

Abstract
The human respiratory syncytial virus (HRSV) genome is composed of a negative-sense single-stranded RNA that is tightly associated with the nucleoprotein (N). This ribonucleoprotein (RNP) complex is the template for replication and transcription by the viral RNA-dependent RNA polymerase. RNP recognition by the viral polymerase involves a specific interaction between the C-terminal domain of the phosphoprotein (P) (PCTD) and N. However, the P binding region on N remains to be identified. In this study, glutathione S-transferase (GST) pulldown assays were used to identify the N-terminal core domain of HRSV N (NNTD) as a P binding domain. A biochemical characterization of the PCTD and molecular modeling of the NNTD allowed us to define four potential candidate pockets on N (pocket I [PI] to PIV) as hydrophobic sites surrounded by positively charged regions, which could constitute sites complementary to the PCTD interaction domain. The role of selected amino acids in the recognition of the N-RNA complex by P was first screened for by site-directed mutagenesis using a polymerase activity assay, based on an HRSV minigenome containing a luciferase reporter gene. When changed to Ala, most of the residues of PI were found to be critical for viral RNA synthesis, with the R132A mutant having the strongest effect. These mutations also reduced or abolished in vitro and in vivo P-N interactions, as determined by GST pulldown and immunoprecipitation experiments. The pocket formed by these residues is critical for P binding to the N-RNA complex, is specific for pneumovirus N proteins, and is clearly distinct from the P binding sites identified so far for other nonsegmented negative-strand viruses.
INTRODUCTION
Human respiratory syncytial virus (HRSV) is the leading cause of severe respiratory tract infections in newborn children worldwide (7). It infects close to 100% of infants within the first 2 years of life and is the main cause of bronchiolitis. Also, bovine respiratory syncytial virus (BRSV), which is very similar to its human counterpart, is a major cause of respiratory disease in calves, resulting in substantial economic losses to the cattle industry worldwide (49). RSV belongs to the genus Pneumovirus of the family Paramyxoviridae and the order Mononegavirales (7). The viral genome consists of a nonsegmented ∼15-kb RNA of negative polarity which encodes 11 proteins. As for all the members of the Mononegavirales, the genomic RNA of RSV is always tightly bound by the viral nucleoprotein (N) and maintained as a helical N-RNA ribonucleoprotein (RNP) complex (9). The RNP is used as the template for transcription and replication by the RNA-dependent RNA polymerase (RdRp) complex, consisting of L (large protein) and its cofactor P (phosphoprotein). Whereas N, P, and L are sufficient to mediate viral replication (15, 50), transcription activity requires L, P, and the M2-1 protein, which functions as a processivity polymerase cofactor (8).
The specific recognition of the viral N-RNA matrix by the RdRp constitutes a prerequisite for viral transcription and replication. This recognition is mediated by P, which interacts with L and N but also with M2-1 (29, 46). By analogy to the Paramyxovirinae and Rhabdoviridae, it is noteworthy that besides its multiple protein interactions within the polymerase complex, RSV P is also believed to play the role of a chaperone to prevent the illegitimate assembly of newly synthesized N on cellular RNA and to keep N in a monomeric form designated N° (39). Thus, it is expected that P–N-RNA and P-N° involve different binding sites on both N and P. The P protein is composed of 241 amino acids and is phosphorylated mainly at Ser-232 (2, 42), although other minor phosphorylation sites have been identified (1, 33). However, nonphosphorylated P efficiently binds to N-RNA in vitro (45), and the precise role of phosphorylation in its activity still remains unclear. P forms homotetramers, and the P oligomerization domain is localized between residues 104 and 163 (5, 26, 27). Except for this domain, the P protein is poorly structured, as the N-terminal (residues 1 to 103) and C-terminal (residues 200 to 241) regions are intrinsically disordered (5, 26, 27, 45). Such intrinsically disordered domains are thought to serve as hubs to promote multiple protein interactions (47). This correlates with the central functions of P within the polymerase complex. The C-terminal domain of P (PCTD) (residues 161 to 241) is engaged in the interaction with the N-RNA complex, and we have previously shown that (i) the last 9 C-terminal residues of P are sufficient for this interaction and (ii) acidic and hydrophobic residues are critical for binding to N-RNA nucleocapsid-like complexes assembled as rings (45).
Recently, the crystal structure of HRSV nucleocapsid-like structures consisting of rings containing 10 N protomers and RNA of 70 nucleotides was determined (44). Each N subunit is organized into four distinct domains, the N- and C-terminal globular domains, termed the NNTD and NCTD, respectively, which are α-helical bundles connected through a hinge region, and the N- and C-terminal extensions, termed N-arm and C-arm, respectively. The RNA binding groove is formed at the NNTD/NCTD interface. Although N-RNA rings used for three-dimensional (3D) structure determinations were cocrystallized with PCTD, no electron densities corresponding to the latter were observed, and the P binding site on the N-RNA complex remained to be determined. Several studies sought to address this point but led to conflicting results (13, 31, 32, 43). More specifically, the implication of the NNTD and/or NCTD in the interaction with P remains to be clarified. In this work, a rational mutational approach based on the structure of N was used to map the domain of the HRSV N protein involved in PCTD binding. The data indicated that the PCTD binding site is located on the NNTD, and this involves critical residues constituting a hydrophobic pocket surrounded by basic residues. These new data open a way to develop antiviral strategies against RSV, targeting an N-P interaction domain.
MATERIALS AND METHODS
Plasmid constructs.
Plasmids pGEX-PCTD and pGEX-P(231-241), containing the sequence of the P C-terminal region (residues 161 to 241 and 231 to 241, respectively), were described previously (5, 45). The full-length N gene or the sequences of N with N-terminal deletions or internal domains of N were PCR amplified (primer sequences are available on request) by using Pfu DNA polymerase (Stratagene, Les Ulis, France) and cloned into pET28a(+) at BamHI-XhoI sites to engineer the pET-N-His plasmids. Point mutations were introduced into pET-N-His by site-directed mutagenesis to replace targeted residues by using the QuikChange site-directed mutagenesis kit (Stratagene). These constructs were used to produce N-derived proteins with a C-terminal poly-His tag. The C-terminal deletion mutants of N were obtained by introducing stop codons at the appropriate site in the coding sequence of pET-N-His to generate an N protein without a poly-His tag. Sequence analysis was carried out to check the integrity of all the constructs.
Plasmids for the eukaryotic expression of the HRSV proteins N, P, M2-1, and L, designated pN, pP, pM2-1, and pL, respectively, were described previously (11, 46). The pM/Luc subgenomic replicon, which encodes the firefly luciferase (Luc) gene under the control of the M-SH gene start sequence, was derived from the pM/SH subgenomic replicon (17) and was described previously (46). Point mutations were introduced into pN and pP by site-directed mutagenesis as described above. To generate plasmid pHA-P, complementary oligonucleotides encoding a hemagglutinin (HA) tag epitope (sequences are available on request) were annealed to generate BamHI-compatible ends. The resulting fragment was inserted into the BamHI restriction site in frame with the P sequence in plasmid pP.
Antibodies.
The following primary antibodies were used for immunofluorescence and/or immunoblotting: a mouse monoclonal anti-N protein antibody (Serotec, Oxford, United Kingdom), a rabbit anti-P antiserum and a rabbit anti-N antiserum described previously (11), a mouse monoclonal anti-α-tubulin antibody (Sigma), and a rat monoclonal anti-HA-peroxidase antibody (Roche). Secondary antibodies directed against mouse and rabbit IgG coupled to Alexafluor-594 or Alexafluor-488 (Invitrogen) were used for immunofluorescence. Secondary antibodies directed against mouse and rabbit IgG coupled to horseradish peroxidase (HRP) (PARIS, Compiègne, France) were used for immunoblotting experiments.
Expression and purification of recombinant proteins.
