

Abstract
Arenaviruses include several causative agents of hemorrhagic fever (HF) disease in humans that are associated with high morbidity and significant mortality. Morbidity and lethality associated with HF arenaviruses are believed to involve the dysregulation of the host innate immune and inflammatory responses that leads to impaired development of protective and efficient immunity. The molecular mechanisms underlying this dysregulation are not completely understood, but it is suggested that viral infection leads to disruption of early host defenses and contributes to arenavirus pathogenesis in humans. We demonstrate in the accompanying paper that the prototype member in the family, lymphocytic choriomeningitis virus (LCMV), disables the host innate defense by interfering with type I interferon (IFN-I) production through inhibition of the interferon regulatory factor 3 (IRF3) activation pathway and that the viral nucleoprotein (NP) alone is responsible for this inhibitory effect (C. Pythoud, W. W. Rodrigo, G. Pasqual, S. Rothenberger, L. Martínez-Sobrido, J. C. de la Torre, and S. Kunz, J. Virol. 86:7728–7738, 2012). In this report, we show that LCMV-NP, as well as NPs encoded by representative members of both Old World (OW) and New World (NW) arenaviruses, also inhibits the nuclear translocation and transcriptional activity of the nuclear factor kappa B (NF-κB). Similar to the situation previously reported for IRF3, Tacaribe virus NP (TCRV-NP) does not inhibit NF-κB nuclear translocation and transcriptional activity to levels comparable to those seen with other members in the family. Altogether, our findings demonstrate that arenavirus infection inhibits NF-κB-dependent innate immune and inflammatory responses, possibly playing a key role in the pathogenesis and virulence of arenavirus.
INTRODUCTION
Arenaviruses are enveloped viruses with a bisegmented negative-stranded RNA genome. Each genomic RNA segment uses an ambisense coding strategy to direct the synthesis of two viral proteins that are encoded in opposite orientations and separated by a noncoding intergenic region (8). The large (L) segment encodes the viral RNA-dependent RNA polymerase (L protein) and a small RING protein (Z) that is the arenavirus counterpart of the matrix (M) protein found in many negative-strand RNA viruses (13, 66, 88). The small (S) segment encodes the viral glycoprotein precursor (GPC) and the viral nucleoprotein (NP). NP is a key component of the viral ribonucleoprotein (RNP) complex that directs viral RNA synthesis and constitutes the minimal unit of infectivity (8). GPC is posttranslationally processed by the cellular protease S1P to produce GP-1 and GP-2, which form the GP glycoprotein complex that makes up the spikes that decorate the surface of the virion structure and mediate receptor recognition and cell entry (5, 8).
Arenaviruses cause chronic infections of rodent species with a worldwide distribution (8). Human infections, which occur via mucosal exposure to aerosols or by direct contact with infectious materials, can cause severe disease, including hemorrhagic fever (HF). Thus, Lassa virus (LASV) and Junin virus (JUNV) are the causative agents of Lassa fever (LF) and Argentine HF disease, respectively, which represent significant public health problems within their geographic regions of endemicity of West Africa (LASV) and Argentina (JUNV) (8, 55, 68, 95). In addition, evidence indicates that the globally distributed prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) is a neglected human pathogen of clinical significance (2, 35, 57) and poses a special threat to immunocompromised individuals (16, 64). Public health concerns posed by arenaviruses are aggravated by the lack of Food and Drug Administration (FDA)-licensed vaccines and current antiarenavirus therapy being limited to an off-label use of the nucleoside analog ribavirin that is only partially effective and associated with significant side effects (37, 56, 60, 76, 83, 94). Therefore, it is important to develop novel and effective antiviral strategies to combat human-pathogenic arenaviruses. Morbidity and mortality associated with LASV infection, and other HF arenavirus infections, have been associated with the failure of the host's innate immune response to restrict virus replication and to facilitate the initiation of an effective adaptive immune response (55). Likewise, chronic infection of the prototypic arenavirus LCMV in mice, the natural reservoir of LCMV, has been shown to be associated with only a very modest increase in production of type I interferon (IFN-I), despite levels of viral RNA being readily detected in most tissues and cell types of the infected host. These findings suggest a virus's ability to counteract the activity of host pattern recognition receptors (PRRs) that sense the presence of viral RNA (78). Accordingly, the arenavirus NP has been shown to inhibit the host's IFN-I response via an early blockade in the IFN-I regulatory factor 3 (IRF3) activation pathway, thereby inhibiting IFN-I production and subsequent induction of IFN-I-stimulated genes (ISGs) (54). The anti-IFN-I activity of LCMV-NP was mapped to its C-terminal region (residues 370 to 553), including the DIEGR motif (amino acid residues 382 to 386) (52). The determination of the X-ray crystal structure of LASV-NP (23, 73) identified a 3′-5′ exonuclease domain within the C-terminal region of the protein, with amino acid residues corresponding to positions D382, E384, D459, H517, and D522 in LCMV-NP contributing to its active site. Mutation-function studies established a link between the exonuclease and anti-IFN-I activities of LASV-NP (23, 52, 73). The exact mechanisms by which arenavirus NPs modulate the IFN-I response still remain unclear, but retinoic acid-inducible gene I (RIG-I)/melanoma-differentiation-associated gene 5 (MDA5) pathway-mediated IFN-I production appears to be involved (97).
Induction of production of the different IFN-I forms upon viral infection involves multiple transcription factors. Early activation of the beta IFN (IFN-β) promoter involves the cooperation of IRF3 with NF-κB and activator protein 1 (AP-1), whereas later induction of IFN-α forms is mainly dependent on IRF7 and, to a lesser extent, IRF3 (20, 28, 31). NF-κB comprises a family of transcription factors found ubiquitously in numerous cell types that play important roles in host inflammatory and immune responses, cell growth and cell differentiation, and apoptosis (28, 31). Under normal conditions, NF-κB is bound to its inhibitor, IκB, resulting in its cytoplasmic retention. Activation of NF-κB signaling requires phosphorylation of IκB by the IκB kinase (IKK) complex (31, 33). The IKK complex contains two catalytic kinase components, IKKα and IKKβ, as well as a nonenzymatic regulatory subunit, NEMO. Two additional members of the family of IKK kinases (TANK-binding kinase 1 [TBK-1] and IKKε) also have the potential to stimulate NF-κB signaling (28, 31). Upon activation, IκB kinase complexes phosphorylate IκB, which is subsequently ubiquitinated and undergoes proteosomal degradation (1). NF-κB is thereby released and enters the nucleus, where it stimulates transcription of genes containing NF-κB-binding sequence elements in their promoters. NF-κB can be activated by various stimuli, including tumor necrosis factor alpha (TNF-α) (28, 31), which is a major proinflammatory cytokine, produced by a variety of cell types, including macrophages, endothelial cells, and epithelial cells (89), and with multiple roles in the regulation of immune response, inflammation, cellular differentiation, apoptosis, and host antiviral defense (27). TNF-α engagement to its receptor leads to signaling cascades that activate IKK complexes (24, 27, 90). NF-κB can also be activated in response to viral infections (14, 91). Because of the importance of NF-κB in the induction of IFN-I and proinflammatory molecules, many viruses have developed mechanisms to evade NF-κB activation (21, 31, 85). Virus-encoded proteins that block NF-κB activation, including influenza virus nonstructural protein 1 (NS1) (91), Hantaan virus (HTNV) nucleocapsid protein (N) (86), and measles virus (MV) V protein (80), have been shown to enhance viral replication in vitro and in vivo and contribute to virus pathogenesis (28, 31).
The HF arenaviruses LASV and JUNV have been shown to inhibit IFN-I and cytokine production, including production of TNF-α, which is controlled by NF-κB (4, 22, 47), suggesting that activation of the NF-κB pathway may be impaired during HF arenaviral infections and that the host's inability to mount an effective inflammatory response may contribute to the pathogenesis of HF arenaviruses. In this work, we provide experimental evidence, for the first time, that NF-κB nuclear translocation and transcriptional activity are inhibited during LCMV infection. Furthermore, we show that expression of LCMV-NP alone is sufficient to inhibit both nuclear translocation and transcriptional activity of NF-κB in a dose-dependent manner. The NP's inhibitory effect on NF-κB activity was shared by other representative members within the Arenaviridae family, with the exception of TCRV. Our mutation-function studies discovered that the same domains and amino acid residues are involved in the NP's ability to interfere with both the IFN-I response and the activation of NF-κB, suggesting a common inhibitory mechanism to mediate efficient evasion of the host IFN-I and inflammatory responses, which may contribute to the failure of the host to control virus multiplication.
MATERIALS AND METHODS
Cell lines and viruses.
Human embryonic kidney epithelial (293T) cells (ATCC CRL-11268), human lung epithelial (A549) cells (ATCC CCL-185), baby hamster kidney fibroblast (BHK-21) cells (ATCC CCL-10), and African green monkey kidney epithelial (Vero) cells (ATCC CCL-81) were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS), supplemented with l-glutamine (2 mM), penicillin (100 units/ml), and streptomycin (100 μg/ml). BHK21 cells constitutively expressing LCMV-NP with a hemagglutinin (HA) tag (54) were generated as described previously (51, 75).
