

Abstract
Herpes simplex virus 1 infection triggers multiple changes in the metabolism of host cells, including a dramatic decrease in the levels of NAD+. In addition to its role as a cofactor in reduction-oxidation reactions, NAD+ is required for certain posttranslational modifications. Members of the poly(ADP-ribose) polymerase (PARP) family of enzymes are major consumers of NAD+, which they utilize to form poly(ADP-ribose) (PAR) chains on protein substrates in response to DNA damage. PAR chains can subsequently be removed by the enzyme poly(ADP-ribose) glycohydrolase (PARG). We report here that the HSV-1 infection-induced drop in NAD+ levels required viral DNA replication, was associated with an increase in protein poly(ADP-ribosyl)ation (PARylation), and was blocked by pharmacological inhibition of PARP-1/PARP-2 (PARP-1/2). Neither virus yield nor the cellular metabolic reprogramming observed during HSV-1 infection was altered by the rescue or further depletion of NAD+ levels. Expression of the viral protein ICP0, which possesses E3 ubiquitin ligase activity, was both necessary and sufficient for the degradation of the 111-kDa PARG isoform. This work demonstrates that HSV-1 infection results in changes to NAD+ metabolism by PARP-1/2 and PARG, and as PAR chain accumulation can induce caspase-independent apoptosis, we speculate that the decrease in PARG levels enhances the auto-PARylation-mediated inhibition of PARP, thereby avoiding premature death of the infected cell.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is an alphaherpesvirus that encodes more than 80 proteins and infects a large percentage of the global human population (36). Like all viruses, HSV-1 depends on the host cell for its replication, and central to this interaction is the viral requirement for macromolecular precursors and chemical energy. Several different human herpesviruses have been examined for their dependence and effect on host metabolism, including cytomegalovirus, Kaposi's sarcoma-associated herpesvirus, and HSV-1 (8, 31, 32, 44). HSV-1-infected cells place a high priority on nucleotide synthesis, anapleurotically feeding the citric acid cycle from pyruvate (44). Specifically, inhibition of pyruvate carboxylase, the enzyme responsible for the conversion of pyruvate to oxaloacetate, significantly decreases HSV-1 titers (44). Infection has also been shown to increase flux from aspartate toward pyrimidine synthesis. An additional, heretofore unexamined, metabolic alteration during HSV-1 infection is the dramatic decrease in the levels of NAD+ (44).
NAD+ is an important cofactor in many of the reduction-oxidation (redox) reactions of central carbon metabolism, but it can also be consumed as a substrate by members of the poly(ADP-ribose) polymerase (PARP) superfamily of enzymes as they catalyze the addition of poly(ADP-ribose) (PAR) chains to proteins (6). PARP-1 is an abundant nuclear enzyme that has been reported to be responsible for more than 99% of the total poly(ADP-ribosyl)ations (PARylations) in the cell. Of the remaining PARP enzymes, only PARP-2 is able to complement a PARP-1 mutation (17), and as a consequence, PARP-1 activity has been reported to have a dominant effect on overall cellular NAD+ levels (12). PARP-1 and PARP-2 (PARP-1/2) are both activated by DNA damage. The resulting PAR polymers, which can be several hundred units long and are highly negatively charged, help recruit DNA damage repair machinery to the sites of single- or double-strand breaks (3). In the case of significant DNA damage, however, cell death usually follows PARP overactivation (42). PARP-1 activity has been implicated in the pathogenesis of several viral infections. It is necessary for efficient integration of the HIV proviral genome (12) as well as lytic infection by Epstein Barr virus (26), but its interactions with alphaherpesviruses are largely unknown.
PARP-1/2 have multiple protein substrates, including many nuclear enzymes such as DNA polymerases, topoisomerases, and p53 (25, 33, 38). The acceptors of the majority of PAR chains (>90%), however, are PARP-1 and PARP-2 themselves (35). This automodification inhibits PARP's catalytic activity, likely by diminishing its DNA binding affinity (19, 48). Removal of the PAR chains occurs via the action of the enzyme poly(ADP-ribose) glycohydrolase (PARG), which possesses both exo- and endoglycosidic activities (7). PARG is the only protein known to cleave PAR chains from protein substrates, and its action on PARP-1/2 effectively restores PARP-1/2 catalytic activity, permitting further PAR polymerization (7). In humans, PARG is a single gene that codes for multiple spliced mRNAs. The full-length mRNA produces a 111-kDa (PARG-111) protein that localizes to the nucleus due to a nuclear localization signal (NLS) present at its N terminus (29). Isoforms of 102 and 99 kDa (PARG-102 and PARG-99, respectively) are found in the cytoplasm but have been shown to shuttle to sites of DNA damage in the nucleus after microirradiation and gamma irradiation (3, 14, 30). Smaller PARG isoforms of low abundance are enriched in mitochondria and do not appear to alter their localization patterns (30, 47).
In this study, we show that HSV-1 replication activates PARP-1/2, depleting cellular pools of NAD+ and increasing total protein PARylation levels. Neither a further decrease nor a rescue of NAD+ levels altered the metabolic effects of HSV-1 on the host cell, suggesting that, to a large extent, HSV-1 infection supersedes other cellular signals to control overall metabolic status. The infection-induced drop in NAD+ was dependent on viral DNA replication, presumably due to DNA damage signals produced during this process. We also found that HSV-1 infection triggered the proteasome-dependent degradation of the 111-kDa isoform of PARG and that this degradation was mediated by the RING finger domain of the immediate-early viral protein ICP0 (41). Depletion of all PARG isoforms by small interfering RNA (siRNA) treatment led to a modest decrease in viral titers, and the block occurred after leaky late gene expression. We conclude that NAD+ levels drop in HSV-1-infected cells due to the activation of PARP-1/2 and that infection actively modulates the balance between PARP and PARG, presumably to modulate the outcome of PARP activation.
MATERIALS AND METHODS
Cells, viruses, and reagents.
Primary human fibroblasts were used between passages 8 to 15. Cells were grown in Dulbecco's modified Eagle medium (DMEM) with 4.5g/liter glucose (Sigma) supplemented with 10% fetal bovine serum, and 100 μg/ml penicillin and streptomycin (Invitrogen). HSV-1 strain F (9) and a C116G/C156A mutant (22) were kindly provided by B. Roizman (University of Chicago) and grown in Vero cells. Stocks were produced by pooling cell-associated virus, obtained by sonication, with cell-free virus. HSV-1 titers were determined by infectious focus assay and are expressed as infectious units (IU). Briefly, fresh fibroblasts were infected with serial dilutions of virus and fixed at 4 h postinfection (hpi) with methanol at −20°C. Foci were identified using a mouse monoclonal antibody to the HSV-1 immediate-early ICP4 protein (39) and goat anti-mouse Alexa Fluor 488-conjugated secondary antibody (Invitrogen).
