose beads by centrifugation at 12,000 × g for 2 min. The fluorescence intensity of the samples was measured using a fluorescence plate reader (Berthold Technologies).
Statistical analysis.
Using Microsoft Excel software, Student's t test for paired data was used to test for statistically significant differences. Data given are means, and bars show standard deviations.
RESULTS
EHV-4-mediated MHC-I downregulation on the surface of infected equine cells.
Confluent equine cells (NBL-6 and FHK) were either mock infected or infected with EHV-4 at an MOI of 5. After 24 h, when infection rates had reached between 80 and 90%, infected cells were harvested, stained with anti-MHC-I MAb H58A, and analyzed by flow cytometry. Cell surface expression of CD44 receptors was measured as a control of cellular protein synthesis, transport, and affection by virus infection. Robust cell surface expression of MHC-I was seen with MAb H58A (recognizing equine MHC-I) on all mock-infected cells (Fig. 1A). On the other hand, we observed a significant downregulation of cell surface MHC-I in infected cells (Fig. 1A), whereas the expression of the cell surface molecule CD44 remained unaffected (data not shown). MHC-I levels on the surface of equine cells were evaluated using another anti-equine MHC-I MAb, CZ3, which confirmed the downregulation in infected but not uninfected cells (data not shown).
Fig 1.

EHV-4 downregulates cell surface MHC-I molecules on equine cells only. Equine NBL-6 and FHK (A) and nonequine MDBK, Vero, and B78HC2 (B) cells were either mock infected or infected with EHV-4 and subjected to flow cytometric analysis. Histograms of MHC-I expression after detection with anti-MHC-I MAb (H58A) are shown. As controls, cells were incubated with irrelevant MAbs of the same IgG isotype. Viable cells (10,000) were analyzed for each sample. Results of one representative experiment are shown.
We further determined whether downregulation of cell surface MHC-I can occur in cells of nonequine origin that can be infected with EHV-4. MDBK and Vero cells were either mock infected or infected with EHV-4 at an MOI of 5 and also analyzed by flow cytometry at 24 h p.i. Expression of MHC-I on the surface of these cells was confirmed using MAb H58A (Fig. 1B) and was not affected on the surface of either MDBK or Vero cells in the absence or presence of virus infection (Fig. 1B).
We reasoned that this result may be attributable to the fact that EHV-4 can bind only to equine MHC-I and not to nonequine MHC-I or that species-specific adaptor proteins are targeted by viral gene products (10). To address these possibilities, murine B78HC2 cells expressing equine MHC-I and conferring EHV-4 entry were also tested for downregulation of MHC-I. Our results showed that equine MHC-I expression on the surface of the murine cell line was not affected, although equine MHC-I was highly expressed on the surface of those cells (Fig. 1B). From our results, we concluded that EHV-4 can downregulate MHC-I on the surface of equine cells only. On the other hand, EHV-4 was not able to downregulate MHC-I on the surface of nonequine cells, regardless of whether or not they expressed equine MHC-I.
EHV-4-induced MHC-I downregulation is dependent on an E gene(s).
To determine the class of viral protein involved in cell surface MHC-I downregulation, UV-inactivated EHV-4 was used to determine whether de novo viral protein synthesis was required for MHC-I downregulation. The results showed that infection of NBL-6 cells with UV-inactivated virus resulted in no loss of MHC-I expression (Fig. 2). To determine whether late viral proteins are involved in MHC-I downregulation, NBL-6 cells were infected for 24 h (infection rates, approximately 90%) in the presence of PAA, which inhibits DNA replication and late viral gene expression. Cell surface MHC-I downregulation was still observed in the presence of PAA (Fig. 2), indicating that a protein(s) expressed with either IE or E kinetics is responsible for cell surface MHC-I downregulation. To further investigate which class of viral proteins was responsible for the activity, we used the protein synthesis inhibitor CX alone or in combination with Act-D. CX treatment followed by Act-D incubation effectively abrogated MHC-I downregulation following virus infection (Fig. 2). Since removal of CX and addition of Act-D should allow translation of IE genes, our results strongly suggest that an EHV-4 early gene, either alone or in association with another gene(s), is responsible for the observed downregulation of cell surface MHC-I.
Fig 2.

An early EHV-4 protein(s) is responsible for MHC-I downregulation. NBL-6 cells were either mock infected or infected with live 4 or UV-inactivated EHV-4 (left panel) or mock infected or infected with EHV-4 in the presence of PAA (middle panel). Cells were infected with EHV-4 in the presence of CX at a concentration of 100 μg/ml for 5 h. The cells were then washed and incubated for 24 h with either fresh medium or medium containing Act-D (right). Histogram overlays of MHC-I expression detected by primary anti-MHC-1 MAb (H58A) and secondary Alexa Fluor 647-conjugated goat anti-mouse IgG are shown. Irrelevant MAbs of the same IgG isotype were used as controls. Results of one representative experiment are shown.
EHV-4 pUL56 is an early viral protein that induces robust MHC-I downregulation in EHV-4-infected cells.
A previous study from our laboratory had identified the EHV-1 UL56 gene product in cell surface MHC-I downregulation (59). Based on the fact that ORF1, the UL56 homolog, (i) is expressed early during viral infection and (ii) is closely similar to its counterpart in EHV-1, we chose to study the possible role of EHV-4 pUL56 in MHC-I downregulation.
To test the hypothesis that EHV-4 pUL56 is responsible for the observed effect on MHC-I, we deleted this gene in the parental virus. Furthermore, EHV-4 was engineered with a point mutation in the start codon of UL49.5 and/or a deletion of UL56. To evaluate the expression of UL56 and UL49.5, FHK cells were infected with the parental, mutant, and revertant viruses and the cell lysates were analyzed by Western blotting. Parental and revertant viruses, but not mutant viruses EHV-4Δ56, EHV-4_49.53A, and EHV-4Δ56_49.53A, expressed pUL56 and pUL49.5 at comparable levels (Fig. 3A). Two different bands with apparent molecular masses of 22 and 28 kDa were detected with the pUL56-specific antibody, indicating that this protein may be modified co- or posttranslationally. The observation was compatible with previous findings on EHV-1 pUL56, which is phosphorylated (59). Treatment of EHV-4-infected cell lysates with phosphatase (λ-PPase) resulted in the absence of the more slowly migrating pUL56 moiety, whereas the 22-kDa pUL56 protein remained unaffected (see Fig. 6C). The results suggested, therefore, that the observed 28-kDa moiety represents a phosphorylated form of pUL56. In addition, standard plaque size measurements and independent single-step growth kinetic experiments were performed using NBL-6 cells. All viruses, parental, mutant, and revertant, grew to similar titers in NBL-6 cells, and there was no significant difference between plaque areas induced by the various recombinant viruses (Fig. 3B and C). We concluded from our analyses that the growth rates of mutant viruses were not significantly affected by the introduced mutations.
