

Abstract
Studies from a number of laboratories have shown that the myeloid lineage is prominent in human cytomegalovirus (HCMV) latency, reactivation, dissemination, and pathogenesis. Existing as a latent infection in CD34+ progenitors and circulating CD14+ monocytes, reactivation is observed upon differentiation to mature macrophage or dendritic cell (DC) phenotypes. Langerhans' cells (LCs) are a subset of periphery resident DCs that represent a DC population likely to encounter HCMV early during primary infection. Furthermore, we have previously shown that CD34+ derived LCs are a site of HCMV reactivation ex vivo. Accordingly, we have utilized healthy-donor CD34+ cells to study latency and reactivation of HCMV in LCs. However, the increasing difficulty acquiring healthy-donor CD34+ cells—particularly from seropositive donors due to the screening regimens used—led us to investigate the use of CD14+ monocytes to generate LCs. We show here that CD14+ monocytes cultured with transforming growth factor β generate Langerin-positive DCs (MoLCs). Consistent with observations using CD34+ derived LCs, only mature MoLCs were permissive for HCMV infection. The lytic infection of mature MoLCs is productive and results in a marked inhibition in the capacity of these cells to promote T cell proliferation. Pertinently, differentiation of experimentally latent monocytes to the MoLC phenotype promotes reactivation in a maturation and interleukin-6 (IL-6)-dependent manner. Intriguingly, however, IL-6-mediated effects were restricted to mature LCs, in contrast to observations with classical CD14+ derived DCs. Consequently, elucidation of the molecular basis behind the differential response of the two DC subsets should further our understanding of the fundamental mechanisms important for reactivation.
INTRODUCTION
Human cytomegalovirus (HCMV) represents an opportunistic pathogen that is a major cause of disease in a number of immunocompromised patient populations (30, 60). A significant contribution to morbidity in the clinical setting results from the reactivation of latent HCMV, particularly in seropositive bone marrow transplant recipients (40). Although the mechanisms that govern HCMV reactivation have not been fully elucidated, work from a number of laboratories has shown that CD34+ hematopoietic cells, and the early granulocyte-macrophage progenitors derived from them, which are normally resident in the bone marrow are an important site for the carriage of HCMV latency in vivo (27, 34, 53) and that reactivation is intrinsically linked with the differentiation to a more mature myeloid cell phenotype (19, 37, 45, 56, 59, 64).
Langerhans' cells (LCs) are a unique population of dendritic cells (DCs) resident in the epidermis and a number of mucosal tissues (e.g., nasal, oral, vaginal, and corneal). They are derived from bone marrow progenitors (26) and exhibit a capacity for self-renewal (11, 36), as well as exhibiting prodigious longevity for a DC, with a half-life of up to 78 days documented (62) and, in one case, a donor's LCs were observed to persist in the recipient for more than 12 months after a skin graft procedure (23). Their generation (and/or survival) both in vitro and in vivo is acutely dependent on transforming growth factor β (TGF-β) (4, 25, 57)—TGF-β knockout mice do not possess LCs—and can be characterized by their (almost) unique expression of the lectin molecule, Langerin (CD207) (6, 15, 42, 61), along with the coexpression of cutaneous leukocyte antigen, E-Cadherin, and class II major histocompatibility complex (MHC) molecules, as well as intracellular Birkbeck granules (reviewed in reference 35). LCs were classically described as potent activators of T cell immunity (50); however, more recent studies with cytolytic viruses argue that the ability of skin resident DCs to respond is subverted specifically by cytolytic viruses and that the major immune response is mediated by cross-presentation by other DC subtypes (2, 5, 21).
In vitro, Langerhans-like cells can be generated from CD34+ cells (CD34+ derived LCs) under certain cell culture conditions, the study of which has been used to ascribe potential LC functions (9, 52, 57, 58). Thus, in the context of infection, it was hypothesized that LCs existing in the periphery encountered infectious agents at epithelial surfaces and represent the first line of defense, which, in part, would involve the activation of a profound CD8 cytotoxic response. Although murine studies suggest that the reality may be more complex than this (for example, cytolytic viruses induce LC apoptosis, and thus the antigen presentation is mediated by a blood-derived CD8+ population of DCs after apoptosis of the infected LCs [2]), it is well established that in humans LCs have an important immunomodulatory role (24).
Studies of HCMV have utilized both CD34+ and CD14+ progenitor cells to analyze the viral interaction with myeloid DCs (22, 28, 29, 39, 45–47). Interestingly, the heterogeneity of the myeloid DC population is also evidenced by the biology of these DCs in the context of HCMV infection. In experimental studies, immature and mature CD14+ derived DCs (MoDCs) are similarly permissive for HCMV lytic infection (47). In contrast, CD34+ derived LCs were previously shown to have an absolute requirement for CD40L-induced maturation for permissive infection (22). Thus, an understanding of the molecular reasons for such differences could impact on our understanding of the regulation of HCMV infection and viral gene expression.
In our own studies of HCMV reactivation, we have relied on both CD34+ and CD14+ derived DC model systems (43, 44, 46). However, it is becoming ever more important to determine whether observations made in experimental latency systems truly reflect events that occur upon the reactivation of naturally latent virus. In our own studies we have focused on the use of CD34+ and CD14+ cells for modeling HCMV latency. Our own experiences have suggested that access to regular supplies of CD34+ material is becoming increasingly difficult. In contrast, CD14+ monocytes are readily available from healthy donors and are present at much higher concentrations in the blood, and thus we hypothesized that we could use MoLCs (MoLCs) as another model for studying the interaction of HCMV with LCs. In the present study, we show that MoLCs exhibit a number of functional similarities with CD34+ LCs regarding the biology of HCMV lytic and latent infection and, pertinently, we have revealed intriguing differences between myeloid DC subsets regarding the biology of HCMV reactivation impacting directly on our understanding of HCMV reactivation from latency.
MATERIALS AND METHODS
Ethical statement.
All research describing studies on primary human material with HCMV were assessed and approved by the Cambridge Local Research Ethics committee. Informed consent was given for the collection of venous blood samples from healthy donors or leukophoresis products from granulocyte-colony-stimulating factor mobilized patients and was performed in accordance with established guidelines for the handling and processing of said tissue by the Cambridge Local Research Ethics committee.
Cells and tissue culture.