Hydrogen peroxide (30% [wt/wt]; Sigma) was used at a concentration of 10 mM. FK866 (Enzo Life Sciences) was dissolved in dimethyl sulfoxide (DMSO) used at 20 nM. Olaparib (LC Laboratories) was dissolved in DMSO and used at 5 to 50 nM. 3-Aminobenzamide (Sigma) was dissolved in DMEM to a stock concentration of 6 mM, filter sterilized (0.22-μm pore size), and used at 3 mM. Acyclovir (Sigma) was dissolved in DMSO and used at 1 μM. MG132 (Cayman Chemicals) was dissolved in DMSO and used at concentrations of 200 nM to 10 μM. Phosphonoacetic acid (Sigma) was dissolved in ethanol and used at 400 μg/ml.
Metabolic analysis.
Fibroblasts were grown to confluence and maintained in the presence of serum for 3 to 5 days. Cells were then washed twice and held in serum-free DMEM for 24 h before infection. At the time of infection, cells were inoculated with virus resuspended in DMEM without serum at a multiplicity of 3 IU/cell. Mock-treated cells were inoculated with the equivalent volume of virus-free DMEM. After a 1-h adsorption period, cells were washed, and fresh medium was added. Following various time intervals, the medium was aspirated from cells and an 80:20 methanol/water (vol/vol) solution at −80°C was added to quench metabolism. Metabolites were then extracted as described previously (49). Extracts were dried under nitrogen gas and resuspended in high-performance liquid chromatography (HPLC)-grade water. Samples were centrifuged at 15,000 × g for 5 min to remove any remaining particulate matter.
To quantify the levels of metabolites in extracts, we utilized an untargeted analysis approach using liquid chromatography (LC) coupled to a stand-alone Orbitrap mass spectrometer (Thermo Fisher Scientific Exactive instrument). This machine performs full scans from 85 to 1,000 m/z at a mass resolution of 100,000 (24). Compound identification is based on retention time on the LC column, and compound mass was measured to within an accuracy of 2 ppm. Peaks were identified and metabolites were quantified with the Metabolomic Analysis and Visualization Engine (MAVEN) software package (27).
For each time point in experiments comparing infected and uninfected cells, an additional plate for each treatment was processed for packed cell volume measurements. Briefly, the cells of one 35-mm plate were added to a packed cell volume tube (Techno Plastic Products), which was centrifuged at 2,000 × g for 5 min before a reading was taken (43). Packed cell volume measurements were used to normalize the metabolite levels between samples. All metabolite levels are averages of duplicate biological experiments.
Protein analysis.
For Western blot assays, fibroblasts were grown to confluence, serum starved for 24 h, and infected or mock treated. After the adsorption period, cells were washed once, and fresh medium was added. At the indicated times postinfection, cells were washed with phosphate-buffered saline (PBS), harvested, and stored at −80°C. To prepare whole-cell lysates, cells were lysed in radioimmunoprecipitation assay (RIPA) light buffer (50 mM Tris-HCl, pH 8.0, 1% NP-40, 0.1% SDS, 150 mM NaCl, 0.1% Triton X-100, 5 mM EDTA) with protease inhibitors (Roche Applied Science). Protein concentrations were determined by Bradford assay (Bio-Rad). Proteins were separated by electrophoresis in an 8% or 10% SDS-containing polyacrylamide gel and transferred to nitrocellulose membranes. Membranes were blocked in PBS–0.1% Tween (PBST) with 5% nonfat dry milk (NFDM). All antibodies were diluted in PBST–5% NFDM. Mouse monoclonal antibodies used in this study included anti-PARG (MABS61; Millipore) (1:1,000), anti-PAR (1020; Tulip Biosciences) (1:1,000), anti-ICP0 (H1A027; Virusys) (1:1,000), anti-ICP4 (1:10; hybridoma supernatant), anti-α-tubulin (T6199; Sigma) (1:5,000), and horseradish peroxidase (HRP)-conjugated anti-β-actin (49900; Abcam) (1:20,000). The rabbit polyclonal antibody used was anti-PARP-1 (9542; Cell Signaling) (1:1000), and the rabbit monoclonal antibody used was anti-phospho-H2AX (9718; Cell Signaling) (1:1,000). The rabbit antiserum used was directed against the N-terminal domain of PARG (29) and was a kind gift of Myron Jacobson (University of North Texas) (1:5,000). The goat polyclonal antibody used was anti-promyelocytic leukemia (PML) protein (sc-9862; Santa Cruz) (1:1,000). Membranes were washed with PBST and subsequently probed with goat anti-rabbit, goat anti-mouse, or mouse anti-goat HRP-coupled secondary antibodies (1:5,000; Jackson ImmunoResearch). Proteins were visualized by chemiluminescence using an ECL detection system (Amersham).
For immunofluorescence assays, fibroblasts on glass coverslips were grown to confluence and serum starved for 24 h before infection. At indicated times, cells were washed in PBS and fixed in methanol/acetone (1:1; −20°C) for 15 min. Samples were then washed in PBS and blocked in PBS–10% human serum–0.1% Triton X-100 for 1 h at room temperature. Primary antibodies were diluted in PBS supplemented with 10% human and 10% goat serum and incubated for 1 h at room temperature. The cells were washed three times with PBS–0.2% Tween between primary and secondary antibody incubations. Secondary antibodies were diluted in PBS–10% goat serum and incubated as described for primary antibodies. Mouse monoclonal antibodies used for indirect immunofluorescence were anti-PARP-1 (WH0000142M1; Sigma) (1:200) and anti-ICP4 (1:20; hybridoma supernatant). Rabbit antibodies used were anti-UL30 (r113; a kind gift of R. Everett, University of Glasgow) (1:3,000) and anti-phospho-H2AX (9718; Cell Signaling) (1:200). Goat anti-mouse or anti-rabbit secondary antibodies conjugated to Alexa Fluor 488 or 546 (Invitrogen) were used at a dilution of 1:1,000. Nuclei were stained with Hoechst 33258 (Invitrogen). Confocal images were obtained using Zeiss LSM510 laser scanning microscope.
Transfections.
For siRNA transfections, fibroblasts were grown to 80% confluence in 24-well dishes and maintained in growth medium without antibiotics. Cells were transfected with 10 pmol of siRNA using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's instructions. For experiments using HSV-1, transfected cells were allowed to incubate for 3 days before being infected with HSV-1 at a multiplicity of 3 IU/cell. Medium and cells were harvested at 18 to 24 hpi as described above. Double-stranded siRNAs used for this experiment were purchased from Sigma and included PARP-1 (Hs02_00332177), PARG (Hs01_00128017), and a universal nontargeting control (SIC0001).
