

Abstract
Halophage CW02 infects a Salinivibrio costicola-like bacterium, SA50, isolated from the Great Salt Lake. Following isolation, cultivation, and purification, CW02 was characterized by DNA sequencing, mass spectrometry, and electron microscopy. A conserved module of structural genes places CW02 in the T7 supergroup, members of which are found in diverse aquatic environments, including marine and freshwater ecosystems. CW02 has morphological similarities to viruses of the Podoviridae family. The structure of CW02, solved by cryogenic electron microscopy and three-dimensional reconstruction, enabled the fitting of a portion of the bacteriophage HK97 capsid protein into CW02 capsid density, thereby providing additional evidence that capsid proteins of tailed double-stranded DNA phages have a conserved fold. The CW02 capsid consists of bacteriophage lambda gpD-like densities that likely contribute to particle stability. Turret-like densities were found on icosahedral vertices and may represent a unique adaptation similar to what has been seen in other extremophilic viruses that infect archaea, such as Sulfolobus turreted icosahedral virus and halophage SH1.
INTRODUCTION
Tailed double-stranded DNA (dsDNA) bacteriophages (order Caudovirales) account for nearly 95% of all documented phages and probably comprise the majority of the viruses on the planet (1). Recent estimations place the global number of virus particles on the order of 1031 to 1032, outnumbering the total number of host cells by a factor of 10 (58). The ubiquity of phages suggests an ecological, predatory role in recycling organic matter, especially in extreme environments, where eukaryotic predators, primarily protozoal grazers, are rare (44). Phages also affect the genetic diversity of their environments by facilitating horizontal gene transfer between host cells (55). The sheer abundance of phages in aquatic environments may supply the greatest selective pressure exerted on host organisms (58).
Bacteriophages of the order Caudovirales are divided into three morphologically distinct families. All consist of isometric or prolate heads that encapsulate linear double-stranded (dsDNA) genomes, but phages of the Myoviridae family have a long, contractile tail (e.g., bacteriophage T4); viruses of the Siphoviridae family consist of a long, noncontractile tail (e.g., bacteriophage lambda); and phages within the Podoviridae family exhibit a short, noncontractile tail (e.g., bacteriophage T7). Recent high-resolution capsid structures of tailed dsDNA phages revealed a conserved fold that is shared among all three phage families. The first of these structures was solved for bacteriophage HK97 (56), and subsequent structures of Caudovirales phages have been shown to adopt the HK97-like fold (28, 30, 35, 42). The limited identity between primary structures of HK97-like capsid proteins is a prototypical example that structure is more conserved than sequence.
Halophage CW02 is a bacteriophage isolated from the south arm of the Great Salt Lake (GSL) and infects a euryhalophilic Salinivibrio costicola-like bacterium, SA50, also isolated from the GSL. At least two other tailed phages of S. costicola, UTAK (22) and G3 (31), have been reported; however, only general features of the structures and genomics of these phage are known. Here we characterized halophage CW02 to determine its relatedness to other dsDNA bacteriophages. Based on negative stain transmission electron microscopy, CW02 is morphologically similar to viruses of the Podoviridae family. Genomic analysis and mass spectrometry (MS) of CW02 genes and proteins revealed the strongest sequence similarities to Pseudomonas phage PA11 (34), roseophage SIO1 (46), vibriophage VpV262 (23), and cyanophage Pf-WMP3 (38), all of which are distant relatives of bacteriophage T7 but members of the T7 supergroup. The 16-Å resolution structure of CW02, solved by cryogenic electron microscopy (cryo-EM) and three-dimensional (3D) reconstruction, permitted fitting of the HK97-like fold into the capsid density, thereby supporting the observation that the capsid proteins of tailed dsDNA phages adopt a common ancestral fold.
(Some of the data in this paper were published in Peter Shen's Ph.D. dissertation.)
MATERIALS AND METHODS
Cultivation and purification.
Host bacteria and bacteriophage were isolated from the GSL in Utah. Sediment and water samples were collected between May and November 2008 in shallow waters along the north shore of Bridger Bay on Antelope Island of the GSL. Salinity was measured at 8% by evaporation. (Salinity is known to vary seasonally due to fresh water inflow and evaporation; see http://ut.water.usgs.gov/greatsaltlake/salinity/.) The pH ranged between 7.5 and 8. The host bacterium, SA50, was first isolated on Halobacteria Medium (per liter of solution, 80 g NaCl, 10 g MgSO4 · 7H2O, 5 g casein hydrolysate, 5 g KCl, 3 g disodium citrate, 1 g KNO3, 1 g yeast extract, and 0.2 g CaCl2 · 6H2O), which was modified from the original 22% NaCl (3) to 8% NaCl and pH 7.5.
The 16S rRNA gene of SA50 was amplified using bacterium-specific primers (27F, 5′ AGA GTT TGA TCM TGG CTC AG 3′; 1492R, 5′ ACG GYT ACC TTG TTA CGA CTT 3′) (36). The reaction mixture contained 200 nM each primer, the deoxynucleoside triphosphates at 250 μM each, 0.2 mg ml−1 bovine serum albumin, 1 U Taq DNA polymerase, and the diluted reaction buffer (Promega, Madison, WI). The amplification parameters were 94°C for 3 min; 25 cycles of 94°C for 45 s, 57°C for 1 min, and 72°C for 2 min; and a final extension step of 72°C for 7 min. Approximately 1,350 bp were sequenced (Molecular Research Core Facility, Idaho State University, Pocatello, ID) and queried against the GenBank database by use of the BLAST search tool. The isolate showed 99% sequence identity with S. costicola subsp. costicola strain ATCC 33508 (NR_027590.1). The isolate was maintained in Halobacteria medium.
CW02 was initially isolated by the use of a plaque assay of filtered GSL water with SA50 as the host strain. CW02 was recovered from plaques by removal from the soft agar overlay with a sterile Pasteur pipette. The agar was transferred to a sterile 8% salt solution, vortexed, and incubated for 30 min to release phage from the matrix. CW02 was then amplified by inoculation into a broth culture of SA50 and incubated on a shaker at 30°C. Phage replication caused lysis of the host cells and clearing of the broth culture after 18 h. After clearing, the broth culture was centrifuged at 5,000 × g for 10 min and passed through a 0.2-μm filter to remove bacteria. Host bacteria and bacteriophage filtrate stocks were stored frozen (−20°C) in 20% glycerol.
The bacteriophage was further purified by isopycnic ultracentrifugation (64% [wt/vol] CsCl) at ∼125,000 × g for 24 h at 4°C. A single viral band was extracted from the gradient. CsCl was removed by dialysis in either pure water or pH 8 buffer containing 1.35 M NaCl, 48 mM MgSO4· 7H2O, 1 mM CaCl2, and 2 mM Tris-Cl. Finally, purified CW02 was washed and concentrated by centrifugal filtration (100-kDa molecular mass cutoff filters; Sartorius Stedim Biotech, Bohemia, NY) to be made suitable for cryo-EM.
Genome isolation and sequencing.
The DNA was isolated as follows from 100 ml of bacteriophage filtrate. The filtrate was concentrated by centrifugal filtration (Amicon Centricon 100,000 molecular weight cutoff filter; Millipore Corp., Billerica, MA) to approximately 2 ml. Contaminant nucleic acids in the concentrated bacteriophage solution were removed by addition of DNase I (1 μg/ml) and RNase A (1 μg/ml) (Sigma-Aldrich Corp., St. Louis, MO), incubated at 37°C for 1 h, and then centrifuged at 110,000 × g for 2 h. The pellet was suspended in 2 ml of SM buffer (5.8 g NaCl, 2 g MgSO4 · 7H2O, 50 ml 1 M Tris [pH 7.5], and 5 ml 2% gelatin per liter of distilled water) and treated with proteinase K (50 μg/ml; Sigma-Aldrich Corp., St. Louis, MO) and 0.5% sodium dodecyl sulfate (SDS; Sigma-Aldrich Corp., St. Louis, MO) at 56°C for 1 h to remove the capsid. The DNA was then extracted through phenol-Tris-Cl, followed by a 50/50 mixture of phenol-chloroform, and finally through pure chloroform. The DNA was ethanol precipitated and resuspended in Tris-EDTA buffer. Genomic DNA was sequenced at the Brigham Young University (BYU) DNA Sequencing Center using a Roche Genome Sequencer FLX instrument and employing the GS FLX Titanium Sequencing XLR70 kit (Roche Diagnostics Corporation, Indianapolis, IN).