Human foreskin fibroblasts (HFFs) were maintained in Eagle minimal essential medium containing 10% fetal calf serum (EMEM-10; Sigma-Aldrich, Poole, United Kingdom) and incubated at 37°C and in 5% CO2 according to the standard procedure for tissue culture. CD14+ mononuclear cells were directly isolated from HCMV-seronegative blood, and immature DCs were generated as described previously. Briefly, peripheral blood mononuclear cells were prepared by centrifugation on a Ficoll-Hypaque (LymphoPrep; Takeda, United Kingdom) density gradient. Magnetic-activated cell sorting using CD14+ antibody-conjugated MicroBeads (Miltenyi Biotec, Surrey, United Kingdom) allowed collection of a CD14+ mononuclear cell-enriched population, which were maintained in X-VIVO 15 serum-free medium (Lonza, Walkersville, MD). To promote differentiation to an MoDC phenotype, cultures were stimulated with interleukin-4 (IL-4; 100 ng/ml) and granulocyte-macrophage colony-stimulating factor (GM-CSF; 100 ng/ml) for 6 days. Alternatively, the cultures were stimulated with IL-4 (10 ng/ml), TGF-β (10 ng/ml), and GM-CSF (100 ng/ml) for 2 days, the medium was changed, and TGF-β (10 ng/ml) and GM-CSF (100 ng/ml) were added for a further 4 days to induce differentiation into immature MoLCs. Loosely adherent cells were collected by moderate aspiration and transferred to fresh wells. Mature DCs and LCs were generated by adding lipopolysaccharide (LPS) in fresh medium (500 ng/ml; Sigma-Aldrich). Alternatively, in studies that analyzed HCMV reactivation, immature LCs (iLCs) were incubated with IL-6 or IL-8 (50 ng/ml) or were coincubated with LPS plus neutralizing antibodies to IL-6, IL-8, or goat IgG isotype control (10 μg/ml; R&D Systems, Minneapolis, MN) in fresh medium. Unless stated otherwise, all cytokines were from Peprotech EC.
Latency establishment and coculture experiments.
First, to ensure that propagated TB40/e and VR1814 strain retained tropism for myeloid cells, all preparations were tested for their ability to infect retinal pigment epithelial cell line and MoDCs prior to use in all studies of HCMV latency. The CD14+ monocytes were cultured for at least 4 h in X-VIVO 15 after isolation and then infected with a preparation of the clinical isolate TB40/e (multiplicity of infection [MOI] of 5). After 3 h, the infected cells were washed and cultured in fresh X-VIVO 15 medium for 7 days. After 7 days, medium was exchanged again, and fresh medium containing cytokines that promote DC and LC differentiation were added as described previously. Reactivation was measured by indirect immunofluorescence for immediate-early (IE) gene expression at 24 h poststimulation of DCs. Alternatively, virus production from reactivating mature MoDCs and MoLCs was assayed after coculture on a confluent monolayer of HFFs with samples of supernatant taken at regular intervals and used to inoculate fresh HFFs to test for infectious virus by indirect immunofluorescence staining.
Indirect immunofluorescence and Western blotting.
Infected cells were rinsed in phosphate-buffered saline (PBS) and fixed for 10 min in 4% paraformaldehyde at room temperature. After permeabilization with 0.1% Triton X-100 in PBS, the cells were incubated with monoclonal mouse anti-IE antibody (Millipore) at a 1:1,000 dilution in PBS for 1 h at room temperature. After being washed with PBS, the bound antibodies were detected using Alexa Fluor 594 (Millipore, Billerica, MA)-conjugated goat anti-mouse immunoglobulins at a 1:1,000 dilution in PBS, together with nuclear stain Hoechst at a 1:1,000 dilution in PBS in the dark for 1 h at room temperature. After a washing step with PBS, the infected cells were visualized using a Nikon immunofluorescence microscope and quantified using ImagePro WCIF ImageJ software (National Institutes of Health). The percent infection was calculated by dividing the number of infected cells (red) by the total number of cells (blue) from at least four fields of view.
Protein expression was detected by Western blotting of denatured protein samples. To detect viral protein expression, mouse anti-IE antibody (1:500; Millipore) or mouse anti-gB antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) was incubated at 4°C overnight with nitrocellulose and incubated with a horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody (1:4,000; Santa Cruz Biotechnology) for 1 h at room temperature. As a loading control, nitrocellulose filters were also incubated with a rabbit anti-GAPDH control (1:1,000; Abcam, Cambridge, United Kingdom), followed by an HRP-conjugated goat anti-rabbit antibody (1:4,000; Santa Cruz Biotechnology); both antibody incubations were performed at room temperature for 1 h. The blots were processed using ECL Plus (GE Life Sciences, Amersham, United Kingdom) and exposed to Kodak autoradiograph film (Sigma-Aldrich).
Nucleic acid isolation and analysis.
Total RNA was extracted from 106 cells using an RNeasy kit as described by the manufacturer (Qiagen, Sussex, United Kingdom). Residual genomic DNA was removed by DNase I digestion (Promega, Madison, WI), followed by the production of first-strand cDNA using the Promega RT system. Standard PCR was carried out using 2× PCR Master Mix (Promega) containing DNA polymerase, MgCl2, and deoxynucleoside triphosphates. Gene- and promoter-specific primers were used to amplify target sequences by PCR under the following cycling conditions: 95°C for 5 min, followed by 20 to 35 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min, and then a final extension at 72°C for 10 min. IE72 was amplified with the sense primer 5′-CAT CCA CAT CTC CCG CTT AT-3′ and the antisense primer 5′-CAC GAC GTT CCT GCA GAC TAT G-3′. The IE product size is 579 bp from DNA and 408 bp from cDNA. A 548-bp actin product was amplified using the sense primer 5′-GCT CCG GCA TGT GCA-3′ and the antisense primer 5′-AGG ATC TTC ATG AGG TAG T-3′ under the same PCR conditions.
For quantitative PCR, primers that amplified the IE region of HCMV were used (31). The following reaction conditions were used in a 96-well plate format with the forward primer AGC GCC GCA TTG AGG A, the reverse primer CAG ACT CTC AGA GGA TCG GCC, and the probe ATC TGC ATG AAG GTC TTT GCC CAG TAC ATT (FAM probe with a TAMRA quencher). PCRs were performed using TaqMan master mix (Applied Biosystems, Foster City, CA) in a 7500HT machine (Applied Biosystems). Actin was amplified using a VIC-actin commercial probe (Applied Biosystems), and statistical analysis and interpretation were performed as described previously (49).
Cell surface phenotype flow cytometry analysis.
