

Abstract
Recent studies in an experimental model of rabies showed major structural changes in the brain involving neuronal processes that are associated with severe clinical disease. Cultured adult rat dorsal root ganglion (DRG) neurons infected with the challenge virus standard-11 strain of rabies virus (CVS) showed axonal swellings and immunostaining for 4-hydroxy-2-nonenal (4-HNE), indicating evidence of lipid peroxidation associated with oxidative stress and reduced axonal growth compared to that of mock-infected DRG neurons. We have evaluated whether nuclear factor (NF)-κB might act as a critical bridge linking CVS infection and oxidative stress. On Western immunoblotting, CVS infection induced expression of the NF-κB p50 subunit compared to that of mock infection. Ciliary neurotrophic factor, a potent activator of NF-κB, had no effect on mock-infected rat DRG neurons and reduced the number of 4-HNE-labeled puncta. SN50, a peptide inhibitor of NF-κB, and CVS infection had an additive effect in producing axonal swellings, indicating that NF-κB is neuroprotective. The fluorescent signal for subunit p50 was quantitatively evaluated in the nucleus and cytoplasm of mock- and CVS-infected rat DRG neurons. At 24 h postinfection (p.i.), there was a significant increase in the nucleus/cytoplasm ratio, indicating increased transcriptional activity of NF-κB, perhaps as a response to stress. At both 48 and 72 h p.i., there was significantly reduced nuclear localization of NF-κB. CVS infection may induce oxidative stress by inhibiting nuclear activation of NF-κB. A rabies virus protein may directly inhibit NF-κB activity. Further investigations are needed to gain a better understanding of the basic mechanisms involved in the oxidative damage associated with rabies virus infection.
INTRODUCTION
Rabies is an acute viral infection of the nervous system that continues to be an important public health problem in humans and animals (15). Rabies is almost invariably fatal, and therapeutic approaches in humans are limited by an incomplete understanding of rabies pathogenesis (13). In natural rabies and in experimental models of rabies using peripheral inoculation with fixed rabies virus strains, there are mild inflammatory changes and generally a paucity of degenerative neuronal changes in the central nervous system (CNS) that do not account for the severity of the disease (11, 24). The lack of degenerative neuronal changes has led to the concept that the CNS disease in rabies is due to neuronal dysfunction rather than to neuronal death (7, 12).
Infection with the challenge virus standard-11 (CVS-11) strain of fixed rabies virus given via hindlimb footpad inoculation in transgenic mice expressing yellow fluorescent protein showed degenerative changes involving neuronal processes with beading of dendrites and axonal swellings, which were associated with a lack of morphological changes involving perikarya (31). In this model, routine histopathological studies were normal aside from mild inflammatory changes. It has been recognized that the axonal degeneration resembles what has been described in diabetic sensory and autoimmune neuropathy (4, 17, 30, 36), and we postulated that the neuronal process degeneration may be mediated by oxidative stress as in diabetic neuropathy (19, 25). Using adult mouse dorsal root ganglion (DRG) cultures, we observed that CVS infection results in axonal swellings and reduced axonal growth compared to that of mock-infected neurons without associated loss of neuronal viability or apoptosis. Immunostaining for adducts of 4-hydroxy-2-nonenal (4-HNE), which is associated with lipid peroxidation (and, hence, oxidative stress), is found at sites of axonal swellings. CVS infection induces oxidative stress in axons and generates abnormal axon morphology (e.g., swellings), and the degenerative changes closely mimic what is seen in dendrites and axons of CVS-infected mice (14).
The inducible transcription factor nuclear factor-κB (NF-κB) plays a key role in mediating transient and sustained changes in gene expression in response to a variety of external changes (10), including viral infections (28). In neurons, NF-κB plays a role in promoting survival as well as degenerative outcomes (6, 8, 23). We have hypothesized that NF-κB activation might act as a critical bridge linking CVS infection and oxidative stress and have investigated its role in CVS-induced oxidative stress in cultured adult rat DRG neurons.
MATERIALS AND METHODS
Virus.
The CVS-11 strain of fixed rabies virus (CVS), which was obtained from William H. Wunner (The Wistar Institute, Philadelphia, PA), was used in these studies. CVS was grown in baby hamster kidney (BHK) cells (C13 clone) in Dulbecco's modified Eagle medium (DMEM) supplemented with 2% newborn calf serum (NCS) (PAA Laboratories, Etobioke, Ontario, Canada) at 37°C in a 5% CO2 incubator. Viral assays of stock virus were performed by counting fluorescent foci on BHK cell monolayers.
DRG neuron cultures.
DRG neurons were isolated from adult male Sprague Dawley rats (210 g) (University of Manitoba, Winnipeg, Manitoba, Canada). Rats were killed by decapitation, the complete vertebral column was removed, and DRG neurons were isolated by dissection. DRG were mechanically and enzymatically dissociated for 45 min at 37°C in 0.125% collagenase type 4 (product no. 47C9497; Worthington, Lakewood, NJ) in Ham's F-12 medium (11765-054; Invitrogen, Carlsbad, CA) and then in 2.5% trypsin (57H9722; Worthington, Lakewood, NJ). Collagenase neutralization was achieved by adding 100% NCS to a final concentration of 33% NCS, cells were washed successively with 10% fetal bovine serum (FBS) and F-12 medium, and then the cells were collected and triturated using a glass pipette in F-12. The cell suspension was filtered through a 70-μm nylon mesh filter to remove nondissociated cells and myelin debris and centrifuged at 300 × g for 10 min. The cells were resuspended in 1 ml F-12 and then spun through a column with 15% essentially fatty-acid-free bovine serum albumin (BSA) (A9205; Sigma-Aldrich) in F-12 medium at 900 × g for 10 min. Cell pellets were collected and resuspended in 2 ml F-12 medium with 2 mM l-glutamine and Bottenstein's N2 supplements without insulin: 0.1 mg/ml transferrin, 20 nM progesterone, 100 μM putrescine, 30 nM sodium selenite, and 0.1 mg/ml fatty-acid-free BSA. For Western blots, cells were plated on six-well plates (Nunclon Surface), whereas for immunocytochemistry 18-mm circle glass coverslips coated with 0.5 mg/ml poly-dl-ornithine (P8638; Sigma-Aldrich) and 2 μg/ml natural mouse laminin (23017-015; Invitrogen) were used. Cultures were maintained in N2–F-12 medium at 37°C in humidified air containing 5% CO2. Two days after plating, viral adsorption of cultured DRG neurons was performed, with CVS at a multiplicity of infection of 10 fluorescent focus-forming units per cell (in N2–F-12 medium with 2% NCS) for 1 h or cultures were mock-infected and then fresh N2–F12 medium was added.