Escherichia coli BL21(DE3) bacteria (Novagen, Madison, WI) transformed with pGEX or pGEX-PCTD were grown at 37°C for 8 h in 100 ml of Luria-Bertani (LB) medium containing 100 μg/ml ampicillin. Bacteria were transformed with pET-N-derived plasmids alone or together with pGEX-PCTD or pGEX-P(231-241) and grown in LB medium containing kanamycin (50 μg/ml) or ampicillin and kanamycin, respectively. The same volume of LB medium was then added, and protein expression was induced by the addition of 80 μg/ml isopropyl-β-d-thiogalactoside (IPTG) to the medium. The bacteria were incubated for 15 h at 28°C and then harvested by centrifugation. For glutathione S-transferase (GST) fusion protein purification, bacterial pellets were resuspended in lysis buffer (50 mM Tris-HCl [pH 7.8], 60 mM NaCl, 1 mM EDTA, 2 mM dithiothreitol [DTT], 0.2% Triton X-100, 1 mg/ml lysozyme) supplemented with a complete protease inhibitor cocktail (Roche, Mannheim, Germany), incubated for 1 h on ice, sonicated, and centrifuged at 4°C for 30 min at 10,000 × g. Glutathione-Sepharose 4B beads (GE Healthcare, Uppsala, Sweden) were added to clarified supernatants and incubated at 4°C for 15 h. The beads were then washed two times in lysis buffer and three times in 1× phosphate-buffered saline (PBS) and then stored at 4°C in an equal volume of PBS. For poly-His fusion protein purification, bacterial pellets were resuspended in lysis buffer (20 mM Tris-HCl [pH 8], 500 mM NaCl, 0.1% Triton X-100, 10 mM imidazole, 1 mg/ml lysozyme) supplemented with a complete protease inhibitor cocktail (Roche). After sonication and centrifugation, lysates were incubated for 1 h with chelating Sepharose Fast Flow beads charged with Ni2+ (GE Healthcare). Finally, beads were successively washed in washing buffer (20 mM Tris-HCl [pH 8], 500 mM NaCl) containing increasing concentrations of imidazole (25, 50, and 100 mM), and proteins were eluted in the same buffer with 800 mM imidazole. Proteins were then dialyzed against 20 mM Tris (pH 8.5)–150 mM NaCl.
Cell culture and transfections.
BHK-21 cells (clone BSRT7/5) constitutively expressing the T7 RNA polymerase (4) were grown in Dulbecco modified essential medium (Lonza, Cologne, Germany) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, and antibiotics. Cells were transfected by using either Lipofectamine 2000 (Invitrogen, Cergy-Pontoise, France) or Fugene HD (Promega, Lyon, France) as described by the manufacturer.
Minigenome replication assay.
Cells at 90% confluence in 24-well dishes were transfected with Lipofectamine 2000 (Invitrogen); a plasmid mixture containing 0.5 μg of pM/Luc, 0.5 μg of pN, 0.5 μg of pP, 0.25 μg of pL, and 0.125 μg of pM2-1 (46), as well as 0.125 μg of p-β-Gal plasmid (Promega) to normalize transfection efficiencies. The dicistronic subgenomic replicon pM/Luc contains the authentic M-SH gene junction and the Luc reporter gene downstream of the gene start sequence present in this gene junction (11, 46, 50). Transfections were done in triplicate, and each independent transfection was performed three times. Cells were harvested at 24 h posttransfection and then lysed in luciferase lysis buffer (30 mM Tris [pH 7.9], 10 mM MgCl2, 1 mM DTT, 1% Triton X-100, and 15% glycerol). Luciferase activities were determined for each cell lysate with an Anthos Lucy 3 luminometer (Bio Advance, Bussy Saint Martin, France) and normalized to β-Gal expression levels.
Fluorescence microscopy.
Immunofluorescence microscopy was performed with cells grown on coverslips and previously transfected with Fugene HD with pN and pP. At 24 h posttransfection, cells were fixed with 4% paraformaldehyde (PFA) for 25 min. Fixed cells were made permeable and blocked for 30 min with PBS containing 0.1% Triton X-100–0.3% bovine serum albumin (BSA). Cells were then successively incubated for 30 min at room temperature with primary and secondary antibody mixtures diluted in PBS containing 0.3% BSA. For nucleus labeling, cells were incubated with Hoechst 33342 (Invitrogen) during incubation with secondary antibodies. Coverslips were mounted with Prolong gold antifade reagent (Invitrogen). Cells were observed with a Nikon TE200 microscope equipped with a CoolSNAP ES2 (Photometrics) camera, and images were processed by using MetaVue (Molecular Devices) and ImageJ software.
Pulldown assays.
Purified recombinant N proteins (wild type and mutants) were incubated in the presence of a 5-fold molar excess of GST or the GST-PCTD fusion protein fixed on beads in a final volume of 100 μl containing 20 mM Tris (pH 8.5)–150 mM NaCl. After 1 h under agitation at 4°C, the beads were extensively washed with 20 mM Tris (pH 8.5)–150 mM NaCl, boiled in 30 μl Laemmli buffer, and analyzed by SDS-PAGE and Coomassie blue staining.
Immunoblotting.
Cells were lysed for 30 min at 4°C by using lysis buffer (10 mM Tris [pH 7.6], 150 mM NaCl, 1 mM EDTA, 1% [vol/vol] Triton X-100, 0.1% deoxycholate) supplemented with a complete protease inhibitor cocktail (Roche). Cell lysates were spun for 10 min at 10,000 × g; supernatants were recovered, mixed with Laemmli buffer, and boiled; and proteins were resolved by SDS-PAGE. Proteins were transferred onto nitrocellulose membranes. The membranes were incubated in blocking solution (PBS–0.05% [vol/vol] Tween 20 supplemented with 5% [wt/vol] milk) for 1 h. Blots were rinsed with PBS containing 0.05% (vol/vol) Tween 20 and incubated with primary antibodies in blocking solution. The membranes were rinsed as described above and incubated for 1 h with the appropriate HRP-conjugated secondary antibodies diluted in blocking solution. The membranes were rinsed, and immunodetections were performed by using an enhanced chemiluminescence (ECL) substrate (GE Healthcare GMB, Saclay, France).
Coimmunoprecipitation assay.
Cells were cotransfected with pHA-P and pN (wild-type or mutant plasmids). After 36 h, transfected cells were lysed for 30 min at 4°C in ice-cold lysis buffer (50 mM Tris HCl [pH 7.4], 2 mM EDTA, 150 mM NaCl, 0.5% NP-40) with a complete protease inhibitor cocktail (Roche), and coimmunoprecipitation experiments were performed on cytosolic extracts. Cell lysates were incubated for 4 h at 4°C with an anti-HA antibody coupled to agarose beads (Euromedex). The beads were then washed 3 times with lysis buffer and 1 time with PBS, and proteins were eluted in Laemmli buffer at 95°C for 5 min and then subjected to SDS-PAGE and immunoblotting as described above.
Circular dichroism spectropolarimetry.
Circular dichroism (CD) experiments were performed with a J-810 spectropolarimeter (Jasco, Tokyo, Japan) in a thermostated cell holder at 20°C. Purified recombinant N proteins or the NNTD was dialyzed against 5 mM phosphate–100 mM NaF (pH 8.5 or pH 6.5, respectively). Far-UV spectra were recorded by using a bandwidth of 1 nm and an integration time of 1 s, with proteins at concentrations of 20 μM. Each spectrum was the average of 10 scans, with a scan rate of 100 nm/min. The spectra were corrected for the blank, smoothed using the FFT filter (Jasco Software, Tokyo, Japan), and treated as previously described (6).
Molecular dynamics simulation.