Stocks of wild-type LCMV (Armstrong strain [ARM]) and Tacaribe virus (TCRV) were prepared as described previously (53, 63, 75). Recombinant LCMV carrying a replacement of an aspartic (D) with an alanine (A) at position 382 of NP, rLCMV/NP* D382A, has been described previously (52). High-titer stocks of rLCMV/NP* D382A were generated in BHK-21 cells constitutively expressing LCMV-NP. Wild-type LCMV and rLCMV/NP* D382A titers were determined by using a focus-forming-unit (FFU) assay and Vero cells with LCMV-NP monoclonal antibody 1.1.3, as described previously (63, 75). TCRV was produced and titrated as described previously (53). All viral infections were performed and maintained in a mixture (1:1) of Opti-MEM I and the corresponding complete medium. Sendai virus (SeV), Cantell strain, was grown in 10-day-old embryonated eggs, as described previously (39, 54).
Plasmids.
The pNF-κB-Fluc reporter plasmid (89) and the pCAGGs green fluorescent protein-p65 (GFP-p65) vector (86) have been previously described and were kindly provided by Adolfo Garcia-Sastre and Megan Shaw, respectively. Hemagglutinin (HA)-tagged wild-type versions of lymphocytic choriomeningitis virus (LCMV), Lassa virus (LASV), Whitewater Arroyo virus (WWAV), Pichinde virus (PICV), Junin virus (JUNV), Machupo virus (MACV), Tacaribe virus (TCRV), and Latino virus (LATV) arenavirus NPs have been previously described (53). LCMV-NP C-terminal (ΔC5, ΔC10, ΔC20, and ΔC200) and internal (ΔDIEG) deletions and single amino acid replacements with alanine (A) in the DIEGR motif (D382A, I383A, E384A, G385A, and R386A) were also previously described (52). Residues on LCMV-NP corresponding to the 3′–5′ exonuclease catalytic site that do not overlap the DIEGR motif (D459, H517, and D522) (23, 73) were replaced with alanine (A) by site-directed mutagenesis (Stratagene) in the pGEM-T vector (Promega) and subcloned into pCAGGs HA-COOH to generate C-terminal HA-tagged versions (61, 63).
pCAGGs plasmids expressing influenza A/PuertoRico/8/34 virus NS1, LCMV-Z, and LCMV-, LASV-, JUNV-, and TCRV-NPs fused to the monomeric red fluorescent protein (mRFP) were generated by subcloning the corresponding open reading frames into a modified pCAGGs multiple cloning site (MCS) expressing mRFP (pCAGGs mRFP). pCAGGs mRFP contains two flanking MCSs allowing N-terminal and C-terminal fusions to mRFP, as described for pCAGGs GFP (51).
NF-κB reporter assays.
SeV or TNF-α induction of the NF-κB-dependent reporter plasmid pNF-κB-Fluc was done as described previously (86). Briefly, 105 293T cells (12-well-plate format) were cotransfected in suspension using calcium phosphate (Stratagene) with 500 ng of pNF-κB-Fluc and the indicated dose of pCAGGs protein expression plasmids, along with 50 ng of an expression plasmid encoding Renilla luciferase (RL) under the control of a simian virus 40 promoter (pSV40-RL) to normalize transfection efficiencies. The total amount of plasmid in each transfection was kept constant by adjusting with empty pCAGGs MCSs. All NF-κB reporter assays were performed in triplicate. For viral infections, 24 h posttransfection (p.t.), cells were mock or SeV infected (multiplicity of infection [MOI] = 3) for 1 h at room temperature in 1× phosphate-buffered saline (PBS). For TNF-α-mediated NF-κB-activation experiments, cells were treated 24 h p.t. with the indicated doses of recombinant human TNF-α (BD Biosciences) for 4 h at 37°C. In both cases, cell lysates were prepared 16 to 18 h posttreatment to determine luciferase reporter activities and protein expression. Luciferase activities were determined using a Promega dual-luciferase reporter assay and a Lumicount luminometer (Hewlett Packard) (63). Reporter gene activation was calculated as fold induction (activation) over the level seen with a noninduced, empty pCAGGs MCS-transfected control. Protein expression was determined by Western blotting.
Nuclear translocation of NF-κB. (i) Nuclear translocation of GFP-p65.
293T cells (104) were cotransfected in suspension using calcium phosphate with 2 μg of the pCAGGs GFP-p65 and 2 μg of the indicated arenavirus (NP and Z) or influenza virus NS1 HA- or mRFP-tagged pCAGGs expression plasmids and plated onto poly-d-lysine-coated tissue culture plates. pCAGGs HA-COOH and pCAGGs mRFP plasmids were used as negative controls. At 24 h p.t., cells were infected with SeV (MOI = 3), and at 12 to 16 h postinfection (p.i.), subcellular localization of GFP-p65 was determined. For HA-tagged versions, cells were fixed with 0.2% (vol/vol) glutaraldehyde containing 2.5% (vol/vol) formaldehyde–1× PBS for 10 min at 4°C and permeabilized using 0.1% (vol/vol) Triton X-100–1× PBS for 10 min at room temperature. Cells were washed with 1× PBS and blocked with 5% (wt/vol) bovine serum albumin (BSA)–1× PBS at 4°C before immunostaining was performed.
(ii) Nuclear translocation of endogenous p65 subunit of NF-κB.
A549 cells (104) at subconfluence were mock or LCMV (MOI = 10) infected for 90 min at 37°C. At 24 h p.i. with LCMV, cells were mock or SeV infected (MOI = 3) or treated with TNF-α (50 ng/ml). At the indicated times postinfection with SeV or 4 h posttreatment with TNF-α, cells were fixed with 4% (vol/vol) formaldehyde–1× PBS for 15 min at room temperature, permeabilized with 0.1% (vol/vol) Triton X-100–1× PBS for 10 min at room temperature, and washed with 1× PBS. Cells were blocked overnight at 4°C with 10% (vol/vol) normal goat serum in 5% (wt/vol) BSA blocking solution before immunostaining.
Immunofluorescence microscopy.
For detection of HA-tagged proteins, cells were incubated with an anti-HA polyclonal antibody (Sigma) diluted in 5% (wt/vol) BSA blocking solution for 1 h at 37°C. Cells were then washed 3 or 4 times with 1× PBS and then incubated with Rhodamine-red-conjugated anti-rabbit immunoglobulin G (IgG) (H+L) (Jackson ImmunoResearch), and 4′,6′-diamidino-2-phenylindole (DAPI; Research Organics) for cellular nucleus staining, in blocking solution for 30 min at 37°C. For the quantification of the nuclear translocation of NF-κB, HA- and mRFP-positive cells (indicative of protein expression) were considered.
For the detection of endogenous NF-κB, A549 cells were incubated with anti-p65 polyclonal antibody (Santa Cruz Biotechnology Inc.) and anti-LCMV NP monoclonal antibody 1.1.3 diluted in 5% (wt/vol) BSA blocking solution for 1 h at 37°C. After incubation, cells were washed 3 or 4 times with 1× PBS and incubated with anti-rabbit IgG Alexa Fluor 488 (Molecular Probes), anti-mouse IgG Texas Red (Jackson ImmunoResearch), and DAPI for cellular nucleus staining and diluted in blocking solution for 30 min at 37°C. Cells were washed 3 or 4 times with 1× PBS. For the LCMV and SeV coinfections, indirect immunofluorescence assays were performed using anti-SeV monoclonal antibodies (54) and anti-LCMV guinea pig polyclonal antibody (75). To calculate percent GFP-p65 or endogenous p65 nuclear translocation, a total of 100 to 150 cells in 3 or 4 nonoverlapping fields were counted under ×20 magnification, in multiple-replicate wells. Representative images of independent transfections or infections were colored using the Adobe Photoshop CS4 (version 11.0) software program.
Protein gel electrophoresis and Western blot analysis.
Proteins (100 μg) from total cell lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto nitrocellulose membranes (Bio-Rad). After blocking in 10% (wt/vol) nonfat dry milk for 3 or 4 h at room temperature, membranes were incubated with anti-HA (Sigma) or anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase; AbCam) polyclonal antibodies overnight at 4°C. After 4 or 5 washes with 1× PBS containing 0.1% (vol/vol) Tween 20, the membranes were incubated for 1 h at room temperature with a horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin IgG secondary antibody (GE, Amersham Biosciences). Protein bands were visualized using a chemiluminescence detection kit (SprayGlo) and autoradiography films (Denville Scientific Inc.) according to manufacturer's instructions. Protein band intensities were quantified using ImageJ 1.45 software (NIH). To assess the relative amount of protein loaded for each sample, the GAPDH band intensity in the first lane was assigned a value of 100% and used to normalize the GAPDH band intensities in the remaining lanes. TNF-α-induced nuclear translocation of p65 in mock- and LCMV-infected cells was also examined by Western blot analysis of cytoplasmic and nuclear lysates as described previously (59).
Statistical analysis.
Determinations of means and standard deviations (SD) and two-tailed, paired Student's t tests were performed using Microsoft Excel software. A P value of less than 0.05 was considered statistically significant.
RESULTS
Effect of LCMV infection on NF-κB-mediated transcriptional activation.