A total of 105 cells were pelleted at 400 × g for 5 min and then resuspended in the residual volume. The cells were incubated with 3 μl of fluorescein isothiocyanate (FITC)-conjugated mouse anti-human CD207, CD14, E-Cadherin, and CD1a antibodies in the dark for 20 min. The appropriate mouse IgG-FITC antibody was used as an isotype control. Alternatively, cells were incubated with 3 μl of allophycocyanin (APC)-conjugated mouse anti-human CD83 or HLA-DR antibody or with the appropriate mouse IgG1-APC isotype control. To detect class I expression, cells were incubated with a mouse anti-human phycoerythrin (PE)-conjugated HLA-ABC antibody or an appropriate isotype-matched control. After washing in 10× volumes of PBS, the cells were pelleted at 400 × g for 5 min and resuspended in 500 μl of phosphate-buffered saline (PBS) before analysis by flow cytometry (BD FACSCalibur or BD FACSsort). The data handling was performed using WinMDI2.9 software. All Antibodies were from BD Life Sciences (Franklin Lakes, NJ).
MLR.
Mixed-leukocyte reaction (MLR) analysis was performed in 96-well round-bottom plates. Different cell densities of mock-infected or TB40/e-infected MoLCs were seeded and then cocultured with 8 × 104 purified allogeneic CD4+ T cells that had been purified from peripheral blood mononuclear cells using a magnetic CD4+ T cell enrichment kit (StemCell Technologies, Vancouver, Canada) for negative selection of CD4+ T cells. MLRs were supplemented with a final concentration of 5 U of IL-2/ml. T cell proliferation and viability was quantified by trypan blue cell counting after 6 days of coculture. Different effector/target (E:T) ratios were set up in triplicate.
RESULTS
CD14+ monocytes differentiated with TGF-β generate a CD207+ population of dendritic cells.
In order to study the function of MoLCs, we isolated CD14+ cells from the peripheral blood of healthy donors and confirmed that they were CD14+ and CD83/CD207− (Fig. 1A). The isolated monocytes were then cultured in X-VIVO 15 medium for 6 days in cytokines that promoted differentiation to either a DC or LC phenotype, resulting in a similar increase in cell size, granularity, and process formation when both cell types were visualized by light microscopy (Fig. 1B). Further characterization was performed alongside CD34+ cells differentiated to an LC phenotype by an analysis of the expression of a panel of a number of phenotypic markers (14, 33). Incubation of monocytes with TGF-β (MoLCs) promoted the formation of a CD207 population (typically, 50 to 70% of the total population) upon differentiation that was also evident in CD34+ LC cultures and also consistent with previous observations (14, 48). Furthermore, the MoLCs were predominantly CD1a (>87%) and E-Cadherin (>74%) positive for both markers and exhibited elevated levels of class I expression compared to classical MoDCs (Fig. 1C) and, as such, resembled the CD34+ LC phenotype rather than the MoDC phenotype. Taken together, these data are in agreement with previous observations that the culture of monocytes in DC differentiation media supplemented with TGF-β promotes a more Langerhans-like phenotype.
Fig 1.

Differentiation of CD14+ monocytes with TGF-β promotes a Langerhans-like phenotype. (A) Freshly isolated monocytes were characterized for CD14+, CD83−, and CD207− expression. (B) Light microscopy of freshly isolated monocytes or monocytes cultured for 6 days in IL-4/GM-CSF or IL-4/GM-CSF/TGF-β to promote the formation of a DC (MoDC)- or an LC (MoLC)-like cell type. (C) MoDCs, MoLCs, or Langerhans cells generated from CD34+ precursors (CD34+ LCs) were analyzed by flow cytometry for class I, class II, CD1a, CD207, and E-Cadherin expression.
MoLCs exhibit maturation dependence for permissive HCMV infection.
Having established we could generate a Langerhans-like cell type from CD14+ derived monocytes, we next sought to determine whether these cells were permissive for HCMV infection. After culture to an immature phenotype, MoLCs or MoDCs were either left unstimulated or incubated with LPS overnight. Then, the cells were infected at an MOI of 5 with the myelotropic TB40/e strain or the laboratory strain, Toledo. Consistent with previous data, staining for IE gene expression showed that both immature and mature MoDCs were permissive for HCMV infection with TB40/e (Fig. 2A). In contrast, efficient infection in MoLCs was only observed after LPS maturation (Fig. 2B). As expected, both DCs and LCs were poorly infected with the laboratory strain, Toledo, which exhibits low myelotropism. The immunofluorescence results observed with TB40/e (Fig. 2A) were consistent with a reverse transcription-PCR (RT-PCR) analysis that showed substantially lower IE72 gene expression in immature MoLCs despite the presence of viral genome in the cells (Fig. 2B).
Fig 2.
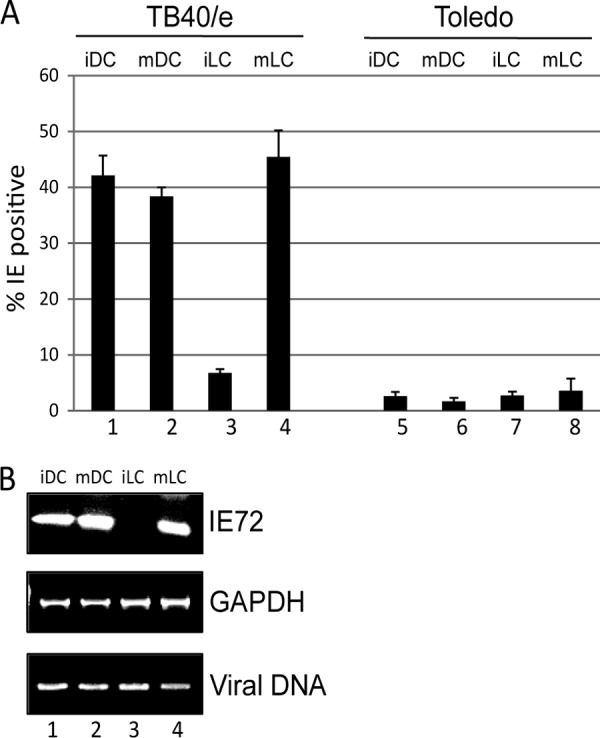
A clinical isolate of HCMV infects immature LCs inefficiently. (A) MoLCs and DCs were infected with TB40/e (columns 1 to 4) or Toledo (columns 5 to 8) before (iDC and iLC) or after (mDC and mLC) LPS stimulation and analyzed for IE gene expression by immunofluorescence microscopy at 24 h postinfection. The percent infection was calculated from four fields per well from an analysis performed in triplicate. (B) MoLCs and DCs were infected with TB40/e before (iDC and iLC) or after (mDC and mLC) LPS stimulation and analyzed by PCR 24 h postinfection for IE72 and GAPDH RNA expression and the presence of viral genomes.