For plasmid transfections, fibroblasts were grown to 70 to 80% confluence and washed with Opti-MEM (Invitrogen) before transfection with 2.0 μg of MTS1 or MTS1-ICP0, kind gifts of Bernard Roizman (University of Chicago), using Lipofectamine LTX with Plus reagent (Invitrogen) according to the manufacturer's instructions. When MG132 was used, drug was added 1 h after transfection in Opti-MEM. Cells were supplemented with growth medium lacking antibiotics 4 h after transfection and were allowed to grow for an additional 24 h. Samples were collected for Western blot assays as described above.
Statistical analysis.
Data are expressed as averages ± standard deviations (SD). Statistical analysis was done using Student's t test, and significance was set at a P value of <0.05.
RESULTS
HSV-1 infection depletes cellular NAD+ pools.
Our laboratories have previously found that NAD+ is markedly depleted following HSV-1 infection (44). To confirm this observation, we initially quantified the levels of NAD+ over the course of HSV-1 (F strain) infection of human fibroblasts. Confluent cells were serum starved for 24 h and then infected or mock infected. Serum starvation synchronizes cells in G0 and decreases variability in the infection environment between cells (4). This treatment also removes extraneous metabolites that would confound mass spectrometry (MS) measurements (31). Cells were sampled from early in infection to the time of maximum virus output at approximately 24 h postinfection (hpi), and NAD+ was measured by liquid chromatography-high resolution mass spectrometry (LC-MS). HSV-1 infection triggered a dramatic, time-dependent decrease in NAD+ levels, such that infected cells had only ∼20% of the NAD+ levels seen in uninfected cells by 24 hpi (Fig. 1A). Levels of NADH also decreased during infection, while overall NAD+/NADH ratios stayed relatively stable. This virus-induced decrease in NAD+ levels could have been due to increased depletion of cellular NAD+ pools or inhibition of NAD+ biosynthesis. To distinguish between these possibilities, we pretreated fibroblasts with 20 nM FK866, a specific and noncompetitive inhibitor of nicotinamide phosphoribosyltransferase (NAMPT), a key enzyme in the salvage-based synthesis of NAD+ from nicotinamide (15). After 24 h of FK866 treatment, uninfected fibroblasts had NAD+ levels depleted by more than 50%. Following this 24-h pretreatment with FK866, cultures were infected with HSV-1, and metabolites were analyzed at 18 hpi. HSV-1 caused a drop in NAD+ levels in FK866-treated cells that was significantly greater than the drop seen in uninfected, drug-treated cells over the same time (Fig. 1B). This argues that the virus-induced depletion of NAD+ was due to increased consumption of existing pools. This effect was not due to residual FK866 increasing viral replication as the drug caused no significant change in viral titers (Fig. 1C).
Fig 1.

HSV-1 infection of human fibroblasts leads to the depletion of cellular NAD+ pools. (A) NAD+ levels over the course of HSV-1 infection (3 IU/cell) of serum-starved fibroblasts, normalized to packed cell volume. Values are averages of duplicate biological experiments (±1 SD). (B) NAD+ levels after pretreatment with 20 nM FK866, an inhibitor of NAD+ biosynthesis, or DMSO. Cells were treated with FK866 for 24 h before being washed and infected with HSV (3 IU/cell) or mock infected. Metabolites were extracted at 18 hpi and normalized to packed cell volume. Values are averages of duplicate experiments (±1 SD). (C) Production of infectious HSV-1 virions in cells pretreated with FK866 for 24 h. Values represent viral yields at 18 hpi and are expressed relative to DMSO-treated cells. Each treatment is the average of triplicate biological experiments (±1 SD). (D) Metabolite abundances after treatment with 20 nM FK866 as described above. Metabolite concentrations are expressed relative to mock-treated cells, and all ratios were log2 transformed. AU, arbitrary units. *, P < 0.05.
In addition to its effects on NAD+, HSV-1 infection of serum-starved fibroblasts also triggers the accumulation of metabolites from upper glycolysis and several intermediates in the synthesis pathways of nucleotides (44). FK866 pretreatment of uninfected cells increased the levels of many of these same compounds but to a much weaker extent than seen after HSV-1 infection (Fig. 1D). When FK866-treated cells were subsequently infected, the metabolic results were similar in magnitude to the changes seen with HSV-1 infection alone.
The depletion of NAD+ levels during infection results from PARP activation.
NAD+ is an essential cofactor in many of the redox reactions of central carbon metabolism, during which NAD+ is converted to and from its reduced form, NADH. Unlike the enzymes that catalyze such reactions, both PARP-1/2 consume NAD+ as a substrate toward the formation of PAR chains. As the primary consumers of NAD+, enzymes of the PARP superfamily, and especially PARP-1, are largely responsible for overall cellular NAD+ levels (6). Given the decrease in NAD+ levels seen during HSV-1 infection, we predicted that the virus might activate PARP-1/2, triggering NAD+ consumption and increasing PAR chain synthesis. We found that over the course of HSV-1 infection, and especially at early time points, the abundance of PARylated proteins increased (Fig. 2A). This was not due to an increase in total PARP-1 or its apoptosis-specific 85-kDa cleavage product (40), which remained constant throughout infection (Fig. 2B). To further test for a role of PARP-1/2, we measured NAD+ in both infected and mock-infected cells after treatment with Olaparib, which specifically and potently inhibits both enzymes (23, 28). Whereas Olaparib had no effect on NAD+ levels in uninfected cells, it completely restored the NAD+ levels of infected cells (Fig. 2C). This effect was not due to the inhibition of HSV-1 replication as drug treatment did not significantly change viral titers (Fig. 2D). The NAD+ rescue phenotype was also recapitulated in the levels of PARylated proteins in infected cells, which decreased dramatically after Olaparib treatment (Fig. 2E). In a control experiment, when fibroblasts were pretreated with Olaparib before exposure to hydrogen peroxide, PARylated protein levels also decreased, consistent with previous data showing that H2O2 activates PARP activity (45, 48). PARP-1/2 inhibition had little effect on the metabolic status of either uninfected or infected cells (Fig. 2F), suggesting that HSV-1 infection supersedes other cellular signals in determining the overall metabolic state of the cell.
Fig 2.