Sequence analysis.
Putative open reading frames (ORFs) of the CW02 genome were determined using GeneMarkS (8) and numbered according to whole-genome homology with bacteriophage PA11 (34). CW02 protein homologues were determined by PSI-BLAST analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi). All protein sequences for pairwise alignments were obtained from GenBank (http://www.ncbi.nlm.nih.gov/GenBank/), including sequences of enterobacterial phage T7 (NC_001604), VpV262 (NC_003907), SIO1 (NC_002519), Pf-WMP3 (NC_009551), and PA11 (NC_007808). Sequence alignments were performed using ClustalW2 (37). Protein secondary structures were predicted using Psi-pred (11), COILS (39), BetaWrapPro (41), or Phyre (32).
Electron microscopy and image reconstruction.
For negative staining, 3.5 μl of purified CW02 was adsorbed onto a glow-discharged, continuous-carbon-coated copper grid, blotted, and then washed. The grid was blotted again, stained with a solution of 1% uranyl acetate, and then blotted a final time and allowed to dry.
For cryo-EM, 3.5 μl of purified CW02 was placed on a glow-discharged, holey-carbon-coated copper grid, blotted, and plunge frozen in liquid ethane with an FEI Vitrobot (FEI, Hillsboro, OR). Specimens were transferred to a Gatan 626 cryoholder (Gatan, Inc., Pleasanton, CA) cooled with liquid nitrogen. Cryo-EM images were acquired at 200 keV and at ×39,000 magnification via low-dose methods at objective lens underfocus levels ranging between 0.2 and 2.8 μm. Focal pairs were taken 1.0 μm apart. Images were recorded either by a Gatan charge-coupled device camera (1,024 by 1,024 pixels) or on Kodak SO-163 film in an FEI Tecnai F30 transmission electron microscope (FEI, Hillsboro, OR). Electron micrographs recorded on film were digitized on a Nikon Super Coolscan 9000 ED scanner.
Particle images were extracted from scanned electron micrographs using X3DPREPROCESS (16). Contrast transfer function (CTF) signal and decay parameters were determined from and applied to the images by the use of Bsoft (bshow and bctf functions) (26) and the algorithm of Conway and Steven (16), except that images within a focal pair were not combined during CTF correction. Origins and orientations of the extracted particles were determined by use of the model-based technique of PFT2 (4), which was adapted to use phase and amplitude information in orientation selection (48). A reference model for PFT2 analysis was generated by the random-model method with imposed icosahedral symmetry (59). The final 3D reconstructions were calculated using EM3DR2 (17, 21). In displayed images, contour levels are given in terms of σ, which was calculated as the number of standard deviations relative to the average map density.
For size calibration, poliovirus (160S form) was used as an internal standard. Poliovirus particles were mixed with CW02. The combined sample was imaged via cryo-EM, and separated images of poliovirus and CW02 were used to compute 3D image reconstructions. A previously calibrated map of 160S poliovirus (5) was used to determine the size of the poliovirus reconstruction (6) and, hence, the sampling size of the CW02 reconstruction. Bsoft (bshow function) (26) was used to measure capsid dimensions.
Handedness determination.
Tilt experiments were performed as described previously (7). Briefly, pairs of micrographs of CW02 were recorded with the same field of view, under the same conditions, but with the specimen at different tilt angles. The first micrographs of the tilt pairs were recorded at 0° tilt, and the second micrographs were recorded at 5° tilt. The origins and orientations of the untilted-particle images were determined using the PFT2 model-based method described above, after which the Bsoft program dhand (14, 26) was used to predict the orientations of the tilted particles (for either handedness), compute tilted projections from enantiomers of the 3D reconstruction, and compare the projections to the experimental images of tilted particles. The correct handedness was the handedness that compared the best.
Structural modeling.
The atomic coordinates of HK97 capsid protein (24) (Protein Data Bank accession no. 1OHG) were fitted as separate pentameric or hexameric units. Only the Cα chain of axial (A) and peripheral (P) domains were used in the fitting. Pentamers and hexamers were first fitted manually as a rigid body into the electron density map of CW02 by use of the program UCSF Chimera (45), after which the fitting was refined by automated fitting using the same program. All surface renderings of the reconstruction were done at a 1-σ contour level, which is defined by the sum of the average and standard deviation of map densities. Segmentation of individual subunits (via UCSF Chimera [45]) was also carried out.
Attempts were made to reconstruct the turrets from cryo-EM images. We used a procedure adapted from a method developed previously by Briggs et al. (10). After icosahedral reconstructions were computed, turrets were re-extracted from the 2D images using only vertices that did not overlap the densities of the capsid shell. Reference-free class averages (SPIDER [50]), and asymmetric and symmetric (C2, C3, C4, C5, C6, C7; Cn is cyclic n-fold symmetry) reconstructions using random models as the initial reference, were computed. Additional class averages were also generated from cryo-EM pictures by masking the capsid shell according to the radius. In addition, we tested if turrets were found on all 5-fold vertices by performing asymmetric and C5-symmetric reconstructions using as the initial reference a CW02 capsid with a cylindrical density on a single vertex.
MS.
CW02 virions were subject to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) to separate individual proteins. The gel was stained using Coomassie blue reagent, after which individual gel bands were excised and then shredded by spinning gel fragments through micropipette tips. To capture all of the proteins in solution, whole virions were also prepared for MS analysis by forgoing SDS-PAGE. Proteins were prepared for analysis by MS by use of a modified filter-aided sample preparation protocol adapted from reference 57. Briefly, the gel fragments were destained using a 1:1 mixture of acetonitrile and 8 M urea, in 0.1 M Tris-HCl (pH 8.5). The denatured proteins were subsequently reduced with 0.1 M dithiothreitol in UA buffer (8 M urea, 0.1 M Tris-Cl, pH 8.5) and then carboamidomethylated with 50 mM iodoacetamide in UA buffer. Each solution was washed by filtration through a 10-kDa molecular mass cutoff filter (Sartorius Stedim Biotech, Bohemia, NY). The urea and iodoacetamide were then replaced with 50 mM ammonium bicarbonate. Proteins were finally digested with sequencing grade trypsin (Promega) and then acidified by the addition of formic acid to 1% of the volume.
Acidified peptide samples were loaded onto a nanoAquity C18 column (75 μm by 15 cm; Waters Corporation, Milford, MA) and eluted by a 100-min binary gradient of Optima grade solvent A (5% acetonitrile, 0.1% formic acid) and Optima grade solvent B (0.1% formic acid, 99.9% acetonitrile) (Thermo Scientific, San Jose, CA) at 425 nl/min composed of the following steps. From a baseline of 95% solvent A, an 8-min gradient to 90% solvent A was performed, followed by a 65-min gradient to 65% solvent A, a 7-min gradient to 5% solvent A for 8 min, and a 3-min return to a 95% solvent A baseline for 9 min.
Column effluent was directed to a nanoelectrospray ionization source on an LTQ-Orbitrap XL (Thermo Scientific, Waltham, MA). Data-dependent acquisition was performed by coupling a 60,000 resolution survey scan in the Orbitrap with up to the top seven subsequent collision-induced dissociation tandem MS (MS/MS) scans acquired in the ion trap (signal threshold of 1,000, normalized collision energy of 35%, isolation width of 2 m/z, with a two-count dynamic exclusion in a 3-min window). A lock mass of a polysiloxane compound at m/z 445.120025 was used as an internal mass calibration standard (43).
MS/MS spectra were converted to the Mascot generic format by the MsConvert program (http://proteowizard.sourceforge.net/pubs.shtml). Mascot searching was queried against a database of ORFs derived from the CW02 genome. The peptide search space was expanded by concatenating the CW02 database with a comprehensive database of the Escherichia coli ATCC 8739 proteome (GenBank accession no. YP_001723016.1). Search results were filtered against a decoy database to a 1% false discovery rate confidence at the peptide level, corresponding to Mascot ion cutoff scores of 22 to 27.
Nucleotide sequence and structural accession numbers.
The CW02 genome, 3D reconstruction, and fit of HK97 coordinates into the CW02 capsid have been deposited in the GenBank database (JQ446452), the EM Data Bank (5388), and the Protein Data Bank (3J1A), respectively.
