

Abstract
Hepatitis C virus (HCV) is one of the most common etiologic agents of chronic liver diseases, including liver cirrhosis and hepatocellular carcinoma. In addition, HCV infection is often associated with extrahepatic manifestations (EHM), including mixed cryoglobulinemia and non-Hodgkin's lymphoma. However, the mechanisms of cell tropism of HCV and HCV-induced EHM remain elusive, because in vitro propagation of HCV has been limited in the combination of cell culture-adapted HCV (HCVcc) and several hepatic cell lines. Recently, a liver-specific microRNA called miR-122 was shown to facilitate the efficient propagation of HCVcc in several hepatic cell lines. In this study, we evaluated the importance of miR-122 on the replication of HCV in nonhepatic cells. Among the nonhepatic cell lines expressing functional HCV entry receptors, Hec1B cells derived from human uterus exhibited a low level of replication of the HCV genome upon infection with HCVcc. Exogenous expression of miR-122 in several cells facilitates efficient viral replication but not production of infectious particles, probably due to the lack of hepatocytic lipid metabolism. Furthermore, expression of mutant miR-122 carrying a substitution in a seed domain was required for efficient replication of mutant HCVcc carrying complementary substitutions in miR-122-binding sites, suggesting that specific interaction between miR-122 and HCV RNA is essential for the enhancement of viral replication. In conclusion, although miR-122 facilitates efficient viral replication in nonhepatic cells, factors other than miR-122, which are most likely specific to hepatocytes, are required for HCV assembly.
INTRODUCTION
More than 170 million individuals worldwide are infected with hepatitis C virus (HCV), and cirrhosis and hepatocellular carcinoma induced by HCV infection are life-threatening diseases (57). Although therapy combining pegylated interferon (IFN) and ribavirin has achieved a sustained virological response in 50% of individuals infected with HCV genotype 1 (37), a more effective therapeutic modality for HCV infection is needed (46). The establishment of in vivo and in vitro infection systems has been hampered by the narrow host range and tissue tropism of HCV. Although the chimpanzee is the only experimental animal susceptible to HCV infection, it is difficult to use the chimpanzee in experiments due to ethical concerns (3). Furthermore, robust in vitro HCV propagation is limited to the combination of cell culture-adapted clones based on the genotype 2a JFH1 strain (HCVcc) and human hepatoma cell lines, including Huh7, Hep3B, and HepG2 (29, 43, 62).
It is well-known that HCV mainly infects hepatocytes. However, the precise mechanism underlying the liver tropism of HCV has not been clarified. Chronic hepatitis C virus infection is often associated with at least one extrahepatic manifestation (EHM), including mixed cryoglobulinemia, non-Hodgkin's lymphoma, lichen planus, thyroiditis, diabetes mellitus, Sjögren syndrome, and arthritis (19). EHMs are frequently more serious than hepatic disease in some patients and sometimes occur even in patients with persistently normal liver functions (19). Mixed cryoglobulinemia is the most-well-characterized HCV-associated disease and is curable by viral clearance through antiviral therapies (6). Although replication of HCV RNA in peripheral blood mononuclear cells (PBMCs) and neuronal cells at a low level was suggested (64), the biological significance of the extrahepatic replication of HCV, particularly in the development of EHMs, is not well understood.
MicroRNAs (miRNAs) are small noncoding RNAs consisting of 20 to 25 nucleotides that modulate gene expression in plants and animals (1, 24). Most miRNAs negatively regulate translation through the interaction with the 3′ untranslated region (UTR) of mRNA in a sequence-specific manner. miRNA 122 (miR-122) is liver specific, is the most abundantly expressed miRNA in the liver, and represses the translation of several mRNAs (5, 7). Jopling et al. reported for the first time that the inhibition of miR-122 dramatically decreased RNA replication in HCV subgenomic replicon (SGR) cells (28). In addition, several reports revealed that a specific interaction between the seed domain of miR-122 and the complementary sequences in the 5′ UTR of HCV RNA is essential for the enhancement of translation and replication of the HCV genome (21, 25, 27, 36). Endogenous expression levels of miR-122 are significantly higher in Huh7 cells than in other hepatic and nonhepatic cell lines (Fig. 1). In addition, previous reports showed that miR-122 expression enhanced the replication of SGR RNA in human embryonic kidney 293 (HEK293) cells and mouse embryonic fibroblasts (MEFs) (8, 35). Furthermore, it was recently shown that exogenous expression of miR-122 facilitates the efficient propagation of HCVcc in Hep3B and HepG2 cells, which are nonpermissive for HCVcc propagation (29, 43). These results suggest that the high susceptibility of Huh7 cells to the propagation of HCVcc is attributable to the high expression level of miR-122 and raise the possibility of expanding the HCV host range through the exogenous expression of miR-122 in nonhepatic cells.
Fig 1.
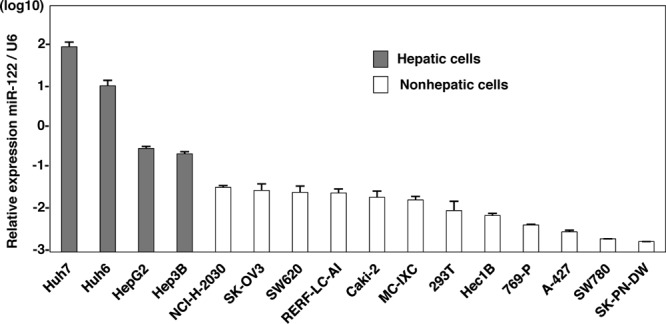
Endogenous expression levels of miR-122 in hepatic and nonhepatic cells. Total miRNAs were extracted from Huh7, Huh6, HepG2, Hep3B, NCI-H-2030, SK-OV3, SW620, RERF-LC-AI, Caki-2, MC-IXC, 293T, Hec1B, 769-P, A-427, SW780, and SK-PN-DW cells, and the expression levels of miR-122 were determined by qRT-PCR.
In this study, we assessed the effect of miR-122 expression on the replication of HCVcc and SGR RNA in several nonhepatic cell lines. Although the exogenous expression of miR-122 in the cell lines facilitates significant RNA replication through a gene-specific interaction between miR-122 and 5′ UTR of HCV RNA, no infectivity was detected in either the cells or the culture supernatants. The current study suggests that the expression of miR-122 plays an important role in HCV cell tropism, as well as in the possible involvement of nonhepatic cells in EHM, through an incomplete propagation of HCV.
MATERIALS AND METHODS
NextBio Body Atlas.
The NextBio Body Atlas application presents an aggregated analysis of gene expression across various normal tissues, normal cell types, and cancer cell lines. It enables us to investigate the expression of individual genes as well as gene sets. Samples for Body Atlas data are obtained from publicly available studies that are internally curated, annotated, and processed (31). Body Atlas measurements are generated from all available RNA expression studies that used Affymetrix U133 Plus or U133A Genechip arrays for human studies. The results corresponding to 128 human tissue samples were incorporated from 1,067 arrays, the results corresponding to 157 human cell types were incorporated from 1,474 arrays, and the results corresponding to 359 human cancer cell lines were incorporated from 376 arrays. Gene queries return a list of relevant tissues or cell types rank ordered by absolute gene expression and grouped by body systems or across all body systems. In the current analysis, we screened for nonhepatic cell lines expressing HCV receptor candidates, including CD81, SR-BI, claudin1 (CLDN1), and occludin (OCLN), or very-low-density lipoprotein (VLDL)-associated proteins, including apolipoprotein E (ApoE), ApoB, and microsomal triglyceride transfer protein (MTTP). A detailed analysis protocol developed by NextBio was described previously (31). The raw data used in this application are derived from the GSK Cancer Cell Line data deposited at the National Cancer Institute website (https://array.nci.nih.gov/caarray/project/woost-00041) and additionally from NCBI Gene Expression Omnibus (GEO) accession number GSE5720 for cell lines SK-OV-3 and SW620.
Sample collection and RNA extraction for microarray analysis.
Total RNAs extracted from cells were purified by using an miRNeasy kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. Eluted RNAs were quantified using a Nanodrop ND-1000 (version 3.5.2) spectrophotometer (Thermo Scientific, Wartham, MA). RNA integrity was evaluated using the RNA 6000 LabChip kit and bioanalyzer (Agilent Technologies, Santa Clara, CA). Each RNA that had an RNA integrity number (RIN) greater than 9.0 was used for the microarray experiments.
Microarray experiment.
Expression profiling was generated using the 4 × 44K whole-human-genome oligonucleotide microarray (version 2.0) G4845A (Agilent Technologies). Each microarray uses 44,495 probes to interrogate 27,958 Entrez gene RNAs. One hundred nanograms of total RNA was reverse transcribed into double-stranded cDNAs by AffinityScript multiple-temperature reverse transcriptase and amplified for 2 h at 40°C. The resulting cDNAs were subsequently used for in vitro transcription by the T7 polymerase and labeled with cyanine-3-labeled cytosine triphosphate (Perkin Elmer, Waltham, MA) for 2 h at 40°C using a low-input Quick-Amp labeling kit (Agilent Technologies) according to the manufacturer's protocol. After labeling, the rates of dye incorporation and quantification were measured with a Nanodrop ND-1000 (version 3.5.2) spectrophotometer (Thermo Scientific), and then the cRNAs were fragmented for 30 min at 60°C in the dark. Differentially labeled samples of 1,650 ng of cRNA were hybridized on Agilent 4 × 44K whole-genome arrays (version 2.0; 026652; Agilent Design) at 65°C for 17 h with rotation in the dark. Hybridization was performed using a gene expression hybridization kit (Agilent Technologies) following the manufacturer's instructions. After washing in gene expression washing buffer, each slide was scanned with the Agilent microarray scanner G2505C. Feature extraction software (version 10.5.1.1) employing defaults for all parameters was used to convert the images into gene expression data. Raw data were imported into a Subio platform (version 1.12) for database management and quality control. Raw intensity data were normalized against GAPDH (glyceraldehyde-3-phosphate dehydrogenase) expression levels for further analysis. These raw data have been accepted by GEO (a public repository for microarray data aimed at storing minimum information about microarray experiments [MIAME]).
