

Abstract
Arthropod-borne flavivirus infection causes serious morbidity and mortality worldwide, but there are currently no effective antiflaviviral chemotherapeutics available for human use. Therefore, it is critical that new therapeutics against virus-specific targets be developed. To identify new compounds that may be used as broadly active flavivirus therapeutics, we have performed a high-throughput screening of 235,456 commercially available compounds for small-molecule inhibitors of the dengue virus NS5 RNA capping enzyme. We identified a family of compounds, the 2-thioxothiazolidin-4-ones, that show potent biochemical inhibition of capping enzyme GTP binding and guanylyltransferase function. During the course of structure-activity relationship analysis, a molecule within this family, (E)-{3-[5-(4-tert-butylbenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]propanoic acid} (BG-323), was found to possess significant antiviral activity in a dengue virus subgenomic replicon assay. Further testing of BG-323 demonstrated that this molecule is able to reduce the replication of infectious West Nile virus and yellow fever virus in cell culture with low toxicity. The results of this study describe the first inhibitor that targets the GTP-binding/guanylyltransferase activity of the flavivirus RNA capping enzyme.
INTRODUCTION
Arthropod-borne virus infections remain a major cause of morbidity and mortality worldwide. More than two billion people are at risk of infection with dengue virus, and 600 million people are at risk of infection with yellow fever virus (5). Globally, an estimated 50 to 100 million cases of dengue and 200,000 cases of yellow fever are reported each year, which result in, respectively, ∼20,000 to ∼30,000 deaths annually (12). There are currently no clinically useable chemotherapeutic options for the treatment of any flavivirus infection, making it essential that new strategies and targets for the treatment of flavivirus infections be identified.
Flaviviruses are small, enveloped, single-stranded, positive-sense RNA viruses with genomes consisting of approximately 11,000 kb of RNA with a 5′ type 1 RNA cap (23). The viral genome is translated as a single open reading frame that encodes a polyprotein precursor that is processed into three structural proteins (the capsid, premembrane, and envelope proteins) and eight nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, 2K, NS4B, and NS5) by viral and cellular proteases (17). Currently, four viral enzymes are being studied as targets for antiviral drug discovery, including the NS3 helicase and protease enzymes and the NS5 RNA-dependent RNA polymerase and capping enzymes (reviewed in reference 9).
In particular, the capping enzyme has received a good deal of attention as a novel antiviral drug target in recent years. The flavivirus capping enzyme has three distinct functions that can be targeted for therapeutic intervention: the N7/2′-O-methyltransferase reactions (2, 3, 7, 10) and the recently discovered guanylyltransferase reaction (11, 14, 15). The formation of the 5′ cap structure is critical to the survival of the virus for several reasons, including direction of viral polyprotein translation and protection of the 5′ end of the genome from cellular exonucleases. It has also been recently shown that a fully mature type 1 cap is a mechanism that cells use to discriminate self from nonself RNAs, and interference with the formation of a mature type 1 cap on the flavivirus genome limits viral replication (6, 28). The flavivirus NS5 N-terminal capping enzyme is highly conserved across the members of the Flavivirus genus, and the GTP and S-adenosylmethionine binding sites, as well as the overall structure of the enzyme, are well conserved (4, 8, 10, 19, 27). The critical nature of the capping enzyme in viral replication and immune evasion and as its conservation across the flavivirus genus position the capping enzyme as an important target for antiviral development efforts. The methyltransferase activity has been the primary capping enzyme target for drug development (16, 21), and ribavirin triphosphate has been observed to bind to GTP and displace it from the enzyme (1).
In this report, we describe the identification and characterization of the 2-thioxothiazolidin-4-one family of compounds as novel inhibitors of the flavivirus RNA capping enzyme guanylyltransferase. We previously reported an analysis of the capping enzyme GTP binding site (10) and a pilot high-throughput screening (HTS) of 46,323 small-molecule compounds using a fluorescence polarization assay (11). Screening for compounds capable of displacing GTP from the dengue virus capping enzyme was performed against 235,456 compounds from the National Screening Laboratory for the Regional Centers of Excellence in Biodefense (NSRB) library located at the Harvard Medical School Longwood Campus. We detail the process of developing structure-activity relationships (SAR) with analogs within this family, which have led to a solid understanding of the parameters of the binding these compounds to the capping enzyme. During this process, we identified a lead compound, (E)-{3-[5-(4-tert-butylbenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]propanoic acid} (referred to as BG-323 here), that competes with GTP binding to the capping enzyme, inhibits capping enzyme protein guanylation activity in vitro, and displays antiviral activity against multiple different flaviviruses in cell culture. These findings demonstrate that the 2-thioxothiazolidin-4-one family is a novel scaffold for developing effective and specific antiviral molecules that target the GTP binding pocket of the flavivirus RNA capping enzyme.
MATERIALS AND METHODS
Expression and purification of flavivirus NS5 capping enzymes.
Recombinant NS5 capping enzyme domains from yellow fever virus (strain 17D, amino acids 1 to 268) and dengue virus type 2 (strain 16681, amino acids 1 to 267) were previously described (10). Briefly, yellow fever virus capping enzyme was produced in Escherichia coli BL21(DE3) Codon Plus cells (Novagen). Dengue virus capping enzyme was produced in E. coli BL21(DE3) pLysS cells (Novagen). Yellow fever virus and dengue virus proteins were induced and purified by the same protocol. Cultures (750 ml) were induced with 400 μM isopropyl-β-d-thiogalactopyranoside (IPTG) overnight at 22°C, and the bacterial pellets were collected and stored at −80°C in low-imidazole lysis buffer. Frozen pellets were thawed and lysed with a Microfluidizer, and the lysate was clarified by centrifugation at 18,000 rpm in an SS-24 rotor. The histidine-tagged proteins were purified from clarified lysates using a nickel-Sepharose column on an AKTA Purifier fast protein liquid chromatography system. The eluted protein was concentrated with Amicon Ultra concentrators (Millipore) with a 10,000 molecular weight cutoff, and the buffer was exchanged for 400 mM NaCl–20 mM Tris (pH 7.5)–0.02% sodium azide–20% glycerol–5 mM Tris (2-carboxyethyl)phosphine (TCEP) hydrochloride on a Superdex 200 gel filtration column (Amersham). Purified proteins were concentrated to 100 μM using Amicon Ultra concentrators with a 10,000 molecular weight cutoff, and the concentrations were determined by measuring absorbance at 280 nm using extinction coefficients obtained from the ExPASy website. Isolated proteins were >99% pure, as estimated by SDS-PAGE and Coomassie blue staining. Purified protein was stored at −80°C in single-use aliquots.