The block to infection of MoLCs is not due to direct TGF-β signaling.
The differences in the permissiveness for infection between immature MoDCs and MoLCs could be attributable to more inherent differences in the different cell types or, alternatively, could be associated with more direct effects of the cytokines in the medium. The major differential between the two subsets of cells is the addition of recombinant TGF-β. Critical for the formation of the LC phenotype (25), TGF-β is also a pleiotropic cytokine that can modulate the activity of a number of cellular signaling pathways (13). To test whether TGF-β signaling was inherently inhibitory to HCMV infection of MoDCs, CD14+ monocytes were cultured in IL-4/GM-CSF for 6 days. Then, 1 h prior to infection, incubated with TGF-β (10 ng/ml) and infected with HCMV. Staining for IE infection at 24 h postinfection indicated that preincubation with TGF-β had no impact on the ability of HCMV to infect MoDCs at both low and high MOIs, suggesting that the long-term effects of promoting differentiation to an MoLC phenotype were responsible for the differential ability of HCMV to infect these different MoDC subsets rather than a direct inhibition initiated by acute TGF-β signaling (Fig. 3).
Fig 3.
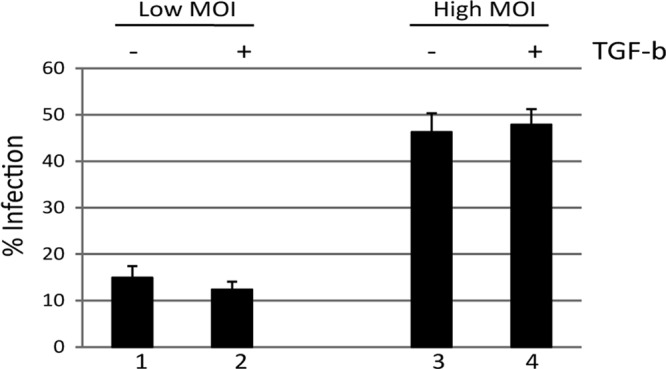
TGF-β does not directly inhibit HCMV infection of immature DCs. CD14+ derived DCs were either not stimulated (columns 1 and 3) or stimulated with TGF-β (columns 2 and 4) for 30 min prior to HCMV infection. The cells were then infected at either a low (columns 1 and 2) or a high (columns 3 and 4) MOI and then assayed for infection by immunofluorescence microscopy at 24 h postinfection.
MoLC are fully permissive, resulting in the production of infectious progeny.
Although our data showed that IE gene expression could be detected in the infected mature MoLCs, we next sought to determine whether the infection was abortive or resulted in the production of infectious progeny. CD14+ cells differentiated to MoLCs were subsequently matured with LPS and then infected with TB40/e at an MOI of 5. Samples of cells were harvested at 24 and 72 h postinfection and analyzed by Western blotting for viral gene expression. IE expression was readily detectable by 24 h, and by 72 h the processed form of glycoprotein B was also detectable (Fig. 4A). Consistent with mature MoLCs being fully permissive for HCMV, low levels of virus was released into the supernatant of infected MoLCs up to 12 days postinfection, with production peaking at day 8, and displayed kinetics similar to those observed with classical MoDCs (Fig. 4B). Furthermore, a freeze-thaw analysis of the MoLC cultures indicated that a significant proportion of infectious virus remained cell bound (Fig. 4C). The data demonstrate that MoLCs supported the completion of the lytic life cycle of HCMV.
Fig 4.

Mature MoLCs support the complete replicative cycle of HCMV. (A) Mature MoLCs were either mock (lanes M) or TB40/e (lanes V) infected and then analyzed by Western blotting for IE, gB, and GAPDH expression at 24 and 72 h postinfection (hpi). (B and C) Mature CD14+ derived DCs (MoDC) or LCs (MoLC) were infected with TB40/e and then analyzed every 2 days for virus production in the supernatant. Alternatively, at 7 days postinfection both the supernatant (cell-free) and the lysed cells (cell-associated) were assayed for infectious virus.
HCMV infection inhibits the ability of MoLCs to induce allogeneic CD4+ T cell responses in a mixed leukocyte reaction.
Previously, it has been shown that the infection of mature CD34+ derived LCs has profound effects on the immune capacity of those cells (22). To determine whether MoLCs were similarly targeted by HCMV, we focused on a functional aspect of DC biology. To do this we performed an MLR. MoLCs mixed with allogeneic CD4+ T cells trigger a robust T cell proliferative response, which is consistent with these cells representing potent activators of the immune response (Fig. 5). However, this activation was significantly diminished after infection with TB40/e (Fig. 5) and was comparable to the phenotype observed for CD34+ derived LCs (22). The most potent effects were observed when the ratio of MoLCs to T cells was highest and support the hypothesis that infection of MoLCs functionally impacts on their ability to activate T cells in vitro. However, it is worth noting that in all our experiments our virus stocks only gave between 30 and 50% infection, suggesting that additional mechanisms, as well as direct effects on the infected cell, may be functioning in these assays. For instance, the fact that the effects were most overt at an E:T ratio of 2:1 suggests that a concentration-dependent factor derived from infected cells was contributing, in part, to HCMV immunosuppression of the MLR reaction. This experiment was repeated four times using four different donor pairs and, in all experiments, HCMV-infected MoLCs induced poor proliferative CD4+ T cell responses compared to uninfected MoLCs stimulated with LPS.
Fig 5.
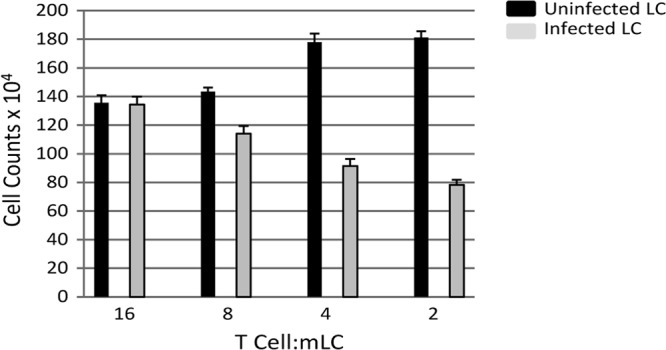
Infected mature MoLCs are impaired in their ability to promote T cell proliferation in a mixed leukocyte reaction. MoLCs were either mock infected (black) or HCMV infected (gray), followed by incubation, at 72 h postinfection, with 8 × 104 T cells at the E:T ratio shown. The T cell proliferation was assayed for the total cell count from triplicate wells. The results shown are representative of four independent repeats.