To assess whether LCMV infection could interfere with NF-κB activation, we transfected LCMV- and mock-infected control 293T cells with a plasmid expressing the Firefly luciferase (Fluc) reporter gene under the control of an NF-κB-dependent promoter (pNF-κB-Fluc), together with a plasmid expressing the Renilla luciferase (RL) reporter gene under the control of a simian virus 40 promoter (pSV40-RL) (54). At 24 h p.t., we either superinfected the cells with SeV (MOI = 3) for 16 to 18 h or treated them with TNF-α (50 ng/ml) for 4 h at 37°C. After SeV infection or TNF-α treatment, we determined levels of luciferase activities in cell lysates. We used levels of RL activity to normalize transfection efficiencies, whereas we assessed NF-κB activation based on levels of Fluc (54) (Fig. 1). Infection with LCMV had a strong inhibitory effect on SeV-induced NF-κB-mediated transcriptional activation (5-fold reduction). In contrast, LCMV infection caused only a very modest, although statistically significant, decrease in TNF-α-induced NF-κB-mediated transcriptional activation (2-fold reduction) (Fig. 1A). Numbers of LCMV-infected cells (Fig. 1B) and production of infectious progeny LCMV (Fig. 1C) were not significantly affected by either SeV infection or treatment with TNF-α.
Fig 1.
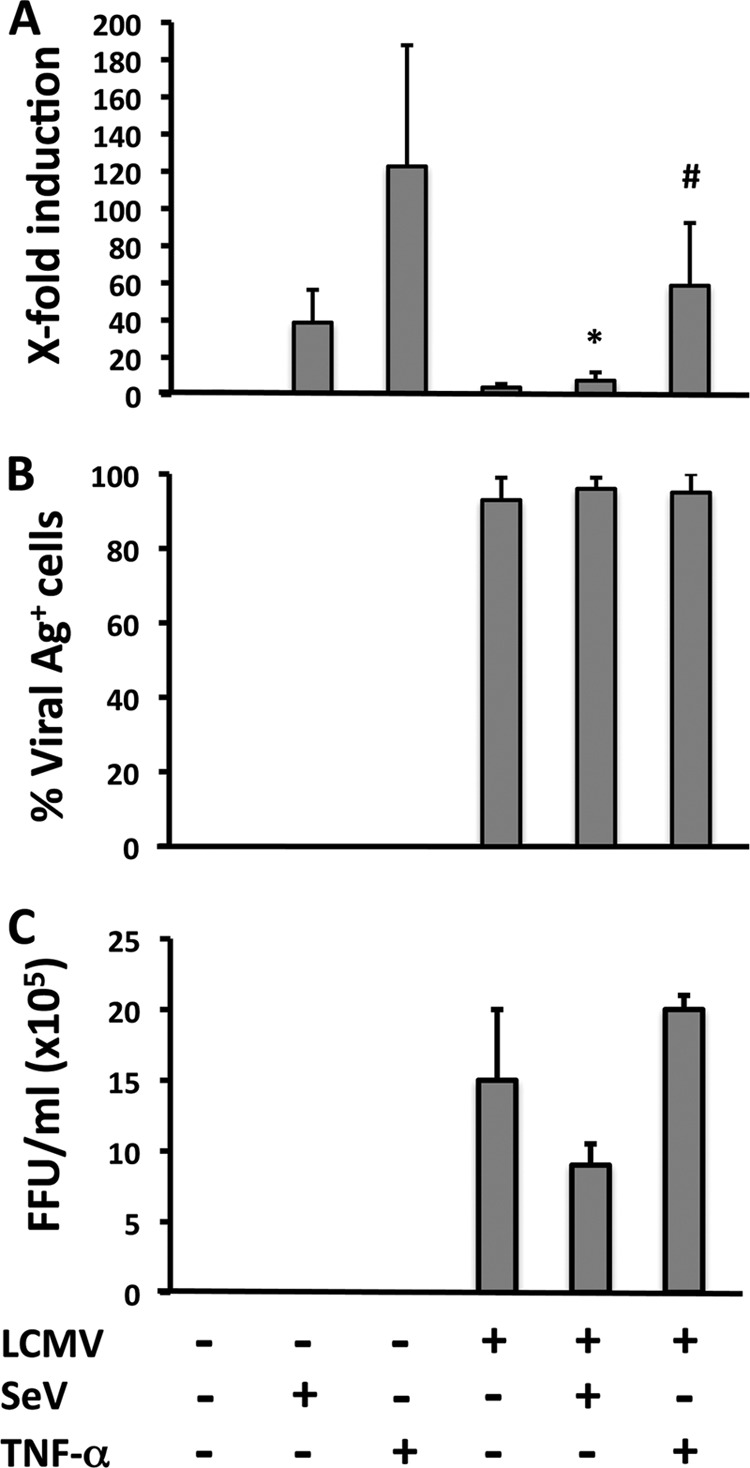
SeV-induced activation of an NF-κB-responsive promoter is inhibited in LCMV-infected cells. (A) A549 cells were mock or LCMV infected (MOI = 0.1) and, 72 h p.i., seeded on 24-well plates (2 × 105 cells/well) prior cotransfection with pNF-κB-Fluc (500 ng) and pSV40-RL (50 ng) plasmids for 5 h, followed by infection with SeV (MOI = 3) or TNF-α treatment (50 ng/ml) for 16 to 18 h or 4 h, respectively, at which time cell lysates were prepared to determine levels of Fluc (NF-κB activation) and RL (normalization of transfection efficiency). Statistical differences in NF-κB-dependent promoter induction between mock- and LCMV-infected cells during SeV infection (*, P = 0.001) and TNF-α treatment (#, P = 0.05) were determined using a 2-tailed paired Student's t test. Reporter gene activation is expressed as fold induction over the level seen with the empty vector-transfected and mock-treated (SeV-uninfected and TNF-α-untreated) control cells. (B and C) From duplicate wells, cells were fixed to determine percentages of LCMV antigen (Ag)-positive cells by immunofluorescence using monoclonal antibody 1.1.3 against LCMV-NP (B), and tissue culture supernatants (TCS) were collected to determine the production of infectious LCMV progeny (in FFU per milliliter) using a focus-forming-unit assay (C). Values shown correspond to averages ± SD of results from two of three independent experiments.
Effect of LCMV-NP and -Z on NF-κB-mediated transcriptional activation.
Both NP (23, 43, 52–54, 73) and Z (15) have been implicated in the arenavirus's ability to counteract the host IFN-I response during infection. We therefore examined whether the same viral gene products also contributed to the virus's ability to interfere with the activation of the NF-κB pathway (Fig. 2A). To that end, we cotransfected 293T cells with pNF-κB-Fluc together with HA-tagged LCMV-NP or -Z expression plasmids (53, 66) and the pSV40-RL plasmid to normalize transfection efficiencies (54). We used empty pCAGGs MCS expression plasmid as a negative control and an HA-tagged influenza (A/Puerto Rico/8/34, H1N1) virus NS1 expression plasmid as a positive control with a viral polypeptide that effectively inhibits the activation of NF-κB (91). At 24 h after transfection, cells were mock or SeV infected to activate NF-κB. At 16 to 18 h p.i., cells were analyzed for luciferase activities (Fig. 2Ai) and for protein expression by Western blotting (Fig. 2Aii). SeV infection strongly induced NF-κB-dependent reporter gene expression in cells transfected with empty or LCMV-Z-expressing pCAGGs plasmids. In contrast, expression of LCMV-NP or influenza virus NS1 resulted in similar levels of inhibition of the NF-κB promoter by SeV infection. All three HA-tagged viral proteins were expressed to similar levels.
Fig 2.

LCMV-NP inhibition of an NF-κB-dependent promoter. (A) Inhibition by LCMV-NP of SeV-mediated induction of an NF-κB-dependent promoter. The NF-κB-responsive plasmid pNF-κB-Fluc (500 ng) was cotransfected with 2 μg of pCAGGs MCS (Empty) or pCAGGs expressing C-terminal HA-tagged versions of LCMV-NP, LCMV-Z, or influenza virus (Flu) NS1 into 293T cells (12-well format, triplicates), together with 50 ng of the Renilla luciferase expression plasmid pSV40-Ren to normalize transfection efficiencies. At 24 h p.t., cells were mock infected (Mock) or infected with SeV (MOI = 3). Luciferase (i) and protein (ii) expression levels were determined 16 to 18 h p.i. (B) Inhibition of TNF-α-mediated induction of an NF-κB-dependent promoter by LCMV-NP. 293T cells (12-well format, triplicates) were cotransfected as described for panel A. At 24 h p.t., cells were treated with the indicated amounts of TNF-α. Activation of the NF-κB reporter plasmid (i) and protein expression levels (ii) were determined 16 to 18 h post-TNF-α treatment. Statistical significance of differences in NF-κB-dependent promoter induction between empty plasmid and LCMV-NP-transfected cells (*, P = 0.002 [TNF-α at 0.5 ng/ml]; **, P = 0.022 [TNF-α at 5 ng/ml]; #, P = 0.006 [TNF-α at 50 ng/ml]) was determined using a 2-tailed paired Student's t test. (C) SeV-mediated activation of the NF-κB-dependent promoter is inhibited by LCMV-NP in a dose-dependent manner. 293T cells (12-well plate format, triplicates) were cotransfected as described for panel A. At 24 h p.t., cells were mock infected (Mock) or infected with SeV (MOI = 3). Luciferase (i) and protein (ii) expression levels were determined 16 to 18 h p.i. (A to C) Reporter gene activation is expressed as fold induction over the level seen with the empty vector-transfected and mock-infected control cells. Cell lysates (100 μg of total protein) from the same transfected cells were used to assess protein expression levels by Western blotting using a polyclonal anti-HA antibody. GAPDH was used as a loading control. The GAPDH band intensity in the first lane (empty plasmid and mock infected) was assigned a value of 100% and used to normalize GAPDH levels in the remaining lanes (bottom numbers). Expression levels of each viral protein were normalized with respect to GAPDH for the same sample. Molecular mass markers (kDa) are indicated on the left and viral proteins on the right.