Depletion of NAD+ during HSV-1 infection is due to PARP-1/2 activation. (A) Analysis of poly(ADP-ribosyl)ation over the course of infection. Serum-starved fibroblasts were infected with HSV-1 (3 IU/cell) or mock infected and harvested at various times postinfection. A 15-min incubation with 10 mM H2O2 at 37°C served as a positive control for PARP activation. Whole-cell lysates were analyzed by Western blotting with an antibody specific to poly(ADP)-ribose. β-Actin was used as a loading control. (B) Analysis of PARP-1 protein levels over the course of infection with HSV-1. Serum-starved fibroblasts were infected with HSV-1 (3 IU/cell) or mock infected. At 1 hpi, cells were washed, and medium was replaced with new medium. At various times postinfection, cells were harvested, and PARP-1 levels were monitored by Western blotting. A caret (∧) indicates a 30-min incubation with 10 mM sorbitol, a positive control for PARP-1 cleavage, an early event in apoptosis. (C) NAD+ levels after treatment with 50 nM Olaparib (Ola) or DMSO. Serum-starved fibroblasts were infected (3 IU/cell) or mock infected, and drug was applied at 1 hpi. Metabolites were extracted at 18 hpi, and NAD+ levels were normalized to packed cell volume. Values are averages of duplicate experiments (±1 SD). (D) Production of infectious HSV-1 virions in cells treated with 50 nM Olaparib as described above. Values are representative of virus yield at 18 hpi and are expressed relative to DMSO-treated cells (±1 SD). (E) Analysis of poly(ADP-ribosyl)ation after PARP-1/2 inhibition. Serum-starved fibroblasts were infected with HSV-1 (3 IU/cell) or mock infected. At 1 hpi, 50 nM Olaparib was applied, and cells were harvested at 18 hpi. H2O2-treated cells were pretreated with 50 nM Olaparib for 1 h before H2O2 application. Whole-cell lysates were analyzed by Western blotting. (F) Metabolite abundances after treatment with 50 nM Olaparib as described above. Metabolite concentrations are expressed relative to equivalent mock-treated cells, and all ratios were log2 transformed. AU, arbitrary units; M, mock infected. *, P < 0.05.
NAD+ levels in infected cells were also rescued after application of 3 mM 3-aminobenzamide (Fig. 3A), a broad-spectrum inhibitor of ADP-ribosylation (42). This drug had a modest (∼30% decrease) effect on virus yield (Fig. 3B), but it could not be ascertained whether this reduction resulted from inhibition of PARP-1/2 or from a general decrease in ADP-ribosylation events. A previous report has shown that inhibition of the ADP-ribosylation activities of tankyrase-1 and tankyrase-2, which would be altered by 3-aminobenzamide but not Olaparib, reduces HSV-1 yields (20).
Fig 3.
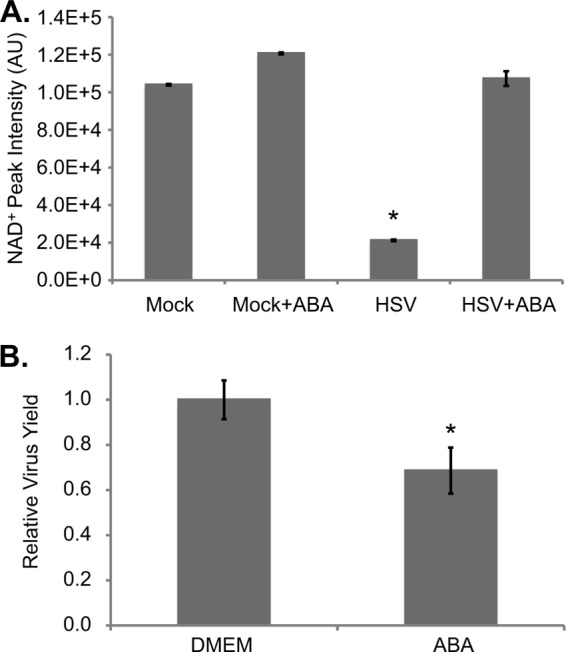
A broad-spectrum ADP-ribosylation inhibitor restores NAD+ levels in HSV-1 infected cells. (A) NAD+ levels after treatment with 3-aminobenzamide (ABA). Serum-starved fibroblasts were infected (3 IU/cell) or mock infected, and 3 mM ABA or DMEM was applied at 1 hpi. Metabolites were extracted at 24 hpi, and NAD+ levels were normalized to packed cell volume. Values are averages of duplicate experiments (±1 SD). (B) Production of infectious HSV-1 virions in cells treated with 3 mM ABA or DMEM as described above. Values are representative of virus yield at 18 hpi and are expressed relative to mock-treated cells (±1 SD).
We conclude that PARP-1/2 activity is strongly activated in HSV-1-infected cells, resulting in the consumption of NAD+ and generating PARylated proteins.
Viral DNA replication is required for PARP activation.
PARP-1/2 activation occurs following DNA damage, and it has been well documented that HSV-1 infection activates certain DNA damage responses (6, 46). Specifically, it has been postulated that the replication and/or resolution of viral genome concatemers mimics double-strand DNA breaks. To determine if viral DNA replication is required for PARP-1/2 activation, we treated cells with acyclovir, a nucleotide analog that is phosphorylated by the viral thymidine kinase and then incorporated into viral DNA, terminating elongation (10). Acyclovir (1 μM) decreased viral yield by ∼200-fold and completely restored NAD+ levels in infected cells (Fig. 4A and B). Acyclovir also reduced the levels of PARylated proteins in infected cells, while having no effect on PARylation following H2O2 treatment (Fig. 4C). Thus, we can conclude that viral DNA replication is necessary for activation of PARP-1/2 and depletion of NAD+ during HSV-1 infection.
Fig 4.