MoLCs support HCMV reactivation in a maturation-dependent manner.
Clearly, the impact of being able to generate LCs from CD14+ precursors will be important for studies of HCMV latency and reactivation on cells isolated from healthy seropositive donors. Thus, it was important to determine whether these cells could be used for studies of HCMV reactivation. First, we used an experimental model of HCMV latency to test this. CD14+ monocytes were infected with TB40/e. An experimental latent infection was confirmed by RT-PCR detection of UL138 in the absence of readily detectable lytic IE gene expression (Fig. 6A), a finding consistent with published data. Subsequently, infected cells were differentiated to classical MoDCs or, alternatively, MoLCs and then analyzed for HCMV reactivation. We have previously observed that the maturation of either MoDCs or CD34+ derived LCs is concomitant with the reactivation of HCMV. We now show here that MoLCs also support the reactivation of IE gene expression upon maturation (Fig. 6B). However, we were struck with the maturation dependence of MoLCs for efficient reactivation. The addition of IL-6, which we have recently shown to promote robust reactivation of HCMV from immature MoDCs (43), did not trigger readily detectable reactivation of IE gene expression from immature MoLCs in a comparable manner. However, the initiation and progression of reactivation observed after LPS treatment was still observed to be IL-6 dependent, with the addition of a neutralizing IL-6 antibody significantly impacting the reactivation of IE gene expression in maturing LCs (Fig. 6B). Furthermore, the kinetics of reactivation of IE gene expression in the presence of different cytokines were also evident in plaque formation and the subsequent production of infectious progeny in cocultures (Fig. 6C and D). Microscopic analysis of the fibroblast monolayers exhibited minimal plaque formation at 10 days after coculture (and at 15 days after reactivation) except in the LPS-stimulated MoLC cultures and, more evidently, in mature MoLCs (LPS) costimulated with IL-6 (Fig. 6C). Furthermore, the observed cytopathic effect correlated with the levels of infectious virus production detectable in the cocultures (Fig. 6D).
Fig 6.

Efficient reactivation of HCMV from MoLCs is maturation dependent and enhanced by IL-6. (A) CD14+ monocytes TB40/e (lanes 1 and 2) or mock (lanes 3 and 4) infected were analyzed for RNA expression at 3 days postinfection. RNA with (+) or without (−) prior RT was amplified in UL138, IE72, and actin-specific PCRs. For the IE72 PCR, an HCMV DNA PCR-positive control was included to confirm that the PCR had worked (lane 5). (B) RNA isolated from immature MoLCs either mock treated (lane 1) or treated with IL-6 (lane 2), IL-8 (lane 3), LPS (lane 4), LPS plus neutralizing IL-6 antibody (lane 5), or LPS plus neutralizing IL-8 antibody (lane 6) was (+) or was not (−) subjected to RT and then amplified in an IE72 or actin PCR. (C and D) MoLCs were cocultured with fibroblasts to assay HCMV reactivation. After 10 days, the cultures were analyzed for evidence of plaque formation (C) and HCMV reactivation by inoculating fresh monolayers of fibroblasts with 50 μl of the supernatant and staining for IE gene expression 24 h postinfection as an indicator of infectious virus in the supernatant (D).
Interestingly, we also observed the same effects in our studies of CD34+ derived LC reactivation. Akin to MoLCs, reactivation was crucially dependent on maturation (LPS), since immature CD34+ derived LCs did not respond to the IL-6 alone (Fig. 7A, lanes 3 and 5). However, IL-6 was observed to be a crucial component of LPS-induced reactivation (Fig. 7B), and the addition of recombinant IL-6 was also observed to have the same enhancing effect on reactivation from mature CD34+ derived LCs (Fig. 7A, lanes 4 and 6).
Fig 7.

Efficient reactivation of HCMV from CD34+ derived LCs is maturation dependent and is responsive to IL-6. (A) CD34+ derived immature LCs either mock treated (column 1) or cultured with LPS (column 2), IL-6 at 5 to 50 ng/ml (columns 3 and 5), or LPS plus IL-6 at 5 to 50 ng/ml (columns 4 and 6) and then analyzed for IE and actin gene expression by real-time quantitative RT-PCR. The results are expressed as the fold change in IE gene expression compared to LPS alone. (B) CD34+ derived immature LCs were either mock treated (column 1) or incubated with LPS (column 2), LPS plus neutralizing IL-6 antibody (IL6; columns 3, 5, and 7), or isotype-matched control (IgG; columns 4, 6, and 8) and analyzed by quantitative RT-PCR for IE and actin gene expression. Reactivation is expressed as a function of LPS alone (100%).
MoLCs support reactivation of naturally latent HCMV from healthy seropositives.
Finally, we have previously observed that the isolation of CD34+ cells from healthy seropositive donors and subsequent differentiation to mature LCs promotes the reactivation of HCMV (45). We show here that MoLCs also support the reactivation of naturally latent HCMV after stimulation with LPS (Fig. 8A). Furthermore, consistent with our data derived from experimental latency analyses, we also observed that IL-6 signaling is an important factor for efficient reactivation of HCMV in maturing CD14+ derived and CD34+ derived LCs and that the effects of IL-6 are only observed in mature LC cultures (Fig. 8) in contrast to previous observations with immature MoDCs (43).
Fig 8.