Plasmids.
The cDNA clones of wild-type (WT) and mutant (MT) pri-miR-122 and Aequorea coerulescens green fluorescent protein (AcGFP) were inserted between the XhoI and XbaI sites of lentiviral vector pCSII-EF-RfA, which was kindly provided by M. Hijikata, and the resulting plasmids were designated pCSII-EF-WT-miR-122, pCSII-EF-MT-miR-122, and pCSII-EF-AcGFP, respectively. Plasmids pHH-JFH1 and pSGR-JFH1, which encode full-length and subgenomic cDNA of the JFH1 strain, respectively, were kindly provided by T. Wakita. pHH-JFH1-E2p7NS2mt contains three adaptive mutations in pHH-JFH1 (53). pHH-JFH1-M1 and pHH-JFH1-M2 were established by the introduction of a point mutation of nucleotide 26 located in site 1 and nucleotide 41 in site 2 of the 5′ UTR of the JFH1 cDNA construct pHH-JFH1. pSGR-Con1, which encodes SGR of the Con1 strain, was kindly provided by R. Bartenschlager. The complementary sequences of miR-122 were introduced into the multicloning site of the pmirGLO vector (Promega, Madison, WI), and the resulting plasmid was designated pmirGLO-compl-miR-122. The plasmids used in this study were confirmed by sequencing with an ABI 3130 genetic analyzer (Applied Biosystems, Tokyo, Japan).
Cell lines.
All cell lines were cultured at 37°C under the conditions of a humidified atmosphere and 5% CO2. The following cells were maintained in Dulbecco modified Eagle medium (DMEM; Sigma-Aldrich, St. Louis, MO) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum (FCS): human hepatocellular carcinoma-derived Huh7, Hep3B, and HepG2; embryonic kidney-derived HEK293 and 293T; lung-derived RERF-LC-AI, NCI-H-2030, and A-427; kidney-derived Caki-2 and 769-P; neuron-derived MC-IXC and SK-PN-DW; uterus-derived Hec1B; ovary-derived SK-OV3; colon-derived SW620; and urinary bladder-derived SW780 cells. RERF-LC-AI (RCB0444) cells were provided by the RIKEN BRC through the National Bio-Resource Project of MEXT, Japan. Hec1B (JCRB1193) cells were obtained from the JCRB Cell Bank. NCI-H-2030, A-427, Caki-2, 769-P, MC-IXC, SK-PN-DW, SK-OV3, and SW780 cells were obtained from the American Type Culture Collection (ATCC). SW620 cells were kindly provided by C. Oneyama. 293T-CLDN cells stably expressing CLDN1 were established by the introduction of the expression plasmids encoding CLDN1 under the control of the CAG promoter of pCAG-pm3. The Huh7-derived cell line Huh7.5.1 was kindly provided by F. Chisari. The Huh7OK1 cell line efficiently propagates HCVcc as previously described (45). Huh7, Hec1B, and HEK293 cells harboring Con1- or JFH1-based HCV SGR were prepared according to the method described in a previous report (47) and maintained in DMEM containing 10% FCS and 1 mg/ml G418 (Nakalai Tesque, Kyoto, Japan).
Antibodies and drugs.
Mouse monoclonal antibodies to HCV nonstructural protein 5A (NS5A) and β-actin were purchased from Austral Biologicals (San Ramon, CA) and Sigma-Aldrich, respectively. Mouse anti-ApoE antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-HCV core protein and NS5A were prepared as described previously (41). Rabbit anti-SR-BI antibody was purchased from Novus Biologicals (Littleton, CO). Rabbit anti-CLDN1 and -OCLN antibodies, Alexa Fluor 488 (AF488)-conjugated anti-rabbit or -mouse IgG antibodies, and AF594-conjugated anti-mouse IgG2a antibodies were purchased from Invitrogen (San Diego, CA). Mouse anti-FKBP8 antibody was described previously (44). Mouse anti-double-stranded RNA (anti-dsRNA) IgG2a (J1 and K2) antibodies were obtained from Biocenter Ltd. (Szirak, Hungary). The HCV NS3/4A protease inhibitor was purchased from Acme Bioscience (Salt Lake City, UT). Human recombinant alpha IFN (IFN-α) and cyclosporine were purchased from PBL Biomedical Laboratories (Piscataway, NJ) and Sigma-Aldrich, respectively. BODIPY 558/568 lipid probe was purchased from Invitrogen. The locked nucleic acid (LNA) targeted to miR-122, LNA-miR-122 (5′-CcAttGTcaCaCtCC-3′), and its negative control, LNA-control (5′-CcAttCTgaCcCtAC-3′) (LNAs are in capital letters, DNAs are in lowercase letters; sulfur atoms in oligonucleotide phosphorothioates are substituted for nonbridging oxygen atoms; the capital C indicates LNA methylcytosine), were purchased from Gene Design (Osaka, Japan) (15).
Preparation of viruses.
pHH-JFH1-E2p7NS2mt was introduced into Huh7.5.1 cells, HCVcc in the supernatant was collected after serial passages (39), and infectious titers were determined by a focus-forming assay and expressed in focus-forming units (FFUs) (62). Mutant HCVcc was produced from Huh7.5.1 cells expressing MT miR-122 according to the method of a previous report with minor modifications (25). HCVpv, a pseudotype vesicular stomatitis virus (VSV) bearing HCV E1 and E2 glycoproteins, was prepared as previously described (61), and infectivity was assessed by luciferase expression on a Bright-Glo luciferase assay system (Promega), following a protocol provided by the manufacturer and expressed in relative light units (RLUs).
Lipofection and lentiviral gene transduction.
Cells were transfected with the plasmids by using Trans IT LT-1 transfection reagent (Mirus, Madison, WI) according to the manufacturer's protocol. LNAs were introduced into cells by Lipofectamine RNAiMAX (Invitrogen). The lentiviral vectors and ViraPower lentiviral packaging mix (Invitrogen) were cotransfected into 293T cells, and the supernatants were recovered at 48 h posttransfection. The lentivirus titer was determined by a Lenti-XTM quantitative reverse transcription-PCR (qRT-PCR) titration kit (Clontech, Mountain View, CA), and the expression levels of miR-122 and AcGFP were determined at 48 h postinoculation.
Quantitative RT-PCR.
HCV RNA levels were determined by the method described previously (18). Total RNA was extracted from cells by using an RNeasy minikit (Qiagen). The first-strand cDNA synthesis and qRT-PCR were performed with TaqMan EZ RT-PCR core reagents and an ABI Prism 7000 system (Applied Biosystems), respectively, according to the manufacturer's protocols. The primers for TaqMan PCR targeted to the noncoding region of HCV RNA were synthesized as previously reported (42). To determine the expression levels of miR-122, total miRNA was prepared by using the miRNeasy minikit, and miR-122 expression was determined by using fully processed miR-122-specific RT and PCR primers provided in the TaqMan microRNA assays according to the manufacturer's protocol. U6 small nuclear RNA was used as an internal control. Fluorescent signals were analyzed with the ABI Prism 7000 system.
Immunoblotting.
Cells were lysed on ice in lysis buffer (20 mM Tris-HCl [pH 7.4], 135 mM NaCl, 1% Triton X-100, 10% glycerol) supplemented with a protease inhibitor mix (Nacalai Tesque). The samples were boiled in loading buffer and subjected to 5 to 20% gradient SDS-PAGE. The proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) and reacted with the appropriate antibodies. The immune complexes were visualized with SuperSignal West Femto substrate (Pierce, Rockford, IL) and detected with an LAS-3000 image analyzer system (Fujifilm, Tokyo, Japan).
Immunofluorescence assay.
Cells cultured on glass slides were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) at room temperature for 30 min, permeabilized for 20 min at room temperature with PBS containing 0.2% Triton, after being washed three times with PBS, and blocked with PBS containing 2% FCS for 1 h at room temperature. The cells were incubated with PBS containing appropriate primary antibodies at room temperature for 1 h, washed three times with PBS, and incubated with PBS containing AF488- or AF594-conjugated secondary antibodies at room temperature for 1 h. For lipid droplet staining, cells incubated in medium containing 20 μg/ml BODIPY for 20 min at 37°C were washed with prewarmed fresh medium and incubated for 20 min at 37°C. Cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). Cells were observed with a FluoView FV1000 laser scanning confocal microscope (Olympus, Tokyo, Japan).
In vitro transcription, RNA transfection, and colony formation.
The plasmids pSGR-Con1 and pSGR-JFH1 were linearized with ScaI and XbaI, respectively, and treated with mung bean exonuclease. The linearized DNA was transcribed in vitro by using a MEGAscript T7 kit (Applied Biosystems) according to the manufacturer's protocol. The in vitro-transcribed RNA (10 μg) was electroporated into Hec1B and HEK293 cells at 107 cells/0.4 ml under conditions of 190 V and 975 μF using a Gene Pulser apparatus (Bio-Rad, Hercules, CA) and plated on DMEM containing 10% FCS. The medium was replaced with fresh DMEM containing 10% FCS and 1 mg/ml G418 at 24 h posttransfection. The remaining colonies were cloned by using a cloning ring (Asahi Glass, Tokyo, Japan) or fixed with 4% PFA and stained with crystal violet at 4 weeks postelectroporation.
Electron microscopy and correlative FM-EM analysis.