Fig 3.

Characterization of recombinant viruses. (A) Expression patterns of pUL56 (top) and pUL49.5 (bottom) in parental, mutant, and revertant viruses were detected by Western blotting. Cell lysates were prepared, and proteins were separated by SDS–10% PAGE before transfer to a polyvinylidene difluoride membrane. The blots were incubated with anti-pUL56 or anti-pUL49.5 polyclonal antibodies. Cell lysates from noninfected FHK cells were included as a control. β-Actin was used as a loading control. (B) For growth kinetics, NBL-6 cells were infected at an MOI of 0.5. Infected cells and supernatants were collected and virus titers were determined at the indicated times p.i. The data presented are means ± SDs of triplicate measurements. (C) Mean ± SD diameters of 50 plaques measured for each virus. The plaque diameter of parental viruses was set to 100%.
Fig 6.

EHV-4 pUL49.5, but not pUL56, can alter MHC-I surface expression in the absence of other viral proteins. Expression of UL49.5 (A) and UL56 (B) proteins in transfected cells was detected by Western blotting. NBL-6 cells were transfected with plasmids expressing pcDNA56, pcDNA49.5, or pcDNA3. Cell lysates were prepared and proteins were separated by SDS–10% PAGE. The blots were incubated with anti-pUL49.5 or anti-pUL56 polyclonal antibodies. Cell lysates from noninfected NBL-6 cells were included as a control. β-Actin was used as a loading control. (C) Lysates of EHV-4-infected cells were either treated with λ-PPase or left untreated. pUL56 was detected by Western blotting using anti-UL56 antibody. β-Actin was used as a loading control. (D) Expression of MHC-I molecules on the surface of transfected NBL-6 cells was determined by flow cytometry. The surface expression of MHC-I molecules of the mock-infected cells was set to 100%. P values for statistical significance are given.
Cell surface expression of MHC-I molecules was determined by flow cytometry, using the anti-MHC-I H58A or CZ3 MAb. EHV-4-mediated surface MHC-I downregulation was observed as early as 4 h p.i. and became more prominent from that time point onwards (Fig. 4A). Surface expression of CD44, used as a control, was not affected, indicating that MHC-I molecules were specifically targeted (Fig. 4A).
Fig 4.
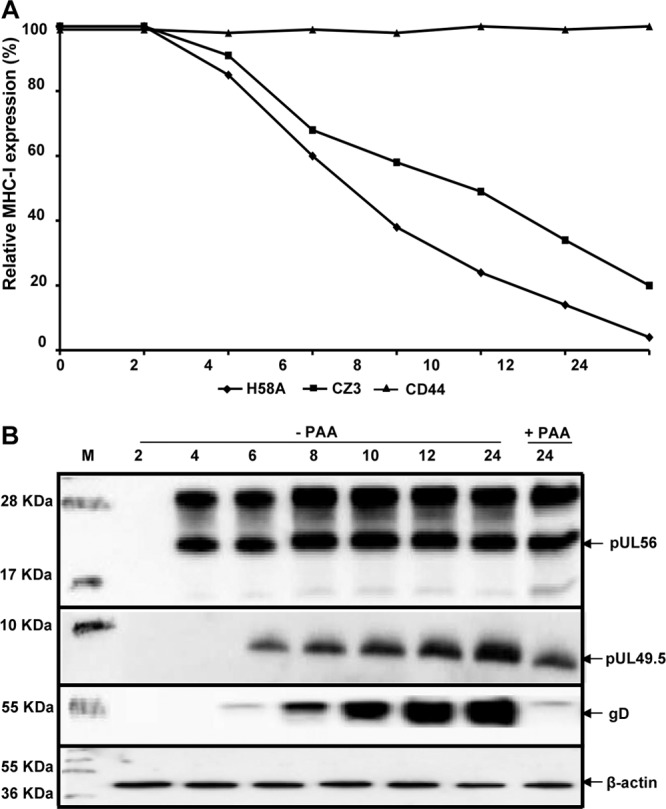
EHV-4 downregulates cell surface MHC-I at early stages of infection. (A) Relative levels of cell surface expression of MHC-I and the cell surface marker CD44 were determined at different time points after EHV-4 infection of NBL-6 cells. At the indicated time points, MHC-I expression was detected with anti-MHC-I MAbs (H58A and CZ3) and analyzed by flow cytometry. The surface expression of both MHC-I and CD44 molecules was set to 100% at time zero. (B) Expression of UL56 and UL49.5 proteins was determined for each time point in panel A by Western blotting in the presence or absence of PAA. Levels of expression of gD and β-actin were determined as temporal and loading controls, respectively.
Western blot analysis was performed with lysates taken at each time point to detect the expression of either pUL56 or pUL49.5 in infected cells in the presence or absence of PAA. Expression of actin and gD, a late viral protein, was assessed to control for the efficiency of PAA treatment. Our results showed that the UL56 protein was detected as early as 4 h p.i. at significant levels, which were not affected by the presence of PAA, in contrast to gD expression (Fig. 4B). On the other hand, pUL49.5 expression started at 6 h p.i. and was slightly affected by the presence of PAA (Fig. 4B).
To analyze a potential involvement of pUL56 and/or pUL49.5 in EHV-4-mediated MHC-I downregulation, mutant EHV-4Δ56, EHV-4_49.53A, or EHV-4Δ56_49.53A was used to infect NBL-6 cells. At the 24-h time point (approximately 88% infected cells), infection with parental EHV-4, EHV-4_49.53A, and all revertant viruses resulted in a significant reduction in cell surface MHC-I expression (Fig. 5A). In contrast, EHV-4Δ56 and EHV-4Δ56_49.53A showed a significantly reduced ability to downregulate MHC-I (Fig. 5A). As observed earlier, cell surface expression of CD44 remained unaffected, indicating that MHC-I molecules were specifically targeted (Fig. 5b). However, when we tested another cell surface molecule, α4β1 integrin, that is also expressed on the surface of NBL-6 cells (10), we found that, similar to the situation with MHC-I, EHV-4 was able to downregulate this surface receptor and that pUL56 is necessary for the function (Fig. 5B). Taken together, our data show that the early UL56 gene, but not UL49.5, is required for downregulation of cell surface MHC-I at early times after EHV-4 infection. However, pUL56 is also able to downregulate another cell surface molecule, α4β1 integrin.
Fig 5.

The UL56 protein is responsible for cell surface MHC-I downregulation in infected cells. (A) NBL-6 cells were either mock infected or infected with parental, mutant, or revertant viruses. MHC-I expression on the cell surface was measured by flow cytometric analysis. Histogram overlays of MHC-I expression detected by anti-MHC-I MAb H58A are given. (B) Expression of other cell surface molecules. Histogram overlays of CD44 and α4β1 integrin expression detected by anti-CD44 and anti-α4β1 integrin (P4C2) MAbs, respectively. Results of one representative experiment are shown.
pUL49.5, but not pUL56, is able to downregulate cell surface MHC-I in the absence of other viral proteins.
