

Abstract
The emergence of HIV-1 drug resistance remains a major obstacle in antiviral therapy. M184I/V and E138K are signature mutations of clinical relevance in HIV-1 reverse transcriptase (RT) for the nucleoside reverse transcriptase inhibitors (NRTIs) lamivudine (3TC) and emtricitabine (FTC) and the second-generation (new) nonnucleoside reverse transcriptase inhibitor (NNRTI) rilpivirine (RPV), respectively, and the E138K mutation has also been shown to be selected by etravirine in cell culture. The E138K mutation was recently shown to compensate for the low enzyme processivity and viral fitness associated with the M184I/V mutations through enhanced deoxynucleoside triphosphate (dNTP) usage, while the M184I/V mutations compensated for defects in polymerization rates associated with the E138K mutations under conditions of high dNTP concentrations. The M184I mutation was also shown to enhance resistance to RPV and ETR when present together with the E138K mutation. These mutual compensatory effects might also enhance transmission rates of viruses containing these two mutations. Therefore, we performed tissue culture studies to investigate the evolutionary dynamics of these viruses. Through experiments in which E138K-containing viruses were selected with 3TC-FTC and in which M184I/V viruses were selected with ETR, we demonstrated that ETR was able to select for the E138K mutation in viruses containing the M184I/V mutations and that the M184I/V mutations consistently emerged when E138K viruses were selected with 3TC-FTC. We also performed biochemical subunit-selective mutational analyses to investigate the impact of the E138K mutation on RT function and interactions with the M184I mutation. We now show that the E138K mutation decreased rates of polymerization, impaired RNase H activity, and conferred ETR resistance through the p51 subunit of RT, while an enhancement of dNTP usage as a result of the simultaneous presence of both mutations E138K and M184I occurred via both subunits.
INTRODUCTION
HIV-1 reverse transcriptase (RT) is crucial for HIV-1 replication and is responsible for converting the single-stranded RNA genome into double-stranded DNA (dsDNA), which becomes integrated into host cell DNA. RT is a multifunctional enzyme that carries out RNA-dependent DNA polymerase (RDDP), DNA-dependent DNA polymerase (DDDP), and RNase H activities (19). HIV-1 RT is a heterodimer composed of p66 (560 amino acid [aa] residues; 66 kDa) and p51 (440 aa residues; 51 kDa). Crystal structure analyses have shown that both subunits contain four common subdomains, designated “fingers” (residues 1 to 85 and 118 to 155), “palm” (residues 86 to 117 and 156 to 236), “thumb” (residues 237 to 318), and “connection” (residues 319 to 426) (30). The nucleic acid binding cleft is formed by the fingers, palm, and thumb subdomains of p66 and the thumb subdomain of p51, which, together with the connection subdomains of both subunits, contributes to the ‘‘floor’’ of the cleft (30, 35). It has been proposed that the p51 subunit simply provides structural support to p66 and does not possess independent enzymatic functions.
Due to its crucial role in the viral replication cycle, HIV-1 RT has been a major target for the development of antiviral therapies. Currently, two classes of RT inhibitors (RTIs) have been approved by multiple regulatory agencies for the treatment of HIV-1 infection, i.e., nucleoside reverse transcriptase inhibitors (NRTIs) and nonnucleoside reverse transcriptase inhibitors (NNRTIs). NRTIs act by causing chain termination, while NNRTIs act allosterically by binding to the NNRTI binding pocket located 10 Å from the polymerase active site (45). Both NRTIs and NNRTIs are key components of highly active antiretroviral therapy (HAART), but both classes of drugs can be compromised by drug resistance, which, in the case of NNRTIs, is due to mutations within the NNRTI binding pocket, often at p66 amino acid positions 100 to 110, 180 to 190, and 220 to 240, that substantially decrease susceptibility to first-generation (older) NNRTIs such as nevirapine (NVP) and efavirenz (EFV) (22). One major characteristic of the first-generation NNRTIs is that they have a low genetic barrier for resistance, as only a single mutation, such as K103N, is sufficient to confer diminished susceptibility to all first-generation NNRTIs. Two second-generation (newer) NNRTIs, etravirine and rilpivirine (RPV), have recently been approved for use in treatment-experienced patients and in drug-naïve patients, respectively. Distinct from the first-generation NNRTIs, both ETR and RPV generally require an accumulation of several mutations in order for resistance to occur (1, 4). A unique feature of these 2 second-generation NNRTIs is that their innate flexibility allows these compounds to adopt multiple conformations such that potent activity can be retained against both wild-type viruses and viruses that are resistant to first-generation NNRTIs (1, 4, 25).