We also examined the effect of TNF-α treatment in cells transfected with pNF-κB-Fluc and LCMV-NP or influenza virus NS1 (Fig. 2B). TNF-α treatment strongly induced NF-κB-dependent reporter gene expression in cells transfected with empty pCAGGs. In contrast, expression of LCMV-NP or influenza virus NS1 resulted in inhibition of activation of the NF-κB promoter by TNF-α treatment, but LCMV-NP was significantly less efficient than influenza virus NS1 in preventing NF-κB activation by TNF-α treatment (Fig. 2Bi). NP and NS1 were readily detected by Western blotting (Fig. 2Bii), indicating that it was unlikely that differences in the magnitude of the inhibition of NF-κB activation were due to differences in expression levels of the different viral proteins. These results suggested that LCMV-NP's ability to interfere with NF-κB activation is influenced by the type of the stimulus used for activation of the NF-κB pathway.
To further confirm this inhibitory effect of LCMV-NP on NF-κB and determine whether it was dose dependent, we cotransfected 293T cells with 10-fold-increasing amounts of LCMV NP-HA expression plasmids and the pNF-κB-Fluc reporter plasmid. We included the pSV40-RL plasmid to normalize transfection efficiencies (Fig. 2C). At 24 h p.t., cells were infected with SeV (MOI = 3), and at 16 to 18 h p.i., cell extracts were prepared for luciferase assays (Fig. 2Ci) and for protein detection by Western blotting (Fig. 2Cii). LCMV-NP exhibited an inhibitory effect on SeV-induced activation of NF-κB reporter gene expression in a dose-dependent manner.
Effects of NPs from representative Old World (OW) and New World (NW) arenaviruses on NF-κB-mediated transcriptional activation.
Our initial findings with LCMV-NP led us to examine the ability of other arenavirus NPs, including those of the OW arenavirus LASV and representative NW arenaviruses from clades A (WWAV and PICV), B (JUNV, MACV, and TCRV), and C (LATV) (53), to block NF-κB-mediated transcriptional activation (Fig. 3A). For this, we cotransfected 293T cells with the indicated NP-HA-expressing pCAGGs plasmids (10 and 100 ng, based on results from Fig. 2C), together with pNF-κB-Fluc (500 ng) and pSV40-RL (50 ng) plasmids. At 24 h p.t., cells were infected with SeV (MOI = 3), and at 16 to 18 h p.i., we prepared cell lysates for luciferase assays (Fig. 3Ai) and for protein detection by Western blotting (Fig. 3Aii). With the exception of TCRV-NP, all OW and NW arenavirus NPs tested similarly inhibited NF-κB transcriptional activity. All NPs, including TCRV-NP, were expressed to similar levels, and we observed a good correlation between the amounts of DNA used in the transfections and the NP expression levels.
Fig 3.

The ability to inhibit NF-κB-mediated transcriptional activation is shared by NPs of OW and NW arenaviruses. (A). Inhibition of an NF-κB-dependent promoter by different arenavirus NPs. 293T cells (12-well plate format, triplicates) were cotransfected with 500 ng of pNF-κB-Fluc together with the indicated amounts (10 and 100 ng, based on results from Fig. 2C) of different arenavirus pCAGGs NP-HA expression plasmids and 50 ng of pSV40-RL expression vector to normalize transfection efficiencies. At 24 h p.t., cells were infected with SeV (MOI = 3) to induce activation of the NF-κB-responsive promoter, and at 16 to 18 h p.i., cell lysates were prepared for luciferase assay (i). Fold inductions were determined with respect to empty plasmid-transfected and mock-infected cells. Statistical significance for differences in SeV-mediated NF-κB promoter activation among cells transfected with different arenavirus NPs versus SeV-infected empty plasmid-transfected cells (*, P = 0.01; **, P = 0.07; #, P = 0.03; ##, P = 0.30) were determined using 2-tailed paired Student's t tests. Expression levels of OW and NW arenavirus NPs were determined from same cell lysates (100 μg of total protein) by Western blotting using an anti-HA polyclonal antibody (ii). GAPDH expression levels were used as a loading control. Protein expression levels were normalized as described for Fig. 2. Molecular mass markers (kDa) are indicated on the left. OW and NW NPs and GAPDH are indicated on the right. (B) Inhibition of nuclear translocation of GFP-p65. 293T cells were cotransfected with plasmids expressing a GFP-tagged p65 protein (pCAGGs GFP-p65; 2 μg), together with 2 μg of the indicated C-terminal mRFP-tagged arenavirus NPs. At 24 h p.t., cells were infected with SeV (MOI = 3), and at 12 to 16 h p.i., the subcellular localization of GFP-p65 was assessed under a fluorescence microscope. As negative controls, cells were transfected with pCAGGs expressing mRFP (Control) and LCMV Z-mRFP. As a positive control, cells were transfected with pCAGGs expressing influenza virus (Flu) NS1-mRFP. Nuclear translocation of the p65 component of NF-κB (green), expression of NPs, Z, or NS1 (red), DAPI nuclear staining (blue), and the corresponding merged images are indicated. (C) Quantification of GFP-p65 nuclear translocation. Percentages of GFP-p65 nuclear translocation were determined by counting 100 to 150 cells in 3 or 4 nonoverlapping fields. Results were evaluated by a two-tailed paired Student's t test for SeV-infected NP-mRFP versus a control with SeV-infected mRFP alone (*, P = 0.047; **, P = 0.243).
The predominant form of NF-κB is a heterodimer of p50 and p65 that is sequestered in the cytoplasm of unstimulated cells by the IκB inhibitor (29). Upon activation, IκB is phosphorylated and NF-κB translocates to the nucleus to bind to NF-κB-responsive elements (28, 31). Thus, nuclear translocation of the NF-κB p65 subunit could be used as a surrogate marker indicative of NF-κB activation. We therefore monitored the effects of the NPs of different arenaviruses on the nuclear translocation of GFP-p65 (Fig. 3B and C). To this end, we cotransfected 293T cells with 2 μg of pCAGGs GFP-p65, together with 2 μg of plasmids encoding NPs of representative OW and NW arenaviruses tagged with the monomeric red fluorescent protein (mRFP). As negative controls, we transfected cells with a plasmid encoding mRFP alone (Control) or fused to LCMV-Z, whereas, as a positive control, we transfected cells with influenza virus (Flu) NS1-mRFP, as influenza virus NS1 has been shown to effectively inhibit nuclear translocation of p65 (91). At 24 h p.t., cells were infected with SeV (MOI = 3), and at 12 to 16 h p.i., the subcellular localizations of p65 (green), NP (red), and cellular nuclei (blue) were determined. All tested arenavirus NPs, with exception of TCRV-NP, inhibited SeV-induced nuclear translocation of GFP-p65. As expected, LCMV-Z did not interfere with nuclear translocation of p65, whereas, consistent with previous findings, influenza virus NS1 exerted a strong inhibitory effect on p65 nuclear translocation (91).
Residues contributing to the anti-IFN-I function of LCMV-NP are critical for inhibition of NF-κB activation.
We previously identified the C-terminal end of LCMV-NP (amino acids 370 to 553) as critically contributing to its IFN-I-counteracting activity (52). Therefore, we examined whether this domain also contributed to the NP-mediated inhibition of NF-κB activation (Fig. 4A). For this, we cotransfected 293T cells with selected C-terminal-truncated LCMV-NP (52) together with pNF-κB-Fluc and pSV40-RL plasmids. We used empty pCAGGs MCS plasmid and wild-type LCMV-NP as negative and positive controls, respectively. At 24 h p.t., we activated NF-κB via SeV (MOI = 3) infection, and at 16 to 18 h p.i., we determined luciferase activities (Fig. 4Ai) and protein expression levels by Western blotting (Fig. 4Aii). As with the anti-IFN-I activity of NP, the five most C-terminal residues of NP were not required to inhibit efficiently SeV-mediated activation of the NF-κB-dependent promoter, but any additional C-terminal deletions resulted in NP forms lacking this inhibitory activity. We observed some minor differences in protein expression levels among the NP mutants compared to wild-type NP, but all NP mutants were readily detected by Western blotting.
Fig 4.

Residues contributing to the anti-IFN activity of LCMV-NP are critical for inhibition of SeV-mediated transcriptional activation of NF-κB. (A and B) Inhibition of SeV-mediated induction of an NF-κB-dependent promoter by LCMV-NP C-terminal deletion (A) and single-amino-acid (B) mutants. 293T cells (12-well format, triplicates) were cotransfected as described for Fig. 2 using two doses (10 and 100 ng) of C-terminal HA-tagged LCMV-NP wild type (WT) and the indicated mutants. NF-κB reporter gene activation (i) is expressed as fold induction over the level seen with the empty vector-transfected and mock-infected control cells. Values were assessed by a two-tailed paired Student's t test versus SeV-infected empty plasmid-transfected cell results (*, P = 0.01 [A]; *, P = 0.049; **, P = 0.002 [B]). Lysates (100 μg of total protein) from same transfected cells were analyzed for NP expression levels by Western blotting using an anti-HA polyclonal antibody (ii). Protein expression levels were normalized with respect to GAPDH as described for Fig. 2. Molecular mass markers (kDa) are indicated on the left. LCMV-NP (wild-type and mutants) and GAPDH are indicated on the right. (C) Effect of LCMV-NP single amino acid substitutions in nuclear translocation of GFP-p65. 293T cells were cotransfected with GFP-p65 together with HA-tagged LCMV-NP wild type or indicated NP mutants as described for Fig. 3B. As a negative control, cells were transfected with an HA-tagged LCMV-Z pCAGGs expression plasmid. At 24 h p.t., cells were infected with SeV (MOI = 3), and subcellular localization of GFP-p65 was assessed at 12 to 16 h p.i. Representative images of GFP-p65 NF-κB (green), LCMV-Z and LCMV-NP wild type (WT) and mutants (red), cellular nuclear staining (blue), and the corresponding merged images are illustrated. (D) Percentage of GFP-p65 nuclear translocation. GFP-p65 nuclear translocation was assessed as described for Fig. 3C. Results were evaluated using a two-tailed paired Student's t test versus SeV-infected LCMV-Z (*, P = 0.171; **, P = 0.034).