To investigate whether the UL49.5 or UL56 proteins are able to alter MHC-I surface expression in the absence of other viral proteins, NBL-6 cells were stably transfected with pcDNA56, pcDNA49.5, or parental pcDNA3, which was used as a control. Furthermore, NBL-6 cells were also transiently transfected with pcDNA49.5 for 72 h. Expression of the UL49.5 and UL56 proteins was verified by Western blot analysis (Fig. 6A and B). Flow cytometric analysis revealed that cells permanently transfected with UL49.5 but not UL56 expression plasmids exhibited a significant reduction of cell surface MHC-I expression compared to the control cells (Fig. 6D). However, after transient expression of UL49.5 protein, we could not detect any change in cell surface expression of MHC-I (data not shown). Western blot analysis showed two bands for the UL56 protein in infected cells (Fig. 3A and 4B), while only a single band was visible in transfected cells (Fig. 6B). Treatment of infected-cell lysates with phosphatase (λ-PPase) resulted in only one band similar to that of UL56 protein in transfected cells (Fig. 6C).
The results indicated that pUL49.5 alone is able to downregulate cell surface MHC-I molecules but that cells need to steadily overexpress the UL49.5 protein to be capable of inhibiting MHC-I levels on the cell surface. On the other hand, pUL56 is unable to reduce MHC-I cell surface expression in transfected NBL-6 cells in the absence of other viral proteins; pUL56 may, therefore, require interaction with (an)other viral protein(s), but not pUL49.5, to exert its effect on MHC-I downregulation. In addition, it appears that phosphorylation of pUL56 also requires the environment of EHV-4 infection.
pUL49.5 is responsible for TAP inhibition in EHV-4-infected cells.
A previous study had shown that UL49.5 proteins of different varicelloviruses, including the UL49.5 protein of EHV-4, have the ability to inhibit TAP when overexpressed in cell lines of the relevant host species (49). However, the role of pUL49.5 in TAP inhibition in infected cells has not yet been studied. To understand the mechanism(s) of MHC-I downregulation, we tested the possibility of TAP inhibition by EHV-4 during infection. NBL-6 cells were either mock infected or infected with mutant virus EHV-4Δ56, EHV-4_49.53A, or EHV-4Δ56_49.53A. After 24 h, TAP-mediated peptide transport was analyzed in permeabilized cells using a fluorescein-conjugated synthetic peptide. We were able to show that parental EHV-4 and EHV-4Δ56 as well as EHV-4_49.5R effectively blocked peptide transport from the cytosol into the ER. In contrast, this inhibition was not seen in cells infected with the EHV-4_49.53A mutant (Fig. 7A). In addition, a similar set of experiments was conducted to determine the onset of TAP inhibition in infected cells, and the results revealed that inhibition of peptide transport activity was observed as early as 6 h p.i. (Fig. 7B). The findings indicate that pUL49.5, but not pUL56, of EHV-4 is responsible for the inhibition of peptide translocation by TAP during virus infection.
Fig 7.
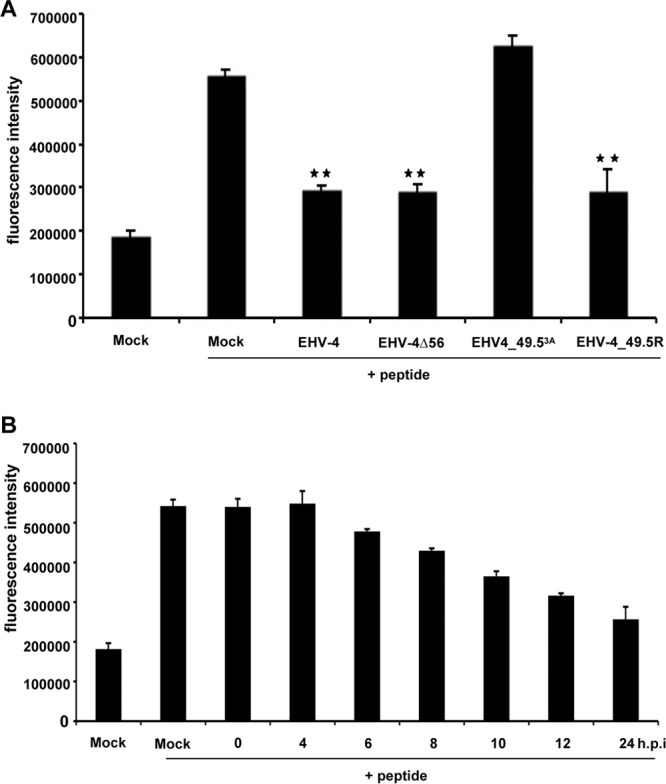
EHV-4 pUL49.5, but not pUL56, significantly inhibits peptide transport into the ER. (A) TAP-dependent peptide transport was determined in NBL-6 cells infected with parental EHV-4, EHV-4Δ56, EHV-4_49.53A, or EHV-4_49.5R. Translocation of the synthetic peptide (CVNKTERAY) carrying a fluorescein-labeled cysteine was evaluated by a fluorescence plate reader. Peptide transport was shown after infection with recombinant viruses and compared to the peptide translocation observed in mock-infected cells. All data represent the means ± SDs of three independent experiments. The asterisks indicate significant differences (P < 0.05) relative to mock-infected cells (+ peptide) means compared to mock-infected cells where peptide was added. (B) EHV-4 inhibits peptide transport activity as early as 6 h p.i. NBL-6 cells were either mock infected or infected with EHV-4 and were used to determine peptide transport at the indicated time points (0, 2, 4, 6, 8, 12, and 24 h p.i.). The data are expressed as fluorescence intensity, which is a measure of peptide translocation into the ER, and related to fluorescence intensities in control cells. The data represent the means ± SDs of two independent experiments.
The UL49.5 protein can affect cell surface MHC-I expression during infection.
We showed that EHV-4_49.53A was able to downregulate surface MHC-I to levels comparable to those for parental virus in infected cells but that EHV-4Δ56_49.53A had a significantly reduced ability to downregulate MHC-I. On the other hand, NBL-6 cells transfected with UL49.5 showed a significant reduction in surface MHC-I expression, likely as a consequence of TAP inhibition. It is noteworthy that MHC-I molecules on the cell surface are constantly internalized by endocytosis and then rerouted and/or degraded. We hypothesized that our inability to detect pUL49.5-mediated downregulation of MHC-I molecules in infected cells might be due to the long half-life of MHC-I molecules on the cell surface and that pUL49.5, unlike pUL56, is not capable of accelerating the internalization rate of MHC-I. To test this hypothesis, we determined the life span of cell surface MHC-I by using CX to inhibit de novo synthesis of cellular proteins. Reduction of cell surface MHC-I started 24 h after the start of treatment, and the rate was significantly increased after 48 h and 72 h, when 28% and 50% of treated cells, respectively, were no longer expressing MHC-I (Fig. 8A). This result was confirmed by using brefeldin A to block transport of the newly synthesized protein to the cell surface, which also demonstrated a significant reduction of cell surface MHC-I molecules after 48 h (Fig. 8A). Furthermore, cells infected with parental or mutant viruses were kept for 48 and 72 h p.i. in order to allow more time for pUL49.5 to inhibit TAP and the cells to remove long-lived cell surface MHC-I. Our results, however, did not show a clear reduction of cell surface levels of MHC-I even after 72 h (Fig. 8B).