RESULTS AND DISCUSSION
CW02 is a member of the T7-like phage supergroup.
The genome of phage CW02, determined by 454 pyrosequencing, had an average read length of 405.47 bp with 44,463 reads and a contig length of 49,391 bp. This corresponded to 366-fold coverage of the genome. The CW02 genome consists of linear dsDNA with a GC content of 47.67%. Analysis of the sequence suggests the genome contains at least 70 ORFs (Table 1) and a single tRNA (Arg; anticodon = AGA). Thirty-six of the ORFs appear to be unique to CW02, as PSI-BLAST analysis did not reveal significant matches.
Table 1.
CW02 putative ORFs and analysis of proteins determined by LC-MS and detected homologues
| ORF | Direction | No. of amino acid residues | Molecular mass (kDa) | Identification by LC-MSa |
BLAST hits and conserved domainsc | E value | Putative function and structure predictiond | |
|---|---|---|---|---|---|---|---|---|
| Unique peptides | % Coverageb | |||||||
| gp1 | + | 78 | 8.7 | |||||
| gp2 | + | 215 | 24.2 | Hypothetical protein ORF001 (Pseudomonas phage PA11) | 7e-45 | Amidotransferase | ||
| Glutamine amidotransferase (Bacillus sp. strain B14905) | 1e-13 | |||||||
| gp3 | + | 437 | 48.8 | Hypothetical protein ORF004 (Pseudomonas phage PA11) | 4e-45 | Amidotransferase | ||
| Glutamine amidotransferase (enterobacterial phage ϕEco32) | 4e-21 | |||||||
| gp4 | + | 64 | 7.2 | |||||
| gp5 | + | 267 | 30.0 | Hypothetical protein ORF005 (Pseudomonas phage PA11) | 1e-85 | |||
| gp6 | + | 77 | 8.7 | |||||
| gp7 | + | 77 | 8.6 | |||||
| gp8 | + | 190 | 21.1 | Hypothetical protein ORF006 (Pseudomonas phage PA11) | 6e-05 | |||
| gp9 | + | 266 | 30.7 | Conserved amidoligase_2 domain | Amidoligase | |||
| Hypothetical protein ORF007 (Pseudomonas phage PA11) | 1e-37 | |||||||
| gp10 | + | 159 | 17.6 | |||||
| gp11 | + | 71 | 7.9 | |||||
| gp12 | + | 252 | 28.6 | Conserved ATP-grasp domain | ATP-grasp protein | |||
| Hypothetical protein ORF008 (Pseudomonas phage PA11) | 1e-63 | |||||||
| ATP-grasp enzyme (enterobacterial phage ϕEco32) | 1e-26 | |||||||
| gp13 | + | 73 | 8.2 | |||||
| gp14 | + | 77 | 8.9 | |||||
| gp15 | + | 60 | 6.9 | |||||
| gp16 | + | 126 | 14.0 | Conserved AIG-2 like domain | AIG-2 protein | |||
| Conserved YtfP/UPF0131 protein (enterobacterial phage ϕEco32) | 8e-25 | |||||||
| gp17 | + | 97 | 10.6 | |||||
| gp18 | + | 82 | 9.8 | |||||
| gp19 | + | 532 | 60.0 | TOPRIM and P-loop_NTPase superfamily | DNA primase/helicase | |||
| Hypothetical protein ORF011 (Pseudomonas phage PA11) | 0.0 | |||||||
| DNA primase/helicase (enterobacterial phage T7) | 1e-23 | |||||||
| gp20 | + | 87 | 10.2 | |||||
| gp21 | + | 161 | 19.3 | Sigma70_r2 superfamily | RNA polymerase sigma factor | |||
| Hypothetical protein ORF012 (Pseudomonas phage PA11) | 1e-15 | |||||||
| RNA polymerase sigma-70 factor (Kordia algicida OT-1) | 2e-07 | |||||||
| gp22 | + | 67 | 7.6 | |||||
| gp23 | + | 629 | 72.6 | DNA polymerase A superfamily | DNA polymerase | |||
| Hypothetical protein ORF013 (Pseudomonas phage PA11) | 0.0 | |||||||
| Putative DNA polymerase (Roseobacter phage SIO1) | 3e-23 | |||||||
| DNA polymerase (enterobacterial phage T7) | 5e-07 | |||||||
| gp24 | + | 69 | 8.0 | |||||
| gp25 | + | 270 | 29.6 | Hypothetical protein ORF015 (Pseudomonas phage PA11) | 3e-36 | |||
| gp26 | + | 215 | 24.7 | PIN_SF superfamily | 5′–3′ exonuclease | |||
| Hypothetical protein ORF018 (Pseudomonas phage PA11) | 3e-65 | |||||||
| 5′–3′ exonuclease (Pseudomonas phage LUZ24) | 1e-25 | |||||||
| gp27 | + | 74 | 8.6 | |||||
| gp28 | + | 117 | 13.2 | Hypothetical protein ORF020 (Pseudomonas phage PA11) | 3e-03 | |||
| gp29 | + | 129 | 15.0 | |||||
| gp30 | + | 256 | 29.7 | Conserved 3′–5′ exonuclease domain of family B DNA polymerases | 3′-5′ exonuclease related to DNA polymerase, epsilon chain | |||
| Hypothetical protein ORF021 (Pseudomonas phage PA11) | 8e-87 | |||||||
| Exonuclease related to epsilon chain of DNA polymerase (Xanthomonas phage Xp10) | 3e-34 | |||||||
| gp31 | + | 115 | 12.8 | Hypothetical protein ViPhICP2p65 (Vibrio phage ICP2) | 1e-25 | Transcriptional repressor | ||
| Transcriptional repressor (Pseudoalteromonas phage H105/1) | 3e-16 | |||||||
| gp32 | + | 63 | 7.2 | |||||
| gp33 | + | 191 | 21.6 | Conserved poly(A) polymerase head domain | Poly(A) polymerase | |||
| gp34 | + | 62 | 7.2 | |||||
| gp35 | − | 608 | 64.9 | 19/18 | 49/43 | Conserved phage tail repeat-like domain | Possible tail fiber | |
| Collagen triple helix repeat protein (Victivallis vadensis ATCC BAA-548) | 8e-06 | |||||||
| Putative tail fiber (Acinetobacter baumannii 1656-2) | 6e-05 | |||||||
| gp36 | − | 102 | 11.5 | Hypothetical protein (Vibrio phage ICP2) | 2e-13 | |||
| gp37 | − | 118 | 13.3 | |||||
| gp38 | − | 144 | 15.4 | ORF-B (predicted transmembrane helix, suspected holin; VpV262) | 8e-29 | Transmembrane helix (Psipred) | ||
| gp39 | − | 684 | 73.7 | 24/25 | 41/44 | Coiled coils (COILS) | ||
| gp40 | − | 1413 | 156.4 | 38/42 | 42/47 | ORF-U (VpV262) | 2e-34 | Putative tail sheath |
| gp26 (Roseobacter phage SIO1) | 2e-13 | |||||||
| gp41 | − | 153 | 16.0 | 1/2 | 9/19 | |||
| gp42 | − | 98 | 11.8 | |||||
| gp43 | − | 741 | 82.0 | 9/11 | 16/19 | Hypothetical protein ORF038 (Pseudomonas phage PA11) | 0.0 | Putative tail appendage, 2 right-handed β-helices (BetaWrapPro) |
| gp27 (Roseobacter phage SIO1) | 8e-74 | |||||||
| ORF-P (VpV262) | 3e-24 | |||||||
| gp44 | − | 64 | 6.9 | Hypothetical protein ORF038 (Pseudomonas phage PA11) | 9e-05 | |||
| gp45 | − | 251 | 29.2 | 3/4 | 17/27 | Hypothetical protein ORF040 (Pseudomonas phage PA11) | 1e-109 | |
| gp28 (Roseobacter phage SIO1) | 7e-26 | |||||||
| ORF-Q (VpV262) | 9e-10 | |||||||
| gp46 | − | 114 | 12.0 | 2 | 41 | Head fiber protein (Bacillus phage B103) | 6e-05 | Possible head fiber |
| gp47 | − | 150 | 15.4 | 5/3 | 36/25 | Putative tail fiber, β-structure (Psipred), right-handed β-helix (BetaWrapPro) | ||
| gp48 | − | 312 | 34.7 | 25/5 | 92/32 | Hypothetical protein ORF044 (Pseudomonas phage PA11) | 2e-137 | Major capsid protein |
| ORF-K (VpV262) | 2e-36 | |||||||
| P22 gp6-like protein (Clostridium phage CP26F and 39-O) | 1e-02 | |||||||
| gp49 | − | 264 | 29.1 | Hypothetical protein ORF045 (Pseudomonas phage PA11) | 1e-27 | Scaffold protein, helical structure (Psipred) | ||
| gp30 (Roseobacter phage SIO1) | 6e-09 | Coiled coil (COILS) | ||||||
| ORF-J (coiled coil; scaffold protein; VpV262) | 8e-09 | |||||||