Immunocytochemistry for tubulin, rabies virus antigen, p50 subunit of NF-κB, and 4-hydroxy-2-nonenal adducts.
CVS- and mock-infected cultured DRG neurons at 72 h postinfection (p.i.) were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) (pH 7.4) for 15 min and permeabilized with 0.3% Triton X-100 in PBS for 3 min. The slides were blocked for 1 h at room temperature in blocking reagent (11 096 176 001; Roche, Mannheim, Germany), NCS, and PBS in a ratio of 1:1:3. Cultures were evaluated with fluorescent staining after incubation overnight at 4°C with the following primary antibodies: mouse monoclonal anti-β-tubulin isotype III (T8660; Sigma-Aldrich) (1:300) as a pan-neuronal marker to label cell bodies and neurites, mouse anti-rabies virus nucleocapsid protein monoclonal antibody (5DF12) (1:80) (obtained from A. I. Wandeler, Canadian Food Inspection Agency, Ottawa, Ontario, Canada), rabbit monoclonal anti-p50 NF-κB p105/50 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), and polyclonal rabbit anti-4-hydroxy-2-nonenal adducts (4-HNE) (ALX-210-767-R100; Alexis Biochemicals, San Diego, CA) (1:100). Secondary antibodies included cyanine 3 (Cy3)-conjugated donkey anti-mouse IgG (711-095-152; Jackson ImmunoResearch Laboratories, West Grove, PA) (1/250) and fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG (711-095-152; Jackson ImmunoResearch Laboratories) (1:250), which were incubated in blocking buffer for 1 h at room temperature. Coverslips were mounted on slides with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (H-1200; Vector Laboratories, Burlingame, CA) and examined using a Carl Zeiss Axioskop 2 mot microscope equipped with fluorescein isocyanate, Cy3, and DAPI filters and coupled to an AxioCam camera with Axo-Vision 4.8 software. Quantitative assessments of 4-HNE puncta were performed on masked images, so that the evaluator was not aware if samples were from CVS- or mock-infected groups, and the number of labeled puncta were analyzed relative to axonal length (in μm).
Evaluation of neurite (axon) outgrowth and 4-HNE punta.
The level of total neurite (axon) outgrowth of DRG neurons from CVS- and mock-infected cultures was analyzed after staining for β-tubulin. Images of cultures were taken from at least 10 random fields from each well. The total number of neuronal cell bodies and the number of intersects of their neurites with a vertical grid were counted using a morphometric approach using ImageJ 1.42 software (available at http://rsbweb.nih.gov/ij/) on masked images. The total number of intersects per neuron was taken as the parameter of total axon outgrowth as previously described (36).
Assessment of 4-HNE puncta and axonal outgrowth with CNTF, SN50, and SN50M.
For ciliary neurotrophic factor (CNTF) experiments, 10 ng/ml recombinant human CNTF (57-NT; R&D Systems, Inc., Minneapolis, MN) was added 24 h prior to viral (or mock) absorption and was continued after viral adsorption, and neurons were refed every 24 h with CNTF to ensure continuous activation. For SN50 experiments, 30 μg/ml SN50 (481480; EMD-Calbiochem, San Diego, CA) or 30 μg/ml mutated SN50 (SN50M) (481486; EMD-Calbiochem) were added to the medium of DRG neurons after viral adsorption, and neurons were refed every 24 h with the same additives. At 72 h postinfection, cells were fixed with 4% paraformaldehyde, and immunostaining was performed for detection of β-tubulin and 4-HNE (see above).
Western blotting for p50, p65, HNE, and SOD2 protein expression.
CVS- and mock-infected DRG neurons 72 h p.i. were lysed using ice-cold neurofilament stabilization buffer containing 0.1 M PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], 5 mM MgCl2, 5 mM EGTA, 0.5% Triton X-100, 20% glycerol, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail (5). Protein amounts were determined by detergent-compatible (DC) protein assay (Bio-Rad, Mississauga, Ontario, Canada). Equivalent amounts of the extracted proteins were loaded (5 μg/lane) onto a 10% SDS-PAGE gel and electrotransferred after electrophoresis onto a nitrocellulose membrane at 100 V for 1 h. Membrane was blocked with 5% milk and 0.05% Tween in PBS for 1 h and then incubated with specific primary antibodies—rabbit monoclonal anti-p50 (1559-1; Epitomics, Burlingame, CA) (1/500), rabbit polyclonal anti-p65 (sc-372; Santa Cruz Biotechnology) (1/500), rabbit polyclonal anti-HNE Michael adducts (393207; EMD Millipore, Temecula, CA) (1/1,000), mouse monoclonal anti-manganese superoxide dismutase (SOD2) (MAB4081; Millipore) (1/1,000), and rabbit polyclonal antibody anti-extracellular signal-regulated kinase (ERK) (sc-93; Santa Cruz Biotechnology)—for protein quantification and normalization, overnight in 5% milk-0.05% Tween in PBS. Proteins were detected using horseradish peroxidase (HRP)-linked anti-rabbit or anti-mouse IgG (Jackson ImmunoResearch Laboratories) that were applied for 1 h, and proteins were detected by Western blotting luminol reagent (sc-2048; Santa Cruz Biotechnology) and imaged by a Bio-Rad Fluor-S image analyzer. Membrane was stripped with Restore TM Western blot stripping buffer (21059; Thermo Scientific, Ontario, Canada) to be reprobed with another primary antibody. Quantity One software was used for densitometeric scan of the blots. Data are expressed as the ratio of the protein scan value obtained divided by the protein scan value of ERK.