HTS.
HTS was performed at the NRSB laboratory located at the Harvard Medical School Longwood campus (Institute of Chemistry and Cell Biology [ICCB] Longwood Screening Facility). To perform the screening, 500 nM purified dengue virus capping enzyme was complexed with 10 nM GTP-BODIPY γ-phosphate-labeled analog (Invitrogen catalog number G22183) in binding buffer (50 mM Tris base [pH 7.5], 0.01% NP-40, 2 mM dithiothreitol). Volumes of 30 μl were dispensed into low-binding opaque black 384-well plates (catalog number 3654; Corning, Corning, NY) with a Matrix WellMate liquid handler (Thermo Fisher Scientific, Waltham, MA). One column of 10 μM (final concentration) GTP was used as a positive control on each plate, and one column was treated with dimethyl sulfoxide (DMSO) as a negative control. Screening compounds were added to each plate with an Epson compound transfer robot fitted with a 100-nl 384-pin transfer array. Plates treated with 100 nl of compound (5-mg/ml stock concentration) were allowed to incubate for 1 h at 23°C, and then total fluorescence and fluorescence polarization signals were detected on an Envision 2103 Multimode plate reader with a plate stacker attachment (Perkin-Elmer, Waltham, MA). Each compound was tested in duplicate. The overall Z′ score of the screening was >0.7. Compounds that reduced both the total fluorescence and fluorescence polarization signals by greater than 50% were cherry-picked and retested on a Victor 3V plate reader.
Determination of apparent Ki values.
Compounds were obtained from ChemDiv and Hit2Lead. All small molecules were diluted in DMSO to 10 mM and stored in a −20°C freezer. All compounds were stored in small aliquots in a desiccator to prevent freeze-thaw cycles. Ki values for each compound were determined based on the equation detailed in reference 18 using a fluorescence polarization assay as previously described (10, 11). Compounds were tested at least three times each, and standard deviations are reported for each Ki value.
Guanylation inhibition assay.
Capping enzyme protein guanylation was performed as described previously (11). Briefly, 3 μM dengue virus capping enzyme was incubated with 1 μM GTP–ATTO-680 (catalog number NU-830-680; Jena Bioscience, Jena, Germany), 500 nM MgCl2, 0.1% NP-40, and 1 μM TCEP. Reaction mixtures were treated with compounds at final concentrations of 100 μM, 50 μM, 25 μM, 10 μM, and 2.5 μM or mock-treated with DMSO as controls for 4 h at 37°C. At the end of the incubation period, samples were quenched with 1 μl of 1 M EDTA and 6× Laemmli buffer was added. Samples were boiled for 15 min and resolved by 12% SDS-PAGE. The gels were imaged for the ATTO-680 signal on a Licor Odyssey UV scanner (Licor, Lincoln, NE), and then the gels were stained with Coomassie blue to verify protein equivalence. Coomassie-stained gels were analyzed with the NIH ImageJ software package. ATTO-680 signals of experimental samples were normalized for protein concentration compared to on-gel control samples. Inhibition values were determined using nonlinear regression analysis in the Prism Software package (GraphPad Software Inc., La Jolla, CA). Average 50% effective concentrations (EC50s) and standard errors of the mean values are reported. Each experiment was performed three times.
Antiviral (replicon) assay.
BHK cells harboring a stable dengue virus type 2 subgenomic replicon (BHK-pD2hRucPac) expressing Renilla luciferase have been previously described (26). BHK-pD2hRucPac cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 3 μg/ml puromycin to maintain the stability of the replicon. Working stocks of BHK-pD2hRucPac cells were used for 2 months and then replaced with fresh stocks to maintain the sensitivity of the replicon to antiviral drugs. Antiviral activity assays were performed by seeding white opaque 96-well cell culture plates (catalog number T-3021-13; Bioexpress, Kaysville, UT) with 2,000 cells/well in 100 μl DMEM supplemented with 10% FBS but without puromycin. Cells were allowed to attach overnight at 37°C. The next day, test compounds were diluted in DMSO and 1 μl of each dilution was added to appropriate wells (200 μM to 78 nM final concentrations). Each plate included a control column of DMSO with no drug and several rows of ribavirin (200 μM to 78 nM final concentrations) to verify the sensitivity of the replicons to the drug. Cells were incubated at 37°C for 72 h and then tested for Renilla luciferase activity and cell viability. The medium was replaced with 25 μl of DMEM containing a 1:1,000 dilution of Viviren Live Cell Renilla luciferase reagent (catalog number E6492; Promega, Madison, WI), and the plates were incubated for 10 min at 37°C. The Renilla luciferase signal was detected on a Victor Multi-Mode plate reader (Perkin-Elmer). After reading of the Renilla luciferase signal, 25 μl of CellTiter-Glo cell viability reagent (catalog number G7571; Promega, Madison, WI) was added to each well and the cells were incubated for 10 min at 37°C. The CellTiter-Glo signal was detected in the same manner as the Viviren signal. Renilla and CellTiter-Glo experimental samples were converted to percentages of the averaged on-plate DMSO controls, and nonlinear regression curves (variable slope) were generated with the Prism software. EC50 values were calculated for Renilla luciferase, and 50% cytotoxic concentrations (CC50) were calculated for the CellTiter-Glo curves, and averaged values and standard deviations over at least three experiments for each are reported. The therapeutic index (TI) was calculated as CC50/EC50.
Viral assays.