| gp50 | − | 82 | 9.6 | Hypothetical protein ORF046 (Pseudomonas phage PA11) | 4e-10 | |||
| gp51 | − | 592 | 67.5 | 20/19 | 33/39 | Hypothetical protein ORF047 (Pseudomonas phage PA11) | 0.0 | Portal protein |
| ORF-H (VpV262) | 1e-78 | |||||||
| gp3, gp5 (Roseobacter phage SIO1) | 1e-63 | |||||||
| gp8 (Phormidium phage Pf-WMP3) | 4e-09 | |||||||
| gp52 | − | 535 | 61.6 | Hypothetical protein ORF049 (Pseudomonas phage PA11) | 0.0 | DNA packaging enzyme/terminase | ||
| ORF-G (VpV262) | 1e-113 | |||||||
| gp1, gp2, gp4 (Roseobacter phage SIO1) | 1e-97 | |||||||
| Terminase (Pf-WMP3) | 6e-16 | |||||||
| Terminase (enterobacterial phage T7) | 4e-06 | |||||||
| gp53 | − | 165 | 19.5 | Hypothetical protein ORF050 (Pseudomonas phage PA11) | 3e-62 | |||
| gp54 | − | 636 | 68.1 | 21/28 | 53/66 | |||
| gp55 | − | 191 | 21.1 | 1 | 11 | Hypothetical protein ORF052 (Pseudomonas phage PA11) | 7e-37 | |
| gp56 | − | 68 | 8.0 | |||||
| gp57 | − | 97 | 11.1 | |||||
| gp58 | + | 210 | 23.4 | |||||
| gp59 | + | 76 | 8.2 | |||||
| gp60 | + | 116 | 13.2 | |||||
| gp61 | + | 69 | 7.8 | |||||
| gp62 | + | 58 | 6.6 | |||||
| gp63 | + | 158 | 18.7 | Conserved hypothetical protein Acear_1218 (Acetohalobium arabaticum DSM 5501) | 3e-21 | |||
| gp64 | + | 74 | 8.2 | |||||
| gp65 | + | 73 | 8.4 | |||||
| gp66 | + | 125 | 14.2 | |||||
| gp67 | + | 121 | 14.3 | Conserved hydrolase_2 domain | Cell wall hydrolase | |||
| Cell wall hydrolase, SleB (alphaproteobacterium BAL199) | 4e-09 | |||||||
| gp68 | + | 144 | 16.4 | |||||
| gp69 | + | 152 | 16.9 | |||||
| gp70 | + | 503 | 56.2 | Hypothetical protein ORF070 (Pseudomonas phage PA11) | 3e-03 | |||
The first value in each column corresponds to data from SDS-PAGE band excision (see Fig. 2). The second value, if present, corresponds to data from whole, non-SDS-PAGE-treated virions.
Percentage of amino acid sequence observed.
Summary of hits found via PSI-BLAST analysis. Protein sources are in parentheses. Conserved domains are underlined.
Secondary structure prediction programs are underlined.
The CW02 genome is similar to the genomes of T7-like phages. PSI-BLAST analysis of putative CW02 ORFs resulted in matches to T7-like phages, including Pseudomonas phage PA11 (34), roseophage SIO1 (46), cyanophage Pf-WMP3 (38), and vibriophage VpV262 (23). The genomes of CW02 and PA11 have at least 24 homologous ORFs in common (Table 1), though the functions of the PA11 gene products have not yet been determined. A gene for a T7-like DNA polymerase (gp23) was identified in the CW02 genome by PSI-BLAST analysis, suggesting an evolutionary relationship between CW02 and T7. However, the genus definition of “T7-like phages” includes the presence of an RNA polymerase-encoding gene (54), which was not detected in the CW02 genome. The genomes of SIO1 and VpV262 also lack an RNA polymerase gene (23). Another attribute of the “T7-like phage” genus is the presence of terminal repeats on either end of the viral genome (54), though SIO1 lacks terminal redundancy (46). The CW02 genome also lacks terminal redundancy. The discrepancies of VpV262 and SIO1 with the definition of the “T7-like phage” genus led to the proposal of a broader, ancestral T7-like supergroup that predates the divergence of phages with and without an RNA polymerase gene (23), here referred to as “T7-like viruses” and “VpV262-like viruses,” respectively.
The arrangement of structural genes suggests that CW02 belongs to the VpV262-like cluster within the T7 supergroup (23). A characteristic feature of the T7 supergroup is the presence of a conserved module of late structural genes involved in the assembly of the capsid head (23). The head structure module of the VpV262-like cluster is arranged such that the genes encoding the terminase, portal, scaffold, and capsid proteins are consecutively ordered and separated by few, if any, gaps (Fig. 1). The tail structure module is generally encoded leftward of the head module (23). The structure module of T7-like viruses is related to the VpV262-like viruses by inversion, with the portal, scaffold, and capsid proteins sequentially ordered, though the terminase ORF is rearranged. In general, the genomes of T7-like viruses contain early (e.g., replicative genes) and late genes along the same strand, whereas the replicative and structural genes of VpV262-like viruses are on opposite strands (23, 38). Bioinformatic analysis of the CW02 genome revealed the same genomic arrangement as other VpV262-like viruses, including VpV262 (23) and Pf-WMP3 (38)—a conserved terminase (gp52), portal (gp51), scaffold (gp49), and capsid protein (gp48) closely grouped in sequential order (Fig. 1) and in the order opposite from that of replicative genes, including the putative DNA polymerase and helicase genes (Table 1). Several large ORFs (gp35 to gp47) are found leftward of the head module, and by virtue of the module arrangement and sequence analysis, these genes may be involved in tail assembly (Table 1). Based on these observations, CW02 likely belongs to the VpV262-like cluster within the T7 supergroup.
Fig 1.

The conserved structure module arrangement of the T7 supergroup. Genomic segments of phages T7, VpV262, Pf-WMP3, and CW02 are shown. The division of the T7 supergroup (dashed line) is characterized by inverted directionality of the genes in T7 compared to the VpV262-like cluster, as discussed in reference 38. Numbering indicates base pair positions from the 5′ to the 3′ end of the coding strand. Scale bar, 1 kb.
Proteins in the mature CW02 particle.
Twelve unique proteins were identified in the mature CW02 virion (Table 1). Proteins associated with the virion were analyzed by liquid chromatography (LC)-MS following the excision of detected SDS-PAGE bands. Ten bands were excised and then treated by in-gel trypsin digestion (Fig. 2). The gel slice corresponding to the lowest-molecular-weight proteins revealed peptide fragments from four unique ORFs (gp41, gp46, gp47, and gp55). Ten of the 12 proteins identified by gel-excision LC-MS were further confirmed by LC-MS of whole, non-SDS-PAGE-treated virions (Table 1). Three of the gel bands were identified as the same protein (gp48), and one gel band revealed two proteins of similar sizes (gp39 and gp43). Five proteins present in the virion (gp39, gp41, gp47, gp54, and gp55) did not have detectable homologous counterparts via PSI-BLAST analysis, and their functions remain unknown (Table 1).
Fig 2.