Fig 8.
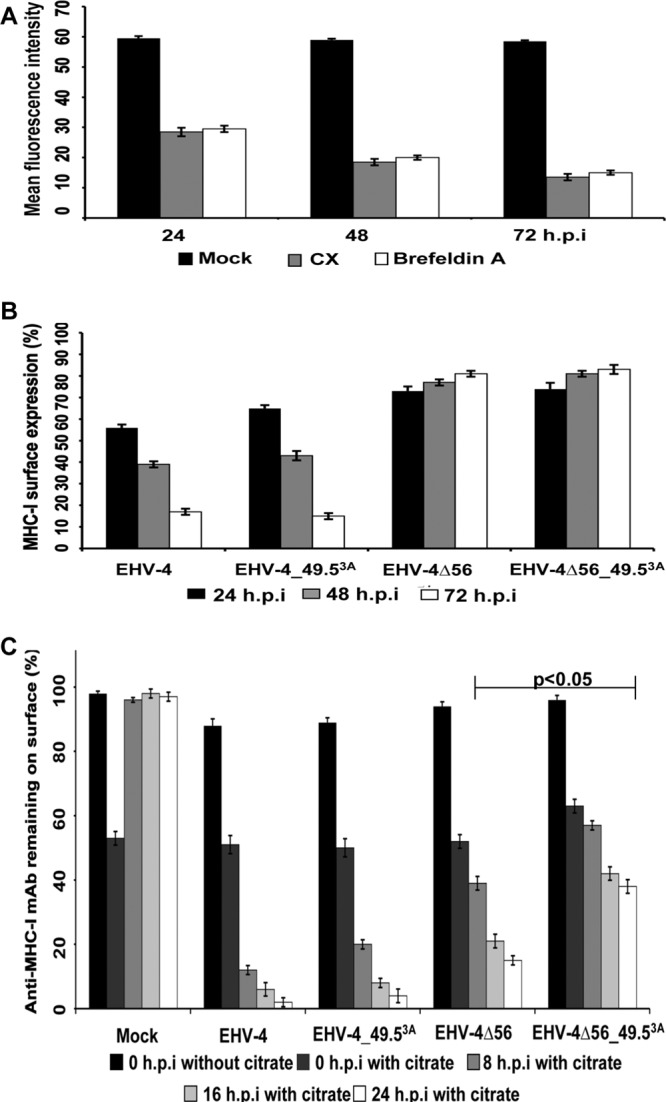
Role of pUL49.5 in MHC-I downregulation on the cell surface. (A) Life span of MHC-I molecules on the cell surface. NBL-6 cells were incubated with either CX or brefeldin A at a concentration of 50 μg/ml and 1 μg/ml, respectively, at the indicated time points. The data represent the means ± SDs of two independent experiments. (B) Downregulation of cell surface MHC-I at different time points. NBL-6 cells were either mock infected or infected with parental or mutant viruses. MHC-I expression on the cell surface was measured by flow cytometry. Expression of MHC-I molecules on the surface of mock-infected cells was set to 100%. The results represent the means ± SDs of two independent experiments. (C) Restoration of cell surface MHC-I in infected cells after citrate treatment. NBL-6 cells were either mock infected or infected with parental or mutant viruses. After 6 h, all cells were treated with citrate buffer (0-h time point). Expression of MHC-I molecules on the cell surface was analyzed at 0, 8, 16, and 24 h after acid treatment. Surface expression of MHC-I molecules on mock-infected cells before citrate treatment was set to 100%. P values for statistical significance are given. The data represent the means ± SDs of two independent experiments.
To further address the question of MHC-I resting times on the cell surface relative to virus infection, we treated mock- or virus-infected cells with citrate buffer (pH 3), which resulted in a significant reduction of cell surface MHC-I in all cases. Eight hours after removal of cell surface MHC-I by acid treatment, mock-infected cells had recovered 100% of the MHC-I molecules on their surfaces. In stark contrast, cells infected with either EHV-4 or EHV-4_49.53A were not able to restore MHC-I and cell surface MHC-I remained low at all time points investigated (8, 16, and 24 h), although a very moderate and not significant increase in MHC-I surface expression was seen in the case of the UL49.5 mutant. In cells infected with either EHV-4Δ56 or the double-deletion mutant EHV-4Δ56_49.53A, MHC-I surface levels clearly increased after citrate treatment compared to those in cells infected with parental virus. In the case of cells infected with EHV-4Δ56_49.53A, a further and additive increase in MHC-I surface levels compared to those for cells infected with the single-deletion mutants was observed, and the increase did reach statistical significance even relative to the levels for cells infected with EHV-4Δ56 alone. From the results, we concluded that (i) pUL49.5 does have an effect on MHC-I downregulation in infected cells that is more pronounced in the absence of pUL56 and (ii) there is at least another viral protein which is able to downregulate MHC-I surface expression and mitigate the role of pUL49.5 on MHC-I expression in infected cells.
DISCUSSION
MHC-I molecules are a particularly attractive target for immune evasion by viruses, as their elimination from the cell surface makes infected cells invisible to CTLs and allows virus to replicate. On the other hand, elimination of cell surface MHC-I as a strategy for deceiving the immune system is not without risk to the virus: natural killer (NK) cells recognize cells deficient in self-molecules, i.e., MHC-I molecules, and destroy them. Inhibition of MHC-I antigen presentation is a hallmark of members of the herpesvirus family, with all viruses analyzed to date having at least one mechanism to accomplish MHC-I downregulation (26, 67, 89). A previous report had identified the EHV-4 UL49.5 protein as such a viral evasion protein that interferes with MHC-I antigen presentation through inhibition of TAP (49). However, this earlier report did not shed light on the ability of EHV-4 to downregulate cell surface MHC-I molecules in infected cells. We show here that EHV-4 can modulate MHC-I expression through two different mechanisms. Besides the ability of the UL49.5 protein in TAP blocking, EHV-4 encodes another protein, pUL56, which can downregulate surface MHC-I at early times after infection, even before expression of pUL49.5 can be detected.