Fluorescence intensity.
CVS- and mock-infected DRG for 24, 48, and 72 h p.i. were stained with p50 antibody (see above). Images of fluorescent cells were collected with standardized parameters of contrast, color, and brightness. A region of interest was drawn around the nucleus or cytoplasm portion of the cell body to measure fluorescence intensity of p50 protein. Mean fluorescence intensity and pixel area values were determined for each selection in the green channel (FITC fluorophore) using NIH Image J software. Data are represented as mean gray multiplied by pixel area values.
Quantitative RT-PCR analysis.
Total RNA was isolated using the mirVana miRNA isolation kit (Ambion, Foster City, CA), and real-time one-step reverse transcription (RT)-PCR was performed using the QuantiTect Probe RT-PCR kit (Qiagen, Toronto, Ontario, Canada). The RAVD1 primer/probe set, which targets the N gene of CVS-11, was used as described by Nadin-Davis et al. (18), and a primer/probe set specific for β-actin was used as described previously (34). Both probes were labeled at the 5′ end with carboxyfluorescein. The lengths of the amplified products for the CVS-11 nucleoprotein (N) gene and β-actin were 115 bp and 138 bp, respectively. The real-time RT-PCR was carried out in final reaction volume of 25 μl containing 2 μl RNA (∼30 ng), 0.8 μM each forward and reverse primer, 0.2 μM fluorescent probe, 12.5 μl of 2× QuantiTect probe RT-PCR master mix (including dNTP mix and reaction buffer), and 0.25 μl QuantiTect RT mix. The thermal profile for the real-time RT-PCR was 50°C for 30 min, 95°C for 15 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60s. Real-time RT-PCR was performed using the SmartCycler II (Cepheid, Sunnyvale, CA). A positive control (RNA of CVS-11) and negative control (water) were included in each run in parallel to unknown samples. β-Actin was used to normalize the CVS-11 RT-PCR results and ensure that all samples contained an equal number of DRG neurons.
Statistical analysis.
Data on CVS- and mock-infected cultures were analyzed using GraphPad Prism version 4 software. Standard two-tailed unpaired Student's t tests with Welch's correction, which did not assume equal variances, were used for evaluating the significance of the difference between the means on assays comparing CVS- and mock-infected cultures. A P value of <0.05 was considered statistically significant.
RESULTS
CVS induced axonal swellings containing 4-HNE protein adducts in adult rat DRG.
CVS- and mock-infected DRG cultures 72 h p.i. were immunostained with the neuron-specific marker β-tubulin and for 4-HNE protein adducts, which are a product of lipid peroxidation and a marker of oxidative stress. Tubulin immunostaining showed healthy mock-infected DRG neurons with neurite outgrowths (Fig. 1A). Axonal swellings of various sizes were induced by CVS infection (Fig. 1B) with staining for 4-HNE protein adducts in the axons and in axonal swellings (Fig. 1D) but not in mock infection (Fig. 1C). These 4-HNE-labeled puncta were colocalized to axonal swellings (Fig. 1F) and were not present in mock-infected cultures (Fig. 1E).
Fig 1.

Formation of axonal swellings containing 4-HNE adducts in DRG cultures. Fluorescence microscopy showing mock-infected (A, C, E) and CVS-infected (B, D, F) cultures. DRG neurons stained for β-tubulin (A, B) showed large axonal swellings in CVS-infected neurons (B). Staining for 4-HNE adducts (C, D) showed that CVS infection induced intense staining for 4-HNE adducts (D) at 72 h postinfection (p.i.) in comparison to mock-infected DRG neurons (C). Merging signals for β-tubulin and 4-HNE (E, F) showed that 4-HNE was expressed in axonal swellings (arrowheads) in CVS infection (yellow) (F).
CVS infection increased protein expression of p50, p65, 4-HNE protein adducts, and manganese SOD2.
In order to evaluate whether NF-κB plays a neuroprotective role in CVS-induced oxidative stress in DRG neurons, we determined the expression of p50 and p65 proteins, the most abundant dimer forms of NF-κB, in CVS and mock-infected DRG cultures 72 h p.i. In addition, we evaluated the expression of 4-HNE protein adducts, an indicator of oxidative stress, and the expression of superoxide dismutase 2 (SOD2), which is known as a primary antioxidant protein and a neuroprotective gene target of NF-κB. Evaluation of the expression of p50, p65, 4-HNE protein adducts, and SOD2 proteins of mock- and CVS-infected DRG neurons by Western blot probing with protein-specific antibodies showed that there was a significant increase of p50 (1.6-fold, P = 0.002) in CVS-infected DRG neurons versus mock-infected DRG neurons. There was also a significantly increased level of expression of p65 protein (2.4-fold, P = 0.0022), 4-HNE adduct protein (2.0-fold, P = 0.0002), and SOD2 proteins (1.3-fold, P = 0.047) (Fig. 2).
Fig 2.