The molecular dynamics (MD) simulations of the NNTD were carried out by using the program NAMD (35) with the CHARMM27 force field (28). The fragment of the crystal structure of the N monomer at positions 31 to 252 (Protein Data Bank [PDB] accession number 2WJ8 [44]) was used as the initial configuration. The N protein was solvated in the explicit molecular water model TIP3P (21). The solvated system was electrostatically neutralized by the addition of 2 chloride ions at coordinates of minimal electrostatic energy. The ionic strength of the solution was set to 0.15 M by the addition of ions of sodium and chloride at random coordinates. The van der Waals interactions were smoothly shifted to zero between 10.0 Å and 12.0 Å. The list of the nonbonded interactions was truncated at 13.5 Å. The lengths of the bonds containing hydrogen atoms were fixed with the SHAKE algorithm (41), and the equations of motion were iterated by using a time step of 2 fs in the velocity Verlet integrator. The electrostatic interactions were calculated with no truncation, using the particle mesh Ewald summation algorithm (10). The pressure and temperature were restrained to 1 atm and 300 K, respectively. One trajectory of 30 ns was produced. The clustering algorithm implemented in the CORREL facility of the CHARMM package (3) was employed to cluster the N structures into groups based on similar values of the ϕ-ψ backbone dihedral angles with a cluster maximum radius of 19.5 Å. The Surflex (version 2.601) molecular docking package (40) was used to define protomols on structures closest to the center of the clusters. A protomol is defined as an ensemble of small fragments that make favorable interactions with the protein surface.
Root mean square fluctuations.
The backbone root mean square fluctuations (RMSFs) were calculated as backbone-averaged root mean square displacements (RMSDs) of the protein atoms from the trajectory-averaged positions after the translation and rotation of the N configurations generated during the MD simulation were initially removed.
RESULTS
Identification of a PCTD binding domain on N.
The N protein of RSV is composed of four structural domains, i.e., the N- and C-terminal arms and the NNTD and NCTD (Fig. 1A) (44). When expressed in bacteria, HRSV N forms nucleocapsid-like structures that interact and can be copurified with GST-PCTD (residues 161 to 241) (45). In a first attempt to identify which domain of N could interact with the PCTD, truncated N proteins with a poly-His tag appended to their C termini (Fig. 1B) were coexpressed with the fusion protein GST-PCTD in E. coli and purified by using either GST or a poly-His tag. Purified complexes were analyzed by SDS-PAGE, and the purification of GST coexpressed in the presence of N was used as a negative control (Fig. 1C, first lane). As shown in Fig. 1C, the deletion of C-terminal residues 1 to 385 of the N protein (N[1–385]) and N[1–379] allowed the recovery of soluble proteins that could still interact with GST-PCTD. However, a further truncation of the C-terminal arm (N[1–359]) resulted in an insoluble protein that could not be purified from cell lysates (data not shown). The deletion of the NCTD (N[1–252] and N[31–252]) or the N-terminal arm of N (N[31–391]) did not impair the interaction with the PCTD (Fig. 1). Although the intensity of the bands corresponding to N[1–252] and N[31–252] was weak after GST purification (Fig. 1C), these interactions were confirmed after His purification (Fig. 1D). Similarly, N[31–391] was not detected after purification by GST-PCTD (Fig. 1C). This result was due to the poor solubility of N[31–391], as the interaction between this domain and GST-PCTD, although weak, was observed after a polyhistidine tag pulldown assay (Fig. 1D). Finally, the recombinant truncated proteins N[31–359], N[253–391], and N[253–361] were insoluble under all the experimental purification conditions that we used (data not shown), and therefore, the interaction between the PCTD and NCTD was not tested.
Fig 1.

Deletion mapping of the PCTD binding domain of N. (A) 3D fold representation of the RSV N protein (PDB accession number 2WJ8) colored according to domains: the N- and C-arms are in blue and green, respectively, and the N- and C-terminal domains (NNTD and NCTD, respectively) are in yellow and red, respectively. Glycines 30, 252, and 361, located at the junction between the domains, are indicated. (B) Schematic illustration of truncated recombinant N proteins. Colors and residue numbers refer to the previously reported N structure (44). N deletion mutants harboring a poly-His tag at the C terminus (pET-N constructs) were coexpressed with GST-PCTD in E. coli and purified by using either GST or poly-His tags. For each deletion mutant, the ability to interact with the GST-PCTD fusion protein or insolubility is summarized on the right. (C and D) SDS-PAGE analysis of the products of copurification between truncated N-His proteins and GST-PCTD, using either GST (C) or polyhistidine (D) purification. Analysis of the products of copurification between GST and N was used as a negative control. (E) Far-UV CD spectra showing that the NNTD presents a mainly α-helical content.
These results show that the domain at residues 31 to 252 corresponding to the NNTD is sufficient to interact with the PCTD in vitro. Dynamic light scattering analysis of the recombinant NNTD also revealed the presence of a single protein population with a 5-nm diameter, corresponding to a hydrodynamic volume of 65,450 Å3 (data not shown). According to data described previously by Uversky (48), this volume corresponds to a theoretical globular protein with 250 residues, which is in agreement with the monomeric form of the His-tagged NNTD that has 231 residues. Far-UV CD spectrum analysis of the NNTD revealed a mainly α-helical content (Fig. 1E), in accordance with the crystal structure of N (44). Altogether, these observations suggest that the target area of the PCTD is present on the surface of the monomeric NNTD.
Further characterization of P residues involved in P–N-RNA interactions.
Previously, P[231–241] fused to GST was shown to be sufficient to copurify N-RNA rings and that hydrophobic residues L238 and F241 were critical for this interaction (45). Furthermore, although single substitutions of each of the 5 acidic residues of P[231–241] with Ala had no apparent impact on N-RNA binding, the simultaneous substitution of the 5 acidic residues abrogated the interaction, suggesting that negative charges also play a role in the P–N-RNA interaction (45). However, such drastic changes in the amino acid sequence could also influence the potential structure of this peptide. To further validate the potential role of acidic residues in the interaction, copurification experiments with the N-RNA complex were performed by using D233A D240A, D233A E239A, and E239A D240A double Ala mutants of GST-P[231–241]. As shown on Fig. 2, similarly to the F241A mutant, the interaction of the N-RNA complex with the E239A D240A and D233A E239A double mutants of the PCTD was abrogated, reinforcing the assumption that acidic residues play a major role in this interaction. We then tried to obtain information on the potential P[231–241] secondary structure required for the interaction with the N-RNA complex. Although previous studies showed that the PCTD is unfolded (26, 45) and is predicted to be structurally disordered by using the PONDR Server, to date, no structural data are available about RSV PCTD folding induced upon N-RNA binding. Intrinsically disordered regions of proteins are thought to adopt secondary structures, in particular α-helices, upon binding to their partners (16, 47). To investigate whether PCTD alpha-helical folding was required for its interaction with the N-RNA complex, residue L236 of P, located in the middle of the sequence of P[231-241], was replaced by proline, and the copurification of the N-RNA complex with GST-P[231-241] was tested. As shown in Fig. 2, the L236P mutation did not affect the copurification of the N-RNA complex, suggesting that the PCTD–N-RNA interaction did not depend on the alpha-helical folding of the PCTD.
Fig 2.

Characterization of PCTD residues involved in the interaction with the N-RNA complex. Shown are data for SDS-PAGE analysis after Coomassie blue staining of the products of copurification between GST-P[231–241] mutant proteins and N-His (left) and a summary of the interactions (right). P[231–241] residues critical for the interaction with N are shown in boldface type in the amino acid sequence.
Search for a potential PCTD binding site on the NNTD surface.