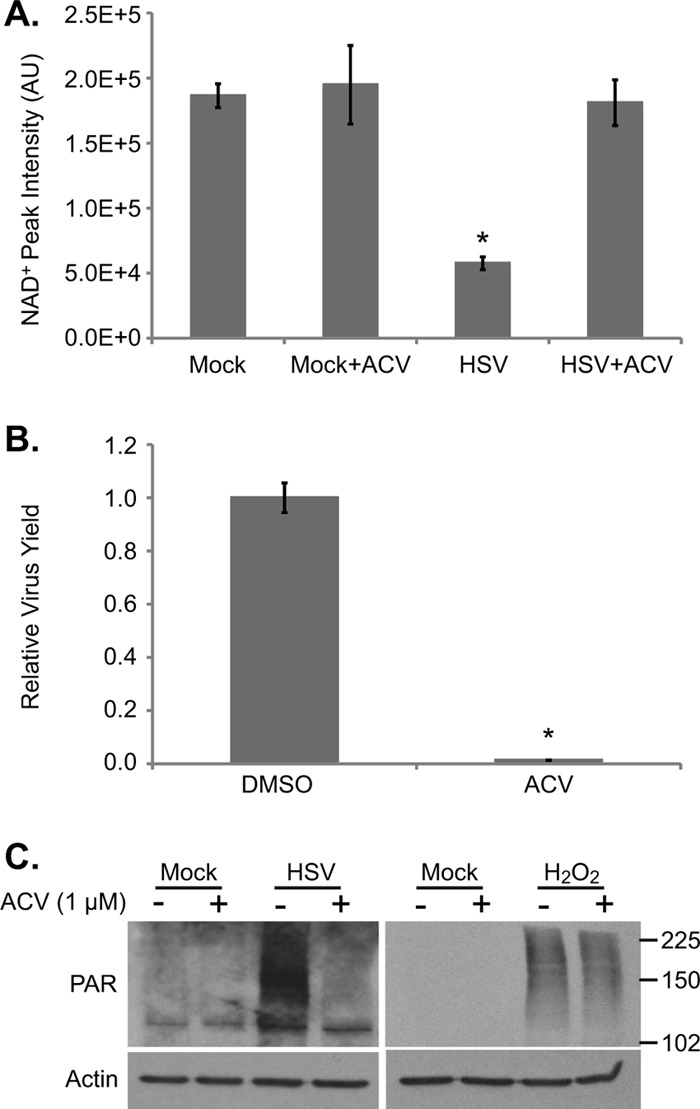
Viral DNA replication is required for PARP activation. (A) NAD+ levels after treatment with 1 μM acyclovir (ACV), an inhibitor of HSV-1 DNA replication, or DMSO. Serum-starved fibroblasts were infected (3 IU/cell) or mock infected, and drug was applied at 1 hpi. Metabolites were extracted at 18 hpi, and NAD+ levels were normalized to packed cell volume. Values are averages of duplicate experiments (±1 SD). (B) Production of infectious HSV-1 virions in cells treated with 1 μM acyclovir as described above. Values are representative of virus yield at 18 hpi and are expressed relative to DMSO-treated cells (±1 SD). (C) Analysis of poly(ADP-ribosyl)ation after inhibition of HSV-1 DNA replication. Serum-starved fibroblasts were infected with HSV-1 (3 IU/cell) or mock infected. At 1 hpi, 1 μM acyclovir was applied, and cells were harvested at 18 hpi. H2O2-treated cells were pretreated with 1 μM acyclovir for 1 h before H2O2 application. Whole-cell lysates were analyzed by Western blotting.
To further examine the relationship between viral DNA replication and PARP-1/2 activation, we measured the phosphorylation of the histone variant H2AX following infection. H2AX phosphorylated on Ser 139 (γH2AX) is a marker of DNA breaks and increases with increased levels of DNA damage (35). As expected, total γH2AX levels increased as HSV-1 infection progressed (Fig. 5A). When infected cells were treated with 400 μg/ml phosphonoacetic acid (PAA), which directly inhibits the viral DNA polymerase (16) at concentrations sufficient to decrease the viral yield by >200-fold, γH2AX levels were markedly decreased at both 8 and 18 hpi (Fig. 5B to D). PAA also decreased total PARylated proteins in infected cells while having no effect on PARylation after application of H2O2 (Fig. 5E). Thus, two drugs that inhibit HSV-1 DNA replication by different mechanisms were both able to reduce infection-induced PARylation, supporting the hypothesis that viral DNA replication is necessary for PARP-1/2 activation during infection. Consistent with this concept, PARP-1 colocalized with the catalytic subunit of the viral polymerase, UL30, at HSV-1 replication centers in greater than 90% of cells undergoing active viral DNA replication (Fig. 5F).
Fig 5.

HSV-1 DNA replication triggers the accumulation of DNA breaks. (A) Images showing the phosphorylation levels of histone H2AX (γH2AX) over the course of HSV-1 infection. Serum-starved fibroblasts were infected (1 IU/cell) and fixed at various times. Protein levels and localization were examined by immunofluorescence using antibodies specific to γH2AX and HSV-1 ICP4. (B and C) Analysis of H2AX phosphorylation after inhibition of the viral DNA polymerase using 400 μg/ml PAA. Serum-starved fibroblasts were infected (0.1 IU/cell for immunofluorescence assay; 3 IU/cell for Western blotting), and drug was applied at 1 hpi. Cells were fixed at 8 hpi (immunofluorescence assay) or harvested at 18 hpi (Western blotting). (D) Production of infectious HSV-1 virions in cells treated with 400 μg/ml PAA as described above. Values are representative of virus yield at 18 hpi and are expressed relative to mock-treated cells. (E) Analysis of poly(ADP-ribosyl)ation after inhibition of HSV-1 DNA replication. Serum-starved fibroblasts were infected with HSV-1 (3 IU/cell) or mock infected and treated with PAA as described above. H2O2-treated cells were pretreated with PAA for 1 h before H2O2 application. Whole-cell lysates were analyzed by Western blotting. (F) PARP-1 localization during HSV-1 infection. Serum-starved fibroblasts were infected with HSV (0.5 IU/cell) and either 400 μg/ml PAA or vehicle was applied at 1 hpi. Cells were fixed at 12 hpi and examined by immunofluorescence using antibodies specific to PARP-1 and HSV-1 UL30. *, P < 0.05.
ICP0 mediates the proteasome-dependent degradation of poly(ADP-ribose) glycohydrolase.
PARP-1 and PARP-2 are both self-inhibited by automodification (6). The enzyme poly(ADP-ribose) glycohydrolase (PARG) can cleave the PAR polymers from PARylated proteins, including PARP-1/2, thus reactivating the enzyme and allowing it to catalyze additional PAR chains (37). To determine if HSV-1 infection alters the balance between PARP and PARG, we first examined total PARG levels in infected cells. In humans, PARG is present in multiple isoforms, including proteins of 111, 102, and 99 kDa (29). Fibroblasts showed two PARG bands at molecular masses matching the 111- and 102/99-kDa isoforms (Fig. 6A). Lower-percentage acrylamide gels separated the bottom band into the 102- and 99-kDa isoforms (data not shown). Upon HSV-1 infection, the levels of the higher-molecular-mass PARG decreased; a similar decrease was also seen upon hydrogen peroxide treatment. To ensure that all the present bands truly represented PARG, an siRNA transfection was done using an siRNA that targeted all isoforms. Both the higher- and lower-molecular-mass bands disappeared with siRNA treatment (Fig. 6A). Further, no change in cell viability, as measured by propidium iodide (PI) uptake, was seen after transfection (nontargeting siRNA, 8.4% ± 1.2% PI positive; PARG siRNA, 8.7% ± 3.2% positive). To determine if the higher-molecular-mass PARG band was the 111-kDa isoform, we used antiserum directed against the N terminus of PARG, which is spliced out in all the smaller isoforms. The single band recognized by this antiserum disappeared with PARG siRNA treatment as well as with infection (Fig. 6B). Knockdown of all PARG isoforms by siRNA treatment resulted in a modest, but significant, decrease in virus yield (Fig. 6C). To locate the block to infection that occurs when all PARG isoforms are knocked down, viral gene products were probed with and without PARG siRNA treatment (Fig. 6D). The levels of VP16, ICP0, and gM remained the same regardless of siRNA treatment, suggesting that the block to infection occurs in a gene not tested or at a step after leaky late gene expression.
Fig 6.