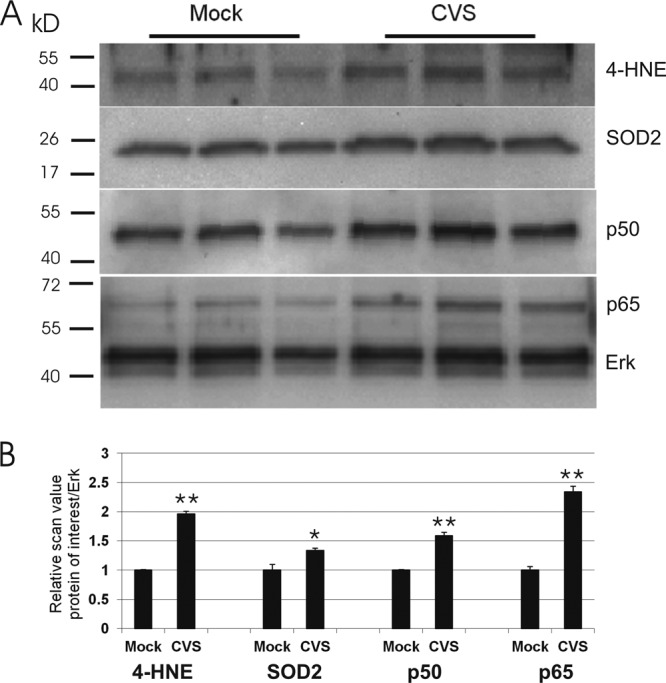
CVS infection altered the protein expression of the p50 and p65 subunits of NF-κB, 4-HNE adducts, and SOD2. Cell protein lysates from mock- and CVS-infected DRG cultures 72 h p.i. were loaded on 10% Tris acrylamide gel. (A) Western blot of p50, p65, 4-HNE, SOD2, and Erk, which served as the total protein loading control. (B) Relative scan values on 3 triplicates show a 1.6-fold increase in p50 (0.75 ± 0.01 versus 1.20 ± 0.06, P = 0.002), 2.4-fold increase in p65 (0.56 ± 0.06 versus 1.34 ± 0.10, P = 0.0022), 2.0-fold increase in 4-HNE (0.65 ± 0.01 versus 1.28 ± 0.05, P = 0.0002), and 1.3-fold increase in SOD2 (0.84 ± 0.11 versus 1.14 ± 0.04, P = 0.047) in CVS infection versus mock infection. Statistical significance is indicated by * (P < 0.05) and ** (P < 0.01).
CNTF reduced axonal swellings of DRG-infected cultures.
We have evaluated the effect of CNTF therapy on DRG neuron cultures because it is known to be a strong inducer of NF-κB, albeit with effects on other pathways as well (27). CVS- or mock-infected DRG neuron cultures 72 h p.i. were treated with 10 ng/ml CNTF for 96 h and stained for β-tubulin and adducts of 4-HNE. Fluorescence microscopy showed that CNTF alone did not result in a significant effect on 4-HNE-labeled puncta in mock-infected DRG neurons (P = 0.84). In comparison to CVS-infected cultures without CNTF, CNTF treatment reduced the axonal swellings in the CVS-infected cultures. Quantification of 4-HNE-labeled puncta in CVS- and mock-infected DRG showed that CVS infection induced a 5.9-fold increase compared to that of mock infection (P < 0.0001), whereas CNTF treatment induced a 2.8-fold decrease in 4-HNE-labeled axonal swellings (P < 0.0001) in CVS-infected neurons. CNTF treatment for a total of 96 h resulted in a 2.0-fold increase in the number of 4-HNE puncta in CVS-infected DRG cultures versus mock-infected cultures (P = 0.01) and, hence, did not completely prevent oxidative stress induced by CVS infection (Fig. 3).
Fig 3.
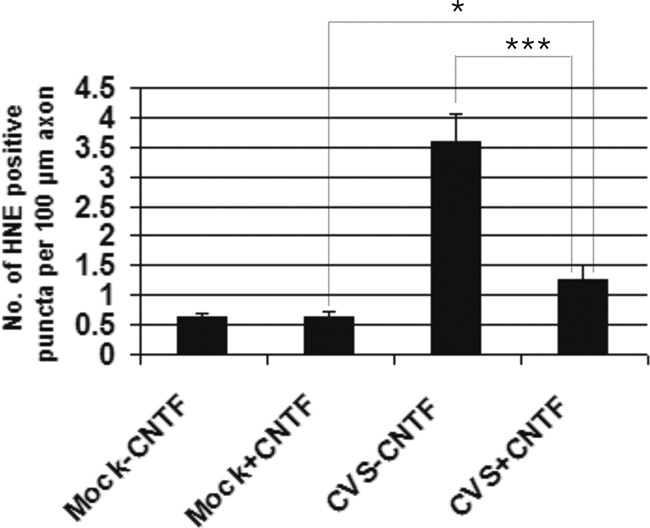
CNTF reduced oxidative stress induced by CVS infection. Evaluation of the number of 4-HNE-labeled puncta per 100 μm of axon (n = 25 to 68) showed that after 96 h of CNTF treatment, there was protection against the oxidative stress phenotype induced by CVS infection at 72 h p.i. In comparison to mock-infected DRG neurons, CVS infection induced a 5.9-fold increase in the number of 4-HNE-labeled puncta (0.612 ± 0.077 versus 3.59 ± 0.45, P < 0.0001). A 2.8-fold reduction in the number of 4-HNE puncta induced by CVS infection was observed when CNTF treatment was applied to CVS-infected DRG cultures (3.59 ± 0.45 versus 1.27 ± 0.23, respectively; ***, P < 0.0001). CVS infection with CNTF treatment resulted in a 2.0-fold increase in 4-HNE-labeled puncta compared to that of mock infection with CNTF treatment (0.64 ± 0.08 versus 1.27 ± 0.23; *, P = 0.01). There was no significant effect with CNTF treatment alone (P = 0.84).
SN50 increased axonal swellings in infected DRG.
In order to assess the role of constitutive and/or inducible expression of NF-κB in axonal injury induced by CVS, we used SN50, a cell-permeable inhibitor peptide of p50 that inhibits translocation of the NF-κB active complex into the nucleus (6). SN50M, a cell-permeable inactive control peptide, was used at the same concentration as SN50 (30 μg/ml) in order to demonstrate the specificity of the action of SN50. With SN50 treatment, axonal swellings were observed in both the mock (Fig. 4A)- and CVS (Fig. 4B)-infected cultures. Immunostaining for 4-HNE protein adducts of mock- and CVS-infected cultures 72 h p.i. treated with SN50 showed that there was an increased expression of 4-HNE in axons and in axonal swellings (Fig. 4C to F). We counted the number of 4-HNE puncta per 100-μm axon in CVS- and mock-infected cultures and determined the effects of SN50 (Fig. 5). In summary, CVS infection (versus mock infection) and SN50 treatment induced the formation of 4-HNE-labeled puncta, and they had an additive effect.
Fig 4.