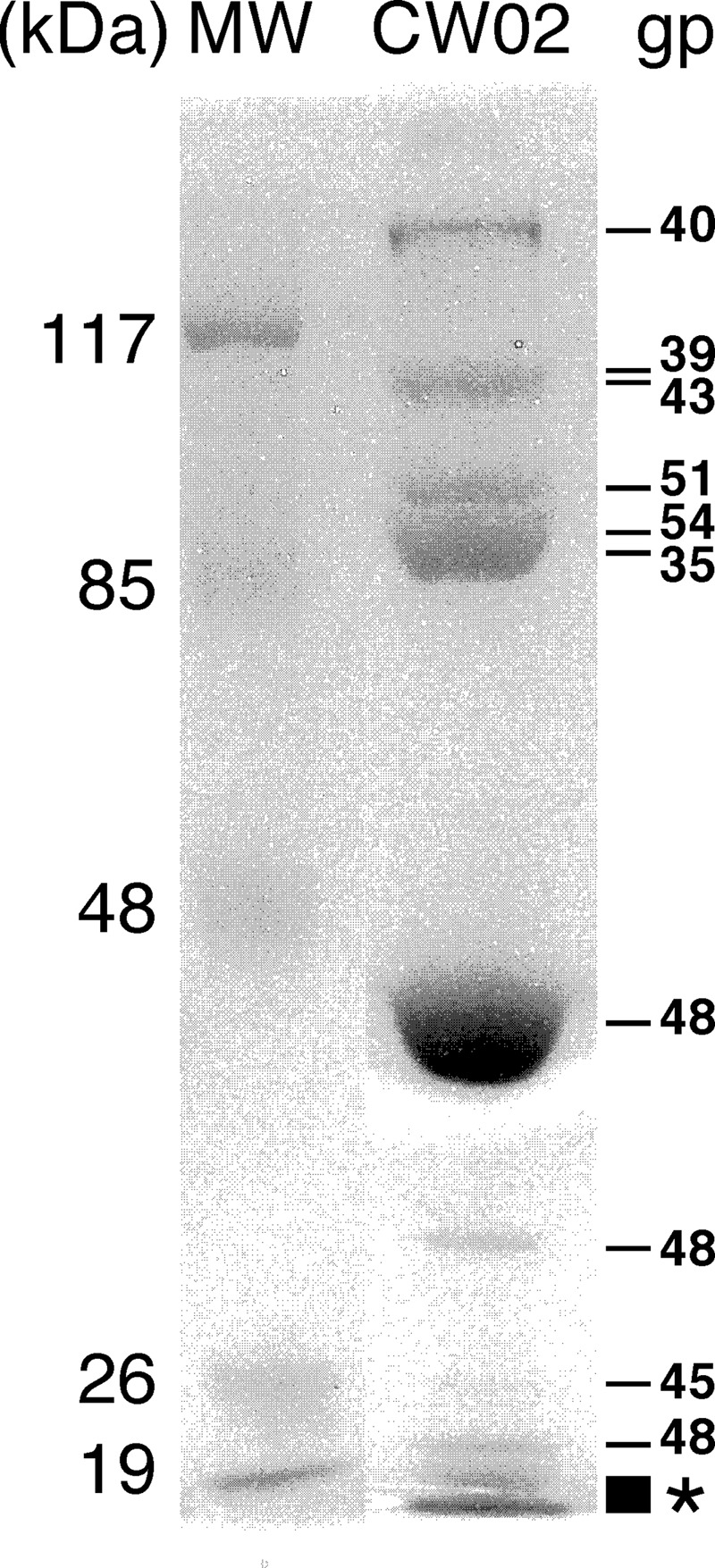
Protein composition of mature wild-type CW02 particles determined by SDS-PAGE and identified by LC-MS. gp39 and gp43 have similar sizes and were not separated in the gel. See Table 1 for properties of gene products. Lane MW contains molecular size markers. Peptide fragments from four unique ORFs (gp41, gp46, gp47, and gp55) were detected in the two lowest MW bands (asterisk).
Proteins in the CW02 head structure module.
We identified gp48 as the major capsid protein because gp48 was clearly the most abundant CW02 protein identified by SDS-PAGE (Fig. 2). PSI-BLAST analysis revealed that gp48 shows some similarities to the putative P22-like coat proteins of Clostridium phages CP26F and 39-O (49), although the E values were only on the order of 10−2 (Table 1). Pairwise sequence alignment of gp48 and the major capsid protein of T7 (gp10) showed just ∼18% identity (55 of 312 residues). Nevertheless, PSI-BLAST analysis revealed that gp48 shows more significant similarities to the putative capsid protein of VpV262 (ORF-K; E value, 3e-31) (23).
Despite poor pairwise sequence alignments, secondary structure prediction methods suggested that gp48 is related to capsid proteins of the T7-like supergroup and other tailed dsDNA phages in general. We used the Phyre Protein Fold Recognition Server (32), which predicted that gp48 adopts the same fold as the major capsid proteins of HK97 and P22 (P < 0.05). This prediction is significant because pairwise alignment of the HK97 and P22 capsid protein sequences showed just 17% identity. Pairwise alignments of gp48 with the major capsid proteins of HK97 and P22 were similar—just 13% and 20% sequence identities, respectively. This is consistent with observations that the HK97-like fold is more conserved than its sequence (28, 30, 35, 42).
Based on sequence comparisons, we propose that gp49 encodes the CW02 scaffolding protein. gp49 lies in the predicted position of the scaffold protein in the head structure module (Fig. 1). PSI-BLAST analysis of gp49 identified similarities to other putative scaffold proteins (Table 1). Many viruses require scaffolding proteins to direct the proper assembly of virus capsids. The proteins are integral components of phage proheads but are not detected in the mature phage particle (25). As expected with phage scaffold proteins, gp49 was not detected by LC-MS of mature virions. Secondary structure prediction algorithms predicted gp49 to consist of an entirely helical structure (data not shown), which is consistent with the crystal structure of phage ϕ29 scaffolding protein (42). Although the sequences of gp49 and the scaffolding protein of bacteriophage T7 share only ∼15% sequence identity (41 of 264 residues), both proteins are predicted to have two or more regions that can form coiled-coil arrangements (Fig. 3). The predictions differ in length and probability, but they suggest that the two proteins have structural similarities.
Fig 3.

Coiled-coil prediction profiles of CW02 gp49 and the scaffold protein of phage T7 (gp9). The probabilities of a coiled coil in either protein were determined using the program COILS.
Based on sequence comparisons, CW02 gp51 appears to be a portal protein. Tailed dsDNA phages encode portal proteins that enable DNA passage during packaging and ejection. The portal also functions as a connector between the capsid and tail proteins. PSI-BLAST analysis of gp51 showed a conserved domain belonging to the head-tail connector superfamily. The sequence of gp51 is strongly conserved with the putative portal proteins of VpV262, SIO1, and Phormidium phage Pf-WMP3/4 (Table 1). We used secondary structure prediction methods to investigate possible similarities between gp51 and the portal protein of phage T7 on the structural level and found the profiles to show some resemblance (data not shown).
Sequence comparisons suggest that CW02 gp52 is the terminase protein. Phage terminases are enzymes that facilitate packing of the viral genome into the phage proheads and, like scaffold proteins, are not found in the mature virion (9). As expected, gp52, was not detected by LC-MS of mature CW02 particles. CW02 gp52 had weak sequence similarity to the terminase protein of phage T7 but stronger sequence similarity to the putative portal proteins of VpV262 and SIO1. Secondary structure prediction of gp52 showed an arrangement similar to that of the terminase of phage T7 (data not shown), suggesting that their functions are conserved.
Proteins in the CW02 tail structure module.
Putative tail proteins were observed leftward of the head structure module. This placement is consistent with tail genes in the known T7-like supergroup (23). A number of large ORFs are found in an appropriate location of the CW02 genome that could reasonably be thought to encode tail-associated proteins. For instance, PSI-BLAST analysis of gp35 revealed a conserved “phage tail-like repeat” domain. Also, gp40, encoded by the largest putative ORF in the CW02 genome, is a 1,413-amino-acid protein—consistent with the large sizes of phage tail sheath proteins. Trimeric structures of coiled coils and β-helices are a common motif in tail-associated proteins, such as the tail spike protein of P22 (homotrimers of β-helices) (51) and tail fibers of bacteriophage T7 (triple-stranded coiled coils) (52). The presence of coiled coils and β-helices in candidate ORFs within the putative tail structure module was examined. The BetaWrapPro program revealed right-handed β-helical motifs in gp43 and gp47, whereas the COILS program detected coiled-coil motifs in gp39 and gp41.
Electron microscopy of CW02 particles.
CW02 virions are morphologically similar to members of the family Podoviridae. Negatively stained CW02 particles were visualized by transmission electron microscopy and revealed particles with icosahedral heads and a short, tail-like feature (Fig. 4A). However, the tail-like structures showed inconsistencies among individual particles. For example, a short, stubby tail was observed in some of the particles, whereas in other particles, a short, thin, fiber-like feature was more apparent. Cryo-EM was used to visualize CW02 particles in the solution state (Fig. 4B). Although similar stubby and thin-fiber tails were observed, tail-like structures were more difficult to discern in cryo-EM micrographs, which may be a consequence of decreased contrast in samples with high salt concentrations (29).