Cells were cultured on a Cell Desk polystyrene coverslip (Sumitomo Bakelite) and were fixed with 2% formaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) containing 7% sucrose. Cells were postfixed for 1 h with 1% osmium tetroxide and 0.5% potassium ferrocyanide in 0.1 M cacodylate buffer (pH 7.4), dehydrated in graded series of ethanol, and embedded in Epon812 (TAAB). Ultrathin (80-nm) sections were stained with saturated uranyl acetate and lead citrate solution. Electron micrographs were obtained with a JEM-1011 transmission electron microscope (JEOL). Correlative fluorescence microscopy (FM)-electron microscopy (EM) allows individual cells to be examined both in an overview with FM and in a detailed subcellular-structure view with EM (51). The NS5A was stained and observed in the Hec1B-derived Con1 SGR cells by the correlative FM-EM method as described previously (44).
Intracellular infectivity.
Intracellular viral titers were determined according to a method previously reported (20). Briefly, cells were extensively washed with PBS, scraped, and centrifuged for 5 min at 1,000 × g. Cell pellets were resuspended in 500 μl of DMEM containing 10% FCS and subjected to three cycles of freezing and thawing using liquid nitrogen and a thermo block set to 37°C. Cell lysates were centrifuged at 10,000 × g for 10 min at 4°C to remove cell debris. Cell-associated infectivity was determined by a focus-forming assay.
Statistical analysis.
The data for statistical analyses are averages of three independent experiments. Results were expressed as means ± standard deviations. The significance of differences in the means was determined by Student's t test.
Microarray data accession number.
Access to data concerning this study may be found under GEO experiment accession number GSE32886.
RESULTS
Nonhepatic cell lines susceptible to HCVcc by expression of miR-122.
Human CD81 (hCD81), SR-B1, CLDN1, and OCLN are crucial for HCV entry (16, 48, 49, 56). First, we examined the expression of these receptor candidates in nonhepatic cell lines by using the web-based NextBio search engine (Cupertino, CA). Multidimensional scaling was used to visualize the differences in expression patterns of molecules of various tissues, cells, and cell lines from those of hepatic cell lines and primary hepatocytes. We selected nine nonhepatic cell lines as possibly being susceptible to HCVcc infection: NCI-H-2030 (lung), Caki-2 (kidney), 769-P (bladder), A-427 (lung), SK-OV3 (ovary), SW780 (bladder), SW620 (colon), RERF-LC-AI (lung), and Hec1B (uterus) (Fig. 2). In addition, three nonhepatic cell lines previously reported to be susceptible to replication of HCV RNA—that is, SK-PN-DW (neuron), MC-IXC (neuron), and 293T-CLDN (kidney)—were included in this study (8, 17). The expression of each receptor molecule in these 12 nonhepatic cell lines was confirmed by fluorescence-activated cell sorter (FACS) analysis and immunoblotting (Fig. 3A and B). To examine the expression of the functional receptors for HCV entry in these cell lines, we inoculated HCVpv into the cells. Ten of the cell lines (A-427 and SW780 being the exceptions) exhibited various degrees of susceptibility to HCVpv infection (Fig. 3C). Therefore, we examined the possibility of propagation of HCVcc by the expression of miR-122 in these 10 cell lines.
Fig 2.
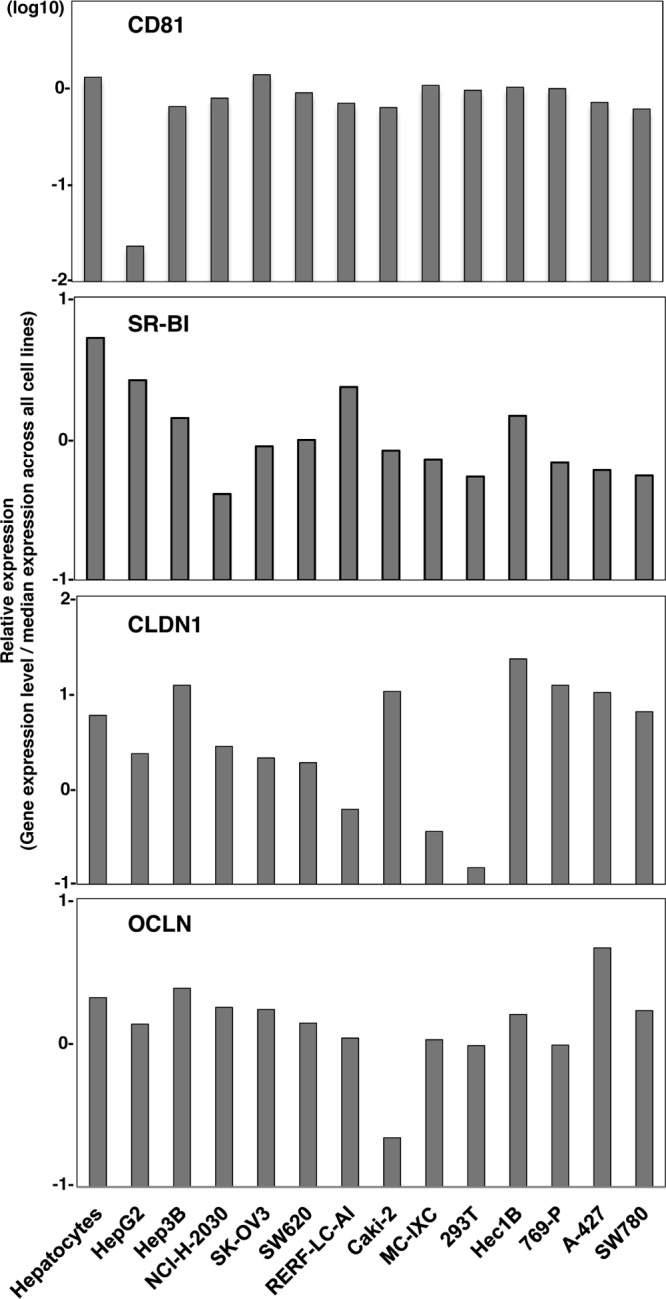
Receptor expression profiling in nonhepatic cells. Relative expression levels of CD81, SR-BI, CLDN1, and OCLN in primary hepatocytes, hepatic cell lines HepG2 and Hep3B, and nonhepatic cells were determined by using the NextBio Body Atlas. Expression levels were standardized by the median expression across all cell lines.
Fig 3.

Expression of functional HCV receptor candidates in nonhepatic cells. (A) Expression of hCD81 in nonhepatic cells was determined by flow cytometry. PE, phycoerythrin. (B) Expression levels of SR-B1, CLDN, and OCLN in the nonhepatic cells were determined by immunoblotting. (C) The nonhepatic cell lines were inoculated with pseudotype VSVs bearing no envelope protein (deltaG), HCV envelope proteins of genotype 1b Con1 strain (HCVpv), or VSV G protein (VSVpv), and luciferase expression was determined at 24 h postinfection.
To introduce miR-122 in the cell lines, we employed a lentiviral vector encoding pri-miR-122, an unprocessed miR-122. To confirm the maturation of pri-miR-122 to form functional RNA-induced silencing complexes (RISCs), suppression of the translation of the target mRNA was determined by a dual reporter assay. Translation of a firefly luciferase mRNA containing the sequences complementary to miR-122 in the 3′ UTR was suppressed by infection with the lentivirus encoding pri-miR-122 but not by infection with a control virus (data not shown), suggesting that the pri-miR-122 is processed into a functionally mature miR-122. By using this lentiviral vector, high levels of miR-122 expression were achieved in the 10 cell lines, comparable to the endogenous expression level of miR-122 in Huh7 cells (Fig. 4A).
Fig 4.

Nonhepatic cell lines susceptible to HCVcc by the expression of miR-122. (A) Exogenous miR-122 was expressed in Huh7, Hec1B, 293T-CLDN, MC-IXC, RERF-LC-AI, SK-OV3, NCI-H-2030, SW620, Caki-2, SK-PN-DW, and 769-P cells by lentiviral vector. Total RNA was extracted from the cells and subjected to qRT-PCR analysis. U6 was used as an internal control. Gray and white bars, endogenous and exogenous levels of miR-122, respectively. (B) HCVcc was inoculated into Huh7 and nonhepatic cell lines expressing (solid lines) or not expressing (dashed lines) exogenous miR-122 at an MOI of 1. Intracellular HCV RNA levels were determined by qRT-PCR at 2, 18, 24, and 48 h postinfection (hpi). (C) Cells were inoculated with HCVcc and simultaneously treated with either 100 U IFN-α or 100 nM HCV protease inhibitor or not treated (control), and intracellular HCV RNA levels were determined by qRT-PCR at 36 h postinfection. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) versus the results for control cells.
To examine the effect of the exogenous expression of miR-122 on HCV replication, the nonhepatic cell lines expressing miR-122 were infected with HCVcc at a multiplicity of infection (MOI) of 1, and intracellular viral RNA was determined (Fig. 4B). The expression of miR-122 significantly increased the amount of the HCV genome in Hec1B, 293T-CLDN, MC-IXC, and RERF-LC-AI cells as well as Huh7 cells and slightly increased it in SK-OV3 and NCI-H-2030 cells. Although the levels of viral RNA in SW620, Caki-2, and SK-PN-DW cells upon expression of miR-122 were higher than those in control cells, no increase of viral RNA was observed. No effect of the expression of miR-122 was observed in 769-P cells. Interestingly, naïve Hec1B cells exhibited a delayed increase in viral RNA from 24 to 48 h postinfection, in contrast to the gradual decrease of viral RNA in other cell lines. Replication of HCV RNA in both naïve and miR-122-expressing Hec1B cells was inhibited by treatment with an inhibitor for HCV protease but not by treatment with IFN-α, due to the lack of an IFN receptor (11), whereas treatments with either IFN-α or the protease inhibitor suppressed the replication of HCV in the other cell lines expressing miR-122 (Fig. 4C). These results indicate that exogenous miR-122 expression enhances the replication of HCV even in nonhepatic cells. Hec1B cells exhibit a delayed replication of HCV, and HCV replication was enhanced by the exogenous expression of miR-122. Therefore, in this study we used Hec1B cells to investigate the biological significance of miR-122 on the replication of HCVcc in nonhepatic cells.