SN50 induced axonal swellings in mock- and CVS-infected cultures. Fluorescence microscopy showing mock (A, C, E)- and CVS (B, D, F)-infected DRG treated with 30 μg/ml SN50 for 72 h p.i. In both mock (A)- and CVS (B)-infected cultures, β-tubulin staining showed axonal swellings and disorganization of the axonal network. 4-HNE staining was increased and swellings were more numerous in CVS-infected DRG (D) than in mock infection (C). Merged tubulin and 4-HNE signals (yellow) (E, F) showed that there was increased 4-HNE staining in axonal swellings of mock (E)- and CVS (F)-infected cultures treated with SN50.
Fig 5.
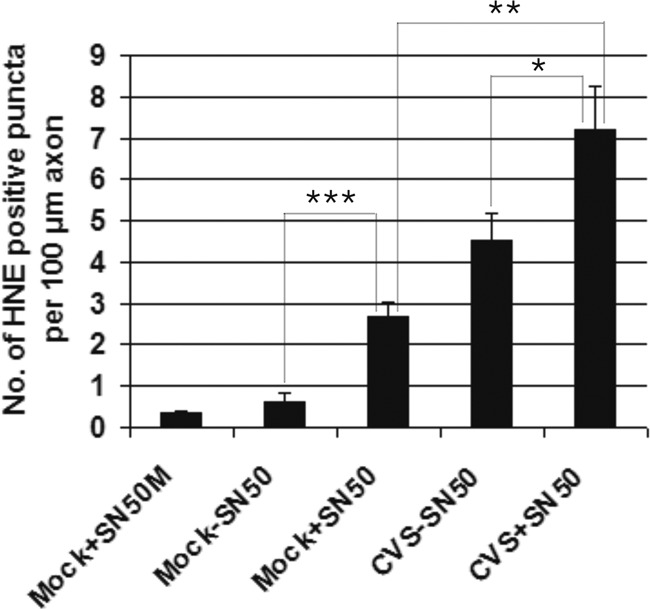
SN50 treatment increased the amount of axonal swellings. Quantification of 4-HNE-labeled puncta (or swellings) per 100-μm axon of mock- and CVS-infected neurons 72 h p.i. (n = 16 to 64) treated with SN50 or without SN50 showed a 4.5-fold increase in the number of puncta in mock-infected cultures treated with SN50 (2.67 ± 0.35, P < 0.0001) compared to that in cultures without SN50 (0.60 ± 0.24, P < 0.0001) and a 7.5-fold increase in CVS infection (4.52 ± 0.66, P < 0.0001) compared to mock infection (0.60 ± 0.24, P < 0.0001). CVS infection induced 1.7-fold more puncta than mock infection with SN50 (2.67 ± 0.34 versus 4.52 ± 0.66, P = 0.0156). CVS infection with SN50 treatment induced 1.6-fold more puncta than CVS infection without SN50 (P = 0.043) and 2.8-fold greater than mock infection with SN50 treatment (P = 0.0008). There were no significant differences in the number of 4-HNE-labeled puncta in mock-infected cells treated without SN50 versus with SN50M (P = 0.213).
SN50 reduced axonal outgrowth in CVS-infected DRG.
We evaluated the effect of CVS infection on axonal growth by determining total axonal outgrowth in mock- and CVS-infected rat DRG neurons 72 h p.i. Infected and uninfected cultures were also treated with SN50 and the combined effect on axonal outgrowth was evaluated (Fig. 6). Both CVS infection and SN50 treatment of mock infection reduced axonal outgrowth, and CVS infection and SN50 treatment had an additive effect in reducing axonal outgrowth. Taken together, the increased number of 4-HNE-labeled puncta and reduced axonal outgrowth with SN50 treatment in CVS infection indicate that NF-κB plays a neuroprotective role in CVS-induced oxidative stress.
Fig 6.
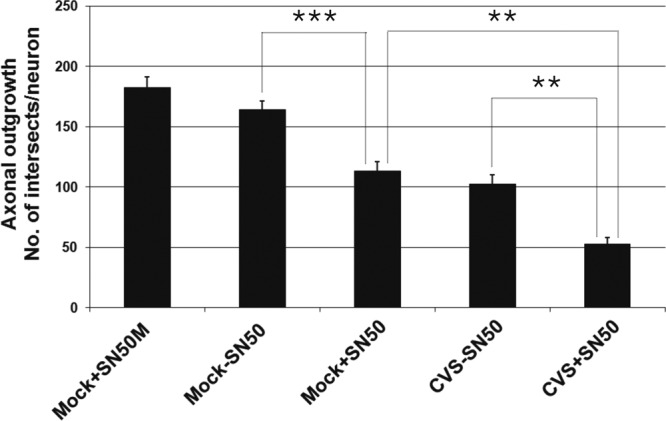
SN50 treatment reduced axonal outgrowth. Mock- and CVS-infected cultures for 72 h (n = 19 to 62) were treated or not treated with SN50 for 72 h. Evaluation of axonal outgrowth by determining the length of total axon outgrowth per neuron shows that, in comparison to mock-infected cells without SN50 (164.2 ± 7.3), SN50 significantly reduced axonal outgrowth in CVS-infected neurons (113.4 ± 7.4; ***, P < 0.0001). Without SN50, CVS-infected neurons significantly reduced outgrowth of their axons at 72 h p.i. (105.6 ± 7.3) compared to that of mock-infected neurons (***, P < 0.0001). There was a greater inhibition of outgrowth with the combination of SN50 and CVS infection (53.0 ± 5.1) than with either CVS infection without SN50 (**, P < 0.001) or mock infection with SN50 (**, P < 0.001). There was no significant difference in axonal outgrowth between mock infection treated without SN50 versus with SN50M (P = 0.12).
Localization of p50 in DRG neurons in CVS infection.
We have shown increased expression of NF-κB proteins, especially p50 protein (Fig. 2), in CVS-infected cultures and that NF-κB is neuroprotective in DRG-infected neurons (Fig. 5 and 6). However, this increase of p50 protein was accompanied by a modest increase of SOD2, which is a primary target of the antioxidant response induced by NF-κB. We addressed whether the CVS-induced NF-κB is functionally active or not by determining whether translocation of p50 into the nucleus occurred with an assessment of the nucleus/cytoplasm ratio of signal for p50.