Fig 4.

Electron microscopy of CW02 by negative stain (A) and cryogenic (B) methods. Stubby or thin tail-like features are marked by white arrowheads. Collapsed, empty particles are shown by black arrowheads. The inset in panel B shows examples of particle images used in computing the 3D reconstruction (black scale bar, 50 nm).
T = 7 levo icosahedral capsid lattice.
Cryo-EM images of CW02 were used to compute a 3D reconstruction. The 3D reconstruction of the CW02 head was determined to a 16-Å resolution using 8,695 particle images (out of a total of 10,677 particle images) and applying icosahedral averaging to maximize the signal-to-noise ratio (Fig. 5A).
Fig 5.

Cryo-EM-based reconstruction of CW02. (A) Stereo image of the CW02 capsid surface rendered at a 1-σ contour level. Structures are shaded from light to dark, corresponding to the radius. (B) The central slice of the reconstruction (perpendicular to a 2-fold symmetry axis). Black represents protein or DNA density. Bar, 25 nm. Black arrowheads indicate turrets. The arrow indicates potential capsid-dsDNA contacts. (C) Plot of average density with respect to the radius of the reconstruction. DNA and capsid density peaks are labeled.
The head of CW02 is arranged in a T = 7 icosahedral lattice (Fig. 5A). T = 7 lattices have two possible arrangements that are mirror images of each other. Because transmission electron microscopy images are 2D projected views, information about the object handedness is lost in its 3D reconstruction. The structures of all currently known T = 7 bacteriophages, including lambda (60), HK97 (56), and T7 (2, 13, 28), are of the levo orientation. We performed tilt experiments to determine the handedness of the CW02 capsid. This method relies on the cross-correlation of images of tilted specimens with projections of the tilted model of either handedness (7). Like that of the other T = 7 phages, the handedness of CW02 was determined to be of the levo orientation. For particles showing a clear difference between left- and right-handed T = 7 lattices (i.e., correlation coefficient difference of ≥0.10), the average correlation coefficient was 0.41 ± 0.06 for T = 7 levo handedness and 0.26 ± 0.06 for T = 7 dextro handedness (n = 40).
Architecture of the CW02 head.
The diameters of the CW02 head measured 691 Å, 627 Å, and 585 Å along the icosahedral 5-fold, 3-fold, and 2-fold axes, respectively. The average thickness of the capsid shell is approximately 37 Å. Sixty hexamers and 12 pentamers comprise the icosahedral head.
We postulate that the CW02 capsid has a conserved structure with that of the T7 capsid and other phages of the Caudovirales order. To date, the capsids of all tailed dsDNA bacteriophages appear to adopt the HK97-like fold. For instance, the pseudoatomic capsid structure of phage T7, solved by cryo-EM and modeling methods, showed the conserved fold (2, 28). The centers between two hexameric capsomeres of the CW02 reconstruction are separated by approximately 140 Å, which is a characteristic feature of the HK97-like fold (15, 20, 30). The fold consists of two domains: the axial (A) domain and the peripheral (P) domain (24, 56) (Fig. 6A). The A domain is a compact, wedge-like structure with two α-helices (α5 and α6) and a four-stranded β-sheet. The P domain is elongated, consisting of a long helix (α3) accompanied by a long β-sheet. Together, the domains form an L-shaped protein with a continuous hydrophobic core (24). The HK97 capsid protein also bears an extended amino-terminal arm and a cross-linking loop (Fig. 6A), but these segments are not as conserved in other tailed dsDNA phages.
Fig 6.

Rigid-body fit of the HK97-like fold (24) into the CW02 capsid. (A) Ribbon diagram of the HK97 protein with pertinent helices and domains labeled. Yellow, N terminus; green, cross-linking arm; blue, A domain; red, P domain (24). Yellow, green, light red, and light blue indicate coordinates omitted from the CW02 fitting. (B and C) Frontal (B) and side (C) views of the HK97 A and P domains (chains A to G) fitted into CW02 hexamers (purple, red, yellow, green, teal, and blue, chains A to F, respectively) and pentamers (orange, chain G). Each subunit is colored according to conventions used previously (24). Solid lines in panel B outline the wedge-like conformation of the A domain. (D) Central section of the CW02 reconstruction fitted by multiple copies of the HK97-like fold. The white arrowhead points to concentric layers of the DNA genome. In all of the panels; representative gpD-like densities are indicated by stars; the A and P domains are labeled in italics; symmetry and hexamer axes (dashed lines) are labeled with pentagons (5-fold), triangles (3-fold), ovals (2-fold), and a hexagon (local 6-fold, in panel C only); and turret densities were omitted.
To further investigate the similarities between CW02 and HK97-like capsids, we performed rigid-body fitting of the HK97 A and P domains into the CW02 reconstruction (Fig. 6B to D). The Cα backbones of the A and P domains were fitted into the density map as either hexamers or pentamers. Flexible loops were omitted from the fitting, leaving 128 to 130 residues in the fitted core structure. Similar results were obtained by fitting hexameric and pentameric capsomeres, e.g., the agreement between the characteristic slants of the α5 and α6 helices into the cryo-EM densities (Fig. 6B, solid lines). The elongated α3 helix and accompanying P domain β-sheet also fit snugly into the CW02 densities (Fig. 6C). The P domains interact in a 3-fold fashion at the 3-fold and quasi-3-fold regions. Our fit showed agreement between the P domain coordinates and the CW02 map, though voluminous triangular densities were found above each of the P domain fits (Fig. 6B and C, star). The overall fit is consistent with previous studies that showed the intracapsomere stabilization role of the A domain and the intercapsomere contacts made by the P domain (24).
Other than the main fold, other elements of the HK97 capsid protein appear to be missing from CW02. The HK97 capsid protein structure consists of a cross-linking loop and extended N-terminal arms that stabilize interactions within each capsomere (18, 56). No evidence of cross-linking is seen in CW02 due to the absence of gp48 oligomers in SDS-PAGE analysis (Fig. 2). The HK97 cross-linking loop and N-terminal arms fit poorly into the intercapsomere space, suggesting that CW02 has different means of intercapsomere contacts (data not shown).
gpD-like densities at the 3-fold and quasi-3-fold axes.
Other densities in the CW02 reconstruction were not accountable by the HK97-like fold. Most prominently, the intersection of every three capsomeres is bridged by a triangular body found on the 3-fold and quasi-3-fold axes (Fig. 5 and stars in Fig. 6B and C). Both types of triangular densities showed consistent structures, despite icosahedral averaging, suggesting that the proteins on the 3-fold and quasi-3-fold axes are identical.
The functions of the triangular densities are unknown, but their structures are reminiscent of the auxiliary protein gpD trimer of bacteriophage lambda (35, 60) (Fig. 7). Similar to the gpD trimer, the positions of the triangular densities in CW02 on the 3-fold and quasi-3-fold axes suggest an intercapsomere stabilization role (35) and its positions coincide with the covalent cross-links formed by the HK97 capsid protein (56). The P domains of the HK97-like fold form intercapsomere contacts directly beneath the triangular densities (Fig. 6). In other HK97-like phages, intercapsomere interactions by the P domain are accompanied by more extensive intercapsomere interactions from additional accessory proteins (including gpD of lambda [60] and hoc of T4 [19]), covalent cross-links (as in HK97 [56]), or additional domains as part of the capsid protein. For instance, the pseudoatomic structure of bacteriophage ϕ29 showed that the major capsid protein consists of the HK97-like domain and an additional immunoglobulin-like domain (42). Bacteriophage P22 also consists of extra globular densities on the outermost surface of the capsid (30). However, unlike the densities seen in CW02, the additional domains in ϕ29 and P22 do not form trimeric structures.
Fig 7.

Segmented views of the subunits that make up the CW02 (A) and lambda (B) (35) capsids. Representative hexameric and pentameric capsomer subunits of the asymmetric unit are colored as in Fig. 6B. Representative trimeric, gpD-like densities at the intersection of every three capsomeres are pink. The turret density of CW02, located along the 5-fold axis, is brown.