Our data demonstrate that EHV-4 is able to significantly downregulate MHC-I molecules on the surface of infected equine (NBL-6 and FHK) cells. We also tested the ability of EHV-4 to downregulate MHC-I molecules on the surface of nonequine cell lines (MDBK and Vero cells). Although MDBK and Vero cells are fully permissive for EHV-4 infection, no modulation of cell surface MHC-I expression was detectable in these nonequine cells. Moreover, EHV-4 was not able to downregulate equine MHC-I molecules expressed on the surface of murine B78HC2 cells. It is noteworthy that EHV-4 utilizes equine MHC-I molecules as receptors to gain entry into equine cells and can enter murine cells only when these cells express equine MHC-I (10). Our finding, therefore, indicates that MHC-I downregulation by EHV-4 is species specific and that the cellular target for EHV-4 likely is not the MHC-I molecule itself but is an adaptor protein(s) that is recognized only if it is of equine origin.
Our finding that EHV-4 can downregulate MHC-I molecules on the surface of infected cells was not surprising, as previous data have shown that the EHV-4 UL49.5 protein can interfere with TAP and induce MHC-I downregulation in transfected cells (49). However, we were surprised by the ability of EHV-4 with the UL49.5 gene deletion to induce MHC-I downregulation at levels similar to those induced by parental virus. This indicated that there is likely at least one additional viral protein that can modulate cell surface MHC-I. The results prompted us to investigate the different mechanisms that the viruses can adopt to downmodulate MHC-I on the surface of infected cells. Downregulation of MHC-I on the surface of infected cells was observed as early as 4 h p.i., increasing to a maximum at 24 h p.i. Lack of MHC-I downregulation in cells infected with UV-inactivated virus clearly indicated that virus replication is essential for the observed effect. The suppression of late viral protein synthesis did not restore MHC-I expression to normal levels, suggesting that MHC-I downregulation largely depends on either IE or E viral proteins. Infection of cells in the presence of CX and Act-D then confirmed that one or more early EHV-4 proteins are responsible for the observed MHC-I downregulation. These findings are in agreement with previous reports on PrV, BHV-1, EHV-1, varicella-zoster virus, feline herpesvirus 1, and HIV-1 (1, 6, 7, 23, 48, 61, 68, 70). Furthermore, a parallel study conducted in our laboratory showed that, in the case of EHV-1, a close relative of EHV-4, the early UL56 protein was able to downregulate cell surface MHC-I (59).
To relate our findings to the role of either pUL49.5 or pUL56, kinetic studies of EHV-4 protein expression were conducted. Our data showed that pUL56 expression started at 4 h p.i. and was not affected by PAA, indicating that pUL56 is an early viral protein. Previous reports have shown that HSV-2 and EHV-1 pUL56 are also expressed at early times after infection (50, 81). On the other hand, UL49.5 protein expression started at 6 h p.i. and was slightly affected by the presence of PAA, which seems to differ from the behavior of pUL49.5 of other varicelloviruses. In the case of PrV, UL49.5 expression was strongly reduced by PAA in different cell types (23), while in BHV-1 it was not (54).
Taking into consideration that MHC-I downregulation started as early as 4 h p.i., the time when UL56 protein expression was first detected, one is tempted to conclude that the EHV-4 UL56 protein is responsible for the observed MHC-I downregulation in infected cells. However, pUL56, in the absence of other viral proteins, was not able to reduce the expression of MHC-I molecules on the surface of transfected cells, indicating that it requires at least another viral protein, which notably is not pUL49.5, to exert its effects on MHC-I cell surface expression. Our findings are in agreement with those of a recent study which also showed that EHV-1 pUL56 is unable to downregulate cell surface MHC-I in the absence of other viral proteins (59). It seems important to point out that cell surface expression of CD44 was not affected by pUL56, while α4β1 integrin expression was. We therefore surmise that EHV-4 pUL56 is able to selectively downregulate certain cell surface molecules and that this downregulation is possibly dependent on their localization in microdomains of cellular membranes. As one of the goals of any virus is to evade immune control (67), integrins may also be a worthwhile target. They can communicate with the cellular environment and engage multiple receptors, including immune receptors (15). Several viruses, including HIV, Ebola virus, simian immunodeficiency virus, HCMV, and Epstein-Barr virus (EBV), were shown to induce downregulation of surface integrins as part of their immune evasion strategy. Modulation of cell surface integrin can influence lymphocyte trafficking, thereby protecting the cell from superinfection and facilitating virus release (15, 31, 71, 86).
The exact mechanism by which the EHV-4 UL56 protein can downregulate surface MHC-I molecules remains to be defined. Our data showed that pUL56 was not able to block TAP during viral infection. On the basis of the properties of pUL56 of related viruses and their interaction partners, it is conceivable that EHV-4 pUL56, in association with cellular and viral proteins, may enhance MHC-I endocytosis. This mechanism has been described before for KSHV and HIV. KSHV encodes two related proteins, K3 and K5, which can cause rapid downregulation and degradation of MHC-I molecules from the plasma membrane by clathrin-dependent endocytosis (19, 43). On the other hand, HIV Nef mediates internalization of MHC-I molecules through clathrin-independent endocytosis and their sequestration in the TGN (30, 70).
Selective modulation of MHC-I allele expression by viruses would be a perfect strategy to allow viruses to walk the fine line between destruction by cytotoxic cells of the adaptive immune response and those of NK cells in the complete absence of MHC-I on the cell surface. Several viruses have evolved strategies to selectively downmodulate HLA-A and HLA-B (MHC-I molecules that are efficient at presenting viral peptides to CTLs) to evade CTL-mediated destruction. On the other hand, HLA-C and HLA-E (the dominant ligands for NK cell-inhibitory receptors) are spared to allow efficient inhibition of NK cells (18, 42, 46, 55). Whether or not EHV-4 has evolved an ability for selective downregulation of surface MHC-I molecules still needs to be elucidated.
In agreement with a previous report, we showed that EHV-4 pUL49.5 was able to inhibit TAP in transfected (49) or infected cells. Such a mechanism would provide an effective way of reducing CTL recognition of infected cells that is used by a number of herpesviruses, including HSV-1, HSV-2, HCMV, BHV-1, EBV, and EHV-1 (2, 25, 37–39, 49). Our observation that UL49.5 protein expression starts at 6 h p.i. and coincides with TAP inhibition suggests that pUL49.5 aids pUL56 in this immune-evasive function and ascertains that viral peptides are not presented by MHC-I molecules. Theoretically, inhibition of TAP does not necessarily result in reduction of MHC-I levels (52), as MHC-I molecules have a long dwell time on the cell surface. This was evidenced by experiments that show a significant reduction of surface MHC-I molecules only 48 h after treatment with either CX or brefeldin A as well as by results showing that EHV-4_49.53A and EHV-4Δ56_49.53A affect cell surface MHC-I expression with efficiencies similar to the efficiency of parental virus. In our attempts to examine the role of pUL49.5 on MHC-I downregulation, we prolonged the time of infection to up to 72 h p.i. Our results showed that EHV-4Δ56 and EHV-4Δ56_49.53A failed to affect cell surface MHC-I expression even after long infection times. However, in cells infected with either EHV-4Δ56 or EHV-4Δ56_49.53A, MHC-I surface levels were clearly restored after citrate treatment compared to those in cells infected with parental virus, and a significant increase in MHC-I levels was observed in cells infected with EHV-4Δ56_49.53A relative to those infected with EHV-4Δ56 alone. In addition, continuous expression of UL49.5 resulted in a significant downregulation of MHC-I molecules, likely because TAP is continuously inhibited in transfected cells due to the effect of the UL49.5 protein. It was previously shown that surface MHC-I was significantly reduced in cells transfected with pUL49.5 of different related viruses, including EHV-4 (49), and here we show that TAP was also significantly blocked in infected cells. Taken together, we concluded that pUL49.5 does have an effect on MHC-I downregulation in both infected and transfected cells that is more pronounced in the absence of pUL56 and that there is at least one other viral protein which is able to downregulate MHC-I surface expression and mask the role of pUL49.5 on MHC-I expression in infected cells.