The recently determined X-ray crystal structure of LASV-NP has revealed a 3′-5′ exonuclease domain within its C-terminal portion (23, 73). Notably, mutations involving residues within the active site of the exonuclease domain of LASV-NP (corresponding to D382, E384, D459, H517, and D522 in LCMV-NP) impaired the anti-IFN-I activity of NP (23, 73). Interestingly, residues D382 and E384 are within the DIEGR motif (residues 382 to 386) that had been previously identified as playing a critical role in the IFN-I-counteracting activity of LCMV-NP (52). To examine whether amino acid residues that play key roles in the NP's ability to counteract the IFN-I response were also involved in NP-mediated inhibition of the transcriptional activity of NF-κB, we cotransfected 293T cells with the pNF-κB-Fluc reporter plasmid, the pSV40-RL control vector, and LCMV-NP wild type or mutants containing alanine (A) substitutions at positions D382, I383, E384, G385, R386, D459, H517, and D522 (Fig. 4B). At 24 h p.t., we activated NF-κB via SeV (MOI = 3) infection, and at 16 to 18 h p.i., we determined luciferase activities (Fig. 4Bi) and protein expression abundances by Western blotting (Fig. 4Bii). LCMV-NP mutants D382A, E384A, G385A, D459A, H517A, and D522A, and, to a lesser extent, also R386A, failed to inhibit SeV-induced transcriptional activity of NF-κB. The exception was I383A, which retained an inhibitory effect similar to that observed with wild-type LCMV-NP. Western blot analysis indicated that all LCMV-NP single amino acid substitutions to alanine (A) were expressed to levels comparable to those of wild-type LCMV-NP, indicating that the differences observed could not be explained by differences in NP expression levels.
We next evaluated the effect of these LCMV-NP mutations on nuclear translocation of NF-κB GFP-p65 (Fig. 4C and D). Consistent with the reporter gene assays, GFP-p65 nuclear translocation was not significantly inhibited in cells expressing NP mutants D382A, E384A, G385A, R386A, D459A, H517A, and D522A. In all cases, we observed cytoplasmic distribution of LCMV-NP mutants, suggesting that altered subcellular distribution of the LCMV-NP mutants could have not contributed to their inability to inhibit the nuclear translocation of GFP-p65. Altogether, these results indicate that the same amino acid residues contribute to the ability of LCMV-NP to counteract both induction of IFN-I and activation of NF-κB, suggesting the possibility of a common inhibitory mechanism.
Effect of TCRV or rLCMV/NP* D382A infection on SeV-induced activation of NF-κB-mediated transcriptional activation.
Our observation that TCRV-NP (Fig. 3) and mutant LCMV-NP D382A (Fig. 4) were unable to inhibit SeV-induced NF-κB-mediated transcriptional activation led us to examine whether, in the context of infected cells, TCRV and rLCMV/NP* D382A were also unable to prevent SeV-induced NF-κB-mediated transcriptional activity (Fig. 5). For this, TCRV- and rLCMV/NP* D382A-infected cells, as well as mock-infected control cells, were cotransfected with pNF-κB-Fluc, together with pSV40-RL, and, 24 h later, infected with SeV (MOI = 3) for 16 to 18 h, at which time we determined levels of luciferase activities (Fig. 5A), as well as numbers of viral antigen-positive cells (Fig. 5B) and production of infectious virus progeny in tissue culture supernatants (Fig. 5C). Compared to LCMV (see Fig. 1), TCRV and rLCMV/NP* D382A exerted only a very modest inhibitory effect on SeV-induced NF-κB-mediated transcriptional activation.
Fig 5.

Infection with TCRV or rLCMV/NP* D382A does not prevent SeV-mediated induction of NF-κB-dependent transcriptional activation. A549 cells were mock infected or infected (MOI = 0.1) with TCRV or rLCMV/NP* D382A and at 72 h p.i. seeded on 24-well plates (2 × 105 cells/well) prior cotransfection with pNF-κB-Fluc (500 ng) and pSV40-RL (50 ng) plasmids for 5 h, followed by infection with SeV (MOI = 3) for 18 h, at which time cell lysates were prepared to determine levels of Fluc (NF-κB activation) and RL (normalization of transfection efficiency). (A) Reporter gene activation is expressed as fold induction over the level seen with the empty vector-transfected and mock-infected control cells. (B and C) From duplicate wells, cells were fixed to determine percentages of antigen-positive cells by immunofluorescence using a mouse hyperimmune serum to TCRV or monoclonal antibody 1.1.3 to LCMV-NP (B), and tissue culture supernatants (TCS) were collected to determine the production of infectious virus (TCRV and rLCMV/NP* D382A) progeny (FFU per milliliter) using a focus-forming-unit assay (C). Values shown correspond to averages ± SD of the results from two of three independent experiments.
Effect of LCMV infection on nuclear translocation of endogenous NF-κB.
To validate our finding that LCMV-NP inhibited nuclear translocation of GFP-p65 in the context of the natural course of an LCMV infection, we used a polyclonal antibody specific for p65 to examine the subcellular distribution of endogenous cellular p65 in LCMV-infected A549 cells in response to SeV infection (Fig. 6A and B) and exogenous TNF-α treatment (Fig. 6C and D).
Fig 6.

SeV-induced but not TNF-α-induced nuclear translocation of endogenous NF-κB p65 is inhibited in LCMV-infected cells. (A) Nuclear translocation of endogenous NF-κB p65 in LCMV-infected cells. Subconfluent monolayers of A549 cells were mock infected (−LCMV) or infected (+LCMV, MOI = 10) with LCMV. At 20 h p.i., cells were mock infected (Mock) or infected with SeV (MOI = 3) and, at 3 h post-SeV infection, fixed, permeabilized, and immunostained with antibodies to endogenous NF-κB p65 (green) and LCMV-NP (red). Cellular nuclei were stained with DAPI (blue). Representative images are shown. (B) Percentages of NF-κB p65 nuclear translocation. Percentages of cells showing nuclear translocation of endogenous NF-κB p65 in mock-infected or LCMV-infected cells in response to SeV infection at the indicated hour postinfection were determined by counting 100 to 150 cells in nonoverlapping fields as described for Fig. 3C. (C) Nuclear translocation of endogenous NF-κB p65 in LCMV-infected cells upon TNF-α treatment. Subconfluent monolayers of A549 cells were mock infected (−LCMV) or infected with LCMV (+LCMV, MOI = 1) and 24 h later mock or TNF-α treated (50 ng/ml) for 4 h, fixed, permeabilized, and immunostained with a polyclonal antibody to endogenous NF-κB p65 and monoclonal antibody 1.1.3 to LCMV-NP. The percentage of cells showing nuclear translocation of endogenous NF-κB p65 in each sample was determined by counting 60 to 100 cells in nonoverlapping fields as described for Fig. 3C. (D) Levels of endogenous NF-κB p65 in cytosolic and nuclear lysates from mock- and LCMV-infected cells treated with TNF-α. Cytoplasmic (C) and nuclear (N) lysates of mock- and LCMV-infected cells treated with TNF-α (50 ng/ml) were analyzed by Western blotting using a polyclonal antibody for endogenous NF-κB p65. Caspase-3 (cytoplasmic) and poly-ADP ribose polymerase (PARP; nuclear) were used to determine purity. Mock-infected and TNF-α mock-treated cell extracts were used as a control.
In mock-infected cells (−LCMV), upon challenge with SeV, we observed a high percentage of cells with accumulation of p65 in the nucleus. In contrast, in cells infected with LCMV (+LCMV) and subsequently left unchallenged or challenged with SeV, we observed a very low percentage of cells with nuclear accumulation of p65 that corresponded with cells showing no detectable levels of LCMV-NP in their cytoplasm. We observed similar levels of SeV infection in cells that had been previously infected with LCMV or left uninfected (data not shown). Consistent with our previous observations that LCMV infection (Fig. 1) or LCMV-NP expression (Fig. 2) was very inefficient in preventing TNF-α-induced NF-κB-mediated transcriptional activation, we observed that nuclear accumulation of endogenous p65 in response to TNF-α treatment was not significantly affected in LCMV-infected cells, as determined by immunofluorescence (Fig. 6C) or Western blot analysis of nuclear and cytosolic cell fractions (Fig. 6D).
DISCUSSION
The IFN-I system serves as a first line of defense against virus infection. Induction of IFN-I during viral infection requires the activation of the transcription factors NF-κB, IRF3, and AP-1 and is essential for the expression of many host genes involved in antiviral defense mechanisms (19, 20, 31). Accordingly, viruses interfere with these pathways via a variety of mechanisms, including targeting of key cellular transcription factors by viral proteins (19, 31, 92, 93), which facilitates virus multiplication and pathogenesis.