The triangular densities likely belong to a separate domain of CW02 gp48 and are not a separate, minor capsid protein. If the triangular densities of CW02 represent separate protein subunits, then 405 to 420 copies of the protein would be required per capsid (i.e., the same number of major capsid protein subunits), as is the case with gpD of lambda (35). Yet, SDS-PAGE analysis of mature CW02 particles did not reveal other proteins as abundant as gp48, arguing against the presence of a separate gpD-like protein (Fig. 2). BLAST analysis of the CW02 genome did not reveal the presence of a gene like that for gpD. Thus, the triangular densities of CW02 most likely represent an additional, non-core domain of gp48, similar to the amino-terminal arm and cross-linking domain in HK97 (24, 56) (Fig. 6A), the Ig-like domain of ϕ29 (42), and the additional globular domains of P22 (30). The additional ∼180 CW02 gp48 residues unmodeled in our fitting not only connect pieces of the core domain but also likely form the additional gpD-like domain observed above the fitted core HK97 structure (Fig. 6).
Lack of tail density in 3D reconstructions.
The presence of a tail structure module in the CW02 genome (Table 1) and apparent tail-like features in the EM images (Fig. 4) suggests that each capsid is associated with a short tail. From these images, the tail appears to be a stubby cylindrical component centered on a thin, short rod.
Yet, despite the use of asymmetry or cyclic 5-fold (C5) symmetry during the reconstruction process, none of our reconstructions produced a tail-like structure. The addition of artificial, cylindrical, tail-like (short and stubby or short and thin) or portal-like densities to a single vertex of the CW02 capsid as an orientation-refining fiducial marker did not produce different structures (data not shown). One noteworthy difference between the pentameric and hexameric capsomeres is the presence of an exterior bulge stemming from the center of the pentamers (Fig. 5). The bulge may correlate with an additional protein complex and may be part of the tail apparatus. Alternatively, the tail-like material observed in negative stain and cryo-EM images (Fig. 4) may be partial discharge of DNA or discharge of protein from the capsid. In many of our preparations of negatively stained and frozen-hydrated particles, tail-like densities were not observed in the electron micrographs (data not shown). Nevertheless, considering all of the evidence, we think that CW02 most likely contains a tail that was “hidden” in our cryo-EM 3D analysis by the decreased contrast of the high-salt buffer (29).
Structure of the encapsidated dsDNA and lack of internal membrane.
HK97-like phages appear to have conserved mechanisms of dsDNA packaging, such that the viral genome is cylindrically spooled within the capsid interior (12, 27). We observed at least eight concentric layers of densities within the icosahedrally averaged head of CW02 (Fig. 5C and D). Each layer is spaced approximately 24 Å apart, similar to the dsDNA densities observed in other bacteriophage structures, including T7 (12, 13) and lambda (27). The layering pattern is consistent with the DNA spooling model established for T7 (12). Weak densities are also observed at the interface between the outermost DNA layer and the capsid (Fig. 5C), indicating possible interactions between the capsid protein and dsDNA core.
Some turreted, extremophilic viruses of the PRD1-like lineage, including Sulfolobus turreted icosahedral virus and SH1, encase an internal lipid membrane that is thought to aid in the injection of the genome into the host cell (33). Reconstructions of CW02 did not show any evidence of material other than dsDNA within the capsid.
Presence of turrets on 5-fold vertices.
On each icosahedral vertex, the CW02 reconstruction showed the presence of turret-like structures that extend ∼40 Å beyond the capsid head. The turret densities appear disconnected from the capsid, suggesting that the linker between the turret and capsid is disordered or incompatible with 5-fold symmetry. To investigate whether the turrets are unique to a single vertex or if they are positioned at each of the 12 5-fold vertices of the capsid, we computed reconstructions of CW02 without any imposed symmetry (in both the orientation finding and 3D image reconstruction processes). These reconstructions also showed the presence of disconnected turrets on all vertices (data not shown). The 3D reconstructions and class averages from the turrets (see Materials and Methods) showed inconclusive results, but control experiments with images of poliovirus complexed with antibodies to the 5-fold region (J. Lin et al., unpublished data) produced correct results. Therefore, CW02 turrets appear to lack a consistent order.
The function of the CW02 turrets and the gene encoding the turret protein remain unknown, but the turrets may be unique appendages similar to those seen in other extremophilic viruses, including the archaeal viruses Sulfolobus turreted icosahedral virus (33, 40) and halovirus SH1 (29). Other icosahedral viruses with protein turrets or spikes on vertices include adenovirus (53) and bacteriophage PRD1 (47). The function of turrets is thought to be to assist in host adsorption (29). Collectively, turreted viruses belong to the PRD1-like lineage and consist of capsids with a single or double β-jellyroll fold, unlike CW02. Thus, the presence of turrets on CW02 could represent structural features conserved between viruses of the PRD1-like and HK97-like lineages.
ACKNOWLEDGMENTS
This research was supported by a BYU Graduate Research Fellowship, a Roland K. Robins Graduate Research Fellowship, and a Jennie R. Swensen Graduate Research Fellowship to P.S.S.; by BYU institutional funds to D.M.B., J.T.P., and D.P.B.; and by grants from the Weber State University Office of Undergraduate Research and Research Scholarship and Professional Growth Committee to M.J.D. The BYU Fulton Supercomputing Lab provided computer resources.
We thank Edward Wilcox and the BYU Sequencing Center for help with genomic sequencing, Robert Swenson and David Eng for help with image processing, Kristin Pande for laboratory assistance, Jeffrey Farrer and Michael Standing for microscopy support, Alasdair Steven and Weimin Wu for discussions of high-salt cryo-EM and gpD, and Zachary Sexton and Cristy Waters for isolation of CW02 from the GSL.
Footnotes
Published ahead of print 16 May 2012
REFERENCES
- 1. Ackermann H-W. 2003. Bacteriophage observations and evolution. Res. Microbiol. 154:245–251 [DOI] [PubMed] [Google Scholar]
- 2. Agirrezabala X, et al. 2007. Quasi-atomic model of bacteriophage T7 procapsid shell: insights into the structure and evolution of a basic fold. Structure 15:461–472 [DOI] [PubMed] [Google Scholar]
- 3. Atlas RM, Parks LC. (ed). 1993. Handbook of microbiological media. CRC Press, Boca Raton, FL [Google Scholar]
- 4. Baker TS, Cheng RH. 1996. A model-based approach for determining orientations of biological macromolecules imaged by cryo-electron microscopy. J. Struct. Biol. 116:120–130 [DOI] [PubMed] [Google Scholar]
- 5. Belnap DM, et al. 2000. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J. Virol. 74:1342–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belnap DM, Grochulski WD, Olson NH, Baker TS. 1993. Use of radial density plots to calibrate image magnification for frozen-hydrated specimens. Ultramicroscopy 48:347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belnap DM, Olson NH, Baker TS. 1997. A method for establishing the handedness of biological macromolecules. J. Struct. Biol. 120:44–51 [DOI] [PubMed] [Google Scholar]
- 8. Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29:2607–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Black LW. 1995. DNA packaging and cutting by phage terminases: control in phage T4 by a synaptic mechanism. Bioessays 17:1025–1030 [DOI] [PubMed] [Google Scholar]
- 10. Briggs JAG, et al. 2005. Classification and three-dimensional reconstruction of unevenly distributed or symmetry mismatched features of icosahedral particles. J. Struct. Biol. 150:332–339 [DOI] [PubMed] [Google Scholar]
- 11. Bryson K, et al. 2005. Protein structure prediction servers at University College London. Nucleic Acids Res. 33:W36–W38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cerritelli ME, et al. 1997. Encapsidated conformation of bacteriophage T7 DNA. Cell 91:271–280 [DOI] [PubMed] [Google Scholar]