West Nile virus (Kunjin subtype) and yellow fever virus (17D) growth curves were determined with BHK cells. Cells were plated into six-well plates at 100,000/well and allowed to attach overnight. The next day, cells were treated with BG-323 or DMSO at the indicated concentrations and concurrently infected with Kunjin or yellow fever virus at a multiplicity of infection (MOI) of 0.01. Medium samples of 250 μl were taken at 4, 12, 24, 36, 48, 72, 96, and 120 h postinfection and stored at −80°C. Sample virus concentrations were determined by plaque assay on BHK cells as previously described (20). Viral growth curves were generated with Prism (GraphPad Inc., La Jolla, CA). Renilla luciferase-expressing Sindbis virus pBG451 was previously described (24). The Renilla signal was detected in infected BHK cells with Viviren Live Cell reagent (Promega), and cell viability was detected with CellTiter-Glo reagent (Promega). Each experiment was performed three times, and the average number of PFU/ml and the standard error of the mean are reported.
Real-time qRT-PCR analysis.
West Nile virus (Kunjin) RNA was extracted from cell culture medium with TRIzol LS (Invitrogen, La Jolla, CA) by following the manufacturer's protocol. Real-time quantitative reverse transcriptase PCRs (qRT-PCRs) for Kunjin virus genomic RNA were performed with the Brilliant III Ultra-Fast SYBR QRT-PCR Master Mix (catalog number 600886; Agilent, Santa Clara, CA) and primers Fwd 6170 (5′-TGGACGGGGAATACCGACTTAGAGG) and Rev 6278 (5′-ACCCCAGCTGCTGCCACCTT). To set up qRT-PCRs, 2 μl of extracted RNA was added to 5 μl of 2× master mix, 1 μl of 5 μM Fwd 6170 primer, 1 μl of 5 μM Rev 6278 primer, and 1 μl of 100 mM dithiothreitol in 96-well PCR plates with optically clear sealing films. No-RNA and no-primer controls were included in each experiment. qRT-PCRs were performed on a Bio-Rad CFX384 real-time PCR thermal cycler under the following cycling conditions: an RT step of 50°C for 10 min, a denaturation step of 95°C for 3 min, and PCR (40 cycles) at 95°C for 5 s and 60°C for 10 s. Melting curves were determined at the end of each run to verify the specificity of the detected SYBR signal. Cycle quantification (Cq) values were determined by setting the threshold to 40 for all experiments. Standard curves were generated from diluted medium containing Kunjin virus at a known titer (PFU/ml), and the Cq values of experimental samples were compared to the standard curve to establish numbers of PFU equivalents/ml for each sample [y = −3.475x + 27.577, y = Cq, x = log10(PFU)]. The limit of detection in these experiments was determined to be 10 PFU equivalents/ml. All experiments were performed three times, and the average and standard error of the mean are reported.
Western blot analysis.
BHK cells were infected with Kunjin virus at an MOI of 0.1 and treated with increasing concentrations of BG-323 or DMSO. At 72 h postinfection, cells were collected and lysates were prepared by boiling in 1× Laemmli buffer. Lysates were resolved by 12% PAGE, and protein was transferred to nitrocellulose membranes. Western blot analysis was performed with anti-NS5 antibody 5D4 (13) and anti-β-actin (Abcam catalog number 6276) on a separate membrane. Bands were detected with an IR-DYE-800 anti-mouse secondary antibody (Rockland Scientific) on an Odyssey UV imaging system.
Modeling analysis.
Each molecular structure was drawn using Maestro and minimized using MacroModel (Schrödinger LLC, New York, NY). For minimization of all small molecules, the dielectric constant was set to 4.0 (aqueous), the maximum number of iterations was set to 1,000, and the convergence threshold was set to 0.05 kJ/Å-mol. The optimized potentials for liquid simulations (OPLS-2005) force field was used, and conjugate gradient methodology was applied.
Docking and scoring.
All small molecules were evaluated with GOLD (25) to determine potential orientations of association with the dengue virus capping enzyme (Protein Data Bank [PDB] code 3EVG) and the yellow fever virus capping enzyme (PDB code 3EVD). Unless discussed below, default parameters were applied. The centroid of the docking sphere is x = 16.37, y = −52.73, and z = 17.934, with an active-site radius of 10 Å. Each run consisted of 50 iterations per small molecule. The fitness function and search settings had annealing parameters such that the van der Waals radius was 4 Å and the hydrogen bonding distance was 2.5 Å. Fifty docked conformations were obtained for each compound, and DSViewerPro 5.0 (Accelrys, San Diego, CA) was used to visualize the properties of binding between the small molecule and the capping enzyme protein. For the majority of compounds, one predominant conformation was obtained and chosen as the physiologically relevant orientation for further analysis. For compounds with multiple orientations (generally observed for compounds with lower binding affinity), the orientation most similar (via visual inspection) to the conserved physiologically relevant orientation was chosen for further analysis. The docking protocol used was previously validated for ligand binding to the dengue virus capping enzyme (11) and can recapitulate the association of GTP, GDP, and GMP within the binding site (data not shown).
RESULTS
HTS.
Based on our previous screening of 46,323 compounds against the yellow fever virus capping enzyme (11), we performed a second screening of the remaining 235,456 compounds in the NRSB library for molecules that were able to displace GTP from the dengue virus capping enzyme. This screening identified 633 compounds that showed >50% inhibition of fluorescence polarization and had a corresponding reduction of total fluorescence intensity similar to what we have previously reported for our GTP-BODIPY binding assay (10, 11). These 633, which were determined to be tractable to medicinal chemistry, were cherry-picked from the NSRB libraries and rechecked for activity using cherry-picked material from the NRSB library on a different plate reader. Of the 633 molecules, 222 repeated, a repeat rate of 35.1%. Among these 222 compounds, we observed a small cluster of compounds with a thioxothiazolidin core and an acid moiety that appeared to displace GTP-BODIPY from the capping enzyme with various strengths based on relative displacement in the HTS assay (Table 1), indicating that SAR may be explored with this family. The thioxothiazolidins were not the strongest “hits” among the 222 compounds that repeated, but because the thioxothiazolidin core is found in known, FDA-approved drugs such as Epalrestat and a number of analogs were commercially available, we chose to pursue this family of inhibitors further.
Table 1.