Several data sets on the role that RT mutations may play in regard to resistance against the 2 second-generation NNRTIs ETR and RPV are now available. For ETR, 20 mutations, including V90I, A98G, L100I, K101E/H/P, V106I, E138A/G/K/Q, V179D/F/T, Y181C/I/V, G190S/A, and M230L, have been identified as resistance-associated mutations (RAMs) (43), while 15 mutations, including K101E/P, E138A/G/K/Q/R, V179L, Y181C/I/V, H221Y, F227C, and M230I/L, have been recognized as RAMs for RPV (26). However, the degree of resistance conferred by each of these mutations can be variable, and studies on recombinant HIV-1 RT enzymes and HIV-1 infectious clones have demonstrated, for example, that the G190A mutation does not affect susceptibility to ETR (53).
The E138K mutation in HIV-1 RT was recently shown to commonly emerge as the first mutation in cell culture selection experiments with ETR (3) and has also been selected in cultures by RPV (4). The phase III DUET clinical trials on the use of ETR in treatment-experienced HIV-1-infected patients showed that substitutions at position V179 were most common among treatment failures, followed by mutations at position E138, among which the E138G and E138Q mutations, but not the E138K mutation, were frequently observed (46). Recently, phase III clinical trials (ECHO and THRIVE) on the use of the combination of RPV-tenofovir disoproxil fumarate (TDF)-emtricitabine (FTC) in drug-naïve patients showed that the most frequent mutations to emerge among virological failures were the mutations E138K and M184I, which are responsible for resistance to FTC and RPV, respectively (34). The M184I mutation was also shown to enhance resistance to RPV and ETR when present together with the E138K mutation (21, 31). These results indicate that the E138K mutation is probably a signature mutation for the second-generation NNRTI RPV and that the role of the E138K mutation in resistance to both RPV and ETR should be further investigated.
Our laboratory recently generated recombinant mutated and wild-type (WT) reverse transcriptase enzymes and HIV-1NL4-3 infectious clones containing these mutations and demonstrated that the E138K mutation compensates for the deficit in deoxynucleoside triphosphate (dNTP) usage by the M184I/V mutations and restores both the RT enzymatic processivity and viral replication capacity of HIV-1 variants harboring the M184I/V mutations (54). The compensatory effect of the M184I/V mutations may have clinical significance in regard to treatment failures involving ETR-RPV as well as the possible presence of these mutations in transmitted resistance. Mutual compensatory fitness was also demonstrated by sensitive growth competition assays involving the E138K and M184I/V mutations (21).
Although viral fitness is one important parameter that determines the evolutionary dynamics of HIV-1-resistant mutants, other factors such as the number of available target cells and mutation or recombination rates can also have impact. In view of the high prevalence of M184I/V mutations due to the clinical use of lamivudine (3TC) and FTC, we wondered why the E138K mutation had not been seen over time in patients who failed 3TC- and/or FTC-based therapies, as might have been expected due to the compensatory effects that we and others have described (21, 54). Obvious questions are whether the pressure of the M184I/V mutations might somehow prevent the emergence of the E138K mutation and whether the E138K mutation might impact the evolutionary dynamics of M184I/V-containing viruses under 3TC-FTC selection pressure. Also, how will different cell types with different dNTP pools respond to drug selection pressure? This study addresses these issues.