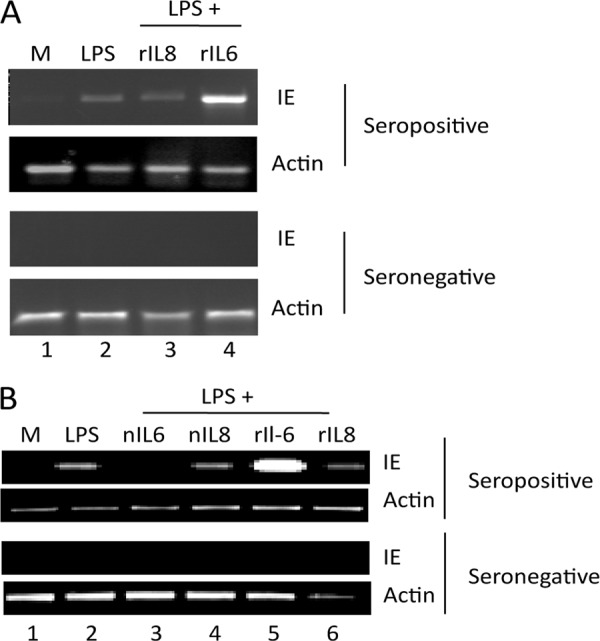
Reactivation of naturally latent HCMV is observed in LCs derived from CD14+ and CD34+ progenitor cells. (A) CD14+ cells isolated from CMV-seropositive and -seronegative donors were differentiated to immature LCs and then either mock treated (lane 1) or incubated with LPS (lane 2), LPS plus 50 ng of IL-8/ml (lane 3), or LPS plus 50 ng of IL-6/ml (lane 4) and analyzed for IE and actin gene expression by RT-PCR (n = 4). (B) CD34+ cells isolated from CMV-seropositive and -seronegative donors were differentiated to immature LCs and then either mock treated (lane 1) or incubated with LPS (lane 2), LPS plus 1 μg of nIL6ab/ml (lane 3), LPS plus 1 μg of nIL8ab/ml (lane 4), LPS plus 50 ng of IL-6/ml (lane 5), or LPS plus 50 ng of IL-8/ml (lane 6) and then analyzed for IE and actin gene expression by RT-PCR (n = 4).
DISCUSSION
A number of laboratories have used experimental latent infection of primary cells to address the mechanisms that govern latency and reactivation of HCMV (16, 17, 19, 20, 37, 43, 44, 55, 64). The use of these models has made it possible to perform large-scale screen based experiments in an attempt to characterize global events that occur during the phases of latent infection (8, 17, 41, 55). Although such exploratory experimentation on naturally latent HCMV is rendered inherently difficult by the low frequency of genome-positive cells in the mononuclear compartment (54), it remains important to confirm findings in experimental systems by studying the natural state of latent HCMV.
A major site of natural HCMV latency in the host is the CD34+ cell compartment that populates the bone marrow (34, 53) and, as such, the use of this tissue has been extensive in the establishment of experimental models of HCMV latency (17, 44, 55, 64). However, in this report we sought to analyze the biology of MoLCs with regard to HCMV and, in particular, how these cells compared to both classical MoDCs and CD34+ LCs. In our experimental models of HCMV latency, the differentiation of CD34+ cells to mature Langerhans DCs—a highly specialized population of DCs resident in the periphery—is important for triggering HCMV reactivation (44, 46). However, since we increasingly wish to understand the interplay between immunology, inflammation, and HCMV reactivation, it is clearly important that we have access to material from fully characterized healthy donors from an immunological viewpoint. This is illustrated by the current debate regarding the contribution of HCMV to the progression of atherosclerosis (1, 7, 12, 18). One postulated explanation for the discrepancies has been attributed to the ability of different individuals to control HCMV infection and disease—and thus a function of the donor-specific immune responses observed in patients (63). Coupled with donor specific immune response outcomes is the observation that the reactivation of HCMV is triggered by cytokines produced by allogeneically stimulated T lymphocytes (56); it thus becomes important to consider the impact of immune mismatching. As such, experiments using a wholly autologous culture system with donors fully characterized from an immunological viewpoint allows us to appreciate these important caveats when studying normal immunological responses to latent and reactivating HCMV in healthy individuals. Consequently, we sought to address whether MoLCs were functionally akin to MoDCs or CD34+ derived LCs in the context of HCMV biology and how these different cell types responded to a proreactivation stimulus.
In the present study, we show that we can generate LCs from monocyte precursors that have strong functional similarities to CD34+ derived LCs with regard to HCMV biology, supporting the premise that LCs exhibit fundamental differences from DCs regarding HCMV biology. CD14+ monocytes isolated from peripheral blood can be differentiated in the presence of TGF-β into LC-like DCs evidenced by the induction of CD207, E-Cadherin, and CD1a surface expression. Furthermore, we show that lytic infection of these cells is dependent on differentiation to a mature phenotype and, once matured, these cells are fully permissive for the complete replication cycle of HCMV, resulting in the production of progeny virus. Consistent with the expression of the complete set of viral genes, a profound effect on the immunostimulatory capacity of the infected MoLCs was observed.
A crucial aspect for our own ongoing studies is the ability of these cells to carry and reactivate HCMV genome in experimental latency. The subtle differences between the LC subtype and other monocyte/DC experimental models (19, 42) observed with regard to their reactivation phenotype could be an important indicator of the fundamental processes involved in HCMV reactivation upon myeloid differentiation through different lineages, assuming that these in vitro-generated cells truly reflect the in vivo populations of myeloid DCs. Furthermore, such HCMV-specific effects could also provide scope for further investigation of the functional differences evident in myeloid DC subsets. The differential response of DC subsets to HCMV is not pathogen specific; culture of monocytes in Th2-mediated inflammatory conditions generates mixed populations of DCs that respond differently to inflammatory stimuli, and thus not all DCs are homogeneous in their normal biology (3). Pertinent to our current studies is the question of why the effects of IL-6 on reactivation are only observed on mature CD34+ derived or MoLCs, whereas immature MoDCs, or monocytes cultured long term in a cytokine cocktail to promote survival (19), reactivate HCMV in response to IL-6 as efficiently as do mature MoDCs (43). One possibility is that immature LCs are less responsive to proinflammatory conditions in general. It has been argued that tolerization of LCs could be an important event to ensure that unnecessary inflammatory immune responses do not occur in the periphery (10). Indeed, it has recently been shown that LCs are less efficient at promoting the development of the effector/memory CD4+ T cell phenotype (51). In the context of HCMV, it has always been proposed that it is a paradox that reactivation occurs in a cell type (i.e., a DC) that represents the archetypal professional antigen-presenting cell. However, in the context of a less-than-robust immune response in the LC subset, reactivation in the periphery could represent an important mechanism that facilitates horizontal dissemination prior to efficient immune surveillance.
Clearly, there is a direct correlation between the permissiveness of differentiated myeloid cells for HCMV infection and the capacity to drive reactivation in these cells upon delivery of inflammatory signals. The triggering of efficient reactivation with inflammatory mediators appears to correlate precisely with the permissiveness of each of the cell types involved for HCMV lytic infection. Indeed, it is possible that we identified these maturation specific events in LCs that are important for reactivation because all our primary cell culture was performed under serum-free conditions. For instance, the culture of CD14+ cells to DCs has been shown to produce a more heterogeneous differentiated myeloid cell phenotype (including macrophage-like cells) in the presence of serum contaminants that could confound analyses attempting to study HCMV reactivation in particular subsets of cells (32, 38). It will be interesting to determine the proteomic differences that exist between immature DCs and LCs that promotes reactivation of HCMV in one subset but not the other in response to inflammatory signaling. It is also anticipated that elucidation of the nature of these inflammatory responsive elements may also impact on our further understanding of the response of different myeloid DC subsets to environmental cues.