Based on these results, we hypothesized that the PCTD binding site on the NNTD should be biochemically complementary to P[231–241] and thus should contain both hydrophobic and basic residues. To identify such candidate regions, the fragment encompassing residues 31 to 252 of N (amino acids 31 to 252) (PDB accession number 2WJ8) was then simulated by molecular dynamics (MD) in explicit water for 30 ns. The root mean square fluctuations (RMSFs), shown in Fig. 3A, reveal three solvent-exposed flexible regions on the NNTD (whose RMSF is higher than 1.5 Å): (i) a turn (residues 103 to 108) that links the strands of the β-hairpin (βI1-βI2), (ii) a bend (residues 142 to 145) that connects the α-helix αI2 and the turn (residues 147 to 149), and (iii) a turn-3/10-helix-coil fragment (residues 171 to 176) that connects the αN3 and αN4 helices (Fig. 4C). The structures generated by the MD simulation were clustered into five similar groups based on ϕ-ψ backbone dihedral angles (Table 1). The structures closest to the center of each cluster were further used to generate protomols on the NNTD by making use of the Surflex docking package. The algorithm of protomol generation consists of spreading small hydrophobic (CH4) and hydrogen bond donor (NH) and acceptor (CO) fragments onto the protein surface and identifying pockets on the surface that make favorable interactions with the small fragments. An inverse image of the protein surface is thus generated by identifying exposed hydrophobic patches and complementary hydrogen bond donors and acceptors. A protomol is defined as the ensemble of the small fragments (CH4, NH, and CO) surrounded by a pocket. Four potential pockets (pocket I [PI] to PIV) were defined as protein atoms within 4.0 Å of the same number of corresponding protomols (Table 1 and Fig. 3B).
Fig 3.
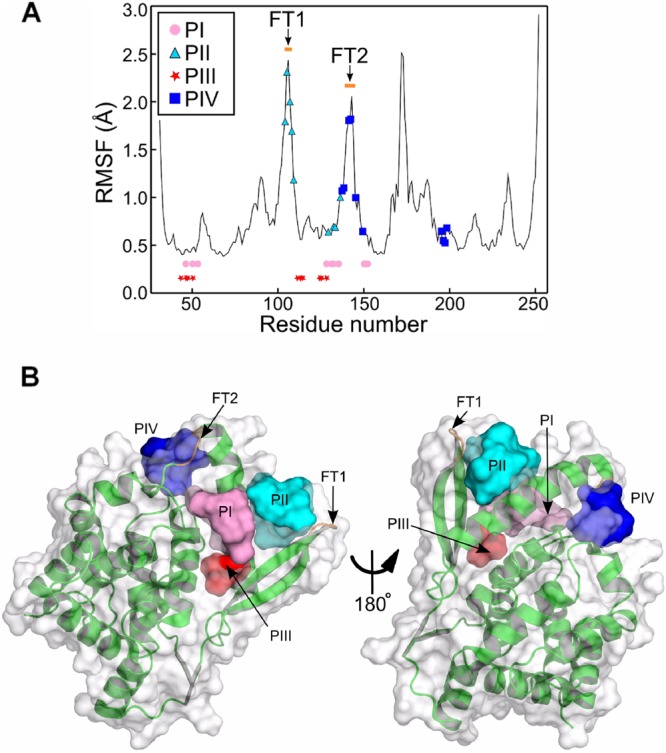
Structural analysis of the NNTD surface. (A) RMSFs of the protein backbone reveal three flexible regions of the core of the fragment encompassing residues 31 to 252 of HRSV N. The putative pockets PI and PIII are defined by members whose backbone is rigid (the RMSF is less than 0.5 Å), while PII and PIV share flexible turn 1 (FT1) and FT2 of the protein, respectively. For clarity, the residues defining PI and PIII are not represented on the RMSF curve but rather on two straight lines. (B) Four pockets were identified by the Surflex molecular docking package. The protomols defining the four pockets are represented through their solvent-accessible surfaces. PI (pink) is surrounded by flat areas, while PII (cyan) and PIV (blue) are located at sharp edges of the protein, having FT1 and FT2 (orange) as components. PIII (red) is an extension of PI.
Fig 4.

Close-up of PI and PII. (A) PI is defined by hydrophobic side chains and hydrogen bond acceptors and donors positioned on rigid fragments of N. Hydrogen bonds and salt bridges are represented by black and red dotted lines, respectively. (B) PII is hosted between an α-helix and the flexible turn of a β-sheet. (C) Sequence alignment of NNTDs of HRSV, BRSV, and PVM by ClustalW2. Strictly conserved residues are indicated by stars, and partially conserved residues are indicated by points under the alignment. Secondary-structure elements are indicated above the sequence, shown in yellow according to the NNTD and in orange for the variable region (44). Residues mutated in this study and potentially implicated in PCTD-N interactions are indicated in boldface type and colored according to their biochemical properties (blue, basic residues; orange, hydrophobic residues; black, neutral residues).
Table 1.
Putative binding pockets defined as protomols on the surface of five representative structures of the NNTD
| Cluster | % of structure | No. of protomols | Presence of pocketa |
|||
|---|---|---|---|---|---|---|
| PI | PII | PIII | PIV | |||
| 1 | 54.3 | 4 | + | + | + | + |
| 2 | 34.0 | 4 | + | + | + | + |
| 3 | 8.3 | 1 | − | − | + | − |
| 4 | 2.8 | 4 | + | + | + | + |
| 5 | 0.6 | 1 | − | − | + | − |
+ and − indicate whether the pocket is observed or not, respectively.
The first pocket (PI) is defined by hydrophobic patches of side chains (SCs) of residues M50, I53, and Y135 and the aliphatic part of the R150 SC; hydrogen bond acceptors of the E128, S131, H151, and D152 SCs; and hydrogen bond donors of the K46, S131, R132, and H151 SCs (Fig. 4A). PI is stabilized by two salt bridges, R150-D152 and E128-R132, and a hydrogen bond between K46 and H151 (Fig. 4A). The second pocket (PII) is settled between the turn of the βI1-βI2 hairpin and the middle of the α-helix αI2 (Fig. 3B and 4B). It is determined by the solvent exposure of the hydrophobic SCs of residues I104, M109, and I129 and the aliphatic part of the K136 SC; hydrogen bond donor SCs of N105, K107, R132, and K133; and the hydrogen bond acceptors of the N105 SC and oxygen backbone atoms of I104 and E108 (Fig. 4B). PII components (I104, N105, K107, E108, and M109) positioned on the turn of the β-hairpin are characterized by higher RMSF values than those for the core of the protein (Fig. 3A). This is in agreement with the high solvent exposure of the β-hairpin turn (Fig. 3B and 4B) and defines a sharp protein edge, which should make an interaction with the P protein unstable.
The third pocket (PIII) is an extension of PI (Fig. 3B), separated by a saddle point made by the exposed SC of M50 and limited by K46 and E128. The two latter residues are not connected by a salt bridge, but rather, they are involved in electrostatic interactions with neighbor residues (E128-R132 and K46-H151) (Fig. 4A). PIII is made by hydrophobic residues L47, M50, F111, V113, L114, and I125 and the aliphatic part of K46; hydrogen bond acceptors of the E128 and Q124 SCs; and the hydrogen bond donor and acceptor of the backbone nitrogen and oxygen atoms of E112.
The fourth pocket (PIV) is defined by hydrophobic SCs of residues M138, M142, V145, Y149, and Y197; hydrogen bond donors of the K137 and K198 side chains; and hydrogen bond acceptors of E141 and the oxygen backbone atoms of residues K195 and R196. PIV components located on the α-helix αI2 C terminus (K137, M138, and E141) and the bend (residues 142 to 145) have high RMSF values (Fig. 3A), and similar to PII, they belong to highly solvated regions that define sharp protein edges, decreasing the strength of the interaction with the P protein. The flexible bend (residues 142 to 145) behaves as an entropic barrier between PI and PIV, making it difficult for the P protein to bind to both PI and PIV at the same time (Fig. 3B).
Effects of targeted N gene mutations on polymerase activity.