Thioxothiazolidin molecules identified by HTS

HTS displacement activity: weak (W), 50 to 75% reduction of fluorescence polarization signal; moderate (M), 76 to 90% reduction; strong (S), >90% reduction.
SAR analysis of thioxothiazolidins.
The four compounds from Table 1 were ordered in 5-mg quantities from ChemDiv and Hit2Lead. Twenty-one additional commercially available compounds with the same core structure as BG-5 (2-thioxothiazolidin-4-one) (Fig. 1A) were also ordered, and apparent Ki values were determined for 24 compounds against the dengue virus and yellow fever virus capping enzymes using 24-point titration curves (10, 11). Compound 2044-5240 caused the protein in the Ki assays to precipitate, and no usable data were obtainable. Otherwise, the thioxothiazolidin-based series of compounds exhibited apparent Ki values ranging from >100 μM to 1.5 μM (Table 2).
Fig 1.

SAR of thioxothiazolidin binding. (A) Three-dimensional rendering of the predicted orientation of BG-5 within the yellow fever virus capping enzyme. In the yellow fever virus protein structure, light blue represents the entire protein, bright green represents the location of hydrophobic subpocket 1, and dark green represents the location of hydrophobic subpocket 2. In the stick form depiction of BG-5, carbon atoms are sea green, iodine atoms are purple, hydrogen atoms are white, oxygen atoms are red, nitrogen atoms are blue, and sulfur atoms are yellow. (B) Summary pharmacophore model based on the Ki values of the dengue virus and yellow fever virus capping enzymes in Table 2. The dash-and-dot red line represents π-π stacking, and the magenta dashed lines represent predicted hydrogen bonds.
Table 2.
SAR analysis of thioxathiazolidinsa
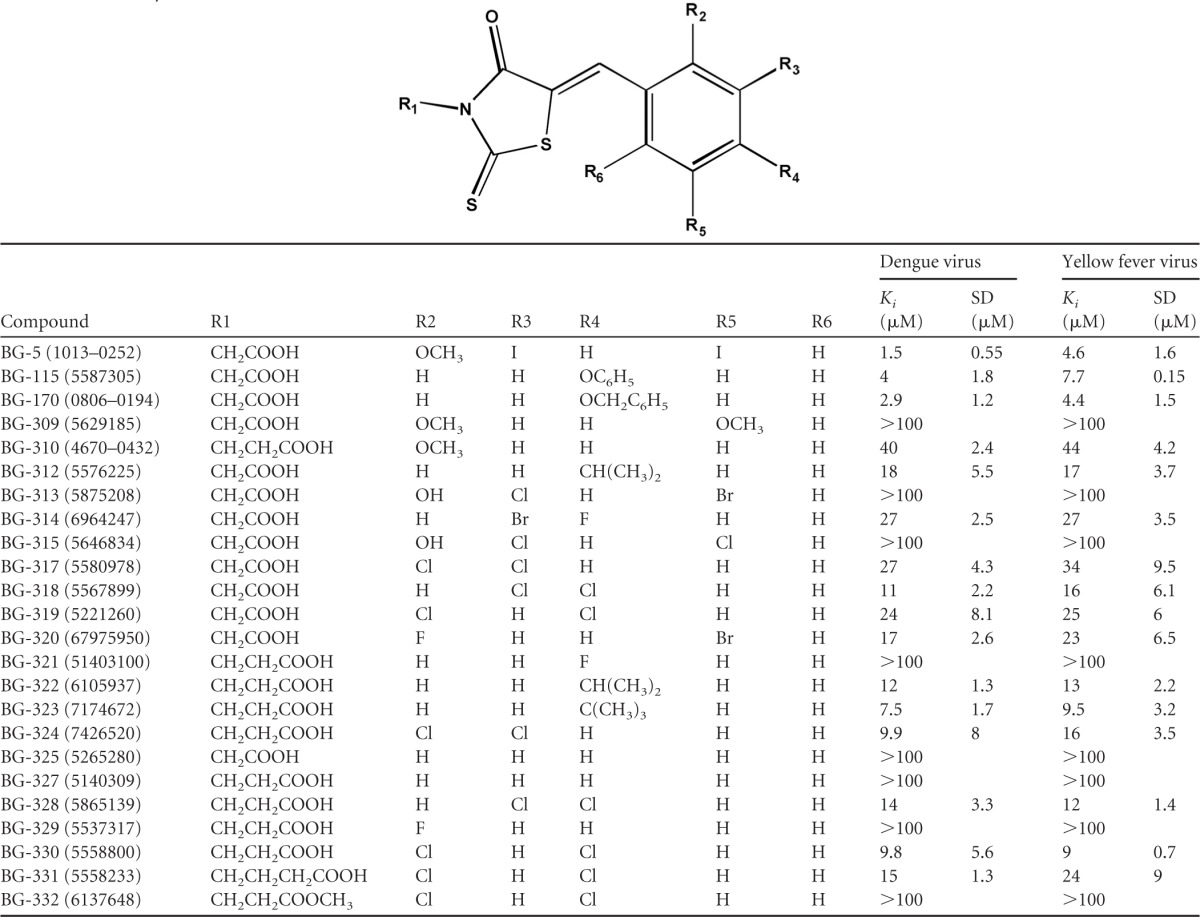
Substitutions are each position are noted. The apparent Ki values of the dengue virus and yellow fever virus capping enzymes were calculated as previously described (11). SD, standard deviation; Ki, inhibition constant.
We visually examined the structures of the small molecules in order to understand how changes in compound structure relate to compound binding affinity. Interestingly, a number of trends were observed. First, substitution on the aromatic ring (positions R2 to R6) appears to be necessary for a small molecule to be able to compete with GTP for the capping enzyme binding site. The aromatic ring of compound BG-327 is not substituted (R2 to R6), and this compound shows no ability to displace GTP (Ki of >100 μM). The addition of an isopropyl group at R4 (BG-322) results in improved affinity (Ki of 12 μM), and replacement of the R4 isopropyl group of BG-322 with a larger t-butyl group (BG-323) further boosts affinity (Ki of 7.5 μM). Both BG-115 (Ki of 4.0 μM) and BG-170 (Ki of 2.9 μM) provide additional evidence of this trend, as both contain bulky groups at position R4. Interestingly, a fluoride atom at R4 is not sufficient for activity (BG-321; Ki, >100 μM). Second, while the compounds examined to date do not allow an exhaustive exploration of the relationship between the position of an aromatic ring substituent and binding affinity, the data do provide some clues. BG-317, BG-318, and BG-319 each have two chlorides on the aromatic ring in various positions. The presence of chlorides at the R3 and R4 positions (BG-318 Ki of 11 μM) results in increased binding compared to molecules with chlorides in the R2 and R3 or R2 and R4 positions, BG-317 (Ki of 27 μM) or BG-319 (Ki of 24 μM), respectively. Additional compounds need to be examined in order to determine whether electronegativity or simply hydrophobic contacts are essential for improved affinity.