Expression of miR-122 is essential for enhancing HCV replication in Hec1B cells.
To confirm the specificity of HCV replication in Hec1B cells, HCVcc was preincubated with an anti-HCV E2 monoclonal antibody, AP-33, or Hec1B/miR-122 and Hec1B/Cont cells were pretreated with anti-hCD81 monoclonal antibody. Replication of HCV RNA was determined upon infection with HCVcc. The antibody treatment significantly inhibited HCV replication in the Hec1B cell line, indicating that HCVcc internalizes into Hec1B cells through a specific interaction between hCD81 and E2 (Fig. 5). Next, we determined the dose dependence of miR-122 expression on the enhancement of HCV replication in Hec1B cells. Huh7.5.1 and Hec1B cells transduced with the lentiviral vector encoding pri-miR-122 were infected with HCVcc at an MOI of 1, and intracellular miR-122 and viral RNA were determined. Expression of miR-122 was increased in Hec1B cells in a dose-dependent manner of the lentivirus, whereas no increase was observed in Huh7.5.1 cells, probably due to the high level of endogenous expression of miR-122 (Fig. 6A, left). HCV RNA replication in Huh7.5.1 and Hec1B cells was correlated with miR-122 expression (Fig. 6A, right), suggesting a close correlation between miR-122 expression and HCV replication.
Fig 5.
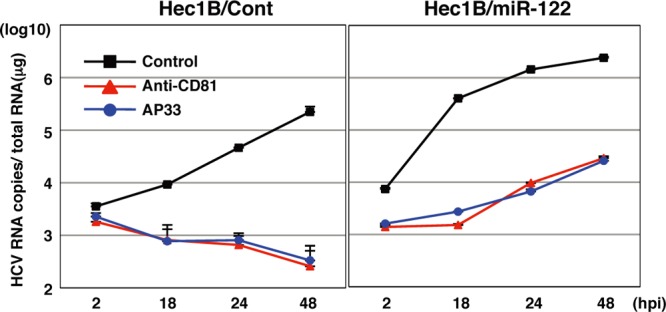
Neutralization of HCVcc infection in Hec1B cells by specific antibodies. HCVcc was preincubated with anti-E2 antibody (AP-33) and inoculated into Hec1B/Cont and Hec1B-miR-122 cells. Cells were preincubated with anti-human CD81 antibody and inoculated with HCVcc. Intracellular HCV RNA levels at 2, 18, 24, and 48 h postinfection were determined by qRT-PCR.
Fig 6.

Expression of miR-122 is essential for the enhancement of HCV replication in the Hec1B cells. (A) Huh7.5.1 and Hec1B cells were transduced with lentiviral vectors expressing miR-122 in a dose-dependent manner and infected with HCVcc at an MOI of 1. Intracellular miR-122 and HCV RNA were determined at 24 h postinfection by qRT-PCR. (B) Huh7.5.1 and Hec1B/miR-122 cells were infected with HCVcc at an MOI of 0.5 or 3 and subjected to immunoblotting and immunofluorescence analyses using anti-NS5A antibodies at 48 h postinfection. The asterisk indicates nonspecific bands. (C) LNAs specific to miR-122 at a final concentration of 5 nM, 20 nM, or 50 nM and control (LNA alone at 50 nM) were introduced into Huh7/Cont, Huh7/miR-122, Hec1B/miR-122, and Hec1B/Cont cells by using Lipofectamine RNAiMAX transfection reagent and infected with HCVcc at an MOI of 1 at 6 h posttransfection. Intracellular HCV RNA levels were determined by qRT-PCR at 12, 24, and 36 h postinfection. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) versus the results for control cells.
Next, we examined the expression of viral proteins in Hec1B/miR-122 cells upon infection with HCVcc by immunoblotting and fluorescence microscopic analyses (Fig. 6B). Expression of NS5A protein was increased in Hec1B/miR-122 cells in an MOI-dependent manner. Expression of NS5A in Hec1B/Cont cells infected with HCVcc at an MOI of 3 was significantly lower than that in Hec1B/miR-122 cells infected with HCVcc at an MOI of 0.5. HCV core and NS proteins were shown to localize mainly on the lipid droplets and cytoplasmic face of the endoplasmic reticulum (ER) in Huh7 and Hep3B/miR-122 cells infected with HCVcc (29, 40). Immunofluorescence analyses revealed that HCV core and NS5A proteins were colocalized with lipid droplets and calnexin, an ER marker, in Hec1B cells infected with HCVcc (Fig. 7).
Fig 7.
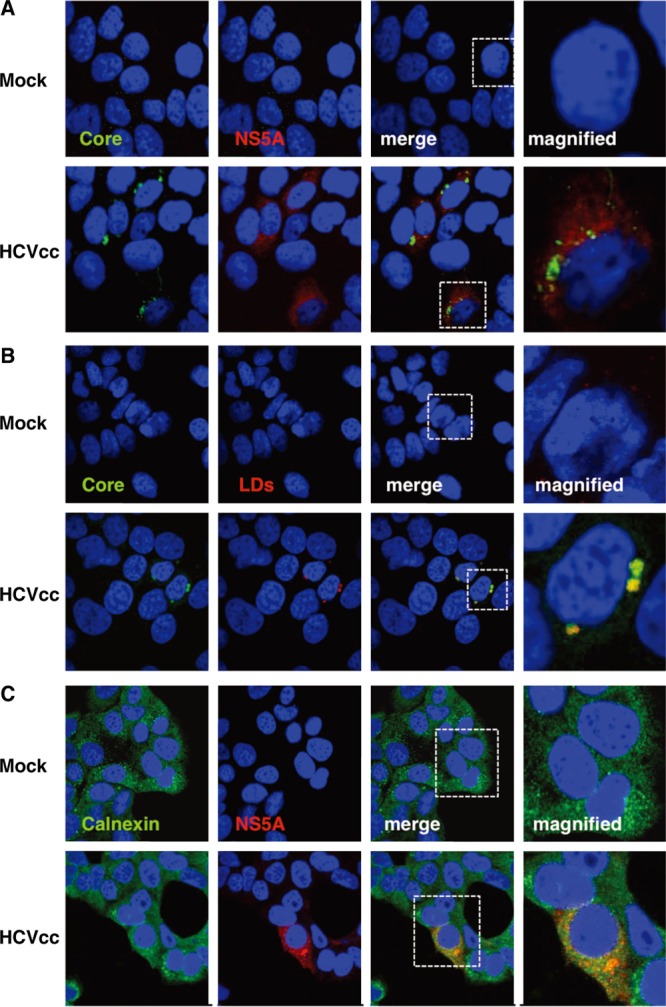
Subcellular localization of core and NS5A proteins in Hec1B/miR-122 cells infected with HCVcc. Hec1B/miR-122 cells infected with or without HCVcc at an MOI of 1 were fixed with 4% PFA at 48 h postinfection and stained with appropriate antibodies to core and NS5A proteins (A), core protein and lipid droplets (B), and NS5A and calnexin (C). The boxes in the merged images were magnified, and the images are displayed on the right.
To further confirm the specificity of the enhancement of HCV replication by the expression of miR-122, Huh7, Hec1B/miR-122, and Hec1B/Cont cells were treated with LNAs that were either specific to miR-122 (LNA-miR-122) or nonspecific (LNA-control) at 6 h before infection with HCVcc. Although the treatment with LNA-miR-122 inhibited the enhancement of viral RNA replication in Huh7 and Hec1B/miR-122 cells in a dose-dependent manner, no inhibition of viral replication was observed in Hec1B/Cont cells (Fig. 6C). These results suggest that Hec1B cells permit HCV replication in a miR-122-independent manner and exogenous expression of miR-122 enhances viral replication.
Specific interaction between miR-122 and the 5′ UTR of HCV is required for HCV replication.
To determine the effect of the specific interaction between miR-122 and the 5′ UTR of the HCV genome on the enhancement of RNA replication, we generated MT pri-miR-122 carrying a substitution of uridine to adenosine in the seed domain and an additional complementary substitution of adenosine to uridine to stabilize the loop structure of pri-miR-122 (Fig. 8A). A high expression level of MT miR-122, comparable to that of WT miR-122, was introduced into Hec1B cells by infection with lentiviral vectors (Fig. 8B). To determine the specificity of miR-122 on the replication of HCV, Hec1B cells expressing either WT or MT miR-122 were inoculated with HCVcc at an MOI of 1. Enhancement of HCV replication was observed in Hec1B cells by the expression of WT but not that of MT miR-122, suggesting that the sequence specificity of miR-122 with the 5′ UTR of HCV is crucial for the efficient replication of HCV (Fig. 8C). To further confirm the effect of the specificity of interaction between miR-122 and the binding sites in the 5′ UTR of HCV on the enhancement of HCV replication, we generated two mutant viruses, HCVcc-M1 and HCVcc-M2, carrying complementary substitutions in the miR-122-binding site 1 alone and in both sites 1 and 2 in the 5′ UTR of HCV, respectively (Fig. 8D). Recently, Jangra et al. demonstrated that the propagation of a mutant HCVcc bearing mutations in sites 1 and 2 in the 5′ UTR was rescued by the expression of MT miR-122 in Huh7.5 cells (25). We confirmed that the propagation of HCVcc-M1 and HCVcc-M2 in Huh7.5.1 cells was rescued by the expression of MT miR-122 but not of WT miR-122, although the recovery of infectious titers of HCVcc-M2 was significantly lower than the recovery of infectious titers of HCVcc-M1 (Fig. 8E). Next, to examine the interaction between miR-122 and the HCV genome in Hec1B cells, we inoculated HCVcc or mutant viruses into Hec1B cells expressing either or both WT and MT miR-122 and determined the replication of HCV RNA by qRT-PCR (Fig. 8F). Expression of WT and MT miR-122 in Hec1B cells permits replication of HCVcc and HCVcc-M2, respectively, although the enhancing effects differed. On the other hand, the expression of both WT and MT miR-122 is required for the replication of HCVcc-M1, because MT and WT miR-122 bind to sites 1 and 2 in the 5′ UTR of this virus, respectively. Interestingly, a low level of HCVcc-M1 replication was also observed in Hec1B cells expressing either WT or MT miR-122, in contrast to the requirement of the corresponding miR-122 for the replication of HCVcc and HCVcc-M2. These results suggest that the specific interaction between miR-122 and the 5′ UTR of HCV is crucial for the replication of HCV.