The MD observations led us to characterize the potential roles of 21 hydrophobic and basic residues of N in P binding by site-directed mutagenesis. As MD data revealed that the putative pockets PI and PII were found in 91% of the total number of structures (Table 1), this mutagenesis study focused on residues found in PI (K46, M50, I53, S131, R132, Y135, R150, and H151) and PII (I104, R107, M109, I129, K133, and K136). The role of PIII, described as an extension of PI, was also investigated by mutating residues F111 and I125. Also, the potential role of PIV, for which the strength of an interaction with P was predicted to be low, was assessed with the K137A mutant. In addition, residues R101, K110, L139, and K140, located close to PI and PII, were included in this study. A sequence alignment of the HRSV, BRSV, and pneumovirus of mice (PVM) N[31–252] domains revealed that, although located within the less conserved segment of the NNTD, most of the residues selected are highly conserved among pneumovirus N proteins (Fig. 4C). All of these residues were replaced by Ala to generate 21 mutant proteins. As assessed by Western blot analysis, all of the mutant N′s presented were expressed in similar amounts in eukaryotic cells (Fig. 5A). First, the functionality of the N mutants was tested with the polymerase complex, using an HRSV plasmid-based minigenome system, as described previously (11, 46). Briefly, the dicistronic subgenomic replicon pM/Luc was cotransfected into BSRT7/5 cells expressing T7 RNA polymerase, together with plasmids pN, pP, pL, and pM2-1, resulting in the replication and transcription of the minigenome. Hence, the production of the Luc protein was dependent on these processes. As shown on Fig. 5B, 10 mutants displayed a reduction of luciferase activity of about 50% compared to the control, which is native N. The R132A mutation had the strongest effect, reducing luciferase activity to near-background levels. Like R132, most of the other critical residues that affected functions are part of PI (K46, M50, S131, Y135, R150, and H151). In addition, the replacement of residues K136, L139, and K140 by Ala strongly reduced the luciferase activity. It is noteworthy that the I53A and I104A mutations led to an increase of the polymerase activity. Altogether, these results suggest that most of the residues of PI are critical for polymerase activity.
Fig 5.

Effects of targeted mutations on viral RNA synthesis. (A) Western blot analysis showing the efficient expression of N mutant proteins in BRST/7 cells. (B and C) Polymerase activity assay in the presence of N (B) and P (C) mutants. BSRT7/5 cells were transfected at 37°C with plasmids encoding the wild-type (WT) or mutant P or N proteins, the M2-1 and L proteins, and the pM/Luc replicon, together with pCMV-βGal for transfection standardization. Viral RNA synthesis was quantified by measuring the luciferase activity after cell lysis 24 h after transfection. Each luciferase minigenome activity value was normalized to β-galactosidase expression values and is the average of data from three independent experiments performed in triplicate. Error bars represent standard deviations calculated based on data from three independent experiments performed in triplicate.
Second, the effect of mutations affecting the interaction between the PCTD and the N-RNA complex on polymerase activity was assayed. The same minigenome assay was then performed with the P protein either deleted of residues 231 to 241 or with single or double mutants of residues 231 to 241 of P. As shown in Fig. 5C, the deletion of residues 231 to 241 totally impaired the polymerase activity, as revealed by the luciferase activity. The mutation of P residue L238 or F241, previously identified as being critical for PCTD–N-RNA interactions, induced a strong or a total inhibition of the luciferase activity, respectively. In contrast, no attenuation of luciferase activity was detected with the D233A, D235A, L236A, and L236P mutations, which did not affect the interaction (Fig. 2) (45). Finally, whereas a single substitution of acidic residues E239 and D240 induced only a slight decrease of the luciferase activity, the E239A D240A double mutant displayed a reduction of luciferase activity of ≥50% compared to the control. Thus, P mutations affecting in vitro PCTD–N-RNA binding also affected RNA polymerase activity, revealing that this interaction is potentially critical for replication and/or transcription steps of the RSV cycle.
Identification of residues of N involved in the interaction with the PCTD.
To determine whether the defect in the polymerase activity associated with mutations of selected residues was associated with a defect of the interaction between P and N, immunoprecipitation assays were performed on cells expressing the N mutants and an HA-tagged P protein. As shown in Fig. 6A, mutations of residues K46, I53, S131, K136, L139, and K140 in N still resulted in an N-P interaction. In contrast, no interaction was detected between HA-P and N with the M50A, R132A, Y135A, R150A, and H151A substitutions. To further investigate the role of these N residues in the specific interaction with the PCTD, a GST pulldown assay was used. N mutant proteins with a C-terminal poly-His tag, GST, and GST-PCTD were independently expressed in E. coli and purified to near homogeneity (Fig. 6B). Before the pulldown experiments were performed, recombinant N proteins were biochemically characterized. When analyzed by native gel electrophoresis, the wild-type N protein migrated as two species corresponding to N-RNA rings comprising 10 and 11 N monomers (Fig. 6C) (44, 45). Despite slight differences in electrophoretic mobilities reflecting changes in the overall charge of these proteins (due to amino acid substitutions), the pattern of migration with two bands was recovered for all of the mutants, revealing that recombinant proteins assembled as N-RNA rings. The N mutants also presented an RNA content similar to that of wild-type N, as assessed by UV spectrum analysis (optical density ratio of 260 nm/280 nm comprised between 1.44 and 1.36 [data not shown]). Having shown that wild-type and mutant N proteins had similar ring organizations, GST pulldown experiments were then performed. Fivefold excesses of GST or GST-PCTD fixed on beads were incubated for 1 h in the presence of either native or mutant N′s. After extensive washes, the presence of N was revealed by Coomassie blue staining following SDS-PAGE. The interaction between GST-PCTD and the N protein was impaired to nearly background levels when residues K46, M50, R132, Y135, R150, and H151 were replaced with Ala (Fig. 6D). Altogether, these data confirmed the direct involvement of residues K46, M50, R132, Y135, R150, and H151 of PI in the interaction with the PCTD. These results also indicate that residues K136, L139, and K140, which are located outside PI and which are critical for viral RNA synthesis, are not critical for this interaction.
Fig 6.

Identification of N residues involved in the N-P interaction. (A) Western blot (WB) analysis of the N-P interaction after an immunoprecipitation assay. Cells were transiently transfected with constructs allowing the expression of the HA-P fusion protein and N mutant proteins. Immunoprecipitations (IP) with whole-cell extracts were performed with an anti-HA antibody. (B) SDS-PAGE analysis of purified recombinant N proteins (left) and GST or GST-PCTD (right). (C) Analysis of N mutants by native PAGE. Arrows indicate the two types of rings (comprising 10 and 11 N monomers) characteristic of the purified recombinant N protein. (D) SDS-PAGE analysis of GST pulldown experiments. Recombinant N-RNA complexes assembled as rings were incubated in the presence of beads coupled with either GST (left) or the GST-PCTD protein (right), and the interaction between GST-PCTD and recombinant N proteins was revealed by Coomassie blue staining. The I104A mutant was used as a control for interactions with PII.
Validation of PI as a site of interaction with the PCTD.
The replacement of residues by Ala may not be sufficient to highlight the role of those residues in the interaction with the PCTD. Therefore, the impact of changes on the biochemical properties of selected PI residues was investigated. This investigation specifically focused on residues I53 and R132, as their replacement by Ala either improved or abrogated the polymerase activity, respectively. To better characterize the role of I53, which is located deep inside PI, the importance of hydrophobicity and steric hindrance at this position was tested by replacing Ile with either Ser or Trp. As with the replacement by Ala, the I53S mutation improved the polymerase complex activity, whereas the I53W mutation abrogated the activity (Fig. 7A). The basic charge of residue 132 may be critical for polymerase activity, and this was investigated by studying the impact of its substitution with either a negatively charged Glu residue or a positively charged Lys residue. As described above for the replacement of this residue with Ala, the R132E substitution impaired polymerase activity, whereas the R132K mutation restored it (Fig. 7A).
Fig 7.