In addition, we recently demonstrated that recombinant HIV-1 RT enzymes containing the E138K mutation are impaired in RNase H activity and have decreased polymerization rates under conditions of high dNTP concentrations (54). While RTs containing either the M184V or M184I mutation, associated with resistance to 3TC and FTC, are impaired in the usage of dNTPs (6, 15, 18), the simultaneous presence of the E138K mutation together with either the M184V or M184I mutation can compensate for this deficit in dNTP usage at low dNTP concentrations through the promotion of tighter dNTP binding. These results indicate that the E138K mutation may also play an important role in the fine-tuning of RT activity. E138 is part of the β7-β8 loop in the p51 subunit at the p66/p51 interface, which is a key structural element for RT dimerization and constitutes the floor of the NNRTI binding pocket (17, 39, 40, 42). Although others have previously described the E138K mutation and have shown that this mutation confers resistance to the [2′,5′-bis-O-(tert-butyldimethylsilyl)-3′-spiro-5″(4″-amino-1″,2″-oxathiole-2″,2″-dioxide)]-β-d-pentofuranosyl (TSAO) family of NNRTIs though the p51 subunit (8, 11), it is still unclear whether the E138K mutation acts in a subunit-specific fashion to impact RT catalytic activities (both polymerase and RNase H) and resistance to second-generation NNRTIs and whether the compensatory effects with the M184I mutation also depend on the presence of the E138K mutation within p51. In the present study, we used subunit-selective mutagenesis to characterize recombinant RT enzymes containing the E138K mutation in either the p66 subunit (p51WT/p66E138K), the p51 subunit (p51E138K/p66WT), or both subunits (p51E138K/p66E138K) and have now demonstrated that ETR resistance, the impairment of RNase H cleavage, and decreased RT polymerization rates are all conferred by the presence of the E138K mutation within the p51 subunit. In contrast, the presence of the E138K mutation within both p51 and p66 is required for the compensatory effects that are mediated by both the E138K and M184I mutations in tandem.
MATERIALS AND METHODS
Chemicals, cells, viruses, and nucleic acids.
Etravirine was a gift of Tibotec (Titusville, NJ). pRT6H-PROT, pRT, and pRT6H51 plasmid DNAs were kindly provided by Stuart F. J. Le Grice, National Institutes of Health, Bethesda, MD.
Cord blood mononuclear cells (CBMCs) were obtained through the Department of Obstetrics, Jewish General Hospital, Montreal, Canada. MT2 cells were obtained from the NIH AIDS Research and Reference Reagent Program. HEK293T cells were obtained from the American Type Culture Collection (ATCC).
HIV-1NL4-3-derived viral clones containing the desired mutations were made as described previously (55). Briefly, fragments spanning RT amino acids 25 to 314 from MscI and PflMI digestion from the RT expression plasmid DNAs harboring the corresponding mutations described below were used to replace the corresponding fragment of pNL4.3PFB proviral DNA (23). Wild-type and mutant viruses were generated by the transfection of proviral plasmid DNAs into HEK293T cells, as described previously, using Lipofectamine 2000 (Invitrogen, Burlington, Ontario, Canada) (55).
A 497-nucleotide (nt) HIV-1 primer binding site (PBS) RNA template spanning the 5′ untranslated region (UTR) to the PBS was transcribed in vitro from AccI-linearized pHIV-PBS DNA (2) by using a T7-MEGAshortscript kit (Ambion, Austin, TX) as described previously (56). The oligonucleotides used in this study were synthesized by Integrated DNA Technologies Inc. (Coralville, IA) and purified by polyacrylamide-urea gel electrophoresis.
Selection of HIV-1 mutants in MT2 cells and CBMCs under drug selection pressure.