Consequently, CVS- and mock-infected cultures 72 h p.i. were immunostained with rabies virus antigen and p50 protein. Immunofluorescence showed that p50 protein was expressed in CVS- and mock-infected DRG neurons. CVS-infected DRG neurons exhibited strong staining for p50 expression in the cytoplasm. Quantification of p50 nuclear expression in the nucleus and in the cytoplasm of CVS-infected and mock-infected neurons showed activation in CVS infection at 24 h p.i. (P < 0.0001), which was followed by decreases at 48 and 72 h p.i. of 1.8-fold (P < 0.002) and 1.6-fold (P < 0.0031), respectively, compared to that of mock-infected cultures (Fig. 7). Hence, there was initial NF-κB activation at 24 h p.i., but at the later time points CVS infection was associated with the development of axonal injury and failure of NF-κB nuclear translocation.
Fig 7.

The total fluorescent signal (green) for the p50 subunit of NF-κB was evaluated in the nucleus and cytoplasm of mock- and CVS-infected DRG neurons. Fluorescent signals were quantitatively assessed by scan densitometry in the nucleus (defined using DAPI) and cytoplasm, and nucleus/cytoplasm ratios were calculated at 72 h p.i. (B) At 24 h p.i., the nucleus/cytoplasm ratio of p50 signal (green) was 0.63 in a CVS-infected DRG neuron; (C) staining for rabies virus antigen (red) in a CVS-infected neuron shows diffuse staining in the cytoplasm with intensely staining inclusion bodies (Negri body-like) that are typical of rabies virus infection; (D) the merged image shows the DAPI-stained nucleus (blue) and yellow-stained viral inclusion bodies in the cytoplasm; (E) kinetics of activity of NF-κB, assessed by using the ratio of p50 in the nucleus/cytoplasm, of CVS- versus mock-infected neurons (n = 54 to 112). CVS infection was associated with a 1.8-fold increase in the expression of p50 protein at 24 h (0.37 ± 0.03 versus 0.66 ± 0.06; ***, P < 0.0001), followed by a decrease of 1.8-fold at 48 h p.i. (0.72 ± 0.04 versus 0.40 ± 0.07; **, P < 0.002), and followed by a decrease of 1.6-fold at 72 h p.i. (0.71 ± 0.05 versus 0.44 ± 0.07; **, P < 0.003) versus mock infection.
Assessment of CVS genomic RNA by using real-time RT-PCR.
In order to determine whether oxidative stress-induced axonal swellings of DRG neurons observed over 72 h p.i. and after treatment with CNTF or SN50 were due to changes in viral replication rather than related to changes in NF-κB activity, CVS-infected DRG neuron cultures were either untreated or treated with CNTF and SN50 as described previously, and the amount of rabies virus genomic RNA was monitored in CVS-infected DRG for 24, 48, and 72 h. There were no significant changes in the amount of genomic RNA at 24, 48, and 72 h p.i. (Fig. 8). Furthermore, CNTF or SN50 had no significant effects on the amounts of genomic RNA, indicating that the observed effects were not due to changes in viral replication (Fig. 8).
Fig 8.
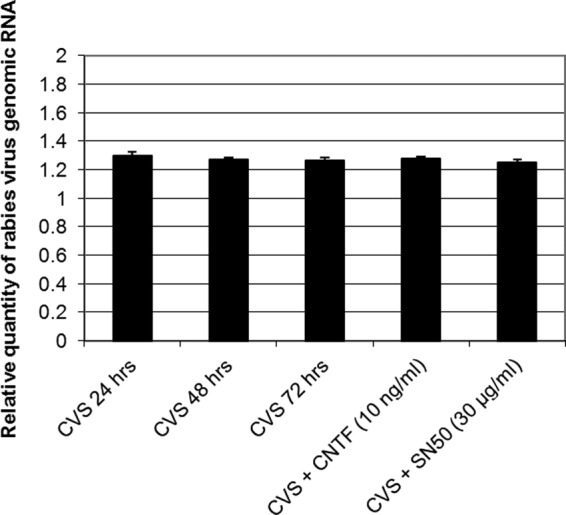
Quantitative RT-PCR for genomic RNA in CVS-infected DRG cultures at 24, 48, and 72 h p.i. CVS-infected DRG neurons at 72 h p.i. also received treatment with SN50 30 μg/ml for 72 h or CNTF 10 ng/ml for 96 h. DRG neurons were infected at an MOI of 10, and total RNA was isolated at 24, 48, and 72 h p.i. The data were normalized using β-actin as an internal control and analyzed using the comparative threshold cycle method. Data are means (±standard errors of the means [SEM]) of values of duplicate samples carried out twice on two independent experiments. There was no significant difference in the amount of N gene of rabies virus genomic RNA over 24 to 72 h p.i. (P > 0.23) or in CVS-infected cells treated without or with SN50 (P > 0.44) or with CNTF (P > 0.44) at 72 h p.i.
DISCUSSION
Morphological features of neuronal process degeneration were observed in CVS-infected adult mice (31). We have investigated the cause of this neuronal process degeneration in cultured DRG neurons because these neurons are permissively infected by CVS, which allows the degenerative effects on their axons to be studied in detail. We have previously demonstrated that oxidative stress occurs in CVS-infected cultured adult mouse DRG neurons (14). The infected neurons maintained viability and demonstrated axonal swellings containing adducts of 4-HNE without evidence of neuronal apoptosis as assessed by terminal deoxynucleotidyl transferase-mediated dUTP-nick end labeling (TUNEL) staining (14). In this study, we have observed the same effects in CVS-infected adult rat DRG neurons under similar experimental conditions. Increases in adducts of 4-HNE reflect lipid peroxidation that occurs in oxidative stress and also have physiological implications due to the capability of 4-HNE to form adducts with key proteins and, in particular, cytoskeletal proteins (21), leading to the development of axonal swellings. It has been demonstrated that formation of 4-HNE adducts coincides with focal aggregation of respiring mitochondria (1, 14), indicating potential modifications of mitochondrial proteins in which the outcome may be an energy deficit due to disruption of the Na/K ATPase pump (35), the mitochondria respiratory chain, and/or alterations to mitochondria motility, dynamics, and turnover, which could lead to elevated intracellular calcium concentrations resulting in degeneration (2, 33).