Levels of a high-molecular-mass PARG isoform decrease during HSV-1 infection. (A) Analysis of PARG protein content in cells transfected with siRNA directed against PARG. Subconfluent fibroblasts were transfected with 10 pmol of siRNA. Three days later, cells were infected with HSV-1 (3 IU/cell), mock infected, or treated with 10 mM H2O2. Whole-cell lysates were analyzed by Western blotting. (B) Levels of the 111-kDa isoform of PARG in infected cells. (Left) Subconfluent fibroblasts were transfected with nontargeting (NT) or PARG (P) siRNA as above. Whole-cell lysates were collected 3 days after transfection. (Right) Serum-starved fibroblasts were infected (1 IU/cell) or mock infected, and whole-cell lysates were collected at 18 hpi. (C) Production of infectious HSV-1 virions in cells treated with PARG siRNA. Values are representative of virus yield at 18 hpi and are expressed relative to nontargeting siRNA-treated cells (±1 SD). (D) Protein content in cells transfected with siRNAs against PARG (in triplicate), VP16, or an NT control and infected 3 days later as described above. Whole-cell lysates were analyzed by Western blotting. M, mock infected. *, P < 0.05.
ICP0 is an immediate-early viral protein with a RING finger domain possessing E3 ubiquitin ligase activity that has been shown to sponsor the proteasome-dependent degradation of a wide range of cellular proteins, including DNA-dependent protein kinase (DNA-PK), PML, and centromere protein B (CENP-B) (13, 34). To determine if the loss of the 111-kDa isoform of PARG during HSV-1 infection was a result of proteasome-dependent degradation, we treated infected cells with MG132, an inhibitor of the 26S proteasome subunit (11) (Fig. 7A). Treatment with the drug substantially increased levels of both PML and the 111-kDa PARG isoform at 12 hpi. To test whether ICP0 was sufficient for PARG-111 degradation, we transfected fibroblasts with an ICP0 expression vector and probed for PARG protein levels. One day after transfection, ICP0-expressing cells expressed lower levels of PARG-111 than cells receiving the empty vector (Fig. 7B), and this decrease was blocked by MG132 treatment.
Fig 7.
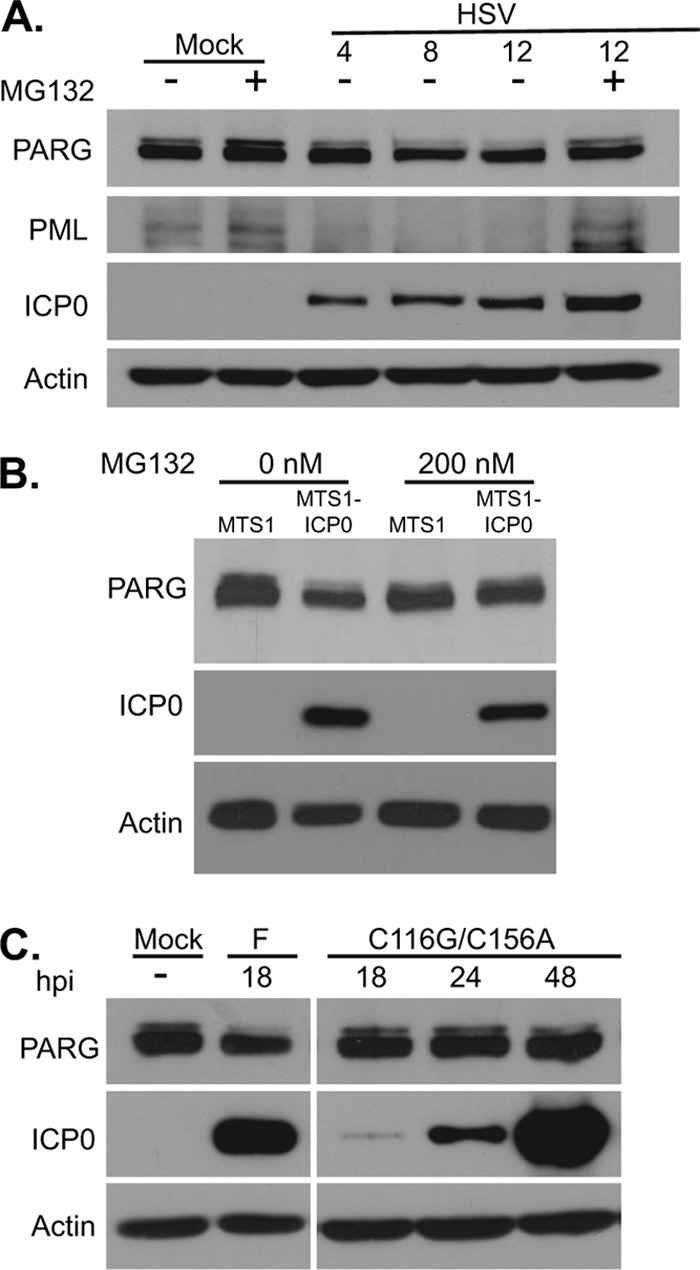
The HSV-1 protein ICP0 mediates the proteasome-dependent degradation of a high-molecular-mass isoform of PARG. (A) Analysis of PARG protein levels after treatment with the proteasome inhibitor MG132. Serum-starved fibroblasts were infected with HSV (3 IU/cell) or mock infected. Cells were washed and treated with DMSO or 10 μM MG132 at 1 hpi. Lysates were harvested at 12 hpi and were analyzed by Western blotting. PML was used as a positive control for HSV-induced, proteasome-dependent degradation of a host cell protein. (B) Subconfluent fibroblasts were transfected with MTS1-ICP0 or the corresponding empty vector MTS1. Then, 200 nM MG132 or DMSO was added to transfected cells at 1 h, and cells were supplemented with growth medium after 4 h. Lysates were harvested at 24 h after transfection. (C) Analysis of PARG protein levels in the C116G/C156A mutant, which contains a mutation in the RING finger E3 ubiquitin ligase domain of ICP0. Serum-starved fibroblasts were infected with HSV F strain or the C116G/C156A mutant (3 IU/cell) or mock infected. Lysates were harvested at the indicated times.
To confirm that ICP0 was necessary for the degradation of PARG-111 by HSV-1, we compared PARG-111 levels in cells after infection with wild-type virus versus infection with C116G/C156A virus, a mutant virus expressing an ICP0 variant lacking E3 ubiquitin ligase activity found in its RING finger domain (22). The C116G/C156A mutant did not induce the degradation of PARG-111, even at late times when ICP0 expression was high (Fig. 7C). Taken together, these results suggest that the RING finger domain of ICP0 is responsible for the decrease in PARG-111 levels seen during infection.