We have established here that CD14+ and CD34+ derived LCs have a similar reactivation phenotype that subtly differs from more classical CD14+ derived DCs. Because we can observe differences between MoLCs and MoDCs, it may be possible to take the same starting cell phenotype, from the same donor, and then differentiate it down two independent pathways that exhibit subtly different phenotypes to perform proteome analyses without the added caveats of donor to donor variability that could occur using unmatched CD34+ and CD14+ cells. As such, one would predict that a greater similarity in proteome profiles may make the identification of the differences responsible for their phenotype with regard to HCMV biology easier to elucidate. Ultimately, study of the molecular kinetics of reactivation in different cell types could provide new clues regarding the differential regulation of mechanisms controlling HCMV reactivation.
ACKNOWLEDGMENTS
This study was funded directly by a Medical Research Council Career Development Fellowship Award to M.B.R. (G:0900466). M.R.W. is supported by grants from the Medical Research Council and Wellcome Trust.
We thank John Sinclair for helpful discussions and critical reading of the manuscript.
Footnotes
Published ahead of print 30 May 2012
REFERENCES
- 1. Adler SP, Hur JK, Wang JB, Vetrovec GW. 1998. Prior infection with cytomegalovirus is not a major risk factor for angiographically demonstrated coronary artery atherosclerosis. J. Infect. Dis. 177:209–212 [DOI] [PubMed] [Google Scholar]
- 2. Allan RS, et al. 2003. Epidermal viral immunity induced by CD8α+ dendritic cells but not by Langerhans cells. Science 301:1925–1928 [DOI] [PubMed] [Google Scholar]
- 3. Bechetoille N, Andre V, Valladeau J, Perrier E, Dezutter-Dambuyant C. 2006. Mixed Langerhans cell and interstitial/dermal dendritic cell subsets emanating from monocytes in Th2-mediated inflammatory conditions respond differently to proinflammatory stimuli. J. Leukoc. Biol. 80:45–58 [DOI] [PubMed] [Google Scholar]
- 4. Borkowski TA, Letterio JJ, Farr AG, Udey MC. 1996. A role for endogenous transforming growth factor β1 in Langerhans cell biology: the skin of transforming growth factor β1-null mice is devoid of epidermal Langerhans cells. J. Exp. Med. 184:2417–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bosnjak L, et al. 2005. Herpes simplex virus infection of human dendritic cells induces apoptosis and allows cross-presentation via uninfected dendritic cells. J. Immunol. 174:2220–2227 [DOI] [PubMed] [Google Scholar]
- 6. Bursch LS, et al. 2007. Identification of a novel population of Langerin+ dendritic cells. J. Exp. Med. 204:3147–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Caposio P, Orloff SL, Streblow DN. 2011. The role of cytomegalovirus in angiogenesis. Virus Res. 157:204–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carlier J, et al. 2011. Paracrine inhibition of GM-CSF signaling by human cytomegalovirus in monocytes differentiating to dendritic cells. Blood 118:6783–6792 [DOI] [PubMed] [Google Scholar]
- 9. Caux C, et al. 1996. CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF+TNF alpha. J. Exp. Med. 184:695–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clausen BE, Kel JM. 2010. Langerhans cells: critical regulators of skin immunity? Immunol. Cell Biol. 88:351–360 [DOI] [PubMed] [Google Scholar]
- 11. Czernielewski JM, Demarchez M. 1987. Further evidence for the self-reproducing capacity of Langerhans cells in human skin. J. Invest. Dermatol. 88:17–20 [DOI] [PubMed] [Google Scholar]
- 12. Danesh J, Collins R, Peto R. 1997. Chronic infections and coronary heart disease: is there a link? Lancet 350:430–436 [DOI] [PubMed] [Google Scholar]
- 13. Dennler S, Goumans MJ, ten Dijke P. 2002. Transforming growth factor beta signal transduction. J. Leukoc. Biol. 71:731–740 [PubMed] [Google Scholar]
- 14. Geissmann F, et al. 1998. Transforming growth factor beta1, in the presence of granulocyte/macrophage colony-stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J. Exp. Med. 187:961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ginhoux F, et al. 2007. Blood-derived dermal Langerin+ dendritic cells survey the skin in the steady state. J. Exp. Med. 204:3133–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. U. S. A. 99:16255–16260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grattan MT, et al. 1989. Cytomegalovirus infection is associated with cardiac allograft rejection and atherosclerosis. JAMA 261:3561–3566 [PubMed] [Google Scholar]
- 19. Hahn G, Jores R, Mocarski ES. 1998. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. U. S. A. 95:3937–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hargett D, Shenk TE. 2010. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. U. S. A. 107:20039–20044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He Y, Zhang J, Donahue C, Falo LD., Jr 2006. Skin-derived dendritic cells induce potent CD8+ T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity 24:643–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hertel L, Lacaille VG, Strobl H, Mellins ED, Mocarski ES. 2003. Susceptibility of immature and mature Langerhans cell-type dendritic cells to infection and immunomodulation by human cytomegalovirus. J. Virol. 77:7563–7574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kanitakis J, Petruzzo P, Dubernard JM. 2004. Turnover of epidermal Langerhans' cells. N. Engl. J. Med. 351:2661–2662 [DOI] [PubMed] [Google Scholar]
- 24. Kaplan DH. 2010. In vivo function of Langerhans cells and dermal dendritic cells. Trends Immunol. 31:446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaplan DH, et al. 2007. Autocrine/paracrine TGF-β1 is required for the development of epidermal Langerhans cells. J. Exp. Med. 204:2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katz SI, Tamaki K, Sachs DH. 1979. Epidermal Langerhans cells are derived from cells originating in bone marrow. Nature 282:324–326 [DOI] [PubMed] [Google Scholar]
- 27. Kondo K, Xu J, Mocarski ES. 1996. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. U. S. A. 93:11137–11142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee AW, et al. 2006. Human cytomegalovirus alters localization of MHC class II and dendrite morphology in mature Langerhans cells. J. Immunol. 177:3960–3971 [DOI] [PubMed] [Google Scholar]