We have shown that, with the exception of TCRV-NP, all arenavirus NPs tested inhibit nuclear translocation and transcriptional activity of IRF3, supporting a critical role of NP in the inhibition of the host IFN-I response in arenavirus-infected cells (53). In this report, we provide evidence that LCMV infection also blocks activation and transcriptional activity of NF-κB induced by SeV infection (Fig. 1). Since several virus-encoded IFN-I-antagonist proteins have been described to counteract two or more of the transcription factors involved in IFN-I production (19, 31, 92, 93), we examined whether LCMV-NP could also inhibit activation of NF-κB. Our results have shown that LCMV-NP, in the absence of any other viral protein, exerts a powerful dose-dependent inhibitory effect on NF-κB-mediated transcriptional activation upon SeV infection. In contrast, LCMV-NP had only a modest inhibitory effect on TNF-α-induced activation of NF-κB (Fig. 2). These results suggest that LCMV-NP is able to interfere with the nonclassical, but not the classical, pathway of NF-κB activation.
Most of the signaling pathways that lead to activation of NF-κB converge on the IKK kinase complex, which acts as a master regulator of NF-κB activation. TNF-α-induced activation of NF-κB is initiated by binding of TNF-α to its receptors, TNFR1 and TNFR2, which results in activation of IKKα and IKKβ of the IKK complex, as well as TBK-1 (11, 42), leading to phosphorylation and subsequent ubiquitination and proteosomal degradation of the IκBα subunit of the inhibitor IκB (31, 33). These events result in the release and nuclear translocation of NF-κB, its binding to NF-κB response elements, and expression of target genes. In contrast, SeV infection stimulates the cellular RNA sensors RIG-I and MDA5 that, via the mitochondrial antiviral signaling protein MAVS (15) (also known as IPS-1/VISA/Cardif) (36, 58, 71, 96), activate IKKα and IKKβ of the IKK complex as well as the IKK-related kinases TBK-1 and IKKε, which have been associated with activation of IRF3/IRF7, as well as the NF-κB pathway (30, 41). Accordingly, we observed that SeV infection induced activation of the IFN-β- and NF-κB-dependent promoters, but the magnitude of the induction was higher with the IFN-β-dependent promoter (Fig. 7A). In contrast, and consistent with the literature, TNF-α induced higher levels of activation of the NF-κB-dependent promoter and weaker activation of the IFN-β-dependent promoter (32, 46, 74) (Fig. 7B). In agreement with previous findings, we observed that transient transfection with either TBK-1 or IKKε promoted activation of both IFN-β (9, 17, 26, 38, 67, 81)- and NF-κB (3, 18, 40, 62, 69, 70, 77, 79, 82, 87)-dependent promoters (Fig. 7C and D). The NF-κB-dependent promoter within the reporter plasmid contains only two NF-κB binding sites, which explains the overall low levels of reporter expression following activation of the promoter. Notably, infection with vesicular stomatitis virus (VSV) was shown to result in recruitment of IKKε, but not TBK-1, by MAVS (45). In the accompanying paper by Pythoud et al. (72), we demonstrate that arenavirus NP strongly binds IKKε but not TBK-1. Moreover, preliminary data revealed a biochemical association of arenavirus NP with IKKα by the use of nonstringent coimmunoprecipitation conditions. Consequently, it is plausible to imagine a situation where arenavirus NPs block efficiently SeV-induced activation of NF-κB while exhibiting only a very limited ability to inhibit TNF-α-mediated activation of NF-κB. A more detailed understanding of arenavirus NP interactions with the various cellular kinases involved in NF-κB activation by viral infection and TNF-α would help to elucidate the mechanisms by which arenavirus NPs can interfere differently with these two key pathways of the host inflammatory response.
Fig 7.

IFN-β and NF-κB promoter activation by TBK-1 and IKKε. (A and B) SeV infection (A) or TNF-α treatment (B) results in activation of both IFN-β- and NF-κB-dependent promoters. Plasmids pNF-κB-GFP (500 ng) and pIFNβ RFP-CAT (500 ng) were cotransfected into 293T cells (12-well format, triplicates). At 24 h p.t., cells were mock infected (Mock) or infected with SeV (MOI = 3) (A) or mock treated (Mock) or treated with 50 ng of TNF-α/ml (B). Reporter plasmid activation was determined 16 to 18 h after infection or TNF-α treatment under a fluorescence microscope (using GFP for NF-κB and RFP for IFN-β). Representative images are shown. (C and D) TBK-1 (C) and IKKε (D) promote activation of both IFN-β- and NF-κB-dependent promoters. 293T cells (12-well format, triplicates) were cotransfected with 500 ng of pNF-κB-GFP (or pNF-κB-Fluc) and 500 ng of pIFNβ RFP-CAT, together with 500 ng of TBK-1 or IKKε expression plasmids, and 50 ng of pSV40-RL expression vector to normalize transfection efficiencies. Activation of the IFN-β and NF-κB reporter plasmids was determined 24 h posttransfection by fluorescence microscopy (using GFP for NF-κB and RFP for IFN-β) and luciferase expression levels (NF-κB Fluc). Representative images are shown. Reporter gene activation is expressed as fold induction over the level seen with the empty vector-transfected control cells.
The ability to inhibit NF-κB activation was shared by many arenavirus NPs (Fig. 3). Interestingly, and as previously observed together with the inhibitory effect of arenavirus NPs on IRF3 activation (53), TCRV-NP was significantly less efficient in inhibiting nuclear translocation of NF-κB and its transcriptional activity. Mutation-function studies using a series of C-terminal and internal deletion mutants, as well as single alanine (A) substitutions (Fig. 4), revealed that the C-terminal region of LCMV-NP, and, specifically, residues within the active site of the NP 3′-5′ exonuclease (D382, E384, D459A, H517A, and D522A), as well as the highly conserved residues G385 and R386 within the DIEGR motif previously shown to be involved in the anti-IFN-I activity of NP (23, 52, 73), also affected the NP's ability to inhibit NF-κB transcriptional activity. Accordingly, both rLCMV/NP* D382A and TCRV exerted only a very modest inhibitory effect on SeV-induced NF-κB transcriptional activity (Fig. 5). Our findings determined using plasmid-based cell transfection assays were also recreated in LCMV-infected cells with endogenous NF-κB after SeV infection or TNF-α treatment (Fig. 6).
The pathogenicity of HF arenaviruses has been linked to an impaired host innate immune response and subsequent deficient adaptive immune responses, which result in uncontrolled high levels of viremia (55). Thus, differences in pathogenicity between the two OW arenaviruses LASV (highly pathogenic) and Mopeia virus (MOPV [nonpathogenic]) have been correlated with LASV's ability to prevent the activation and corresponding cytokine production observed in MOPV-infected macrophages and dendritic cells (DCs) (47, 65). Whether or not MOPV-NP is able to counteract the IFN-I response remains to be determined, but recent evidence suggests that MOPV infection is not able to counteract the IFN-I response at levels comparable to those seen with LASV-infected cells (10). Likewise, in PICV-infected guinea pigs, a surrogate animal model of LF, a lethal (P18) but not an attenuated (P2) variant of PICV caused inhibition of proinflammatory cytokine production early during infection (6). This was found to correlate with the ability of the pathogenic PICV-P18 to induce increased amounts of the transcription-repressing p50/p50 homodimer of NF-κB, whereas infection with the attenuated PICV-P2 led to accumulation of the trans-activating p65/p50 heterodimer of NF-κB (7). PICV-P2 and -P18 have an arginine (R) and a lysine (K), respectively, at position 374 of NP, but the two NPs were equally able to inhibit SeV-mediated IFN-β promoter activation (43), suggesting that this single amino acid substitution in PICV-NP is unlikely to be responsible for the observed differences between the attenuated P2 and the virulent P18 variants of PICV in NF-κB activation. Similarly, infection of human monocytes with the nonpathogenic NW arenavirus TCRV was accompanied by significant upregulation of interleukin-6 (IL-6), IL-10, and TNF-α compared to infection of monocytes with the pathogenic NW arenavirus JUNV (22). These results are in agreement with our observation that TCRV-NP is not as efficient as other arenavirus NPs in blocking IFN-I (53) and NF-κB responses (this work). Intriguingly, although increased production of IFN-α, IFN-β, TNF-α, IL-6, IL-10, or IL-12 was not observed in JUNV-infected human monocytes or macrophages, several pro- and anti-inflammatory cytokines have been shown to occur at elevated levels in patients with fatal or severe JUNV infection (25, 44, 50), a situation similar to that observed in human cases of LASV infection (47). However, studies of LASV infection of cynomolgus monkeys have shown a correlation between an early and strong innate immune response and control of virus multiplication and recovery from LASV-induced disease (34). These findings support a model of arenavirus pathogenesis in which an early robust production of cytokine by infected monocytes and macrophages may be protective. This model is also consistent with gene expression data from a model of LF based on LCMV infection of macaques (12). During previremic early stages of infection, LCMV had mainly an inhibitory effect on host gene expression, including expression of genes within the IFN-I and NF-κB pathways. However, upon the onset of viremia, the trend was reversed, and in macaques infected with the virulent WE strain of LCMV, there was an overall increase in host gene transcription (48, 49). One could envision that, early during infection, expression of NP might contribute to a limited induction of IFN-I and NF-κB activation in the infected cells. Over time, however, as the numbers of infected cells and the overall viral load rise, many noninfected cells could start to respond to the signals provided by infected cells, thus leading to an overall increase in host gene transcription.