PI constitutes the site of interaction with the PCTD. (A) Minigenome assay. Normalized luciferase activity was determined for the I53 and R132 mutants. Error bars represent standard deviations calculated based on data from three independent experiments performed in triplicate. (B) Observation of cellular colocalization between P and N mutants in cytoplasmic inclusion bodies. N and P proteins were coexpressed in BSRT7/5 cells; cells were then fixed at 24 h posttransfection and labeled with anti-P (green) and anti-N (red) antibodies; and the distribution of viral proteins was observed by fluorescence microscopy. Nuclei were stained with Hoechst 33342. Scale bars, 10 μm. (C) Western blot analysis of products of immunoprecipitation assays preformed with an anti-HA antibody on cells coexpressing HA-P and N mutant proteins. (D) Analysis of GST pulldown assays performed with GST-PCTD and I53 and R132 mutant recombinant N proteins assembled as N-RNA rings. (E) Far-UV CD spectra showing the absence of major conformational changes between wild-type N (black) and the R132A (light gray) or I53W (gray) mutant.
The substitution of residues R132 and I53 may have affected subcellular localization when coexpressed with P. As previously described, when expressed individually, P and N present a diffuse cytoplasmic localization, whereas their coexpression leads to the formation of inclusion bodies (IBs) similar to the structures observed during RSV infection, where P and N colocalize (12, 13). A total loss of IB formation was observed with the R132A and R132E N mutants, which also correlated with abrogated polymerase activity (Fig. 7A and B). In contrast, the R132K mutation restored the formation of IBs and also restored polymerase activity. Similarly, although IBs were still observed with the I53A and I53S N mutants, the I53W mutation prevented the formation of IBs. By analogy to IBs that are observed during rabies virus or vesicular stomatitis virus (VSV) infection (18, 25), the correlation between IB formation and efficient polymerase activity strongly suggests that RSV IBs indeed constitute the site of viral replication and transcription. The localization of N mutants in IBs also correlated with the presence (I53S and R132K) of an N-P interaction, whereas the absence of IBs correlated with a loss (I53W and R132E) of the N-P interaction, as revealed by immunoprecipitation (Fig. 7C). These observations were supported by data from in vitro GST pulldown experiments. The interaction between GST-PCTD and the I53W, R132A, or R132E mutant N-RNA rings was totally abrogated (Fig. 7D), although those recombinant proteins were still able to form rings containing RNA (Fig. 6C and data not shown). However, these results may not be due to potential major conformational changes induced upon mutation, as assessed by far-UV CD spectrum analysis, as the N mutant proteins presented secondary structures similar to those of the wild-type N protein, with a mainly α-helical content (Fig. 7E).
Overall, these data validate the major implication of the roles of some residues of PI, particularly I53 and R132, in both the interaction with the PCTD and polymerase activity. These results suggest that the positive charge at position 132 is critical and that, more than hydrophobicity, the lateral-chain length of residue I53 is important for the PCTD–N-RNA interaction.
DISCUSSION
As for all viruses of the order Mononegavirales, the P–N-RNA interaction is critical for RNP recognition by the RSV polymerase complex and, as a consequence, for the synthesis of viral RNA. P is thought to be a connector between L and N. The identification of the sites of interactions between P and its partners thus constitutes a major interest not only for an understanding of the mechanisms of viral RNA synthesis but also for the development of antiviral drug design strategies. The mapping of the P binding site on the RSV N protein was previously investigated by several groups using different approaches (13, 22, 24, 31, 32). However, contradictory results were obtained from those studies, in which the nature of N (N-RNA or N°) was not defined. For example, Krishnamurthy and Samal used a yeast two-hybrid screen coupled to an immunoprecipitation assay with HeLa cells coexpressing the BRSV P and N proteins and found that an N-P interaction was abolished by internal and C-terminal deletions of N (24). Using a similar approach, i.e., immunoprecipitation in HEp-2 cells coexpressing the BRSV P and truncated N proteins, Khattar et al. (22) found that P binding required residues 244 to 290 and 338 to 364 but not C-terminal residues 365 to 391. In the same way, Garcia-Barreno et al. (13) showed that the deletion of the last 39 amino acids of N still resulted in coimmunoprecipitation with P but inhibited the formation of IBs. On the contrary, based on a study of the interaction between P and chimeric PVM and HRSV N proteins, Stokes et al. (43) determined that residues 352 to 369 near the C terminus of N were important for P binding. A characterization of N monoclonal antibodies led to conclusions on the implication of the C terminus of N in the interaction with P (32). Finally, a binding inhibition assay between P and N in the presence of peptides derived from the N sequence revealed a role for the N-terminal extremity of N (31). In summary, although focusing mainly on the C-terminal part of N and particularly on the C-terminal residues, all of those studies did not succeed in identifying a consensus site for P binding on N.
Recently, the determination of the X-ray structure of HRSV N bound to RNA (44) has opened the door to a rational search for the P binding site(s). N-RNA rings were purified based on the specific interaction between N and the PCTD, after the coexpression of recombinant GST-PCTD and N proteins in bacteria. In the present work, the 3D atomic structure of RSV N was used as a basis to search for a P binding site on the N surface. First, the interaction between the PCTD and N structural domains was investigated by coexpressing GST-PCTD with either the N- or C-arm or the NNTD or NCTD of N. Altogether, the data demonstrated that the NNTD (residues 31 to 252) interacted specifically with the PCTD. These results also revealed that the deletion of N-terminal residues up to D31 and C-terminal residues up to K379 in N did not abrogate the PCTD-N interaction, indicating that the most N- and C-terminal residues of N were not necessary for this interaction.
To identify a potential PCTD binding site on the NNTD and the critical residues and potential folding of P[231–241], the minimal domains of interaction with N were investigated initially. As previously reported (45), the data confirmed that hydrophobic and acidic P[231–241] residues are critical for this interaction. Furthermore, as the replacement of L236 by a proline did not impair the interaction with N, P[231–241] binding may not depend on α-helix folding. Based on these observations, MD simulations were used to search for potential complementary sites at the surface of the NNTD. This approach defined four candidate pockets constituted mainly of hydrophobic and basic residues. The potential involvement of 21 residues of N in the N-P interaction was investigated by Ala scanning. As the N-P interaction is required for viral RNA synthesis by the polymerase complex, a functional HRSV minireplicon system was used to screen for the importance of these residues for polymerase activity. This approach highlighted a critical role of all 8 tested residues of PI (K46, M50, I53, S131, R132, Y135, R150, and H151) but also of three nearby residues (K136, L139, and K140) (Fig. 8A). It must be noted that this system does not allow discriminations between the roles of the residues in replication and/or transcription and other off-target effects. Immunoprecipitation and pulldown experiments were then combined to determine whether the effect of mutations on the inhibition of viral RNA synthesis correlated with a defect of the N-P interaction. Above all, using an in vitro pulldown assay between GST-PCTD and mutant N-RNA rings, we confirmed that the Ala substitution of residues K46, M50, R132, Y135, R150, and H151 of PI abrogated the interaction (Fig. 6D), revealing a direct role of these residues in the interaction with the PCTD. On the contrary, a PCTD-N interaction was conserved with K136A, L139A, and K140A N mutants, suggesting that these residues may have another function within the polymerase complex and could possibly be involved in the interaction with either another domain of P, the L protein, or an unidentified cellular partner.
Fig 8.