A final trend suggested by the data is that affinity is affected by the distance between the acid moiety at the R1 position and the thiazolidin core. For example, BG-317 (one carbon) and BG-324 (two carbons) have observed Kis of 27 μM and 9.9 μM, respectively. Similar effects were observed with compound BG-319 (one carbon; Ki of 24 μM) and compound BG-330 (two carbons; Ki of 9.8 μM). There are, however, data within this set that do not support this trend. BG-318 (one carbon; Ki of 11.3 μM) and BG-328 (two carbons; Ki of 13.6 μM) have similar affinities for the enzyme. Elongation of R1 to three carbons (e.g., butyric acid) has a negative effect on the compound's affinity (compare BG-330 [two carbons] to BG-331 [three carbons]), and modification of the propionic acid to methylpropionate ablates its activity. Collectively, these data suggest that there is likely a structural preference for the distance of the acidic group from the thiazolidin core and that the acid moiety plays a role in compound association with the binding site. Interestingly, there is no observed improvement in affinity without substitutions on the aromatic ring (compare BG-325 and BG-327), confirming the necessity of the aromatic substitutions for activity.
Overall, our results suggest that for capping enzyme binding, compounds from the thioxothiazolidin-based series require substitution of the aromatic ring (R2 to R6) and that an acid group is necessary for inhibition. Furthermore, we have preliminary information suggesting that affinity is sensitive to the distance between the aromatic group and the acid moiety. These data, which are summarized in Fig. 1B, may aid in the future optimization of this family of compounds as capping enzyme inhibitors.
Inhibition of guanylyltransferase activity.
To determine if the thioxothiazolidin molecules interfere with guanylyltransferase activity, we tested the ability of analogs with GTP Ki values of less than 10 μM to interfere with the formation of the guanylated intermediate of the dengue virus capping enzyme (Table 3). We observed that the guanylation 50% inhibitory concentration (IC50) of each compound was roughly equivalent to the GTP displacement Ki value, except in the cases of BG-324 and BG-330, where guanylation inhibition was weakened and strengthened, respectively. Chlorines in the R2 and R3 positions appear to weaken the enzymatic inhibition capacity of BG-324 (IC50 of 155 μM), whereas chlorines in the R2 and R4 positions appear to increase enzymatic inhibition (IC50 of 2.6 μM), although the Ki values of BG-324 and BG-330 are identical. How the capping enzyme forms the guanylated intermediate is unknown, as there is no recognizable guanylyltransferase active-site motif present in any flavivirus protein (15). Therefore, without understanding how the reaction occurs, it is unclear how the presence of chlorines at R2, R3, and R4 may differentially affect enzymatic activity. Overall, this finding indicates that of the majority of the compounds within this group that displace GTP with reasonable Ki values interfere with the enzymatic activity of the capping enzyme to roughly similar extents.
Table 3.
Guanylation inhibition and dengue virus replicon antiviral activity assaysa
| Compound | Guanylation IC50 (μM) ± SEM | Replicon EC50 (μM) ± SEM | Replicon CC50 (μM) ± SEM | Replicon TI | ClogPb |
|---|---|---|---|---|---|
| BG5 | 6.9 ± 3.1 | 97.0 ± 17.0 | 270.0 ± 94.0 | 2.8 | 4.14 |
| BG115 | 8.8 ± 4.9 | 58.0 ± 1.2 | 74.0 ± 4.7 | 1.3 | 3.94 |
| BG170 | 7.8 ± 2.4 | 71.0 ± 22.5 | 80.0 ± 32.3 | 1.1 | 4.01 |
| BG323 | 7.3 ± 2.9 | 30.8 ± 1.3 | 184.0 ± 41.7 | 6.0 | 4.22 |
| BG324 | 155.7 ± 56.9 | 8.5 ± 2.1 | 12.0 ± 2.5 | 1.4 | 3.89 |
| BG330 | 2.6 ± 0.1 | 8.7 ± 1.2 | 12.0 ± 2.0 | 1.4 | 3.89 |
Guanylation inhibition assays and replicon assays were performed as described in Materials and Methods. TI was calculated as CC50/EC50.
ClogP, calculated partition coefficient.
Preliminary antiviral testing of thioxothiazolidins.
Concurrently with our biochemical SAR analysis of the thioxothiazolidin analogs, we tested the abilities of the six compounds that displayed Ki values lower than 10 μM to reduce Renilla luciferase expression from a persistent dengue virus replicon in BHK cells (26). Renilla luciferase expression in replicon cells treated with decreasing concentrations of the indicated compound was assayed 72 h after treatment and used to develop EC50 curves. We also calculated the CC50 of each compound and determined the TI of each compound (Table 3). Compound BG-5 showed a weak antiviral effect, with a TI value of 2.8 (EC50 of 97 μM, CC50 of 270 μM). BG-323 demonstrated a TI of 6, which was higher than that of the ribavirin positive control used in the replicon assays (ribavirin TI of 4.1). The EC50 of BG-323 is 30.8 μM, and the CC50 value is 184 μM, indicating that while BG-323 is effective at a higher concentration than ribavirin (EC50 of 12 μM), it is also significantly less toxic than ribavirin (CC50 of 49 μM).
Effects of BG-323 against infectious virus.