Fig 8.

Specific interaction between miR-122 and the 5′ UTR of HCV is required for HCV replication. (A) Structures of pri-miR-122 and the nucleotide sequences of WT and MT miR-122, which has a substitution of uridine to adenosine in the seed domain and an additional complementary substitution of adenosine to uridine for stable expression. (B) WT or MT miR-122 was introduced into Hec1B cells by a lentiviral vector, and miR-122 expression levels were determined by qRT-PCR. (C) HCVcc was inoculated into Hec1B cells expressing either WT or MT miR-122 and control cells at an MOI of 1, and the intracellular HCV RNA levels were determined by qRT-PCR. (D) Diagram of mutant viruses HCVcc-M1 and HCVcc-M2 carrying complementary substitutions in the miR-122-binding site 1 alone and both sites 1 and 2 in the 5′ UTR of HCV, respectively. (E) Viral RNA of HCVcc, HCVcc-M1, or HCVcc-M2 was electroporated into Huh7.5.1 cells expressing either WT or MT miR-122, and infectious titers of the viruses recovered in the culture supernatants at 72 h postinfection of the second passage were determined by a focus-forming assay in cells expressing either WT or MT miR-122. Red, blue, and green bars, infectious titers of HCVcc, HCVcc-M1, and HCV-M2, respectively. (F) HCVcc, HCVcc-M1, or HCV-M2 was inoculated into Hec1B cells expressing either or both WT and MT miR-122 at an MOI of 0.5, and intracellular HCV RNA levels were determined at 12, 24, and 36 h postinfection by qRT-PCR. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) versus the results for control cells.
Viral particle formation in hepatic and nonhepatic cells.
These data suggest that miR-122 expression facilitates replication of HCV RNA in nonhepatic cells. Recently, we have shown that expression of miR-122 facilitates infectious particle formation of HCVcc in a hepatoma cell line, Hep3B (29). To examine the effect of miR-122 expression on particle formation in nonhepatic cells, intracellular and extracellular viral RNA levels in cells infected with HCVcc were determined. Intracellular RNA replication in the hepatic cell lines, including Huh7.5.1 and Hep3B/miR-122, was increased up to 72 h postinfection with HCVcc, whereas in nonhepatic cell lines, including 293T-CLDN/miR-122 and Hec1B/miR-122, such replication was comparable to that in the hepatic cell lines until 24 h postinfection but reached a limit at this point (Fig. 9A). In spite of no clear increase of intracellular HCV RNA in Hep3B/Cont cells upon infection with HCVcc (Fig. 9A), subtle but substantial production of infectious particles was detected in the culture supernatants at 72 h postinfection, in contrast to no production of infectious particles in those of the nonhepatic cell lines (Fig. 9B). Furthermore, no focus formation was observed in Hec1B/miR-122 cells upon infection with HCVcc, in contrast to the many foci in Huh7.5.1 cells (Fig. 9C), and no infectivity was detected even in the lysates of Hec1B/miR-122 cells infected with HCVcc (Fig. 9D). These results suggest that not only the replication efficiency of viral RNA but also other factors are involved in the assembly of HCV and that the viral assembly process is impaired in Hec1B/miR-122 cells infected with HCVcc, in spite of the efficient replication of HCV RNA.
Fig 9.
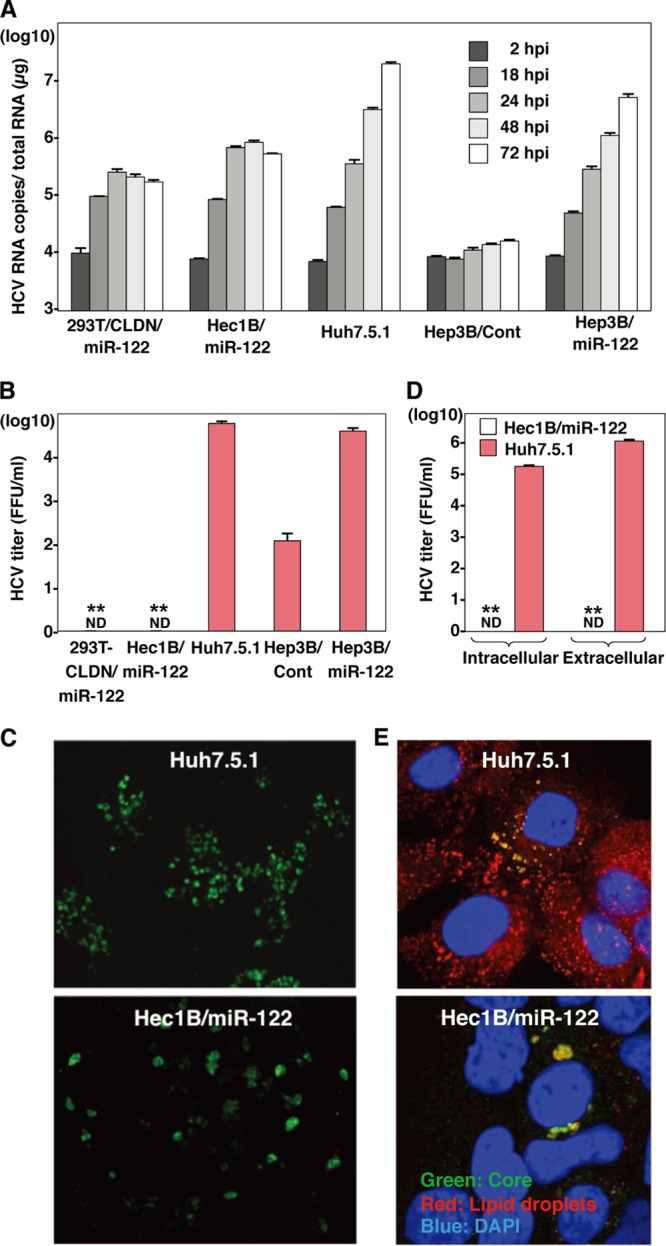
Viral particle formation in hepatic and nonhepatic cells. (A) HCVcc was inoculated into 293T-CLDN/miR-122, Hec1B/miR-122, Hep3B/Cont, and Hep3B/miR-122 cells at an MOI of 1 and into Huh7.5.1 cells at an MOI of 0.1. HCV RNA levels (copies/μg) in cells at 2, 18, 24, 48, and 72 h postinfection were determined by qRT-PCR. (B) HCVcc was inoculated into 293T-CLDN/miR-122, Hec1B/miR-122, Hep3B/Cont, and Hep3B/miR-122 cells at an MOI of 1 or into Huh7.5.1 cells at an MOI of 0.1, and infectious titers in the culture supernatants were determined at 72 h postinfection by a focus-forming assay in Huh7.5.1 cells. ND, not determined. (C) Huh7.5.1 and Hec1B/miR-122 cells were infected with HCVcc at MOIs of 0.1 and 1, respectively, incubated with 1% methylcellulose in DMEM containing 10% FCS for 72 h, fixed with 4% PFA, and subjected to immunofluorescence analysis using anti-NS5A antibody. (D) Hec1B/miR-122 and Huh7.5.1 cells were infected with HCVcc at MOIs of 1 and 0.1, respectively, and infectious titers in cells and supernatants were determined by a focus-forming assay at 72 h postinfection. (E) Huh7.5.1 and Hec1B/miR-122 cells were infected with HCVcc at MOIs of 0.1 and 1, respectively, fixed with 4% PFA, and subjected to immunofluorescence assay using anti-core protein antibody (green). Lipid droplets and cell nuclei were stained with BODIPY (red) and DAPI (blue), respectively. Asterisks indicate significant differences (**, P < 0.01) versus the results for Huh7.5.1 cells.
It was previously shown that lipid droplets, diacylglycerol O-acyltransferase 1 (DGAT1), and apolipoproteins B and E play an important role in the assembly of HCV particles (10, 22, 40). To understand the molecular mechanisms underlying the low efficiency of infectious particle formation in nonhepatic cells, we first examined the subcellular localization of lipid droplets and HCV core protein in Hec1B/miR-122 cells infected with HCVcc. Although the core protein was detected around the lipid droplets, as seen in Huh7.5.1 cells, only a small amount of lipid droplets was detected in Hec1B/miR-122 cells infected with HCVcc compared with the amount detected in Huh7.5.1 cells (Fig. 9E), suggesting that the low level of lipid droplet formation is involved in the impairment of infectious particle formation in nonhepatic cells.