We now hypothesize that NF-κB, a redox-sensitive protein, which plays critical roles in neuronal survival, axonal growth, and antiviral responses (28, 29, 32), might play a pivotal role in CVS-induced oxidative stress. First, we used a potent activator of NF-κB (and also other signal transduction pathways), CNTF, to determine whether it would exert a neuroprotective effect in CVS-infected cultures of DRG neurons. CNTF (10 ng/ml) markedly reduced, albeit incompletely, the number of 4-HNE-labeled puncta in CVS-infected cultures over a period of 96 h. This result suggests that NF-κB pathways may be important but lack specificity because of other potential non-NF-κB-mediated signaling pathways, including the JAK2/STAT3 and phosphoinositide-3 kinase (PI-3K)/Akt signaling pathways (27). Next, we performed studies by inhibiting NF-κB transcriptional activity with the peptide inhibitor SN50, which blocks movement of the p50 active transcription complex of NF-κB into the nucleus. It is known in embryonic and adult DRG neurons that survival and axonal growth require activation of the p50 and p65 dimers of NF-κB (6, 9, 26). We have shown that CVS infection and SN50 (30 μg/ml) exert an additive effect in producing axonal swellings with 4-HNE-labeled puncta and in inhibiting axonal outgrowth in the cultures. This effect on CVS infection may be acting on the p50 subunit or on other subunits of NF-κB. However, we can conclude that NF-κB exerts a neuroprotective function in CVS-induced oxidative stress in preventing the development of axonal swellings and in promoting axonal growth.
Western immunoblotting studies demonstrated a mild increase in the expression of the p50 and p65 subunits of NF-κB in CVS infection versus that in mock infection at 72 h p.i. (1.6 and 2.4, respectively). However, detailed quantitative studies of the p50 subunit with cellular localization (in nucleus versus cytoplasm) showed that NF-κB activation (1.8-fold increase) with nuclear translocation of p50 occurred only at 24 h p.i., possibly as a response to stress. Conversely, at 48 and 72 h p.i., there was inhibition of p50 translocation into the nucleus with 1.8- and 1.6-fold decreases, respectively, in the nucleus/cytoplasm ratio of signal. We have previously reported the observation of the development of axonal swellings, an important morphological feature of early axonal degeneration in oxidative stress in mouse DRG neurons, at 48 and 72 h p.i (14). There is a temporal association with the loss of functionally active NF-κB and the development of morphological features of oxidative stress. Hence, in CVS-induced oxidative stress, NF-κB does not remain functionally active and is unable to exert a neuroprotective effect in the infected neuron. We have shown that these changes are not due to effects on rabies virus replication because there were no significant changes in levels of rabies virus genomic RNA determined with quantitative RT-PCR.
It is unknown how rabies virus (CVS) interferes with the neuroprotective activation of NF-κB in infected neurons. However, we postulate that one or more viral proteins may directly interact with the NF-κB pathway. For example, in the case of hepatitis C virus (HCV) infection, the hepatitis C virus type 1a (but not type 1b) core protein suppresses NF-κB activation by direct interaction with IκB kinase β in macrophages (16). Two open reading frames of vaccinia virus (designated A52R and A46R) interfere with NF-κB activation by potently blocking both IL-1- and Toll-like receptor 4-mediated activation and partially inhibiting IL-2-mediated activation of NF-κB, respectively (3). A52R produces a protein that is analogous in structure to the MyD88 scaffolding protein involved in the IL-1-mediated activation of NF-κB (3). Cowpox, raccoonpox, and some strains of vaccinia virus (e.g., Copenhagen strain) prevent the degradation of the NF-κB inhibitor, IκBα (20). As a result, NF-κB remains bound to IκBα at its prenuclear localization site and is unable to enter the nucleus. Using an alternative mechanism, the African swine fever virus produces a protein that mimics IκBα and similarly prevents entry of NF-κB into the nucleus (22).
In summary, rabies virus blocks activation of the NF-κB pathway in CVS-infected neurons, which reduces host defenses against oxidative stress and promotes the development of axonal degeneration. This occurs without any associated significant effects on rabies virus replication. Because viral spread in the host occurs relatively early in the course of infection, this neuronal damage in the host is inconsequential with respect to viral transmission between hosts and the “success” of rabies virus as a virus. In the future, experimental studies could be performed in natural hosts using street (wild-type) rabies virus strains. Unfortunately, although the NF-κB pathway is involved in diverse diseases, effective pharmacological approaches still need to be developed for therapeutic use in these diseases, including rabies.
ACKNOWLEDGMENTS
This work was supported by the Canadian Institutes of Health Research/Manitoba Regional Partnership Program with the Manitoba Health Research Council (to A. C. Jackson and P. Fernyhough) and the St. Boniface Hospital Research Foundation.
We thank Thamir Alandijany for technical assistance.