- 13. Cerritelli ME, Conway JF, Cheng N, Trus BL, Steven AC. 2003. Molecular mechanisms in bacteriophage T7 procapsid assembly, maturation, and DNA containment, p 301–323 In Chiu W, Johnson JE. (ed), Advances in protein chemistry: virus structure, vol 64 Academic Press, San Diego, CA: [DOI] [PubMed] [Google Scholar]
- 14. Cheng N, et al. 2002. Handedness of the herpes simplex virus capsid and procapsid. J. Virol. 76:7855–7859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi KH, et al. 2008. Insight into DNA and protein transport in double-stranded DNA viruses: the structure of bacteriophage N4. J. Mol. Biol. 378:726–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Conway JF, Steven AC. 1999. Methods for reconstructing density maps of “single” particles from cryoelectron micrographs to subnanometer resolution. J. Struct. Biol. 128:106–118 [DOI] [PubMed] [Google Scholar]
- 17. Crowther RA, Amos LA, Finch JT, DeRosier DJ, Klug A. 1970. Three dimensional reconstructions of spherical viruses by Fourier synthesis from electron micrographs. Nature 226:421–425 [DOI] [PubMed] [Google Scholar]
- 18. Duda RL. 1998. Protein chainmail. Cell 94:55–60 [DOI] [PubMed] [Google Scholar]
- 19. Fokine A, et al. 2004. Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. U. S. A. 101:6003–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fokine A, et al. 2005. A three-dimensional cryo-electron microscopy structure of the bacteriophage φKZ head J. Mol. Biol. 352:117–124 [DOI] [PubMed] [Google Scholar]
- 21. Fuller SD, Butcher SJ, Cheng RH, Baker TS. 1996. Three-dimensional reconstruction of icosahedral particles—the uncommon line. J. Struct. Biol. 116:48–55 [DOI] [PubMed] [Google Scholar]
- 22. Goel U, Kauri T, Ackermann H-W, Kusher DJ. 1996. A moderately halophilic Vibrio from a Spanish saltern and its lytic bacteriophage. Can. J. Microbiol. 42:1015–1023 [Google Scholar]
- 23. Hardies SC, Comeau AM, Serwer P, Suttle CA. 2003. The complete sequence of marine bacteriophage VpV262 infecting Vibrio parahaemolyticus indicates that an ancestral component of a T7 viral supergroup is widespread in the marine environment. Virology 310:359–371 [DOI] [PubMed] [Google Scholar]
- 24. Helgstrand C, et al. 2003. The refined structure of a protein catenane: the HK97 bacteriophage capsid at 3.44 Å resolution. J. Mol. Biol. 334:885–899 [DOI] [PubMed] [Google Scholar]
- 25. Hendrix RW, Casjens SR. 1975. Assembly of bacteriophage lambda heads: protein processing and its genetic control in petit lambda assembly. J. Mol. Biol. 91:187–199 [DOI] [PubMed] [Google Scholar]
- 26. Heymann JB, Belnap DM. 2007. Bsoft: image processing and molecular modeling for electron microscopy. J. Struct. Biol. 157:3–18 [DOI] [PubMed] [Google Scholar]
- 27. Hud NV, Downing KH. 2001. Cryoelectron microscopy of λ phage DNA condensates in vitreous ice: the fine structure of DNA toroids. Proc. Natl. Acad. Sci. U. S. A. 98:14925–14930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ionel A, et al. 2011. Molecular rearrangements involved in the capsid shell maturation of bacteriophage T7. J. Biol. Chem. 286:234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jäälinoja HT, et al. 2008. Structure and host-cell interaction of SH1, a membrane-containing, halophilic euryarchaeal virus. Proc. Natl. Acad. Sci. U. S. A. 105:8008–8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang W, et al. 2003. Coat protein fold and maturation transition of bacteriophage P22 seen at subnanometer resolutions. Nat. Struct. Biol. 10:131–135 [DOI] [PubMed] [Google Scholar]
- 31. Kauri T, Ackermann H-W, Goel U, Kushner DJ. 1991. A bacteriophage of a moderately halophilic bacterium. Arch. Microbiol. 156:435–438 [Google Scholar]
- 32. Kelley LA, Sternberg MJE. 2009. Protein structure prediction on the web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 33. Khayat R, et al. 2005. Structure of an archaeal virus capsid protein reveals a common ancestry to eukaryotic and bacterial viruses. Proc. Natl. Acad. Sci. U. S. A. 102:18944–18949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kwan T, Liu J, DuBow M, Gros P, Pelletier J. 2006. Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J. Bacteriol. 188:1184–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lander GC, et al. 2008. Bacteriophage lambda stabilization by auxiliary protein gpD: timing, location, and mechanism of attachment determined by cryoEM. Structure 16:1399–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175 In Stackebrandt E, Goodfellow M. (ed), Nucleic acid techniques in bacterial systematics. John Wiley and Sons, New York, NY [Google Scholar]
- 37. Larkin MA, et al. 2007. ClustalW and ClustalX version 2. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 38. Liu X, et al. 2008. Genomic analysis of freshwater cyanophage Pf-WMP3 infecting cyanobacterium Phormidium foveolarum: the conserved elements for a phage. Microb. Ecol. 56:671–680 [DOI] [PubMed] [Google Scholar]
- 39. Lupas A, Van Dyke M, Stock J. 1991. Predicting coiled coils from protein sequences. Science 252:1162–1164 [DOI] [PubMed] [Google Scholar]
- 40. Maaty WSA, et al. 2006. Characterization of the archaeal thermophile Sulfolobus turreted icosahedral virus validates an evolutionary link among double-stranded DNA viruses from all domains of life. J. Virol. 80:7625–7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McDonnell AV, et al. 2006. Fold recognition and accurate sequence-structure alignment of sequences directing beta-sheet proteins. Proteins Struct. Funct. Bioinform. 63:976–985 [DOI] [PubMed] [Google Scholar]
- 42. Morais MC, et al. 2005. Conservation of the capsid structure in tailed dsDNA bacteriophages: the pseudoatomic structure of φ29. Mol. Cell 18:149–159 [DOI] [PubMed] [Google Scholar]
- 43. Olsen JV, et al. 2005. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4:2010–2021 [DOI] [PubMed] [Google Scholar]
- 44. Oren A, Bratbak G, Heldal M. 1997. Occurrence of virus-like particles in the Dead Sea. Extremophiles 1:143–149 [DOI] [PubMed] [Google Scholar]
- 45. Pettersen EF, et al. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 46. Rohwer F, et al. 2000. The complete genomic sequence of the marine phage Roseophage SIO1 shares homology with nonmarine phages. Limnol. Oceanogr. 45:408–418 [Google Scholar]
- 47. Rydman PS, et al. 1999. Bacteriophage PRD1 contains a labile receptor-binding structure at each vertex. J. Mol. Biol. 291:575–587 [DOI] [PubMed] [Google Scholar]
- 48. Sanz-García E, Stewart AB, Belnap DM. 2010. The random-model method enables ab initio three-dimensional reconstruction of asymmetric particles and determination of particle symmetry. J. Struct. Biol. 171:216–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Seal BS, et al. 2011. Clostridium perfringens bacteriophages φCP39O and φCP26F: genomic organization and proteomic analysis of the virions. Arch. Virol. 156:25–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shaikh TR, et al. 2008. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat. Protoc. 3:1941–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Steinbacher S, et al. 1996. Crystal structure of phage P22 tailspike protein complexed with Salmonella sp. O-antigen receptors. Proc. Natl. Acad. Sci. U. S. A. 93:10584–10588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Steven AC, et al. 1988. Molecular substructure of a viral receptor-recognition protein. The gp17 tail-fiber of bacteriophage T7. J. Mol. Biol. 200:351–365 [DOI] [PubMed] [Google Scholar]
- 53. van Raaij MJ, Mitraki A, Lavigne G, Cusack S. 1999. A triple beta-spiral in adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401:935–938 [DOI] [PubMed] [Google Scholar]
- 54. van Regenmortel MHV, et al. 2000. Virus taxonomy: the classification and nomenclature of viruses. VIIth Report of the International Committee on Taxonomy of Viruses Academic Press, San Diego, CA [Google Scholar]
- 55. Van Valen L. 1973. A new evolutionary law. Evol. Theor. 1:1–30 [Google Scholar]
- 56. Wikoff WR, et al. 2000. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289:2129–2133 [DOI] [PubMed] [Google Scholar]
- 57. Wiśniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat. Methods 6:359–362 [DOI] [PubMed] [Google Scholar]
- 58. Wommack KE, Colwell RR. 2000. Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64:69–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yan X, Dryden KA, Tang J, Baker TS. 2007. Ab initio random model method facilitates 3D reconstruction of icosahedral particles. J. Struct. Biol. 157:211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang F, et al. 2000. Novel fold and capsid-binding properties of the lambda-phage display platform protein gpD. Nat. Struct. Biol. 7:230–237 [DOI] [PubMed] [Google Scholar]