Next we examined the expression patterns of molecules involved in lipid metabolism by using cDNA microarray and qPCR analyses. Although expression levels of low-density lipoprotein receptor (LDLR), sterol regulatory element-binding protein 1c (SREBP1c), SREBP2, and DGAT1 in nonhepatic Hec1B and 293T cells were comparable to those in hepatic Huh7 and Hep3B cells, those of VLDL-associated proteins, including ApoE, ApoB, and MTTP, in nonhepatic cells were significantly lower than those in hepatic cells (Fig. 10). Collectively, these results suggest that intracellular functional lipid metabolism, including the biosynthesis of lipid droplets and the production of VLDL, participates in the assembly of HCV.
Fig 10.
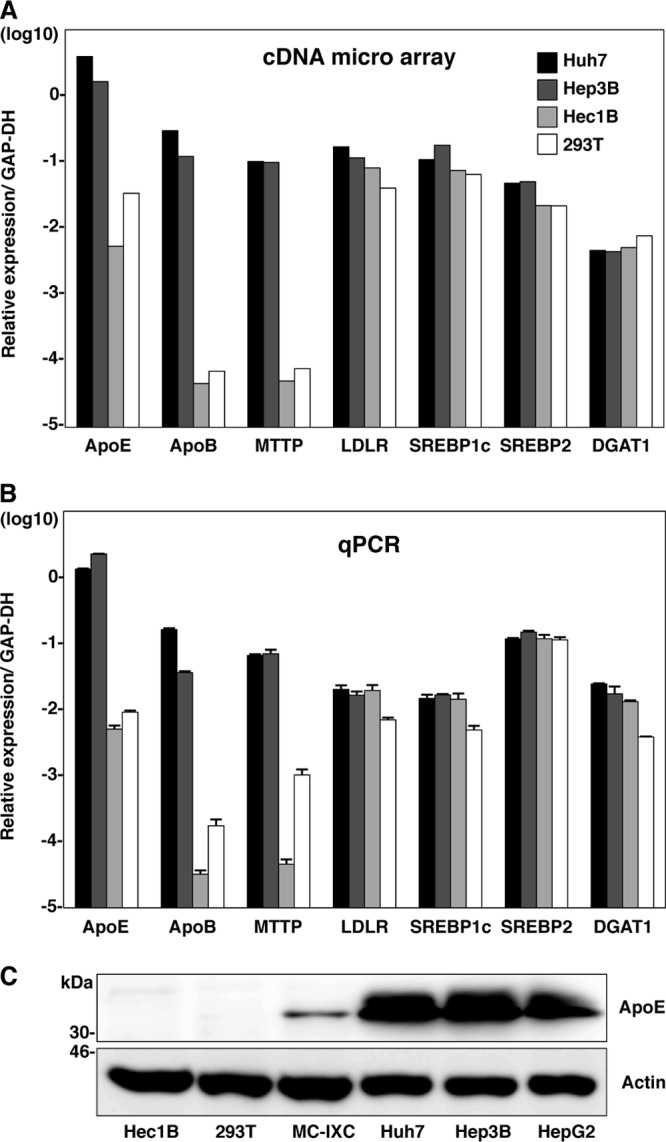
Expression of lipid metabolism-associated proteins in hepatic and nonhepatic cells. (A) Expression levels of ApoE, ApoB, MTTP, LDLR, SREBP1c, SREBP2, and DGAT1 were compared among hepatic (Huh7 and Hep3B) and nonhepatic (Hec1B and 293T) cells using cDNA microarray analyses. (B) Total RNA was extracted from the cells, and expression levels of ApoE, ApoB, MTTP, LDLR, SREBP1c, SREBP2, and DGAT1 gene were determined by qPCR. (C) Nonhepatic (Hec1B, 293T, and MC-IXC) and hepatic (Huh7, Hep3B, and HepG2) cells were subjected to immunoblotting using anti-ApoE antibody.
Establishment of HCV replicon in Hec1B/miR-122 cells.
It was previously shown by using RNA replicon cells based on the JFH1 strain that expression of miR-122 enhanced the translation of HCV RNA in HEK293 cells and MEFs (8, 35). We tried to establish HCV replicon cells based on genotype 1b Con1 and genotype 2a JFH1 strains in Hec1B/miR-122 and HEK293 cells stably expressing miR-122 (HEK293/miR-122). To examine the colony formation efficiency of the HCV RNAs of the Con1 and JFH1 strains, SGR RNA was electroporated into the cell lines and selected by G418 for 3 weeks. Expression of miR-122 in Hec1B cells significantly enhanced the colony formation of SGR of the Con1 strain (Fig. 11A), suggesting that the expression of miR-122 in Hec1B cells supports the efficient replication of SGR. HCV replication in 20 replicon clones established by the transfection with SGR RNA of the Con1 strain in Hec1B/miR-122 cells was examined by qRT-PCR and immunoblotting. All clones contained high levels of HCV RNA (3 × 106 to 5 × 107 copies per μg of total RNA) (Fig. 11B), and expression of NS5A was well correlated with the levels of HCV RNA in the clones (Fig. 11C). Two replicon clones (clones 2 and 10) in Hec1B/miR-122 cells exhibiting high levels of RNA replication and NS5A expression further confirmed the high level of expression of NS5A by immunofluorescent microscopy (Fig. 11D). These results suggest that expression of miR-122 facilitates the efficient replication of SGR of at least two HCV genotypes in Hec1B cells.
Fig 11.

Establishment of Con1-based HCV replicon cells by using Hec1B cells. (A) WT or replication-defective SGR RNA of the HCV Con1 strain was electroporated into Hec1B/Cont, Hec1B/miR-122, and Huh7 cells, and the medium was replaced with DMEM containing 10% FCS and 1 mg/ml G418 at 24 h postelectroporation. Colonies were stained with crystal violet after 3 weeks of selection with G418. (B) Total RNAs of 20 selected clones were extracted and subjected to qRT-PCR. (C) The 20 SGR clones were subjected to immunoblotting using anti-NS5A antibody. Huh7-derived Con1-based SGR cells were used as a positive control. (D) NS5A proteins in SGR clones 2 and 10 were stained with appropriate antibodies and examined by fluorescence microscopy. Huh7-derived Con1-based SGR and parental Hec1B cells were used for positive and negative controls, respectively.
Our previous reports showed that HCV NS proteins were colocalized with dsRNA and cochaperone molecules, FK506-binding protein 8 (FKBP8), in dot-like structures on the ER membrane of Huh7 replicon cells (59). Colocalization of NS5A with dsRNA or FKBP8 was observed in the dot-like structures in not only Huh7 SGR cells but also Hec1B/miR-122 SGR cells (Fig. 12A), suggesting that the dot-like structure required for efficient viral replication is also generated in Hec1B/miR-122 replicon cells. It has been shown that HCV replication induces the formation of convoluted membranous structures, called membranous webs, in Huh7 cells (13, 45). FM-EM techniques revealed the localization of NS5A on the convoluted structures in Hec1B/miR-122 replicon cells (Fig. 12B). These results suggest that the replication complex required for viral replication was also generated in the Hec1B/miR-122 replicon cells, as was seen in the Huh7 replicon cells.
Fig 12.

Replication complex in Hec1B/miR-122 replicon cells. (A) Huh7 and Hec1B/miR-122 cells harboring the Con1 SGR RNA were fixed, permeabilized, and stained with antibodies to NS5A and dsRNA or FKBP8. The boxed areas in the merged images were magnified, and the images are displayed on the right. (B) Hec1B-derived Con1 SGR cells were stained with anti-NS5A antibody. Identical fields were observed under EM by using the correlative FM-EM technique. The boxed areas are magnified, and the images displayed on the right.
miR-122 is a crucial determinant of HCVcc propagation.
It has been shown that the infectivity of HCVcc in cured cells, established when IFN treatment induces the elimination of the viral genome from the Huh7 replicon cells harboring an HCV RNA, is significantly higher than that in parental Huh7 cells (2, 66). Therefore, we tried to establish Hec1B-based cured cells from the Con1 SGR clones harboring a high copy number of HCV RNA. Treatment with cyclosporine and the protease inhibitor of HCV suppressed NS5A expression in Hec1B/miR-122 SGR clone 2 in a dose-dependent manner (Fig. 13A), whereas no reduction was observed by the IFN treatment due to a lack of an IFN receptor, as shown in Fig. 4C. It was reported that monotherapy by the HCV protease inhibitor induces the emergence of resistant breakthrough viruses (34, 55). Therefore, we treated five Hec1B/miR-122 SGR clones (clones 2, 5, 10, 14, and 16) with 1 μg/ml cyclosporine and 100 nM protease inhibitor for HCV. Viral RNA was determined by qRT-PCR every 5 days posttreatment. Elimination of viral RNA was achieved in four clones (clones 2, 5, 10, and 14) within 20 days posttreatment (Fig. 13B). Replication of HCV RNA in the cured cells infected with HCVcc at an MOI of 0.5 was 2- to 30-fold higher than that in parental cells at 24 h postinfection (Fig. 13C). In addition, replication of HCV RNA in cured clone 2 infected with HCVcc at an MOI of 0.1 was comparable to that in Huh7.5.1 cells until 24 h postinfection (Fig. 13D). Expression of NS5A was significantly increased in cured clone 2 compared to that in the parental Hec1B/miR-122 cells (Fig. 13E and F). It was previously shown that the increased permissiveness of Huh7-derived cured cells, Huh7.5 cells, is attributable to a mutation in the RIG-I gene (58). To examine the innate immune response in the parental and cured Hec1B/miR-122 cells, induction of IFN-stimulated gene 15 (ISG15) was determined upon stimulation with IFN-α or VSV. Although induction of ISG15 was not observed in either parental or cured cells upon stimulation with IFN-α due to a lack of an IFN receptor (11) (Fig. 14A), it was detected in both cells infected with VSV (Fig. 14B). Therefore, other mechanisms should be involved in the enhancement of permissiveness of Hec1B-derived cured cells. Ehrhardt et al. showed that the expression levels of miR-122 in Huh7-derived cured cells, including Huh7.5, Huh7.5.1, and Huh7-Lunet cells, are significantly higher than those in parental Huh7 cells (14). In addition, our recent study indicated that levels of expression of miR-122 in the cured Huh7 and Hep3B/miR-122 cells were higher than those in parental cells (29). Levels of expression of miR-122 in the Hec1B-based cured cell clones are also higher than those in parental Hec1B/miR-122 cells (Fig. 13G). These results suggest that a high level of miR-122 expression is a crucial determinant of high susceptibility to HCVcc propagation in the cured cells.