In conclusion, we show that EHV-4 has evolved two different and likely complementary mechanisms to modulate cell surface MHC-I molecules. Early after infection, the virus induces downregulation of surface MHC-I through its UL56 protein, a process that is followed by TAP inhibition to ensure complete hiding of the virus from the lethal effects of CTLs. Understanding the mechanisms by which EHV-4 can modulate MHC-I may have important implications in the design of improved vaccines and/or antiviral drugs.
ACKNOWLEDGMENTS
This work was supported by DFG grant 143/4-1 and an unrestricted grant from the Freie Universität to N.O. A.S. was supported by a grant from the Egyptian Ministry of Higher Education, W.A. is an Alexander von Humboldt postdoctoral fellow.
Footnotes
Published ahead of print 23 May 2012
REFERENCES
- 1. Abendroth A, Lin I, Slobedman B, Ploegh H, Arvin AM. 2001. Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells. J. Virol. 75:4878–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahn K, et al. 1997. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 6:613–621 [DOI] [PubMed] [Google Scholar]
- 3. Ahn K, et al. 1996. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 15:3247–3255 [PMC free article] [PubMed] [Google Scholar]
- 4. Aisenbrey C, et al. 2006. Structure and dynamics of membrane-associated ICP47, a viral inhibitor of the MHC I antigen-processing machinery. J. Biol. Chem. 281:30365–30372 [DOI] [PubMed] [Google Scholar]
- 5. Allen GP, Bryans JT. 1986. Molecular epizootiology, pathogenesis, and prophylaxis of equine herpesvirus-1 infections. Prog. Vet. Microbiol. Immunol. 2:78–144 [PubMed] [Google Scholar]
- 6. Ambagala AP, Gopinath RS, Srikumaran S. 2004. Peptide transport activity of the transporter associated with antigen processing (TAP) is inhibited by an early protein of equine herpesvirus-1. J. Gen. Virol. 85:349–353 [DOI] [PubMed] [Google Scholar]
- 7. Ambagala AP, Hinkley S, Srikumaran S. 2000. An early pseudorabies virus protein down-regulates porcine MHC class I expression by inhibition of transporter associated with antigen processing (TAP). J. Immunol. 164:93–99 [DOI] [PubMed] [Google Scholar]
- 8. Azab W, Akashi H. 2011. Recent advances in equine herpesvirus 4 genetics using BAC technology. In Leffhalm JE. (ed), Horses: biology, domestication, and human interactions. Nova Science Publishers Inc., Hauppauge, NY [Google Scholar]
- 9. Azab W, et al. 2009. Cloning of the genome of equine herpesvirus 4 strain TH20p as an infectious bacterial artificial chromosome. Arch. Virol. 154:833–842 [DOI] [PubMed] [Google Scholar]
- 10. Azab W, Osterrieder N. 2012. Glycoproteins D of equine herpesvirus type 1 (EHV-1) and EHV-4 determine cellular tropism independently of integrins. J. Virol. 86:2031–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Azab W, et al. 2010. Characterization of a thymidine kinase-deficient mutant of equine herpesvirus 4 and in vitro susceptibility of the virus to antiviral agents. Antiviral Res. 85:389–395 [DOI] [PubMed] [Google Scholar]
- 12. Azab W, et al. 2010. Glycoprotein C of equine herpesvirus 4 plays a role in viral binding to cell surface heparan sulfate. Virus Res. 151:1–9 [DOI] [PubMed] [Google Scholar]
- 13. Blevitt JM, et al. 1999. A fluorescence-based high throughput screen for the transporter associated with antigen processing. J. Biomol. Screen. 4:87–91 [DOI] [PubMed] [Google Scholar]
- 14. Breathnach CC, Yeargan MR, Sheoran AS, Allen GP. 2001. The mucosal humoral immune response of the horse to infective challenge and vaccination with equine herpesvirus-1 antigens. Equine Vet. J. 33:651–657 [DOI] [PubMed] [Google Scholar]
- 15. Budde ML, Lhost JJ, Dudley DM, Rakasz EG, O'Connor DH. 2010. Integrin alpha4beta7 is downregulated on the surfaces of simian immunodeficiency virus SIVmac239-infected cells. J. Virol. 84:6344–6351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cadwell K, Coscoy L. 2008. The specificities of Kaposi's sarcoma-associated herpesvirus-encoded E3 ubiquitin ligases are determined by the positions of lysine or cysteine residues within the intracytoplasmic domains of their targets. J. Virol. 82:4184–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cadwell K, Coscoy L. 2005. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science 309:127–130 [DOI] [PubMed] [Google Scholar]
- 18. Cohen GB, et al. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671 [DOI] [PubMed] [Google Scholar]
- 19. Coscoy L, Ganem D. 2000. Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. U. S. A. 97:8051–8056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crabb BS, Studdert MJ. 1995. Equine herpesviruses 4 (equine rhinopneumonitis virus) and 1 (equine abortion virus). Adv. Virus Res. 45:153–190 [DOI] [PubMed] [Google Scholar]
- 21. Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. 2005. Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol. Rev. 207:145–157 [DOI] [PubMed] [Google Scholar]
- 22. Davison AJ, et al. 2009. The order Herpesvirales. Arch. Virol. 154:171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deruelle MJ, Van den Broeke C, Nauwynck HJ, Mettenleiter TC, Favoreel HW. 2009. Pseudorabies virus US3- and UL49.5-dependent and -independent downregulation of MHC I cell surface expression in different cell types. Virology 395:172–181 [DOI] [PubMed] [Google Scholar]
- 24. Friedman HM. 2003. Immune evasion by herpes simplex virus type 1, strategies for virus survival. Trans. Am. Clin. Climatol. Assoc. 114:103–112 [PMC free article] [PubMed] [Google Scholar]
- 25. Fruh K, et al. 1995. A viral inhibitor of peptide transporters for antigen presentation. Nature 375:415–418 [DOI] [PubMed] [Google Scholar]
- 26. Fruh K, Gruhler A, Krishna RM, Schoenhals GJ. 1999. A comparison of viral immune escape strategies targeting the MHC class I assembly pathway. Immunol. Rev. 168:157–166 [DOI] [PubMed] [Google Scholar]
- 27. Furman MH, Loureiro J, Ploegh HL, Tortorella D. 2003. Ubiquitinylation of the cytosolic domain of a type I membrane protein is not required to initiate its dislocation from the endoplasmic reticulum. J. Biol. Chem. 278:34804–34811 [DOI] [PubMed] [Google Scholar]