To test if BG-323 could interfere with the replication of infectious viruses, we performed multistep growth curve determinations with West Nile virus (Kunjin) and yellow fever virus in BHK cells with various concentrations of BG-323 added at the time of infection (Fig. 2A). We observed that BG-323 reduced virus titers by close to 3 logs at several times during Kunjin growth, and we also saw a slightly less robust effect with yellow fever virus. The antiviral effect was most pronounced at the highest concentration of BG-323 (100 μM), which is significantly less than the CC50 of BG-323 (184 μM). To determine if BG-323 had activity against nonflaviviruses, we treated BHK cells with BG-323 and infected the cells for 24 h with a Sindbis virus that expresses Renilla luciferase (24). Only a very minor reduction in the Renilla signal was observed at a 100 μM concentration of BG-323 with a corresponding minor reduction in cell viability (Fig. 2B). Therefore, BG-323 does not appear to interfere with the replication of a nonflavivirus and the findings suggest that BG-323 has specificity in inhibiting flavivirus infection.
Fig 2.

(A) Growth curve analysis of Kunjin and yellow fever viruses with increasing concentrations of BG-323. BHK cells were infected with Kunjin or yellow fever virus at an MOI of 0.01, and the indicated concentration of BG-323 was added at time zero. Medium samples were collected at the indicated times and stored at −80°C. Samples virus titers were determined by plaque assay. Curves were generated in Prism. n = 3. (B) Sindbis virus is insensitive to BG-323 treatment. BHK cells were infected with a Renilla luciferase-expressing Sindbis virus (pBG451) (24) at an MOI of 0.01, and BG-323 was added at the indicated concentrations. The Renilla luciferase (white bars) and CellTiter-Glo (black bars) signal levels measured are shown in relative light units (RLU). n = 3.
To monitor if BG-323 was affecting viral protein accumulation in infected cells, we treated Kunjin virus-infected cells (MOI of 0.01) with increasing concentrations of BG-323 and assayed the cells for the presence of the viral NS5 protein at 48 h postinfection by Western blot analysis (Fig. 3A). We observed a significant decrease in NS5 with increasing BG-323 concentrations and noted that a base level of NS5 was present even at high concentrations of BG-323, likely because of translation of the input viral RNA. The significant decrease in NS5 levels indicates that BG-323 may interfere with the translation of viral proteins from de novo synthesized genomic RNAs or alters the stability of the translated NS5 protein in infected cells. Next we sought to determine if BG-323 was also reducing the amount of viral RNA being released into the culture medium. We infected BHK cells with Kunjin virus at an MOI of 0.01 and treated the cells with DMSO or 50 or 100 μM BG-323 or mock treated them with DMSO. At 72 h after infection, we collected medium and extracted viral RNA, which was used in real-time qRT-PCRs. We observed that DMSO treatment had minimal effects on viral RNA in the medium, whereas 50 μM or 100 μM BG-323 significantly reduced the amount of viral RNA detectable in the medium (Fig. 3B).
Fig 3.

(A) Western blot analysis of Kunjin virus-infected BHK cells treated with BG-323. BHK cells were infected at an MOI of 0.01 with Kunjin virus and treated with the indicated concentration of BG-323. At 48 h postinfection, cells were collected and NS5 and β-actin proteins were detected by Western blotting. (B) Detection of viral RNA in BG-323-treated medium. BHK cells were infected with Kunjin virus at an MOI of 0.01, and cells were mock treated or treated with DMSO, 50 μM BG-323, or 100 μM BG-323 at the time of infection. Cells were incubated for 48 h, and medium samples were collected and stored at −80°C. Samples were thawed, RNA was extracted, and RNA was quantified by qRT-PCR. qRT-PCR Cq values were compared to those of a Kunjin virus RNA standard curve and converted to log10 PFU equivalents/ml. n = 3.
We observed that the viral titers of BG-323-treated cultures reached the same level as mock-treated controls at later times during infection, indicating that BG-323 may not be stable throughout the time course. To assess the activity of BG-323 over time, we incubated BG-323 in medium at 37°C for 3, 2, 1, or 0 days. We then added the preincubated medium to BHK cells, added Kunjin virus at an MOI of 0.01, and took medium samples 72 h later. We determined viral RNA content in medium collected from the infected cells by qRT-PCR (Fig. 4A). The amount of viral RNA present in samples with pretreated medium was significantly less than that in medium treated with BG-323 at the time of infection, indicating that BG-323 becomes less active over longer incubation periods. To determine if replacement of BG-323 during infection could overcome this effect, we replaced BG-323-containing medium on Kunjin virus-infected cells every 24 h for 72 h and monitored the viral RNA in the culture medium at each time point. We observed that adding fresh BG-323 to the cells significantly reduced the amount of viral RNA in the medium over time (Fig. 4B), indicating that the virus remained sensitive to BG-323 and was not becoming resistant to BG-323 over the course of the experiment.
Fig 4.

(A) Stability of BG-323 at 37°C. DMSO or 50 μM (final concentration) BG-323 was added to the culture medium (DMEM), and medium samples were incubated in a CO2 incubator at 37°C for 48 or 24 h prior to infection. At time zero, preincubated medium or fresh medium and BG-323 were added to BHK cells and the cells were subsequently infected with Kunjin virus at an MOI of 0.01. At 72 h postinfection, medium samples were collected and stored at −80°C. RNA was prepared and quantified by qRT-PCR as described in Materials and Methods. n = 3. (B) Replacement of BG-323 reduces Kunjin virus replication. BHK cells were infected with Kunjin virus at an MOI of 0.01 and treated with 100 μM BG-323 or DMSO. At 24, 48, and 72 h postinfection, medium samples were collected and medium on infected cells was replaced with fresh medium with 100 μM BG-323 or DMSO as indicated. Viral RNA at 24, 48, and 72 h postinfection (PI) was quantified by qRT-PCR as PFU equivalents/ml. n = 3.
DISCUSSION
This report describes the discovery of a group of novel inhibitors of flavivirus replication that may provide pharmacological benefits for the medical treatment of flaviviruses. We have identified a series of compounds able to inhibit an enzyme essential for viral replication and that show cell culture activity similar to that of the known antiviral ribavirin.