Fig 13.

miR-122 is a crucial determinant for the efficient replication of HCVcc and replicon RNA. (A) Hec1B Con1 replicon clone 2 was treated with stepwise concentrations of cyclosporine (CsA; 0.2, 1, and 5 μg/ml), NS3/4A protease inhibitor (10, 100, and 1,000 nM), or IFN-α (10, 100, and 1,000 IU) and subjected to immunoblotting using anti-NS5A antibody at 48 h posttreatment. (B) Five Con1-based SGR clones were treated with the combination of 1 μg/ml cyclosporine and 100 nM HCV protease inhibitor to eliminate the HCV genome. Intracellular HCV RNA levels at 5, 10, 15, 20, 25, and 30 days posttreatment were determined by qRT-PCR analysis. (C) HCVcc was inoculated with Hec1B/Cont, parental Hec1B/miR-122, and Hec1B-based cured cells (clones 2, 5, 10, and 14) at an MOI of 1. Intracellular HCV RNA levels at 2, 12, 18, 24, and 36 h postinfection were determined by qRT-PCR analysis. (D) HCVcc was inoculated into Huh7.5.1 and Hec1B/miR-122 cured clone 2 cells at an MOI of 1. Intracellular HCV RNA levels were determined by qRT-PCR at 12, 24, 36, and 48 h postinfection. (E and F) Hec1B/Cont, parental Hec1B/miR-122, and cured cells of clone 2 were infected with HCVcc at an MOI of 0.5. After 48 h, the cells were subjected to immunoblotting and immunofluorescence analyses using appropriate antibodies. (G) Total miRNAs were extracted from Hec1B/Cont (white), parental Hec1B-miR-122 cells (gray), and four cured cell clones (black). miR-122 expression levels in these cells were determined by qRT-PCR analysis. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) versus the results for parental Hec1B/miR-122 cells.
Fig 14.
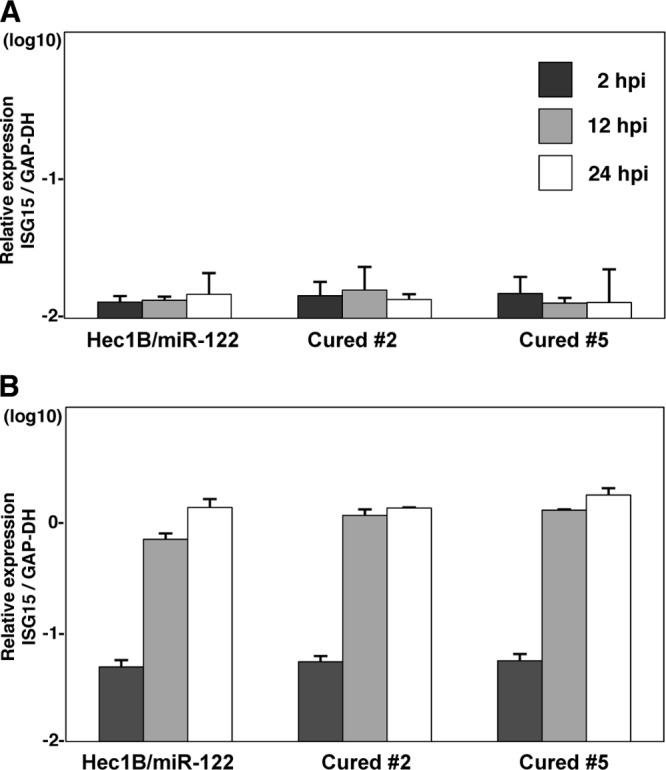
Innate immune responses in parental and cured Hec1B/miR-122 cells. Parental and cured Hec1B/miR-122 cells were stimulated with 100 U of IFN-α (A) or VSV (B). The expression levels of IFN-stimulated gene 15 (ISG15) were determined by qRT-PCR at 2, 12, and 24 h posttreatment.
DISCUSSION
Although multiple epidemiological studies have revealed that HCV infection induces several EHMs, they have not well elucidated the molecular mechanisms of the EHMs induced by HCV infection (19). Indeed, HCVcc does not infect PBMCs (38). It has been shown that two neuroepithelioma cell lines permit HCVcc infection at low levels (17) and lymphotropic strains or quasispecies of HCV exist in infected individuals (12, 52). Furthermore, many molecules involved in the entry, replication, and assembly of HCVcc have been identified, although these molecules are not sufficient to explain the liver tropism of HCV. Recently, a liver-specific microRNA, miR-122, was shown to facilitate the efficient replication of HCV through a specific interaction with the complementary sequences in the 5′ UTR of HCV RNA (21, 25, 27, 36). In addition, exogenous expression of miR-122 facilitates the replication of SGR of the JFH1 strain in HEK293 cells (8) and the propagation of HCVcc in HepG2 and Hep3B nonpermissive hepatoma cells (29, 43), suggesting that the expression of miR-122 is required for the efficient replication of HCV. However, HCV replicon cells have also been established in HeLa and LI90 cells derived from stellate cells in which no exogenous miR-122 is expressed (30, 63). In this study, naïve Hec1B cells also exhibited a low level of replication upon infection with HCVcc (Fig. 4B), and this replication was resistant to treatment with an inhibitor of miR-122, LNA-miR-122 (Fig. 6C), suggesting that miR-122 expression is not a necessary condition but is required for the enhancement of HCV replication and that HCV is capable of replicating in nonhepatic cells in an miR-122-independent manner. Although the application of miR-122-specific LNAs to chronic hepatitis C patients is now in progress (32), further studies are needed to clarify the mechanisms underlying the miR-122-independent replication of HCV in more detail.
Although the importance of receptor-mediated entry in the cell tropism of HCV has been evaluated (16, 65), cDNA microarray databases, including the NextBio search engine, revealed that HCV receptor candidates, including hCD81, SR-BI, CLDN1, and OCLN, are highly expressed in many nonhepatic tissues. In addition, our current data and previous reports demonstrated that many nonhepatic cells permitted the entry of the pseudotype viruses bearing HCV envelope proteins, suggesting that other host factors must be involved in the cell tropism of HCV to human hepatocytes (4, 17, 54, 61). The data in this study suggest that miR-122 expression and functional lipid metabolism play crucial roles in the determination of an efficient propagation of HCV in vitro. On the other hand, previous studies showed the compartmentalization of genetic variation in HCV between hepatic and nonhepatic tissues, suggesting that HCV is capable of replicating in nonhepatic tissues expressing either miR-122, ApoE, ApoB, or MTTP (52). Collectively, these results suggest that entry receptors, miR-122, and functional lipid metabolism are mainly involved in the regulation of internalization, RNA replication, and assembly of HCV, respectively, and are important factors in determining the cell tropism of HCV to hepatocytes. On the other hand, it might be feasible to speculate that EHMs observed in chronic hepatitis C patients are caused by an incomplete miR-122-independent propagation of HCV in nonhepatic cells.
In spite of the efficient replication of HCV in Hec1B and 293T-CLDN cells expressing miR-122, no infectious particle was detected, in contrast to the case with hepatic cells (Fig. 9), suggesting the involvement of liver-specific host factors and/or machineries in the assembly of infectious particles. In general, the liver plays a major role in lipid metabolism, such as in fatty acid and lipoprotein syntheses (9), and many reports have indicated the involvement of lipid metabolism, especially triglyceride metabolism, in the assembly and budding of HCV particles. Lipid droplets, MTTP, ApoB, and ApoE have been shown to participate in the assembly and secretion of infectious particles of HCVcc in Huh7 cells (20, 23, 26, 40). In the current analyses, there were fewer lipid droplets and the expression levels of ApoE, ApoB, and MTTP were lower in nonhepatic cells than in hepatic cells. Although minus-strand HCV RNA and viral proteins were detected in nonhepatic cells (33, 60), it was shown that the recurrence of HCV after liver transplantation for patients with HCV-induced liver diseases was mainly caused by HCV variants generated in the liver but not in nonhepatic tissues (50). These results support the notion that replication of HCV RNA in nonhepatic cells is unlikely to be a reservoir for persistent infection, due to the lack of infectious particle formation.
The endogenous expression of miR-122 is hardly detected in Hec1B cells, in contrast to the abundant expression of miR-122 in Huh7 cells. Therefore, more accurate analyses of the biological significance of the interaction between miR-122 and the 5′ UTR on the replication of HCVcc in Hec1B could be possible by introducing mutations not only into viruses but also into miR-122. Replication of HCVcc and a mutant virus bearing two mutations in the 5′ UTR (HCVcc-M2) was observed in Hec1B cells expressing WT and MT miR-122, respectively, although the level of replication was lower in cells infected with HCVcc-M2 than in those infected with HCVcc, probably due to the mutations in the 5′ UTR (Fig. 8F). In contrast, a mutant virus (HCVcc-M1) bearing a mutation in site 1 alone exhibited efficient replication in Hec1B cells expressing both WT and MT miR-122 comparable to the replication level of the wild-type virus in cells expressing WT miR-122. Furthermore, the replication level of HCVcc-M1 was low in Hec1B cells expressing either WT or MT miR-122, suggesting that interaction between miR-122 and either of the seed sequence-binding sites in the 5′ UTR has an equal ability to enhance the replication of the HCV genome. However, it was shown that the ability of miR-122 to promote the growth of a laboratory strain of HCV (HJ3-5) is dependent upon its direct interaction with both seed sequence-binding sites in the 5′ UTR and that the binding to site 1 is more important for efficient replication than the binding to site 2 (25). Recently, it was shown that the binding of miR-122 to the 5′ UTR of the HCV genome masks the 5′-terminal sequences of the viral genome through the 3′ overhanging nucleotides of miR-122 (36). It is necessary to evaluate the importance of this enhancement mechanism on mutant HCVcc infection in Hec1B cells.