- 28. Furman MH, Ploegh HL, Tortorella D. 2002. Membrane-specific, host-derived factors are required for US2- and US11-mediated degradation of major histocompatibility complex class I molecules. J. Biol. Chem. 277:3258–3267 [DOI] [PubMed] [Google Scholar]
- 29. Galocha B, et al. 1997. The active site of ICP47, a herpes simplex virus-encoded inhibitor of the major histocompatibility complex (MHC)-encoded peptide transporter associated with antigen processing (TAP), maps to the NH2-terminal 35 residues. J. Exp. Med. 185:1565–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Greenberg ME, Iafrate AJ, Skowronski J. 1998. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 17:2777–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gregory CD, Murray RJ, Edwards CF, Rickinson AB. 1988. Downregulation of cell adhesion molecules LFA-3 and ICAM-1 in Epstein-Barr virus-positive Burkitt's lymphoma underlies tumor cell escape from virus-specific T cell surveillance. J. Exp. Med. 167:1811–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griffin BD, Verweij MC, Wiertz EJ. 2010. Herpesviruses and immunity: the art of evasion. Vet. Microbiol. 143:89–100 [DOI] [PubMed] [Google Scholar]
- 33. Halenius A, et al. 2006. Physical and functional interactions of the cytomegalovirus US6 glycoprotein with the transporter associated with antigen processing. J. Biol. Chem. 281:5383–5390 [DOI] [PubMed] [Google Scholar]
- 34. Hansen TH, Bouvier M. 2009. MHC class I antigen presentation: learning from viral evasion strategies. Nat. Rev. Immunol. 9:503–513 [DOI] [PubMed] [Google Scholar]
- 35. Heldens JG, et al. 2001. Clinical and virological evaluation of the efficacy of an inactivated EHV1 and EHV4 whole virus vaccine (Duvaxyn EHV1,4). Vaccination/challenge experiments in foals and pregnant mares. Vaccine 19:4307–4317 [DOI] [PubMed] [Google Scholar]
- 36. Hewitt EW. 2003. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology 110:163–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hewitt EW, Gupta SS, Lehner PJ. 2001. The human cytomegalovirus gene product US6 inhibits ATP binding by TAP. EMBO J. 20:387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hill A, et al. 1995. Herpes simplex virus turns off the TAP to evade host immunity. Nature 375:411–415 [DOI] [PubMed] [Google Scholar]
- 39. Hislop AD, et al. 2007. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J. Exp. Med. 204:1863–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Honess RW, Watson DH. 1977. Herpes simplex virus resistance and sensitivity to phosphonoacetic acid. J. Virol. 21:584–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Horst D, Verweij MC, Davison AJ, Ressing ME, Wiertz EJ. 2011. Viral evasion of T cell immunity: ancient mechanisms offering new applications. Curr. Opin. Immunol. 23:96–103 [DOI] [PubMed] [Google Scholar]
- 42. Ishido S, et al. 2000. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity 13:365–374 [DOI] [PubMed] [Google Scholar]
- 43. Ishido S, Wang C, Lee BS, Cohen GB, Jung JU. 2000. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74:5300–5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jackson MR, Cohen-Doyle MF, Peterson PA, Williams DB. 1994. Regulation of MHC class I transport by the molecular chaperone, calnexin (p88, IP90). Science 263:384–387 [DOI] [PubMed] [Google Scholar]
- 45. Joyce S. 1997. Traffic control of completely assembled MHC class I molecules beyond the endoplasmic reticulum. J. Mol. Biol. 266:993–1001 [DOI] [PubMed] [Google Scholar]
- 46. Kavanagh DG, Gold MC, Wagner M, Koszinowski UH, Hill AB. 2001. The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J. Exp. Med. 194:967–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koppers-Lalic D, et al. 2005. Varicelloviruses avoid T cell recognition by UL49.5-mediated inactivation of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. U. S. A. 102:5144–5149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koppers-Lalic D, et al. 2001. The UL41-encoded virion host shutoff (vhs) protein and vhs-independent mechanisms are responsible for down-regulation of MHC class I molecules by bovine herpesvirus 1. J. Gen. Virol. 82:2071–2081 [DOI] [PubMed] [Google Scholar]
- 49. Koppers-Lalic D, et al. 2008. Varicellovirus UL 49.5 proteins differentially affect the function of the transporter associated with antigen processing, TAP. PLoS Pathog. 4:e1000080 doi:10.1371/journal.ppat.1000080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koshizuka T, et al. 2002. Identification and characterization of the UL56 gene product of herpes simplex virus type 2. J. Virol. 76:6718–6728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kurtz BM, Singletary LB, Kelly SD, Frampton AR., Jr 2010. Equus caballus major histocompatibility complex class I is an entry receptor for equine herpesvirus type 1. J. Virol. 84:9027–9034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lankat-Buttgereit B, Tampe R. 2002. The transporter associated with antigen processing: function and implications in human diseases. Physiol. Rev. 82:187–204 [DOI] [PubMed] [Google Scholar]
- 53. Lilley BN, Ploegh HL. 2005. Viral modulation of antigen presentation: manipulation of cellular targets in the ER and beyond. Immunol. Rev. 207:126–144 [DOI] [PubMed] [Google Scholar]
- 54. Lipinska AD, et al. 2006. Bovine herpesvirus 1 UL49.5 protein inhibits the transporter associated with antigen processing despite complex formation with glycoprotein M. J. Virol. 80:5822–5832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lopez-Botet M, Llano M, Ortega M. 2001. Human cytomegalovirus and natural killer-mediated surveillance of HLA class I expression: a paradigm of host-pathogen adaptation. Immunol. Rev. 181:193–202 [DOI] [PubMed] [Google Scholar]
- 56. Lorenzo ME, Ploegh HL, Tirabassi RS. 2001. Viral immune evasion strategies and the underlying cell biology. Semin. Immunol. 13:1–9 [DOI] [PubMed] [Google Scholar]
- 57. Lybarger L, Wang X, Harris M, Hansen TH. 2005. Viral immune evasion molecules attack the ER peptide-loading complex and exploit ER-associated degradation pathways. Curr. Opin. Immunol. 17:71–78 [DOI] [PubMed] [Google Scholar]