Crystal structures of the capping enzyme with GTP bound have been previously solved (10), and based on mutational studies, GTP association is shown to require π-π stacking interactions with Phe 24 and the formation of hydrogen bonds with Lys 13, Leu 16, Asn 17, Leu 19, Lys 28, Ser 150, Arg 213, and Ser 215. A water bridge also forms between the nitrogenous base and the backbone oxygen of Leu 19 that is suggested to play a role in affinity.
Based on the computational docking of BG-5 (Fig. 1A), the compound in this series with the highest affinity for the enzyme, we suggest that the members of the thioxothiazolidin family of compounds interact with the enzyme in ways that mimic the association of GTP. Specifically, this compound appears to hydrogen bond with Lys 28 and Ser 150 and π-π stack with the aromatic side chain of Phe 24. In addition, BG-5, GTP, and other compounds with improved binding affinity appear to interact with two subpockets within the capping enzyme GTP binding site. Subpocket 1 allows for interactions between the small molecules and Phe 24 and Lys 21, while subpocket 2 allows for interactions between the small molecules and Asn 17, Leu 18, and Leu 19. Interaction with the two subpockets provides a rationale for the SAR shown in Table 2. First, compounds with increased bulk in the R2 to R6 positions are predicted to have increased interactions within one or both of the subpockets (for example, BG-322 and BG-323). Second, the addition of a carbon-carbon bond linking the acid moiety in the R1 position suggests that these compounds could protrude into these subpockets to a greater extent while still maintaining the ability of compounds to form hydrogen bonds with Ser 150 and Lys 28. Interestingly, the two subpockets also provide a structural explanation for the similar inhibitory effects of BG-318 and BG-328: based on docking studies, both compounds appear to interact with subpocket 2 to similar (nonoptimal) extents, and for this reason, affinity is not increased by altering the distance between the thiazolidin core and the acid moiety. Currently, BG-5 has the highest affinity for the capping enzyme, and its proposed orientation within the binding site (Fig. 1A) supports our hypothesis that compounds able to interact with either or both subpockets have an increased affinity for the binding site. Additional analogs need to be tested to further clarify how the thioxothiazolidins bind to the capping enzyme, although our preliminary pharmacophore presented in Fig. 1B provides a sound basis for further SAR development.
Two compounds in this study, BG-5 and BG-323, displayed antiviral activity in the dengue virus replicon assay (Table 3). BG-323 displayed a TI of 6, which indicates that they are more efficacious than our ribavirin positive controls. Further testing of BG-323 showed that it was able to suppress the replication of West Nile (Kunjin) and yellow fever viruses by plaque assay, qRT-PCR analysis of medium samples, and Western blot analysis. These data suggest that BG-323 is able to reduce the amount of infectious virus that is released from infected cells and indicate that BG-323 has significant antiviral activity in culture. This finding is exciting, as BG-323 (shown docked to the yellow fever virus capping enzyme in Fig. 5) has a structure similar to those of known FDA-approved drugs, such as the antidiabetes drug Epalrestat (22), indicating that an optimized thioxothiazolidin may be developed into a drug clinically usable for the treatment of flavivirus infection. One concern is that thioxothiazolidins are considered to be somewhat promiscuous in HTS assays. However, the observation that BG-323 demonstrates antiviral activity against flaviviruses but not alphaviruses (Fig. 2B) suggests that BG-323 is not merely a “sticky” molecule but has some specificity against flavivirus replication, making it an interesting candidate for further development. Additionally, we have previously identified other core structures that may be substituted for the thioxothiazolidin core to mitigate any liabilities that the moiety may present (11).
Fig 5.

Molecular docking of BG-323 with the capping enzyme. On the left, a three-dimensional rendering of the predicted orientation of BG-323 within the yellow fever virus capping enzyme is presented. In the yellow fever virus protein, light blue represents the entire protein, bright green represents the location of hydrophobic subpocket 1, and dark green represents the location of hydrophobic subpocket 2. In the stick form depiction of BG-323, carbon atoms are cyan, hydrogen atoms are white, sulfur atoms are yellow, oxygen atoms are red, and nitrogen atoms are blue. On the right, a flatland model of predicted interactions between of BG-323 and the yellow fever virus capping enzyme is presented. BG-323 is depicted in line form surrounded by capping enzyme residues. Light green represents the location of hydrophobic subpocket 1, and dark green represents the location of hydrophobic subpocket 2. The dash-and-dot red line represents the predicted π-π stacking interactions, and the pink dashed lines represent predicted hydrogen bonds.
The biochemical data we present demonstrate that BG-323 has activity against the capping enzyme guanylyltransferase in vitro, although it is possible that part of the antiviral effect observed in cells may be due to BG-323 interfering with the methyltransferase activity of the capping enzyme or interfering with a cellular protein such as aldose reductase (the target of Epalrestat). Further testing is needed to definitively show the mechanism of action of this series of compounds in cell culture. We have also tested if BG-323 interferes with other enzymatic assays, such as PCR, but have not observed any significant effect (data not shown), indicating that BG-323 is not broadly reactive toward enzymes.
While BG-323 does show promise as a novel anti-flaviviral therapeutic, there are a few issues that need to be addressed to increase its efficacy. The BG-323 TI of 6 is somewhat low, and increasing this value will be critical to further development. BG-323 binds to the capping enzyme with a Ki of ∼10 μM and has an EC50 of ∼30 μM, indicating that the molecule is able to pass cellular membranes relatively effectively and interfere with viral replication at a concentration not much higher than is inhibitory concentration in biochemical assays. Therefore, increasing binding affinity while maintaining cellular permeability may help lower the effective EC50 of the compound series to increase the TI of the molecule and bring its inhibitory activity into a more therapeutically useful range. In addition, BG-323 possesses a Michael acceptor group that may increase reactivity and lead to the weak toxicity we observed in cell culture. Several approaches to mitigate these issues and potentially increase the TI of BG-323 are currently being investigated, such as replacement of the propanoic acid group with the bioisostere tetrazol, conversion of the propanoic acid to an ethyl ester that may act as a prodrug, and/or reduction of the Michael acceptor to avoid undesired reactivity. Additionally, BG-323 does appear to lose activity during the course of longer experiments, suggesting that the compound may degrade over time or may bind albumin. Medicinal chemistry and pharmacokinetic studies are necessary to identify analogs or formulations that may increase the stability of the compound over time in vitro and in vivo. Further SAR development, including mutagenesis analysis of the capping enzyme and cocrystallization of BG-323 and the capping enzyme, will help clarify critical interactions and increase binding affinity.