- 29. Lee AW, et al. 2011. Human cytomegalovirus decreases constitutive transcription of MHC class II genes in mature Langerhans cells by reducing CIITA transcript levels. Mol. Immunol. 48:1160–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Legendre C, Pascual M. 2008. Improving outcomes for solid-organ transplant recipients at risk from cytomegalovirus infection: late-onset disease and indirect consequences. Clin. Infect. Dis. 46:732–740 [DOI] [PubMed] [Google Scholar]
- 31. Leruez-Ville M, et al. 2003. Monitoring cytomegalovirus infection in adult and pediatric bone marrow transplant recipients by a real-time PCR assay performed with blood plasma. J. Clin. Microbiol. 41:2040–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lutz MB, Rossner S. 2007. Factors influencing the generation of murine dendritic cells from bone marrow: the special role of fetal calf serum. Immunobiology 212:855–862 [DOI] [PubMed] [Google Scholar]
- 33. MacAry PA, et al. 2001. Mobilization of MHC class I molecules from late endosomes to the cell surface following activation of CD34-derived human Langerhans cells. Proc. Natl. Acad. Sci. U. S. A. 98:3982–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mendelson M, Monard S, Sissons P, Sinclair J. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 77(Pt 12):3099–3102 [DOI] [PubMed] [Google Scholar]
- 35. Merad M, Ginhoux F, Collin M. 2008. Origin, homeostasis and function of Langerhans cells and other Langerin-expressing dendritic cells. Nat. Rev. Immunol. 8:935–947 [DOI] [PubMed] [Google Scholar]
- 36. Merad M, et al. 2002. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat. Immunol. 3:1135–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Minton EJ, Tysoe C, Sinclair JH, Sissons JG. 1994. Human cytomegalovirus infection of the monocyte/macrophage lineage in bone marrow. J. Virol. 68:4017–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mitani H, et al. 2000. Activity of interleukin 6 in the differentiation of monocytes to macrophages and dendritic cells. Br. J. Haematol. 109:288–295 [DOI] [PubMed] [Google Scholar]
- 39. Moutaftsi M, Mehl AM, Borysiewicz LK, Tabi Z. 2002. Human cytomegalovirus inhibits maturation and impairs function of monocyte-derived dendritic cells. Blood 99:2913–2921 [DOI] [PubMed] [Google Scholar]
- 40. Peggs KS, Mackinnon S. 2004. Cytomegalovirus: the role of CMV post-haematopoietic stem cell transplantation. Int. J. Biochem. Cell Biol. 36:695–701 [DOI] [PubMed] [Google Scholar]
- 41. Poole E, McGregor Dallas SR, Colston J, Joseph RS, Sinclair J. 2011. Virally induced changes in cellular microRNAs maintain latency of human cytomegalovirus in CD34 progenitors. J. Gen. Virol. 92:1539–1549 [DOI] [PubMed] [Google Scholar]
- 42. Poulin LF, et al. 2007. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J. Exp. Med. 204:3119–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reeves MB, Compton T. 2011. Inhibition of inflammatory interleukin-6 activity via ERK-MAPK signaling antagonizes human cytomegalovirus reactivation from dendritic cells from latency. J. Virol. 85:12750–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reeves MB, Lehner PJ, Sissons JG, Sinclair JH. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodeling. J. Gen. Virol. 86:2949–2954 [DOI] [PubMed] [Google Scholar]
- 45. Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. U. S. A. 102:4140–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reeves MB, Sinclair JH. 2010. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 91:599–604 [DOI] [PubMed] [Google Scholar]
- 47. Riegler S, et al. 2000. Monocyte-derived dendritic cells are permissive to the complete replicative cycle of human cytomegalovirus. J. Gen. Virol. 81:393–399 [DOI] [PubMed] [Google Scholar]
- 48. Sallusto F, Lanzavecchia A. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179:1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 50. Schuler G, Steinman RM. 1985. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J. Exp. Med. 161:526–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shklovskaya E, et al. 2011. Langerhans cells are precommitted to immune tolerance induction. Proc. Natl. Acad. Sci. U. S. A. 108:18049–18054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Siena S, et al. 1995. Massive ex vivo generation of functional dendritic cells from mobilized CD34+ blood progenitors for anticancer therapy. Exp. Hematol. 23:1463–1471 [PubMed] [Google Scholar]
- 53. Sindre H, et al. 1996. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 88:4526–4533 [PubMed] [Google Scholar]
- 54. Slobedman B, Mocarski ES. 1999. Quantitative analysis of latent human cytomegalovirus. J. Virol. 73:4806–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Slobedman B, et al. 2004. Impact of human cytomegalovirus latent infection on myeloid progenitor cell gene expression. J. Virol. 78:4054–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Soderberg-Naucler C, Fish KN, Nelson JA. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119–126 [DOI] [PubMed] [Google Scholar]
- 57. Strobl H, et al. 1997. flt3 ligand in cooperation with transforming growth factor-β1 potentiates in vitro development of Langerhans-type dendritic cells and allows single-cell dendritic cell cluster formation under serum-free conditions. Blood 90:1425–1434 [PubMed] [Google Scholar]
- 58. Strunk D, et al. 1996. Generation of human dendritic cells/Langerhans cells from circulating CD34+ hematopoietic progenitor cells. Blood 87:1292–1302 [PubMed] [Google Scholar]
- 59. Taylor-Wiedeman J, Sissons P, Sinclair J. 1994. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 68:1597–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Torres-Madriz G, Boucher HW. 2008. Immunocompromised hosts: perspectives in the treatment and prophylaxis of cytomegalovirus disease in solid-organ transplant recipients. Clin. Infect. Dis. 47:702–711 [DOI] [PubMed] [Google Scholar]
- 61. Valladeau J, et al. 2000. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 12:71–81 [DOI] [PubMed] [Google Scholar]
- 62. Vishwanath M, et al. 2006. Development of intravital intermittent confocal imaging system for studying Langerhans cell turnover. J. Invest. Dermatol. 126:2452–2457 [DOI] [PubMed] [Google Scholar]
- 63. Zhu J, et al. 2001. Discordant cellular and humoral immune responses to cytomegalovirus infection in healthy blood donors: existence of a Th1-type dominant response. Int. Immunol. 13:785–790 [DOI] [PubMed] [Google Scholar]
- 64. Zhuravskaya T, et al. 1997. Spread of human cytomegalovirus (HCMV) after infection of human hematopoietic progenitor cells: model of HCMV latency. Blood 90:2482–2491 [PubMed] [Google Scholar]