- 58. Lybarger L, Wang X, Harris MR, Virgin HW, IV, Hansen TH. 2003. Virus subversion of the MHC class I peptide-loading complex. Immunity 18:121–130 [DOI] [PubMed] [Google Scholar]
- 59. Ma G, Feineis S, Osterrieder N, Van de Walle GR. 2012. Identification and characterization of equine herpesvirus type 1 pUL56 and its role in virus-induced downregulation of major histocompatibility complex class I. J. Virol. 86:3554–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Minke JM, Audonnet JC, Fischer L. 2004. Equine viral vaccines: the past, present and future. Vet. Res. 35:425–443 [DOI] [PubMed] [Google Scholar]
- 61. Montagnaro S, et al. 2009. Feline herpesvirus-1 down-regulates MHC class I expression in an homologous cell system. J. Cell. Biochem. 106:179–185 [DOI] [PubMed] [Google Scholar]
- 62. Nataraj C, et al. 1997. Bovine herpesvirus 1 downregulates the expression of bovine MHC class I molecules. Viral Immunol. 10:21–34 [DOI] [PubMed] [Google Scholar]
- 63. Neumann L, Kraas W, Uebel S, Jung G, Tampe R. 1997. The active domain of the herpes simplex virus protein ICP47: a potent inhibitor of the transporter associated with antigen processing. J. Mol. Biol. 272:484–492 [DOI] [PubMed] [Google Scholar]
- 64. Patel JR, et al. 2003. Equid herpesvirus (EHV-1) live vaccine strain C147: efficacy against respiratory diseases following EHV types 1 and 4 challenges. Vet. Microbiol. 92:1–17 [DOI] [PubMed] [Google Scholar]
- 65. Peaper DR, Cresswell P. 2008. Regulation of MHC class I assembly and peptide binding. Annu. Rev. Cell Dev. Biol. 24:343–368 [DOI] [PubMed] [Google Scholar]
- 66. Petersen JL, Morris CR, Solheim JC. 2003. Virus evasion of MHC class I molecule presentation. J. Immunol. 171:4473–4478 [DOI] [PubMed] [Google Scholar]
- 67. Ploegh HL. 1998. Viral strategies of immune evasion. Science 280:248–253 [DOI] [PubMed] [Google Scholar]
- 68. Rappocciolo G, Birch J, Ellis SA. 2003. Down-regulation of MHC class I expression by equine herpesvirus-1. J. Gen. Virol. 84:293–300 [DOI] [PubMed] [Google Scholar]
- 69. Reed SM, Toribio RE. 2004. Equine herpesvirus 1 and 4. Vet. Clin. North Am. Equine Pract. 20:631–642 [DOI] [PubMed] [Google Scholar]
- 70. Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. 1996. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 2:338–342 [DOI] [PubMed] [Google Scholar]
- 71. Simmons G, Wool-Lewis RJ, Baribaud F, Netter RC, Bates P. 2002. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J. Virol. 76:2518–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Slater JD, et al. 2006. Report of the equine herpesvirus-1 Havermeyer Workshop, San Gimignano, Tuscany, June 2004. Vet. Immunol. Immunopathol. 111:3–13 [DOI] [PubMed] [Google Scholar]
- 73. Stevenson PG, Efstathiou S, Doherty PC, Lehner PJ. 2000. Inhibition of MHC class I-restricted antigen presentation by gamma 2-herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 97:8455–8460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stokes A, Corteyn AH, Murray PK. 1991. Clinical signs and humoral immune response in horses following equine herpesvirus type-1 infection and their susceptibility to equine herpesvirus type-4 challenge. Res. Vet. Sci. 51:141–148 [DOI] [PubMed] [Google Scholar]
- 75. Stroh T, Erben U, Kuhl AA, Zeitz M, Siegmund B. 2010. Combined pulse electroporation—a novel strategy for highly efficient transfection of human and mouse cells. PLoS One 5:e9488 doi:10.1371/journal.pone.0009488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sugawara S, Abo T, Kumagai K. 1987. A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. J. Immunol. Methods 100:83–90 [DOI] [PubMed] [Google Scholar]
- 77. Tai SH, et al. 2010. Complete genomic sequence and an infectious BAC clone of feline herpesvirus-1 (FHV-1). Virology 401:215–227 [DOI] [PubMed] [Google Scholar]
- 78. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 79. Tomazin R, et al. 1996. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J. 15:3256–3266 [PMC free article] [PubMed] [Google Scholar]
- 80. Townsend A, Bodmer H. 1989. Antigen recognition by class I-restricted T lymphocytes. Annu. Rev. Immunol. 7:601–624 [DOI] [PubMed] [Google Scholar]
- 81. Van de Walle GR, et al. 2010. A vectored equine herpesvirus type 1 (EHV-1) vaccine elicits protective immune responses against EHV-1 and H3N8 equine influenza virus. Vaccine 28:1048–1055 [DOI] [PubMed] [Google Scholar]
- 82. Verweij MC, et al. 2008. The varicellovirus UL49.5 protein blocks the transporter associated with antigen processing (TAP) by inhibiting essential conformational transitions in the 6+6 transmembrane TAP core complex. J. Immunol. 181:4894–4907 [DOI] [PubMed] [Google Scholar]
- 83. von Einem J, Smith PM, Van de Walle GR, O'Callaghan DJ, Osterrieder N. 2007. In vitro and in vivo characterization of equine herpesvirus type 1 (EHV-1) mutants devoid of the viral chemokine-binding glycoprotein G (gG). Virology 362:151–162 [DOI] [PubMed] [Google Scholar]
- 84. Vossen MT, Westerhout EM, Soderberg-Naucler C, Wiertz EJ. 2002. Viral immune evasion: a masterpiece of evolution. Immunogenetics 54:527–542 [DOI] [PubMed] [Google Scholar]
- 85. Wang X, Lybarger L, Connors R, Harris MR, Hansen TH. 2004. Model for the interaction of gammaherpesvirus 68 RING-CH finger protein mK3 with major histocompatibility complex class I and the peptide-loading complex. J. Virol. 78:8673–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Warren AP, Owens CN, Borysiewicz LK, Patel K. 1994. Down-regulation of integrin alpha 1/beta 1 expression and association with cell rounding in human cytomegalovirus-infected fibroblasts. J. Gen. Virol. 75(Pt 12):3319–3325 [DOI] [PubMed] [Google Scholar]
- 87. Wiertz EJ, et al. 1996. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84:769–779 [DOI] [PubMed] [Google Scholar]
- 88. Williams A, Peh CA, Elliott T. 2002. The cell biology of MHC class I antigen presentation. Tissue Antigens 59:3–17 [DOI] [PubMed] [Google Scholar]
- 89. Yewdell JW, Hill AB. 2002. Viral interference with antigen presentation. Nat. Immunol. 3:1019–1025 [DOI] [PubMed] [Google Scholar]