BG-323 represents a valuable platform for the further development of antiviral inhibitors of flavivirus RNA replication. Future studies will focus on decreasing the EC50 of BG-323, improving bioavailability, and testing this family of molecules for in vitro and in vivo efficacy against a number of additional flaviviruses.
ACKNOWLEDGMENTS
HTS capability was provided by the NSRB within the New England Regional Center for Excellence (NIAID U54 AI057159). This work was supported by a grant from the Rocky Mountain Regional Center for Excellence (RMRCE) (NIH/NIAID U54 AI065357) and a grant from NIH/NIAID (4R01AI046435-11).
We thank Su Chiang and the staff at the NSRB for assistance in performing the screening and postscreening analysis. We thank Hamid Gari and Kevin Haworth for their assistance in the HTS and Brian McNaughton and Ritwik Burai for resynthesizing BG-323 for validation testing. We also thank Stephanie Moon for help with the Kunjin virus qRT-PCR assay, Alexander Khromykh for providing the Kunjin virus used in this study, Roy Hall for providing NS5 antibody 5D4, and Jordan Steel for performing Sindbis virus assays.
Footnotes
Published ahead of print 6 June 2012
REFERENCES
- 1. Benarroch D, et al. 2004. A structural basis for the inhibition of the NS5 dengue virus mRNA 2′-O-methyltransferase domain by ribavirin 5′-triphosphate. J. Biol. Chem. 279:35638–35643 [DOI] [PubMed] [Google Scholar]
- 2. Bhattacharya D, Ansari I, Striker R. 2009. The flaviviral methyltransferase is a substrate of casein kinase 1. Virus Res. 141:101–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhattacharya D, et al. 2008. Phosphorylation of yellow fever virus NS5 alters methyltransferase activity. Virology 380:276–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bollati M, et al. 2009. Recognition of RNA cap in the Wesselsbron virus NS5 methyltransferase domain: implications for RNA-capping mechanisms in flavivirus. J. Mol. Biol. 385:140–152 [DOI] [PubMed] [Google Scholar]
- 5. Centers for Disease Control and Prevention 2009. Division of Vector-Borne Diseases (DVBD). Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/ncezid/dvbd/index.html [Google Scholar]
- 6. Daffis S, et al. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Egloff M, et al. 2007. Structural and functional analysis of methylation and 5′-RNA sequence requirements of short capped RNAs by the methyltransferase domain of dengue virus NS5. J. Mol. Biol. 372:723–736 [DOI] [PubMed] [Google Scholar]
- 8. Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. 2002. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 21:2757–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geiss B, Stahla H, Hannah A, Gari H, Keenan S. 2009. Focus on flaviviruses: current and future drug targets. Future Med. Chem. 1:327–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geiss B, et al. 2009. Analysis of flavivirus NS5 methyltransferase cap binding. J. Mol. Biol. 385:1643–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Geiss BJ, et al. 2011. A high-throughput screening assay for the identification of flavivirus NS5 capping enzyme GTP-binding inhibitors: implications for antiviral drug development. J. Biomol. Screen. 16:852–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guzman A, Isturiz RE. 2010. Update on the global spread of dengue. Int. J. Antimicrob. Agents 36(Suppl 1):S40–S42 [DOI] [PubMed] [Google Scholar]
- 13. Hall RA, et al. 2009. Monoclonal antibodies to the West Nile virus NS5 protein map to linear and conformational epitopes in the methyltransferase and polymerase domains. J. Gen. Virol. 90:2912–2922 [DOI] [PubMed] [Google Scholar]
- 14. Henderson BR, Saeedi BJ, Campagnola G, Geiss BJ. 2011. Analysis of RNA binding by the dengue virus NS5 RNA capping enzyme. PLoS One 6:e25795 doi:10.1371/journal.pone.0025795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Issur M, et al. 2009. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA 15:2340–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lim SP, et al. 2011. Small molecule inhibitors that selectively block dengue virus methyltransferase. J. Biol. Chem. 286:6233–6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lindenbach B, Thiel H, Rice C. 2007. Flaviviridae: the viruses and their replication, p 1101–1152 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 1 Lippincott-Raven Publishers, Philadelphia, PA [Google Scholar]
- 18. Martin RL, Renosto F, Segel IH. 1991. A simple method for calculating the dissociation constant of a receptor (or enzyme).unlabeled ligand complex from radioligand displacement measurements. Arch. Biochem. Biophys. 284:26–29 [DOI] [PubMed] [Google Scholar]
- 19. Mastrangelo E, et al. 2007. Structural bases for substrate recognition and activity in Meaban virus nucleoside-2′-O-methyltransferase. Protein Sci. 16:1133–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paredes A, Alzuru M, Mendez J, Rodriguez-Ortega M. 2003. Anti-Sindbis activity of flavanones hesperetin and naringenin. Biol. Pharm. Bull. 26:108–109 [DOI] [PubMed] [Google Scholar]
- 21. Puig-Basagoiti F, et al. 2009. Identification and characterization of inhibitors of West Nile virus. Antiviral Res. 83:71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramirez MA, Borja NL. 2008. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy 28:646–655 [DOI] [PubMed] [Google Scholar]
- 23. Rice C, et al. 1985. Nucleotide sequence of yellow fever virus: implications for flavivirus gene expression and evolution. Science 229:726–733 [DOI] [PubMed] [Google Scholar]
- 24. Steel JJ, Henderson BR, Lama SB, Olson KE, Geiss BJ. 2011. Infectious alphavirus production from a simple plasmid transfection. Virol. J. 8:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. 2003. Improved protein-ligand docking using GOLD. Proteins 52:609–623 [DOI] [PubMed] [Google Scholar]
- 26. Whitby K, et al. 2005. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J. Virol. 79:8698–8706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Y, et al. 2007. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 81:3891–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Züst R, et al. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and nonself mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12:137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]