MT2 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine per milliliter, 100 U of penicillin per milliliter, and 100 μg of streptomycin per milliliter. Cord blood mononuclear cells (CBMCs) stimulated by phytohemagglutinin A (PHA) were cultured in 10% RPMI 1640 medium supplemented with 10% qualified FBS, 20 U of human interleukin-2 (IL-2)/ml, 5 μg of hydrocortisone/ml, 2 mM l-glutamine/ml, 100 U of penicillin/ml, and 100 μg of streptomycin/ml. Cells in 24-well tissue culture plates were infected with recombinant viral clones at a similar multiplicity of infection (MOI). Selection for viral resistance mutations was performed by using increasing concentrations of RT inhibitors at starting concentrations below the 50% effective concentration (EC50), as described previously (3, 38). As controls, all viruses were simultaneously passaged without drugs. Virus-containing culture media were harvested and kept at −80°C for subsequent standard genotypic analyses. Selections for resistance were performed over 9 weeks and 19 weeks for MT2 cells and CBMCs, respectively.
Recombinant reverse transcriptase expression and purification.
The p51 and p66 sequences of RT were amplified by PCR from pNL43-derived infectious clones (55) and cloned into the pRSFDuet-1 vector (Novagen) and the pCDFDuet-1 vector (Novagen), respectively, using NcoI and SalI restriction sites for both subunits. Sequences coding for a hexahistidine (His6) tag were added at the N terminus of p51. There was no tag added to either terminus of the p66 subunit. To produce mutant RT, mutations were introduced into specific subunits by using the QuikChange mutagenesis kit (Agilent Technologies Canada Inc., Mississauga, Ontario, Canada). DNA sequencing was performed across the RT coding region to verify the absence of spurious mutations and the presence of desired mutations. RT enzymes were expressed in Escherichia coli BL21 cells (Invitrogen, Burlington, Ontario, Canada) and purified by nickel affinity chromatography and Q Sepharose ion-exchange chromatography, as described previously (33). The polymerase activity of each recombinant RT preparation was evaluated in triplicate by using the synthetic homopolymeric poly(rA)/p(dT)12–18 template/primer (T/P) (Midland Certified Reagent Company) as described previously (41). An active unit of RT was defined as the amount of enzyme that incorporates 1 pmol of dTTP in 10 min at 37°C.
RT inhibitor susceptibility assays.
Susceptibility to ETR was assayed by using recombinant RT enzymes and a heterodimeric HIV-1 PBS RNA T/P system as described previously (56). Briefly, RT reaction buffer containing 50 mM Tris-HCl (pH 7.8), 6 mM MgCl2, 60 mM KCl, and dNTPs (5 μM each) with 2.5 μCi of [3H]dTTP (70 to 80 mCi/mmol), 30 nM heterogeneous HIV-1 RNA template/primer, the same activity of RT enzymes, and variable amounts of RT inhibitors were included in 50-μl reaction volumes. In the reaction mixtures, the final concentrations of ETR were 0, 0.01, 0.03, 0.10, 0.30, 1.00, 3.00, and 10.00 μM. Reaction mixtures were incubated at 37°C for 30 min, the reactions were terminated by the addition of 0.2 ml of 10% cold trichloroacetic acid (TCA) and 20 mM sodium pyrophosphate to the mixtures, and the mixtures were incubated for at least 30 min on ice. The precipitated products were filtered through a 96-well MutiScreen HTS FC filter plate (Millipore) and sequentially washed with 200 μl of 10% TCA and 150 μl of 95% ethanol. The radioactivity of incorporated products was analyzed by liquid scintillation spectrometry using a 1450 MicroBeta TriLux microplate scintillation and luminescence counter (Perkin-Elmer). The 50% inhibitory concentration (IC50) of each RTI was determined by nonlinear regression analysis using GraphPad Prism5.01 software.
RT-catalyzed RNase H activity.