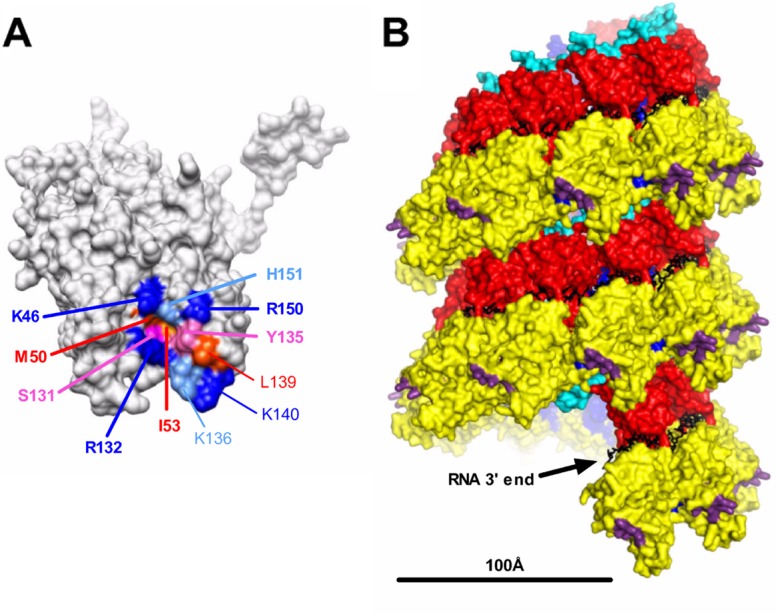
Localization of the PI region on the nucleocapsid. (A) Localization at the surface of the N monomer of residues identified in this study as being critical for RNA synthesis. Residues of PI are indicated in boldface type. Residues are colored according to their biochemical properties (blue, basic residues; red, hydrophobic residues; pink, neutral residues). (B) Side view of the helical nucleocapsid modeled from the ring using a pitch of 70 Å according data reported previously by Tawar et al. (44). Structural domains are colored; i.e., the NCTD and NNTD are in red and yellow, respectively, and the C-arm and N-arm are in light blue and blue, respectively. Residues critical for the interaction with the PCTD are shown in purple, and RNA is shown in black. The bar indicates 100 Å. The structure of the RSV N protein bound to RNA (44) was obtained from the Protein Data Bank (PDB accession number 2WJ8) and visualized by using UCSF Chimera molecular graphics software (34).
Most of the residues identified are located in the region formed by the αN1 helix (residues 37 to 54), the β-hairpin (residues 97 to 114) followed by the αI2 helix (residues 121 to 139), and the αI2-αN3 loop (residues 140 to 154) (Fig. 4C) and form a continuous surface on N, at the tip of the spike projection formed by the NNTD (Fig. 8A). A comparison of the 3D structures of Mononegavirales N proteins of RSV, Borna disease virus, and VSV also revealed that this region is highly specific for the RSV N protein (44). A representation of this surface on the modeled helical nucleocapsid (44) shows that it should be located at the periphery of the helical nucleocapsid and thus should be easily accessible for P binding (Fig. 8B), supporting the results presented here. It is noteworthy that this site is spatially close to the NCTD located down in the same axis of the subsequent turn of the helix. This observation could explain the results reported previously by other groups who have investigated P-N interactions, as deletions inside the NCTD could destabilize the PI orientation and, thus, the association between the PCTD and N. Based on these observations, and in order to obtain information on the potential way of insertion of the most C-terminal residues of P within PI, the role of the two residues I53 and R132 in the interaction with the PCTD was characterized. These residues were of particular interest, as their replacement by Ala led to an increase (I53) or a total defect (R132) in polymerase activity. Furthermore, with I53 being located inside the cavity of the pocket and R132 being located at the periphery, the substitution of these residues aided in discriminating the orientation of PCTD residues with regard to residues of PI. The mutation of these residues affected PCTD binding without inducing major conformational changes of N. As the replacement of I53 by Trp abrogated the PCTD-N interaction, this suggested that, more than hydrophobicity, steric hindrance inside PI is critical for the interaction with P. In the same way, a positive charge at position 132 is also necessary for the N-P interaction. These results correlate with the critical role of the most C-terminal residues F241, D240, E239, and L238 of P in the interaction with N-RNA rings. We can thus hypothesize that F241 or L238 could insert inside PI of N and that the surrounding electrostatic interactions between N positive charges and P negative charges should play a major role in driving this interaction. PI of N thus constitutes a good target for the development of RSV-specific antiviral drugs that interfere with the P–N-RNA interaction.
Mechanisms of interaction between P and N-RNA complexes have already been characterized for other viruses belonging to the order Mononegavirales. For measles virus (MV) and Sendai virus (SeV), which belong to the genera Morbillivirus and the Respirovirus, respectively, of the family Paramyxoviridae, the C-terminal end of P forms a small and dynamic three-helical bundle that stabilizes upon binding to the disordered C-terminal domain of N, termed NTAIL (19, 20, 23). For the Rhabdoviridae (rabies virus and VSV), the C-terminal domain of P that binds to N is much larger and more structured than those of MV and SeV (30, 36). Moreover, for both VSV and rabies virus, the PCTD lies on top of an N protomer and is pinched by NCTD loops from two adjacent N monomers (14, 37). In contrast, for mumps virus, which belongs to the genus Rubulavirus of the family Paramyxoviridae, it was shown previously that the P binding region of N is not located at the C terminus of N but rather in the N-terminal region of N, suggesting that the sites of attachment for the polymerase vary among paramyxoviruses (23). However, by analogy with MV and SeV, it was proposed previously that the poorly folded C-terminal arm of RSV N constitutes the binding site of RSV P (38, 39). The identification of the P binding site of RSV on the NNTD reveals that for this virus, the short (10 residues) and intrinsically disordered C terminus of P interacts with a highly structured domain of N. Nevertheless, further investigations will be necessary to determine if the PCTD could fold upon binding to N. It is noteworthy that two kinds of N-P complexes may exist in cells and may play a specific role. For all negative-strand viruses, nucleoproteins have to be kept soluble by interacting with P to prevent its interaction with cellular RNAs. This complex is designated the N°-P complex. Therefore, several P binding sites may exist on the surface of N, which might depend on different conformational states and engage other domains of P.
ACKNOWLEDGMENTS
We thank Bernard Delmas for discussion and Julian Hiscox and Alejandra Tortorici for critical reading of the manuscript.
Molecular graphic images were produced by using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH grant P41 RR-01081). This work was carried out with the financial support of the Agence Nationale de la Recherche, specific program ANR Blanc 2011 (ANR 11 BSV8 024 01), and was granted access to the high performance computing resources of CCRT/CINES/IDRIS under the allocation 2012-i2012076378 made by GENCI (Grand Equipement National de Calcul Intensif).