NF-κB is also involved in the regulation of cellular apoptosis. In cultured cells, infection with TCRV, but not JUNV, has been shown to induce a robust cytopathic effect (22). Interestingly, we observed that infection of A549 cells with rLCMV/NP* D382A, a virus whose NP lacked the ability to counteract induction of IFN-I, resulted in a high degree of cytopathic effect than was not seen in cells infected with wild-type rLCMV (data not shown). A similar situation has been observed with recombinant influenza viruses lacking the NS1 protein, which is known to counteract both induction of IFN-I and NF-κB activation (84). It is plausible that the cytopathic effect observed with TCRV and rLCMV/NP* (D382A) could be related to their NPs being unable to modulate the activation of the NF-κB pathway. Further studies would be required to determine the role of arenavirus NP in cellular apoptosis.
Our results have uncovered a previously unknown function of arenavirus NP, namely, its ability to interfere with NF-κB activation, which could contribute to the multiple mechanisms by which arenaviruses counteract the host initial innate defenses and subsequent adaptive immune responses. A better knowledge of the mechanisms underlying the inhibitory activity of arenavirus NPs on the IFN-I and inflammatory responses would lead to a better understanding of the pathogenic and immunogenic properties of arenaviruses. Insights into arenavirus virulence may also open new avenues for the generation of highly attenuated arenavirus that could be evaluated as vaccine candidates and may suggest new antiviral targets for therapeutic modulation in the treatment of arenavirus infections.
ACKNOWLEDGMENTS
We thank members of the laboratory of L.M.-S. for their discussions and S. Dimitrova for expert technical assistance. We thank A. Garcia-Sastre and M. Shaw (Mount Sinai School of Medicine, New York, NY) for providing us with plasmids pNF-κB-Fluc and pCAGGs GFP-p65, respectively. We thank M. Kiebala and S. Maggirwar for providing us with the antibody to the p65 subunit of NF-κB. We thank B. Ward for his advice and support with the use of fluorescence microscopy.
W.W.S.I.R. is a Rochester Vaccine Fellowship recipient (2009). E.O.-R. is a Fullbright-Conicyt Fellowship recipient (BIO 2008). C.P. and S.K. were supported by a grant of the VonTobel Foundation. Research in the laboratory of L.M.-S. was partially funded by NIH grants RO1 AI077719 and HHSN272201000055C. Research in the laboratory of J.C.D.L.T. was supported by grants RO1 AI047140, RO1 AI077719, and RO1 AI079665 from NIH/NIAID.
Footnotes
Published ahead of print 23 May 2012
REFERENCES
- 1. Alkalay I, et al. 1995. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. U. S. A. 92:10599–10603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barton LL. 1996. Lymphocytic choriomeningitis virus: a neglected central nervous system pathogen. Clin. Infect. Dis. 22:197 doi:10.1093/clinids/22.1.197 [DOI] [PubMed] [Google Scholar]
- 3. Bonnard M, et al. 2000. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 19:4976–4985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borrow P, Martinez-Sobrido L, de la Torre JC. 2010. Inhibition of the type I interferon antiviral response during areanvirus infection. Viruses 2:2443–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borrow P, Oldstone MB. 1994. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology 198:1–9 [DOI] [PubMed] [Google Scholar]
- 6. Bowick GC, et al. 2007. Identification of differentially activated cell-signaling networks associated with pichinde virus pathogenesis by using systems kinomics. J. Virol. 81:1923–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bowick GC, et al. 2009. Attenuated and lethal variants of Pichinde virus induce differential patterns of NF-kappaB activation suggesting a potential target for novel therapeutics. Viral Immunol. 22:457–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buchmeier MJ, de la Torre JC, Peters CJ. 2007. Arenaviridae: The viruses and their replication, p 1791–1827 In Knipe PD, Howley P, Griffin D, Lamb R, Martin M, Roizman B, Straus S. (ed), Fields virology, 5th ed, vol II Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 9. Cárdenas WB, et al. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 80:5168–5178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carnec X, et al. 2011. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J. Virol. 85:12093–12097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Delhase M, et al. 2012. TANK-binding kinase 1 (TBK1) controls cell survival through PAI-2/serpinB2 and transglutaminase 2. Proc. Natl. Acad. Sci. U. S. A. 109:E177–E186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Djavani MM, et al. 2007. Early blood profiles of virus infection in a monkey model for Lassa fever. J. Virol. 81:7960–7973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eichler R, et al. 2004. Characterization of the Lassa virus matrix protein Z: electron microscopic study of virus-like particles and interaction with the nucleoprotein (NP). Virus Res. 100:249–255 [DOI] [PubMed] [Google Scholar]
- 14. Elco CP, Guenther JM, Williams BR, Sen GC. 2005. Analysis of genes induced by Sendai virus infection of mutant cell lines reveals essential roles of interferon regulatory factor 3, NF-kappaB, and interferon but not toll-like receptor 3. J. Virol. 79:3920–3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fan L, Briese T, Lipkin WI. 2010. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 84:1785–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fischer SA, et al. 2006. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 354:2235–2249 [DOI] [PubMed] [Google Scholar]
- 17. Fitzgerald KA, et al. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496 [DOI] [PubMed] [Google Scholar]
- 18. Fujita F, et al. 2003. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol. Cell. Biol. 23:7780–7793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. GarcíA-Sastre A. 2004. Identification and characterization of viral antagonists of type I interferon in negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 283:249–280 [DOI] [PubMed] [Google Scholar]
- 20. GarcíA-Sastre A. 2002. Mechanisms of inhibition of the host interferon alpha/beta-mediated antiviral responses by viruses. Microbes Infect. 4:647–655 [DOI] [PubMed] [Google Scholar]
- 21. GarcíA-Sastre A, Sansonetti PJ. 2010. Host-pathogen interactions. Curr. Opin. Immunol. 22:425–427 [DOI] [PubMed] [Google Scholar]
- 22. Groseth A, et al. 2011. Tacaribe virus but not junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Negl. Trop. Dis. 5:e1137 doi:10.1371/journal.pntd.0001137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO. 2011. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. U. S. A. 108:2396–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hayden MS, Ghosh S. 2008. Shared principles in NF-kappaB signaling. Cell 132:344–362 [DOI] [PubMed] [Google Scholar]
- 25. Heller MV, Saavedra MC, Falcoff R, Maiztegui JI, Molinas FC. 1992. Increased tumor necrosis factor-alpha levels in Argentine hemorrhagic fever. J. Infect. Dis. 166:1203–1204 [DOI] [PubMed] [Google Scholar]
- 26. Hemmi H, et al. 2004. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199:1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herbein G, O'Brien WA. 2000. Tumor necrosis factor (TNF)-alpha and TNF receptors in viral pathogenesis. Proc. Soc. Exp. Biol. Med. 223:241–257 [DOI] [PubMed] [Google Scholar]
- 28. Hiscott J. 2007. Convergence of the NF-kappaB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 18:483–490 [DOI] [PubMed] [Google Scholar]
- 29. Hiscott J, et al. 2003. Convergence of the NF-kappaB and interferon signaling pathways in the regulation of antiviral defense and apoptosis. Ann. N. Y. Acad. Sci. 1010:237–248 [DOI] [PubMed] [Google Scholar]
- 30. Hiscott J, Lin R, Nakhaei P, Paz S. 2006. MasterCARD: a priceless link to innate immunity. Trends Mol. Med. 12:53–56 [DOI] [PubMed] [Google Scholar]
- 31. Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. 2006. Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene 25:6844–6867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360 [DOI] [PubMed] [Google Scholar]
- 33. Israel A. 2010. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2:a000158 doi:10.1101/cshperspect.a000158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jahrling PB, et al. 1980. Lassa virus infection of rhesus monkeys: pathogenesis and treatment with ribavirin. J. Infect. Dis. 141:580–589 [DOI] [PubMed] [Google Scholar]
- 35. Jahrling PB, Peters CJ. 1992. Lymphocytic choriomeningitis virus. A neglected pathogen of man. Arch. Pathol. Lab. Med. 116:486–488 [PubMed] [Google Scholar]
- 36. Kawai T, et al. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988 [DOI] [PubMed] [Google Scholar]
- 37. Kilgore PE, et al. 1997. Treatment of Bolivian hemorrhagic fever with intravenous ribavirin. Clin. Infect. Dis. 24:718–722 [DOI] [PubMed] [Google Scholar]
- 38. Kishore N, et al. 2002. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J. Biol. Chem. 277:13840–13847 [DOI] [PubMed] [Google Scholar]
- 39. Kochs G, Garcia-Sastre A, Martinez-Sobrido L. 2007. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 81:7011–7021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koop A, et al. 2011. Novel splice variants of human IKKepsilon negatively regulate IKKepsilon-induced IRF3 and NF-kB activation. Eur. J. Immunol. 41:224–234 [DOI] [PubMed] [Google Scholar]
- 41. Koshiba T, Bashiruddin N, Kawabata S. 2011. Mitochondria and antiviral innate immunity. Int. J. Biochem. Mol. Biol. 2:257–262 [PMC free article] [PubMed] [Google Scholar]
- 42. Kuai J, et al. 2004. NAK is recruited to the TNFR1 complex in a TNFalpha-dependent manner and mediates the production of RANTES: identification of endogenous TNFR-interacting proteins by a proteomic approach. J. Biol. Chem. 279:53266–53271 [DOI] [PubMed] [Google Scholar]