RNase H activity was assayed by using a 41-mer 5′-32P-labeled heteropolymeric RNA template, kim40R, that was annealed to a complementary 32-nucleotide DNA oligomer termed kim32D at a 1:4 molar ratio, as described previously (54). Reactions were conducted at 37°C with mixtures containing an RNA-DNA duplex substrate (20 nM) with RT enzymes (∼200 nM) in assay buffer (50 mM Tris-HCl [pH 7.8], 60 mM KCl, 5 mM MgCl2) in the presence of a heparin trap (2 mg/ml). Aliquots were removed at different time points after the initiation of reactions and quenched by using an equal volume of formamide sample loading buffer (96% formamide, 0.1% each bromophenol blue and xylene cyanol FF, and 20 mM EDTA). The samples were heated to 90°C for 3 min, cooled on ice, and electrophoresed through 6% polyacrylamide–7 M urea gels. The gels were analyzed by phosphorimaging. The efficacy of the heparin trap was verified by preincubation experiments performed by a 10-min preincubation of enzymes with the substrate and various concentrations of the heparin trap, followed by the initiation of RNase H activity in the presence of magnesium (see below).
RNA-dependent DNA polymerase activity.
The same 497-nt RNA and 5′-end 32P-labeled D25 primers described previously (54) were used to assess the polymerization rates of recombinant RT enzymes in time course experiments. Final reaction mixtures contained 20 nM T/P, 400 nM RT enzyme, 50 mM Tris-HCl (pH 7.8), and 50 mM NaCl. Reactions were initiated by the addition of 6 mM MgCl2 and dNTPs at 200 μM to the mixtures, and the mixtures were sampled at 30 s and 60 s, respectively, and mixed with 2 volumes of stop solution. Reaction products were separated by 6% denaturing polyacrylamide gel electrophoresis and analyzed as described previously (54).
Processivity assays.
The processivity of recombinant RT proteins was analyzed as described previously, using a heteropolymeric RNA template in the presence of a heparin enzyme trap to ensure a single processive cycle, i.e., a single round of binding and of primer extension and dissociation (54). The T/P was prepared by annealing the 497-nt HIV PBS RNA with the 32P-5′-end-labeled 25-nt DNA primer D25 at a molar ratio of 1:1, denatured at 85°C for 5 min, and then slowly cooled to 55°C for 8 min and 37°C for 5 min to allow the specific annealing of the primer to the template. RT enzymes with equal amounts of activity and 40 nM T/P were preincubated for 5 min at 37°C in a buffer containing 50 mM Tris-HCl (pH 7.8), 50 mM NaCl, and 6 mM MgCl2. Reactions were initiated by the addition of dNTPs at 0.5 μM and a heparin trap (final concentration, 3.2 mg/ml) to the mixtures, and the mixtures were incubated at 37°C for 30 min; 2 volumes of stop solution (90% formamide, 10 mM EDTA, and 0.1% each of xylene cyanol and bromophenol blue) were then added to stop the reaction. Reaction products were denatured by heating at 95°C and analyzed by using 6% denaturing polyacrylamide gel electrophoresis and phosphorimaging. The effectiveness of the heparin trap was verified in control reactions in which the trap was preincubated with a substrate before the addition of RT enzymes and dNTP.
Analysis of steady-state kinetics.
Kinetics studies were carried out by a modification of a previously described method using homopolymeric poly(rA)/p(dT)12–18 and complementary dTTP as the nucleotide substrate (54). The reaction mixture (10 μl) contained 50 mM Tris-HCl (pH 7.8), 60 mM KCl, 6 mM MgCl2, 5 mM dithiothreitol (DTT), 0.5 U/ml poly(rA)/p(dT)12–18, RT enzymes, variable concentrations of the tracer [3H]dTTP, and cold dTTP (0.2 to 200 μM). Reactions were run at 37°C and quenched by the addition of 0.2 ml of 10% cold trichloroacetic acid and 20 mM sodium pyrophosphate to the mixtures; products were filtered onto Millipore 96-well MutiScreen HTS FC filter plates (catalog number MSFCN6B) and sequentially washed with 200 μl of 10% TCA and 150 μl of 95% ethanol. The radioactivity of incorporated products was analyzed by liquid scintillation spectrometry using a Perkin-Elmer 1450 MicroBeta TriLux microplate scintillation and luminescence counter. The steady-state kinetic parameter Km for nucleotide substrates was determined by nonlinear regression analysis of the substrate concentration and initial velocity data using the Michaelis-Menten equation with the program GraphPad Prism5.01 according to the manufacturer's instructions.