DISCUSSION
Interactions between herpesviruses and their host cells, including changes in metabolic status upon infection, are varied and complex. NAD+ plays a significant role as a cofactor in multiple reactions of central carbon metabolism, and its levels help regulate the overall energy state of the cell (21). Here, we show that HSV-1 infection dramatically decreases the levels of NAD+ in the cell (Fig. 1) and increases protein PARylation (Fig. 2A). Treatment with inhibitors proved that both of these phenotypes were secondary to PARP-1/2 activation (Fig. 2C to F) and dependent on viral DNA replication (Fig. 4 and 5E). The levels of γH2AX increase during infection but are reduced when the viral DNA polymerase is inhibited (Fig. 5A to C), which suggests that inhibition of viral genome replication decreases at least some types of DNA damage typically induced by infection. As PARP-1/2 is activated by DNA damage (37), the depletion of NAD+ during HSV-1 infection may be due to virus-induced DNA damage activating PARP activity.
PARG counteracts the activity of PARP-1/2, and the balance in the activity of these proteins is critical as removal of PAR automodifications from PARP-1/2 can restore its catalytic activity (7). Fibroblasts expressed the 111-, 102-, and 99-kDa isoforms of PARG, and their relative abundances agreed with endogenous PARG levels previously seen in HeLa cells (29). Knockdown of all PARG isoforms by siRNA (Fig. 6A) resulted in moderately decreased HSV titers (Fig. 6B), suggesting that some PARG activity facilitates HSV-1 replication. It was also shown that the viral protein ICP0 directed degradation of PARG-111 but not its smaller isoforms (Fig. 7).
As the relative roles of PARP-1/2 and PARG must be tightly regulated for the proper response to DNA damage (50) and as HSV-1 infection alternately activates and represses different components of damage repair (46), it is intriguing that ICP0 mediates the degradation of the 111-kDa isoform but not the 102- and 99-kDa isoforms of PARG. The differences between these isoforms are not well characterized although their overexpression in HEK293 cells suggests that PARG-111 is nuclear whereas PARG-102 and -99 are located in the cytoplasm (29). It has also been suggested that these smaller isoforms may shuttle to the nucleus under certain conditions (14). PARG-null animals are embryonic lethal (18), and PARG siRNA treatment reducing all PARG isoforms lowered virus yields (Fig. 6B), raising the possibility that selective degradation of PARG-111 allows for optimal viral growth. Selective PARG-111 degradation in mouse astrocytes has been shown to slow the rate of nuclear PAR degradation and protect against PARP-dependent cell death, which is termed parthanatos (5). In this pathway, PARP-generated PAR polymers translocate from the nucleus to the cytoplasm, where they bind with high affinity to apoptosis-inducing factor (AIF) located at the mitochondrial membrane (45). This binding releases AIF into the cytoplasm, and it eventually enters the nucleus, where it induces cell death. Thus, ICP0-mediated degradation of PARG-111 could represent a viral strategy to protect infected cells from premature death due to excessive production of PAR polymer by preventing the removal of inhibitory PAR chains from PARP, thereby reducing its activity at nuclear replication compartments. Retention of the cytoplasmic 102- and 99-kDa PARG isoforms during infection could also help prevent parthanatos as overexpression of cytoplasmic PARG in mouse neurons has been shown to decrease cytoplasmic PAR levels and inhibit the release of AIF from the mitochondria after parthanatos-inducing treatments (1). It remains unclear whether PARP-1/2 activity contributes positively to the viral life cycle or if they alternatively represent a host defense strategy. In this regard, it should be noted that the HSV-1 protein ICP4 is PARylated but to unknown effect (2).
HSV-1 infection triggers a specific set of changes to metabolite levels during infection, many of which differ from those seen with the related betaherpesvirus human cytomegalovirus (HCMV) (44). It is interesting that HCMV does not trigger the loss of NAD+. The substantial difference in infection kinetics between these viruses, with HSV-1 replicating its genome earlier after infection and completing its replication cycle much faster than HCMV, may be the reason for this contrast. Maintaining NAD+ levels would be especially important to HCMV as infection triggers increased flux through glycolysis, which requires NAD+ as a cofactor (44).
Drug-induced depletion of NAD+ levels of uninfected cells by FK866 gave a metabolic output similar to that seen with infection alone. However, inhibiting the loss of NAD+ levels during HSV-1 infection with Olaparib did not modify the global metabolic state induced by infection. So while the metabolic alterations induced by HSV-1 infection are similar to the alterations seen when NAD+ is depleted in uninfected cells, the virus-induced changes are not themselves dependent on the depletion of NAD+ during infection. Thus, despite the large number of metabolic reactions that utilize NAD+ as a cofactor, HSV-1 infection appears to have a more dominant role than NAD+ levels in determining the overall metabolic status of the cell.
In summary, we have found that HSV-1 infection activates PARP-1/2 and that this results in the massive depletion of NAD+. This change, however, is not responsible for the other metabolic effects of infection, nor does it significantly impact viral replication. By late in infection, PARG-111 is degraded in an ICP0-dependent manner, but complete degradation of all PARG isoforms is inhibitory to the virus. This work provides an explanation for the profound changes in NAD+ levels that occur subsequent to HSV-1 infection, and it demonstrates that HSV-1 infection actively alters the fine-tuned balance between PARP and PARG in infected cells.
ACKNOWLEDGMENTS
We gratefully acknowledge critical commentary and technical advice provided by B. Roizman, L. Terry, and E. O'Keefe.
This work was supported by grants from the National Institutes of Health to T.S. (CA82396) and J.D.R. (AI078063). S.L.G. is supported by a National Science Foundation Graduate Research Fellowship (DGE-0646086).