In summary, we demonstrated that HCV is capable of replicating at a low level in nonhepatic cells and that exogenous expression of miR-122 facilitates efficient viral replication but not the production of infectious particles, probably due to the lack of hepatocytic lipid metabolism in nonhepatic cell lines. These results suggest that miR-122 plays a crucial role in determination of the cell tropism of HCV and the possible involvement of incomplete propagation of HCV in the development of EHM in hepatitis C patients.
ACKNOWLEDGMENTS
We thank M. Tomiyama for her secretarial work. We also thank C. Oneyama, M. Hijikata, T. Wakita, and F. Chisari for providing experimental materials. We also thank H. Ohmori for her excellent technical assistance.
This work was supported in part by grants-in-aid from the Japanese Ministry of Health, Labor, and Welfare (Research on Hepatitis); the Japanese Ministry of Education, Culture, Sports, Science, and Technology; and the Osaka University Global Center of Excellence Program.
Footnotes
Published ahead of print 16 May 2012
REFERENCES
- 1. Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bukh J. 2004. A critical role for the chimpanzee model in the study of hepatitis C. Hepatology 39:1469–1475 [DOI] [PubMed] [Google Scholar]
- 4. Burgel B, et al. 2011. Hepatitis C virus enters human peripheral neuroblastoma cells—evidence for extra-hepatic cells sustaining hepatitis C virus penetration. J. Viral Hepat. 18:562–570 [DOI] [PubMed] [Google Scholar]
- 5. Burns DM, D'Ambrogio A, Nottrott S, Richter JD. 2011. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 473:105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Calleja JL, et al. 1999. Sustained response to interferon-alpha or to interferon-alpha plus ribavirin in hepatitis C virus-associated symptomatic mixed cryoglobulinaemia. Aliment. Pharmacol. Ther. 13:1179–1186 [DOI] [PubMed] [Google Scholar]
- 7. Castoldi M, et al. 2011. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J. Clin. Invest. 121:1386–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang J, et al. 2008. Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J. Virol. 82:8215–8223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coppack SW, Jensen MD, Miles JM. 1994. In vivo regulation of lipolysis in humans. J. Lipid Res. 35:177–193 [PubMed] [Google Scholar]
- 10. Cun W, Jiang J, Luo G. 2010. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J. Virol. 84:11532–11541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daly C, Reich NC. 1993. Double-stranded RNA activates novel factors that bind to the interferon-stimulated response element. Mol. Cell. Biol. 13:3756–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Di Liberto G, et al. 2006. Clinical and therapeutic implications of hepatitis C virus compartmentalization. Gastroenterology 131:76–84 [DOI] [PubMed] [Google Scholar]
- 13. Egger D, et al. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ehrhardt M, et al. 2011. Profound differences of microRNA expression patterns in hepatocytes and hepatoma cell lines commonly used in hepatitis C virus studies. Hepatology 54:1112–1113 [DOI] [PubMed] [Google Scholar]
- 15. Elmen J, et al. 2008. LNA-mediated microRNA silencing in non-human primates. Nature 452:896–899 [DOI] [PubMed] [Google Scholar]
- 16. Evans MJ, et al. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805 [DOI] [PubMed] [Google Scholar]
- 17. Fletcher NF, et al. 2010. Hepatitis C virus infection of neuroepithelioma cell lines. Gastroenterology 139:1365–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fukuhara T, et al. 2011. Intracellular delivery of serum-derived hepatitis C virus. Microbes Infect. 13:405–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Galossi A, Guarisco R, Bellis L, Puoti C. 2007. Extrahepatic manifestations of chronic HCV infection. J. Gastrointestin. Liver Dis. 16:65–73 [PubMed] [Google Scholar]
- 20. Gastaminza P, et al. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82:2120–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henke JI, et al. 2008. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 27:3300–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Herker E, et al. 2010. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 16:1295–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hishiki T, et al. 2010. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J. Virol. 84:12048–12057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huntzinger E, Izaurralde E. 2011. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 12:99–110 [DOI] [PubMed] [Google Scholar]
- 25. Jangra RK, Yi M, Lemon SM. 2010. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J. Virol. 84:6615–6625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang J, Luo G. 2009. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 83:12680–12691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jopling CL, Schutz S, Sarnow P. 2008. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581 [DOI] [PubMed] [Google Scholar]
- 29. Kambara H, et al. 2012. Establishment of a novel permissive cell line for propagation of hepatitis C virus by the expression of microRNA miR122. J. Virol. 86:1382–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kato T, et al. 2005. Nonhepatic cell lines HeLa and 293 support efficient replication of the hepatitis C virus genotype 2a subgenomic replicon. J. Virol. 79:592–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kupershmidt IQ, et al. 2010. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One 5:e13066 doi:10.1371/journal.pone.0013066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lanford RE, et al. 2010. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lerat H, et al. 1996. Specific detection of hepatitis C virus minus strand RNA in hematopoietic cells. J. Clin. Invest. 97:845–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin C, et al. 2004. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J. Biol. Chem. 279:17508–17514 [DOI] [PubMed] [Google Scholar]
- 35. Lin LT, et al. 2010. Replication of subgenomic hepatitis C virus replicons in mouse fibroblasts is facilitated by deletion of interferon regulatory factor 3 and expression of liver-specific microRNA 122. J. Virol. 84:9170–9180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Machlin ES, Sarnow P, Sagan SM. 2011. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. U. S. A. 108:3193–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Manns MP, et al. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958–965 [DOI] [PubMed] [Google Scholar]
- 38. Marukian S, et al. 2008. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48:1843–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Masaki T, et al. 2010. Production of infectious hepatitis C virus by using RNA polymerase I-mediated transcription. J. Virol. 84:5824–5835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miyanari Y, et al. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 41. Moriishi K, et al. 2010. Involvement of PA28gamma in the propagation of hepatitis C virus. Hepatology 52:411–420 [DOI] [PubMed] [Google Scholar]
- 42. Morris T, Robertson B, Gallagher M. 1996. Rapid reverse transcription-PCR detection of hepatitis C virus RNA in serum by using the TaqMan fluorogenic detection system. J. Clin. Microbiol. 34:2933–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Narbus CM, et al. 2011. HepG2 cells expressing microRNA miR-122 support the entire hepatitis C virus life cycle. J. Virol. 85:12087–12092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Okamoto T, et al. 2006. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 25:5015–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Okamoto T, et al. 2008. A single-amino-acid mutation in hepatitis C virus NS5A disrupting FKBP8 interaction impairs viral replication. J. Virol. 82:3480–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pawlotsky JM, Chevaliez S, McHutchison JG. 2007. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 132:1979–1998 [DOI] [PubMed] [Google Scholar]
- 47. Pietschmann T, et al. 2002. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 76:4008–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pileri P, et al. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941 [DOI] [PubMed] [Google Scholar]
- 49. Ploss A, et al. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ramirez S, et al. 2009. Hepatitis C virus compartmentalization and infection recurrence after liver transplantation. Am. J. Transplant. 9:1591–1601 [DOI] [PubMed] [Google Scholar]
- 51. Rieder CL, Bowser SS. 1985. Correlative immunofluorescence and electron microscopy on the same section of Epon-embedded material. J. Histochem. Cytochem. 33:165–171 [DOI] [PubMed] [Google Scholar]
- 52. Roque-Afonso AM, et al. 2005. Compartmentalization of hepatitis C virus genotypes between plasma and peripheral blood mononuclear cells. J. Virol. 79:6349–6357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Russell RS, et al. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 105:4370–4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sainz B, Jr, et al. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 18:281–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sarrazin C, et al. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777 [DOI] [PubMed] [Google Scholar]
- 56. Scarselli E, et al. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Seeff LB. 2002. Natural history of chronic hepatitis C. Hepatology 36:S35–S46 doi:10.1002/hep.1840360706 [DOI] [PubMed] [Google Scholar]
- 58. Sumpter RJ, et al. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Taguwa S, et al. 2009. Cochaperone activity of human butyrate-induced transcript 1 facilitates hepatitis C virus replication through an Hsp90-dependent pathway. J. Virol. 83:10427–10436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takyar ST, Li D, Wang Y, Trowbridge R, Gowans EJ. 2000. Specific detection of minus-strand hepatitis C virus RNA by reverse-transcription polymerase chain reaction on polyA(+)-purified RNA. Hepatology 32:382–387 [DOI] [PubMed] [Google Scholar]
- 61. Tani H, et al. 2007. Replication-competent recombinant vesicular stomatitis virus encoding hepatitis C virus envelope proteins. J. Virol. 81:8601–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wakita T, et al. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Watanabe N, et al. 2011. Hepatitis C virus RNA replication in human stellate cells regulates gene expression of extracellular matrix-related molecules. Biochem. Biophys. Res. Commun. 407:135–140 [DOI] [PubMed] [Google Scholar]
- 64. Wilkinson J, Radkowski M, Laskus T. 2009. Hepatitis C virus neuroinvasion: identification of infected cells. J. Virol. 83:1312–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang W, et al. 2008. Correlation of the tight junction-like distribution of claudin-1 to the cellular tropism of hepatitis C virus. J. Biol. Chem. 283:8643–8653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhong J, et al. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]