- 43. Lan S, McLay L, Aronson J, Ly H, Liang Y. 2008. Genome comparison of virulent and avirulent strains of the Pichinde arenavirus. Arch. Virol. 153:1241–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Levis SC, et al. 1985. Correlation between endogenous interferon and the clinical evolution of patients with Argentine hemorrhagic fever. J. Interferon Res. 5:383–389 [DOI] [PubMed] [Google Scholar]
- 45. Lin R, et al. 2006. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 80:6072–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lomvardas S, Thanos D. 2002. Modifying gene expression programs by altering core promoter chromatin architecture. Cell 110:261–271 [DOI] [PubMed] [Google Scholar]
- 47. Lukashevich IS, et al. 1999. Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: different effects on IL-8 and TNF-alpha gene expression. J. Med. Virol. 59:552–560 [PMC free article] [PubMed] [Google Scholar]
- 48. Lukashevich IS, et al. 2004. LCMV-mediated hepatitis in rhesus macaques: WE but not ARM strain activates hepatocytes and induces liver regeneration. Arch. Virol. 149:2319–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lukashevich IS, et al. 2003. Arenavirus-mediated liver pathology: acute lymphocytic choriomeningitis virus infection of rhesus macaques is characterized by high-level interleukin-6 expression and hepatocyte proliferation. J. Virol. 77:1727–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marta RF, et al. 1999. Proinflammatory cytokines and elastase-alpha-1-antitrypsin in Argentine hemorrhagic fever. Am. J. Trop. Med. Hyg. 60:85–89 [DOI] [PubMed] [Google Scholar]
- 51. Martínez-Sobrido L, et al. 2010. Hemagglutinin-pseudotyped green fluorescent protein-expressing influenza viruses for the detection of influenza virus neutralizing antibodies. J. Virol. 84:2157–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Martínez-Sobrido L, et al. 2009. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 83:11330–11340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martínez-Sobrido L, Giannakas P, Cubitt B, Garcia-Sastre A, de la Torre JC. 2007. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 81:12696–12703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martínez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC. 2006. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 80:9192–9199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McCormick JB, Fisher-Hoch SP. 2002. Lassa fever. Curr. Top. Microbiol. Immunol. 262:75–109 [DOI] [PubMed] [Google Scholar]
- 56. McKee KT, Jr, Huggins JW, Trahan CJ, Mahlandt BG. 1988. Ribavirin prophylaxis and therapy for experimental argentine hemorrhagic fever. Antimicrob. Agents Chemother. 32:1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mets MB, Barton LL, Khan AS, Ksiazek TG. 2000. Lymphocytic choriomeningitis virus: an underdiagnosed cause of congenital chorioretinitis. Am. J. Ophthalmol. 130:209–215 [DOI] [PubMed] [Google Scholar]
- 58. Meylan E, et al. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172 [DOI] [PubMed] [Google Scholar]
- 59. Meylan E, et al. 2009. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 462:104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mistry N, Shapero J, Crawford RI. 2009. A review of adverse cutaneous drug reactions resulting from the use of interferon and ribavirin. Can. J. Gastroenterol. 23:677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Muñoz-Jordán JL, et al. 2005. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 79:8004–8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nomura F, Kawai T, Nakanishi K, Akira S. 2000. NF-kappaB activation through IKK-i-dependent I-TRAF/TANK phosphorylation. Genes Cells 5:191–202 [DOI] [PubMed] [Google Scholar]
- 63. Ortiz-Riaño E, Cheng BYH, de la Torre JC, Martínez-Sobrido L. 2011. The C-terminal region of lymphocytic choriomeningitis virus nucleoprotein contains distinct and segregable functional domains involved in NP-Z interaction and counteraction of the type I interferon response. J. Virol. 85:13038–13048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Palacios G, et al. 2008. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 358:991–998 [DOI] [PubMed] [Google Scholar]
- 65. Pannetier D, Faure C, Georges-Courbot MC, Deubel V, Baize S. 2004. Human macrophages, but not dendritic cells, are activated and produce alpha/beta interferons in response to Mopeia virus infection. J. Virol. 78:10516–10524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Perez M, Greenwald DL, de la Torre JC. 2004. Myristoylation of the RING finger Z protein is essential for arenavirus budding. J. Virol. 78:11443–11448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. 2004. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 199:1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Peters CJ. 2002. Human infection with arenaviruses in the Americas. Curr. Top. Microbiol. Immunol. 262:65–74 [DOI] [PubMed] [Google Scholar]
- 69. Peters RT, Liao SM, Maniatis T. 2000. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell 5:513–522 [DOI] [PubMed] [Google Scholar]
- 70. Pomerantz JL, Baltimore D. 1999. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18:6694–6704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Potter JA, Randall RE, Taylor GL. 2008. Crystal structure of human IPS-1/MAVS/VISA/Cardif caspase activation recruitment domain. BMC Struct. Biol. 8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pythoud C, et al. 2012. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKε. J. Virol. 86:7728–7738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Qi X, et al. 2010. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 468:779–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Reis LF, Ho Lee T, Vilcek J. 1989. Tumor necrosis factor acts synergistically with autocrine interferon-beta and increases interferon-beta mRNA levels in human fibroblasts. J. Biol. Chem. 264:16351–16354 [PubMed] [Google Scholar]
- 75. Rodrigo WW, de la Torre JC, Martinez-Sobrido L. 2011. Use of single-cycle infectious lymphocytic choriomeningitis virus to study hemorrhagic fever arenaviruses. J. Virol. 85:1684–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rodríguez M, McCormick JB, Weissenbacher MC. 1986. Antiviral effect of ribavirin on Junin virus replication in vitro. Rev. Argent Microbiol. 18:69–74 [PubMed] [Google Scholar]
- 77. Sankar S, Chan H, Romanow WJ, Li J, Bates RJ. 2006. IKK-i signals through IRF3 and NFkappaB to mediate the production of inflammatory cytokines. Cell. Signal. 18:982–993 [DOI] [PubMed] [Google Scholar]
- 78. Saron MF, Riviere Y, Hovanessian AG, Guillon JC. 1982. Chronic production of interferon in carrier mice congenitally infected with lymphocytic choriomeningitis virus. Virology 117:253–256 [DOI] [PubMed] [Google Scholar]
- 79. Sasai M, Matsumoto M, Seya T. 2006. The kinase complex responsible for IRF-3-mediated IFN-beta production in myeloid dendritic cells (mDC). J. Biochem. 139:171–175 [DOI] [PubMed] [Google Scholar]
- 80. Schuhmann KM, Pfaller CK, Conzelmann KK. 2011. The measles virus V protein binds to p65 (RelA) to suppress NF-kappaB activity. J. Virol. 85:3162–3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–1151 [DOI] [PubMed] [Google Scholar]
- 82. Shimada T, et al. 1999. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int. Immunol. 11:1357–1362 [DOI] [PubMed] [Google Scholar]
- 83. Snell N. 1988. Ribavirin therapy for Lassa fever. Practitioner 232:432. [PubMed] [Google Scholar]
- 84. Stasakova J, et al. 2005. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J. Gen. Virol. 86(Pt 1):185–195 [DOI] [PubMed] [Google Scholar]
- 85. Symeonidou I, et al. 2010. Modulation of NF-kappaBeta signalling pathways by parasites. J. Biol. Regul. Homeost. Agents 24:471–479 [PubMed] [Google Scholar]
- 86. Taylor SL, Frias-Staheli N, Garcia-Sastre A, Schmaljohn CS. 2009. Hantaan virus nucleocapsid protein binds to importin alpha proteins and inhibits tumor necrosis factor alpha-induced activation of nuclear factor kappa B. J. Virol. 83:1271–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tojima Y, et al. 2000. NAK is an IkappaB kinase-activating kinase. Nature 404:778–782 [DOI] [PubMed] [Google Scholar]
- 88. Urata S, Yasuda J, de la Torre JC. 2009. The Z protein of the new world arenavirus tacaribe virus has bona fide budding activity that does not depend on known late domain motifs. J. Virol. 83:12651–12655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wajant H, Pfizenmaier K, Scheurich P. 2003. Tumor necrosis factor signaling. Cell Death Differ. 10:45–65 [DOI] [PubMed] [Google Scholar]
- 90. Wajant H, Scheurich P. 2001. Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling. Int. J. Biochem. Cell Biol. 33:19–32 [DOI] [PubMed] [Google Scholar]
- 91. Wang X, et al. 2000. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 74:11566–11573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Weber F, Kochs G, Haller O. 2004. Inverse interference: how viruses fight the interferon system. Viral Immunol. 17:498–515 [DOI] [PubMed] [Google Scholar]
- 93. Weber F, Kochs G, Haller O, Staeheli P. 2003. Viral evasion of the interferon system: old viruses, new tricks. J. Interferon Cytokine Res. 23:209–213 [DOI] [PubMed] [Google Scholar]
- 94. Weissenbacher MC, Calello MA, Merani MS, McCormick JB, Rodriguez M. 1986. Therapeutic effect of the antiviral agent ribavirin in Junin virus infection of primates. J. Med. Virol. 20:261–267 [DOI] [PubMed] [Google Scholar]
- 95. Weissenbacher MC, Laguens RP, Coto CE. 1987. Argentine hemorrhagic fever. Curr. Top. Microbiol. Immunol. 134:79–116 [DOI] [PubMed] [Google Scholar]
- 96. Xu LG, et al. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727–740 [DOI] [PubMed] [Google Scholar]
- 97. Zhou S, et al. 2010. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 84:9452–9462 [DOI] [PMC free article] [PubMed] [Google Scholar]