Footnotes
Published ahead of print 23 May 2012
REFERENCES
- 1. Akude E, Zherebitskaya E, Chowdhury SKR, Girling K, Fernyhough P. 2010. 4-Hydroxy-2-nonenal induces mitochondrial dysfunction and aberrant axonal outgrowth in adult sensory neurons that mimics features of diabetic neuropathy. Neurotox. Res. 17:28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. 2007. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J. Neurosci. 27:422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bowie A, et al. 2000. A46R and A52R from vaccinia virus are antagonists of host IL-1 and Toll-like receptor signaling. Proc. Natl. Acad. Sci. U. S. A. 97:10162–10167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ebenezer GJ, et al. 2007. Denervation of skin in neuropathies: the sequence of axonal and Schwann cell changes in skin biopsies. Brain 130:2703–2714 [DOI] [PubMed] [Google Scholar]
- 5. Fernyhough P, et al. 1999. Aberrant neurofilament phosphorylation in sensory neurons of rats with diabetic neuropathy. Diabetes 48:881–889 [DOI] [PubMed] [Google Scholar]
- 6. Fernyhough P, et al. 2005. Activation of nuclear factor-κB via endogenous tumor necrosis factor α regulates survival of axotomized adult sensory neurons. J. Neurosci. 25:1682–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fu ZF, Jackson AC. 2005. Neuronal dysfunction and death in rabies virus infection. J. Neurovirol. 11:101–106 [DOI] [PubMed] [Google Scholar]
- 8. Grilli M, Memo M. 1999. Nuclear factor-kappaB/Rel proteins: a point of convergence of signalling pathways relevant in neuronal function and dysfunction. Biochem. Pharmacol. 57:1–7 [DOI] [PubMed] [Google Scholar]
- 9. Gutierrez H, O'Keeffe GW, Gavalda N, Gallagher D, Davies AM. 2008. Nuclear factor kappa B signaling either stimulates or inhibits neurite growth depending on the phosphorylation status of p65/RelA. J. Neurosci. 28:8246–8256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hayden MS, Ghosh S. 2012. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26:203–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iwasaki Y, Tobita M. 2002. Pathology, p 283–306 In Jackson AC, Wunner WH. (ed), Rabies. Academic Press, San Diego, CA [Google Scholar]
- 12. Jackson AC. 2007. Pathogenesis, p 341–381 In Jackson AC, Wunner WH. (ed), Rabies. Elsevier Academic Press, London, United Kingdom [Google Scholar]
- 13. Jackson AC. 2011. Therapy of human rabies. Adv. Virus Res. 79:365–375 [DOI] [PubMed] [Google Scholar]
- 14. Jackson AC, Kammouni W, Zherebitskaya E, Fernyhough P. 2010. Role of oxidative stress in rabies virus infection of adult mouse dorsal root ganglion neurons. J. Virol. 84:4697–4705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jackson AC, Wunner WH. 2007. Rabies, 2nd ed Elsevier Academic Press, London, United Kingdom [Google Scholar]
- 16. Joo M, et al. 2005. Hepatitis C virus core protein suppresses NF-κB activation and cyclooxygenase-2 expression by direct interaction with IκB kinase β. J. Virol. 79:7648–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lauria G, et al. 2003. Axonal swellings predict the degeneration of epidermal nerve fibers in painful neuropathies. Neurology 61:631–636 [DOI] [PubMed] [Google Scholar]
- 18. Nadin-Davis SA, Sheen M, Wandeler AI. 2009. Development of real-time reverse transcriptase polymerase chain reaction methods for human rabies diagnosis. J. Med. Virol. 81:1484–1497 [DOI] [PubMed] [Google Scholar]
- 19. Obrosova IG, et al. 2005. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes 54:234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oie KL, Pickup DJ. 2001. Cowpox virus and other members of the orthopoxvirus genus interfere with the regulation of NF-κB activation. Virology 288:175–187 [DOI] [PubMed] [Google Scholar]
- 21. Petersen DR, Doorn JA. 2004. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic. Biol. Med. 37:937–945 [DOI] [PubMed] [Google Scholar]
- 22. Revilla Y, et al. 1998. Inhibition of nuclear factor κB activation by a virus-encoded IκB-like protein. J. Biol. Chem. 273:5405–5411 [DOI] [PubMed] [Google Scholar]
- 23. Ridder DA, Schwaninger M. 2009. NF-kappaB signaling in cerebral ischemia. Neuroscience 158:995–1006 [DOI] [PubMed] [Google Scholar]
- 24. Rossiter JP, Jackson AC. 2007. Pathology, p 383–409 In Jackson AC, Wunner WH. (ed), Rabies. Elsevier Academic Press, London, United Kingdom [Google Scholar]
- 25. Russell JW, et al. 2002. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 16:1738–1748 [DOI] [PubMed] [Google Scholar]
- 26. Saleh A, et al. 2011. Tumor necrosis factor-α elevates neurite outgrowth through an NF-κB-dependent pathway in cultured adult sensory neurons: diminished expression in diabetes may contribute to sensory neuropathy. Brain Res. 1423:87–95 [DOI] [PubMed] [Google Scholar]
- 27. Sango K, Yanagisawa H, Komuta Y, Si Y, Kawano H. 2008. Neuroprotective properties of ciliary neurotrophic factor for cultured adult rat dorsal root ganglion neurons. Histochem. Cell Biol. 130:669–679 [DOI] [PubMed] [Google Scholar]
- 28. Santoro MG, Rossi A, Amici C. 2003. NF-κB and virus infection: who controls whom. EMBO J. 22:2552–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sarnico I, et al. 2009. NF-kappaB dimers in the regulation of neuronal survival. Int. Rev. Neurobiol. 85:351–362 [DOI] [PubMed] [Google Scholar]
- 30. Schmidt RE, Beaudet LN, Plurad SB, Dorsey DA. 1997. Axonal cytoskeletal pathology in aged and diabetic human sympathetic autonomic ganglia. Brain Res. 769:375–383 [DOI] [PubMed] [Google Scholar]
- 31. Scott CA, Rossiter JP, Andrew RD, Jackson AC. 2008. Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome in experimental rabies in yellow fluorescent protein-expressing transgenic mice. J. Virol. 82:513–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Teng FY, Tang BL. 2010. NF-kappaB signaling in neurite growth and neuronal survival. Rev. Neurosci. 21:299–313 [DOI] [PubMed] [Google Scholar]
- 33. Verkhratsky A, Fernyhough P. 2008. Mitochondrial malfunction and Ca2+ dyshomeostasis drive neuronal pathology in diabetes. Cell Calcium 44:112–122 [DOI] [PubMed] [Google Scholar]
- 34. Wakeley PR, et al. 2005. Development of a real-time, TaqMan reverse transcription-PCR assay for detection and differentiation of lyssavirus genotypes 1, 5, and 6. J. Clin. Microbiol. 43:2786–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Waxman SG. 2006. Ions, energy and axonal injury: towards a molecular neurology of multiple sclerosis. Trends Mol. Med. 12:192–195 [DOI] [PubMed] [Google Scholar]
- 36. Zherebitskaya E, Akude E, Smith DR, Fernyhough P. 2009. Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes 58:1356–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]