Footnotes
Published ahead of print 23 May 2012
REFERENCES
- 1. Asenjo A, Rodriguez L, Villanueva N. 2005. Determination of phosphorylated residues from human respiratory syncytial virus P protein that are dynamically dephosphorylated by cellular phosphatases: a possible role for serine 54. J. Gen. Virol. 86:1109–1120 [DOI] [PubMed] [Google Scholar]
- 2. Barik S. 1992. Transcription of human respiratory syncytial virus genome RNA in vitro: requirement of cellular factor(s). J. Virol. 66:6813–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brooks BR, et al. 2009. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30:1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Castagne N, et al. 2004. Biochemical characterization of the respiratory syncytial virus P-P and P-N protein complexes and localization of the P protein oligomerization domain. J. Gen. Virol. 85:1643–1653 [DOI] [PubMed] [Google Scholar]
- 6. Chenal A, et al. 2002. Does fusion of domains from unrelated proteins affect their folding pathways and the structural changes involved in their function? A case study with the diphtheria toxin T domain. Protein Eng. 15:383–391 [DOI] [PubMed] [Google Scholar]
- 7. Collins PL, Crowe JE. 2007. Respiratory syncytial virus and metapneumovirus, p 1601–1646 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 8. Collins PL, et al. 1995. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc. Natl. Acad. Sci. U. S. A. 92:11563–11567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cowton VM, McGivern DR, Fearns R. 2006. Unravelling the complexities of respiratory syncytial virus RNA synthesis. J. Gen. Virol. 87:1805–1821 [DOI] [PubMed] [Google Scholar]
- 10. Darden T, York DM, Pedersen L. 1993. Particle mesh Ewald: An N × log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092 [Google Scholar]
- 11. Fix J, Galloux M, Blondot ML, Eleouet JF. 2011. The insertion of fluorescent proteins in a variable region of respiratory syncytial virus L polymerase results in fluorescent and functional enzymes but with reduced activities. Open Virol. J. 5:103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garcia J, Garcia-Barreno B, Vivo A, Melero JA. 1993. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 195:243–247 [DOI] [PubMed] [Google Scholar]
- 13. Garcia-Barreno B, Delgado T, Melero JA. 1996. Identification of protein regions involved in the interaction of human respiratory syncytial virus phosphoprotein and nucleoprotein: significance for nucleocapsid assembly and formation of cytoplasmic inclusions. J. Virol. 70:801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Green TJ, Luo M. 2009. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor, P. Proc. Natl. Acad. Sci. U. S. A. 106:11713–11718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grosfeld H, Hill MG, Collins PL. 1995. RNA replication by respiratory syncytial virus (RSV) is directed by the N, P, and L proteins; transcription also occurs under these conditions but requires RSV superinfection for efficient synthesis of full-length mRNA. J. Virol. 69:5677–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Habchi J, Longhi S. 2012. Structural disorder within paramyxovirus nucleoproteins and phosphoproteins. Mol. Biosyst. 8:69–81 [DOI] [PubMed] [Google Scholar]
- 17. Hardy RW, Wertz GW. 1998. The product of the respiratory syncytial virus M2 gene ORF1 enhances readthrough of intergenic junctions during viral transcription. J. Virol. 72:520–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog. 6:e1000958 doi:10.1371/journal.ppat.1000958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Houben K, Marion D, Tarbouriech N, Ruigrok RW, Blanchard L. 2007. Interaction of the C-terminal domains of Sendai virus N and P proteins: comparison of polymerase-nucleocapsid interactions within the paramyxovirus family. J. Virol. 81:6807–6816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jensen MR, et al. 2008. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: application to the molecular recognition element of Sendai virus nucleoprotein. J. Am. Chem. Soc. 130:8055–8061 [DOI] [PubMed] [Google Scholar]
- 21. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Physiol. 79:926–935 [Google Scholar]
- 22. Khattar SK, Yunus AS, Collins PL, Samal SK. 2000. Mutational analysis of the bovine respiratory syncytial virus nucleocapsid protein using a minigenome system: mutations that affect encapsidation, RNA synthesis, and interaction with the phosphoprotein. Virology 270:215–228 [DOI] [PubMed] [Google Scholar]
- 23. Kingston RL, Baase WA, Gay LS. 2004. Characterization of nucleocapsid binding by the measles virus and mumps virus phosphoproteins. J. Virol. 78:8630–8640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krishnamurthy S, Samal SK. 1998. Identification of regions of bovine respiratory syncytial virus N protein required for binding to P protein and self-assembly. J. Gen. Virol. 79(Pt 6):1399–1403 [DOI] [PubMed] [Google Scholar]
- 25. Lahaye X, et al. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J. Virol. 83:7948–7958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Llorente MT, et al. 2006. Structural analysis of the human respiratory syncytial virus phosphoprotein: characterization of an alpha-helical domain involved in oligomerization. J. Gen. Virol. 87:159–169 [DOI] [PubMed] [Google Scholar]
- 27. Llorente MT, et al. 2008. Structural properties of the human respiratory syncytial virus P protein: evidence for an elongated homotetrameric molecule that is the smallest orthologue within the family of paramyxovirus polymerase cofactors. Proteins 72:946–958 [DOI] [PubMed] [Google Scholar]
- 28. MacKerell AD, Jr, Banavali N, Foloppe N. 2000. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 56:257–265 [DOI] [PubMed] [Google Scholar]
- 29. Mason SW, et al. 2003. Interaction between human respiratory syncytial virus (RSV) M2-1 and P proteins is required for reconstitution of M2-1-dependent RSV minigenome activity. J. Virol. 77:10670–10676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mavrakis M, McCarthy AA, Roche S, Blondel D, Ruigrok RW. 2004. Structure and function of the C-terminal domain of the polymerase cofactor of rabies virus. J. Mol. Biol. 343:819–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murphy LB, et al. 2003. Investigations into the amino-terminal domain of the respiratory syncytial virus nucleocapsid protein reveal elements important for nucleocapsid formation and interaction with the phosphoprotein. Virology 307:143–153 [DOI] [PubMed] [Google Scholar]
- 32. Murray J, Loney C, Murphy LB, Graham S, Yeo RP. 2001. Characterization of monoclonal antibodies raised against recombinant respiratory syncytial virus nucleocapsid (N) protein: identification of a region in the carboxy terminus of N involved in the interaction with P protein. Virology 289:252–261 [DOI] [PubMed] [Google Scholar]
- 33. Navarro J, Lopez-Otin C, Villanueva N. 1991. Location of phosphorylated residues in human respiratory syncytial virus phosphoprotein. J. Gen. Virol. 72(Pt 6):1455–1459 [DOI] [PubMed] [Google Scholar]
- 34. Pettersen EF, et al. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 35. Phillips JC, et al. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ribeiro EA, Jr, et al. 2008. Solution structure of the C-terminal nucleoprotein-RNA binding domain of the vesicular stomatitis virus phosphoprotein. J. Mol. Biol. 382:525–538 [DOI] [PubMed] [Google Scholar]
- 37. Ribeiro EA, Jr, et al. 2009. Binding of rabies virus polymerase cofactor to recombinant circular nucleoprotein-RNA complexes. J. Mol. Biol. 394:558–575 [DOI] [PubMed] [Google Scholar]
- 38. Ruigrok RW, Crepin T. 2010. Nucleoproteins of negative strand RNA viruses; RNA binding, oligomerisation and binding to polymerase co-factor. Viruses 2:27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruigrok RW, Crepin T, Kolakofsky D. 2011. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 14:504–510 [DOI] [PubMed] [Google Scholar]
- 40. Ruppert J, Welch W, Jain AN. 1997. Automatic identification and representation of protein binding sites for molecular docking. Protein Sci. 6:524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ryckaert JP, Ciccotti G, Berendsen HJC. 1977. Numerical-integration of Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Phys. 23:327–341 [Google Scholar]
- 42. Sanchez-Seco MP, Navarro J, Martinez R, Villanueva N. 1995. C-terminal phosphorylation of human respiratory syncytial virus P protein occurs mainly at serine residue 232. J. Gen. Virol. 76(Pt 2):425–430 [DOI] [PubMed] [Google Scholar]
- 43. Stokes HL, Easton AJ, Marriott AC. 2003. Chimeric pneumovirus nucleocapsid (N) proteins allow identification of amino acids essential for the function of the respiratory syncytial virus N protein. J. Gen. Virol. 84:2679–2683 [DOI] [PubMed] [Google Scholar]
- 44. Tawar RG, et al. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283 [DOI] [PubMed] [Google Scholar]
- 45. Tran TL, et al. 2007. The nine C-terminal amino acids of the respiratory syncytial virus protein P are necessary and sufficient for binding to ribonucleoprotein complexes in which six ribonucleotides are contacted per N protein protomer. J. Gen. Virol. 88:196–206 [DOI] [PubMed] [Google Scholar]
- 46. Tran TL, et al. 2009. The respiratory syncytial virus M2-1 protein forms tetramers and interacts with RNA and P in a competitive manner. J. Virol. 83:6363–6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Uversky VN. 2011. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 43:1090–1103 [DOI] [PubMed] [Google Scholar]
- 48. Uversky VN. 2002. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11:739–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Valarcher JF, Taylor G. 2007. Bovine respiratory syncytial virus infection. Vet. Res. 38:153–180 [DOI] [PubMed] [Google Scholar]
- 50. Yu Q, Hardy RW, Wertz GW. 1995. Functional cDNA clones of the human respiratory syncytial (RS) virus N, P, and L proteins support replication of RS virus genomic RNA analogs and define minimal trans-acting requirements for RNA replication. J. Virol. 69:2412–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]