Footnotes
Published ahead of print 23 May 2012
REFERENCES
- 1. Andrabi SA, et al. 2006. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. U. S. A. 103:18308–18313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blaho JA, et al. 1992. Differences in the poly(ADP-ribosyl)ation patterns of ICP4, the herpes simplex virus major regulatory protein, in infected cells and in isolated nuclei. J. Virol. 66:6398–6407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blenn C, Wyrsch P, Althaus FR. 2011. The ups and downs of tannins as inhibitors of poly(ADP-Ribose)glycohydrolase. Molecules 16:1854–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Browne E, Wing B, Coleman D, Shenk T. 2001. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J. Virol. 75:12319–12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burns DM, Ying W, Kauppinen TM, Zhu K, Swanson RA. 2009. Selective down-regulation of nuclear poly(ADP-ribose) glycohydrolase. PLoS One 4:e4896 doi:10.1371/journal.pone.0004896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D'Amours D, Desnoyers S, D'Silva I, Poirier GG. 1999. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 342:249–268 [PMC free article] [PubMed] [Google Scholar]
- 7. Davidovic L, Vodenicharov M, Affar EB, Poirier GG. 2001. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp. Cell Res. 268:7–13 [DOI] [PubMed] [Google Scholar]
- 8. Delgado T, et al. 2010. Induction of the Warburg effect by Kaposi's sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 107:10696–10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357–364 [DOI] [PubMed] [Google Scholar]
- 10. Elion GB. 1982. Mechanism of action and selectivity of acyclovir. Am. J. Med. 73:7–13 [DOI] [PubMed] [Google Scholar]
- 11. Giuliano M, D'Anneo A, De Blasio A, Vento R, Tesoriere G. 2003. Apoptosis meets proteasome, an invaluable therapeutic target of anticancer drugs. Ital. J. Biochem. 52:112–121 [PubMed] [Google Scholar]
- 12. Ha HC, et al. 2001. Poly(ADP-ribose) polymerase-1 is required for efficient HIV-1 integration. Proc. Natl. Acad. Sci. U. S. A. 98:3364–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hagglund R, Roizman B. 2003. Herpes simplex virus 1 mutant in which the ICP0 HUL-1 E3 ubiquitin ligase site is disrupted stabilizes cdc34 but degrades D-type cyclins and exhibits diminished neurotoxicity. J. Virol. 77:13194–13202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haince JF, Ouellet ME, McDonald D, Hendzel MJ, Poirier GG. 2006. Dynamic relocation of poly(ADP-ribose) glycohydrolase isoforms during radiation-induced DNA damage. Biochim. Biophys. Acta 1763:226–237 [DOI] [PubMed] [Google Scholar]
- 15. Hasmann M, Schemainda I. 2003. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 63:7436–7442 [PubMed] [Google Scholar]
- 16. Honess RW, Watson DH. 1977. Herpes simplex virus resistance and sensitivity to phosphonoacetic acid. J. Virol. 21:584–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huber A, Bai P, de Murcia JM, de Murcia G. 2004. PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair (Amst.) 3:1103–1108 [DOI] [PubMed] [Google Scholar]
- 18. Koh DW, et al. 2004. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc. Natl. Acad. Sci. U. S. A. 101:17699–17704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krishnakumar R, Kraus WL. 2010. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell 39:8–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Z, et al. 2012. Herpes simplex virus requires PARP activity for efficient replication and induces ERK-dependent phosphorylation and ICP0-dependent nuclear localization of tankyrase 1. J. Virol. 86:492–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin SJ, Guarente L. 2003. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 15:241–246 [DOI] [PubMed] [Google Scholar]
- 22. Lium EK, Silverstein S. 1997. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential α27 gene. J. Virol. 71:8602–8614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loh VM, Jr, et al. 2005. Phthalazinones. Part 1: the design and synthesis of a novel series of potent inhibitors of poly(ADP-ribose)polymerase. Bioorg. Med. Chem. Lett. 15:2235–2238 [DOI] [PubMed] [Google Scholar]
- 24. Lu W, et al. 2010. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone Orbitrap mass spectrometer. Anal. Chem. 82:3212–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Malanga M, Pleschke JM, Kleczkowska HE, Althaus FR. 1998. Poly(ADP-ribose) binds to specific domains of p53 and alters its DNA binding functions. J. Biol. Chem. 273:11839–11843 [DOI] [PubMed] [Google Scholar]
- 26. Mattiussi S, et al. 2007. Inhibition of Poly(ADP-ribose)polymerase impairs Epstein Barr virus lytic cycle progression. Infect. Agents Cancer 2:18 doi:10.1186/1750-9378-2-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Melamud E, Vastag L, Rabinowitz JD. 2010. Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 82:9818–9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Menear KA, et al. 2008. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phth alazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 51:6581–6591 [DOI] [PubMed] [Google Scholar]
- 29. Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK. 2004. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp. Cell Res. 297:521–532 [DOI] [PubMed] [Google Scholar]
- 30. Mortusewicz O, Fouquerel E, Ame JC, Leonhardt H, Schreiber V. 2011. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res. 39:5045–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. 2006. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2:e132 doi:10.1371/journal.ppat.0020132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Munger J, et al. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26:1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohashi Y, et al. 1986. Poly(ADP-ribosyl)ation of DNA polymerase beta in vitro. Biochem. Biophys. Res. Commun. 140:666–673 [DOI] [PubMed] [Google Scholar]
- 34. Paulus C, Nitzsche A, Nevels M. 2010. Chromatinisation of herpesvirus genomes. Rev. Med. Virol. 20:34–50 [DOI] [PubMed] [Google Scholar]
- 35. Redon CE, et al. 2010. Histone γH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin. Cancer Res. 16:4532–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed Lippincott Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 37. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. 2010. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer. 10:293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Satoh MS, Lindahl T. 1992. Role of poly(ADP-ribose) formation in DNA repair. Nature 356:356–358 [DOI] [PubMed] [Google Scholar]
- 39. Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 34:684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Simbulan-Rosenthal CM, Rosenthal DS, Iyer S, Boulares AH, Smulson ME. 1998. Transient poly(ADP-ribosyl)ation of nuclear proteins and role of poly(ADP-ribose) polymerase in the early stages of apoptosis. J. Biol. Chem. 273:13703–13712 [DOI] [PubMed] [Google Scholar]
- 41. Smith MC, Boutell C, Davido DJ. 2011. HSV-1 ICP0: paving the way for viral replication. Future Virol. 6:421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sodhi RK, Singh N, Jaggi AS. 2010. Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic implications. Vascul. Pharmacol. 53:77–87 [DOI] [PubMed] [Google Scholar]
- 43. Stettler M, et al. 2006. New disposable tubes for rapid and precise biomass assessment for suspension cultures of mammalian cells. Biotechnol. Bioeng. 95:1228–1233 [DOI] [PubMed] [Google Scholar]
- 44. Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. 2011. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 7:e1002124 doi:10.1371/journal.ppat.1002124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Y, et al. 2011. Poly(ADP-Ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (Parthanatos). Sci. Signal. 4:ra20 doi:10.1126/scisignal.2000902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weitzman M, Weller S. 2011. Interactions between HSV-1 and the DNA damage response. In Weller S. (ed), Alphaherpesviruses. Caister Academic Press, Norfolk, VA [Google Scholar]
- 47. Whatcott CJ, Meyer-Ficca ML, Meyer RG, Jacobson MK. 2009. A specific isoform of poly(ADP-ribose) glycohydrolase is targeted to the mitochondrial matrix by a N-terminal mitochondrial targeting sequence. Exp. Cell Res. 315:3477–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ying W, Sevigny MB, Chen Y, Swanson RA. 2001. Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc. Natl. Acad. Sci. U. S. A. 98:12227–12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan J, Bennett BD, Rabinowitz JD. 2008. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat. Protoc. 3:1328–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou Y, Feng X, Koh DW. 2011. Activation of cell death mediated by apoptosis-inducing factor due to the absence of poly(ADP-ribose) glycohydrolase. Biochemistry 50:2850–2859 [DOI] [PubMed] [Google Scholar]