RESULTS
Evolution of HIV-1 mutants selected in MT2 cells and CBMCs under drug pressure.
It is well known that HIV-1 harboring the M184I/V mutations has a low viral fitness because of deficient dNTP usage, especially in cell types with low dNTP pools. We recently demonstrated that the E138K mutation improved dNTP binding and compensated for the deficit in dNTP usage associated with the M184I/V mutations. Intracellular dNTP pools may impact the evolution of resistant variants, and MT2 cells contain inherently higher concentrations of natural dNTPs than primary cells such as cord blood mononuclear cells (CBMCs). We therefore studied these two different cell types in selection experiments to investigate whether they impact the evolutionary dynamics of viruses containing the E138K or M184I/V substitutions (Table 1). When recombinant HIV-1 containing the E138K mutation was selected with 3TC or FTC, it was observed that the M184I mutation emerged in MT2 cells in almost all cases, while the M184V mutation was selected in CBMCs. In the case of M184I/V viruses selected with ETR, the E138K mutation emerged in almost all cases. These selections were performed on at least three different occasions, with similar results being obtained each time. These results indicate that the presence of the M184I/V mutations does not prevent the emergence of the E138K mutation, nor does the presence of the E138K mutation prevent the emergence of the M184I/V mutations. The more frequent presence of the M184V mutation in CBMCs may be due to a higher dNTP usage than that with the M184I mutation, which is more impaired in dNTP usage than the M184V mutation. Interestingly, WT viruses grown in MT2 cells did not develop the E138K mutation under ETR pressure but instead developed the E138G/Q mutation, whereas the E138K mutation was consistently selected under ETR pressure in CBMCs. These differences might also result from different dNTP pool sizes in the two cell types.
Table 1.
Mutations emerging in HIV-1 viral mutants selected with RT inhibitors in MT2 cells and in CBMCs
| Virus | Drug(s) | Mutation(s) selected |
|
|---|---|---|---|
| MT2 cellsa | CBMCsb | ||
| WT | 3TC | M184M/I/V | ND |
| FTC | M184I | ND | |
| ETR | E138E/G, Y181C | L100I, E138K | |
| 3TC-ETR | V90I, M184I | E138K | |
| FTC-ETR | E138Q, M184I | E138K | |
| TDF-ETR | Y181C | ND | |
| E138K | 3TC | M184I | M184V |
| FTC | M184M/I/T | M184V | |
| 3TC-TDF | M184I | ND | |
| FTC-TDF | None | ND | |
| M184I | ETR | E138K | E138K, M230M/L |
| TDF-ETR | E138E/K, M230M/L | ND | |
| M184V | ETR | L100I, E138K | E138K |
| TDF-ETR | E138E/K | ND | |
Selection experiments with MT2 cells were performed over a period of 9 weeks.
Selection experiments with CBMCs were performed over a period of 19 weeks. ND, not determined.
Purification of recombinant HIV-1 heterodimeric RT enzyme.
We purified reconstituted RT heterodimers after separately expressing each subunit by the cotransformation of E. coli with two individual plasmid DNAs. The recombinant WT RT heterodimer (p66/p51) and RT enzymes containing the E138K mutation in either or both subunits were purified to >95% homogeneity; the RT p66 and p51 subunits were purified at similar molar ratios based on SDS-PAGE analysis (Fig. 1A), showing that the mutations introduced did not interfere with either heterodimer formation or enzyme purification. No significant differences in specific activity were observed among the purified mutant RT enzymes compared to WT RT (Fig. 1B). We have found that this system produces more efficient yields in our hands than the use of M15 bacterial cells (20).
Fig 1.
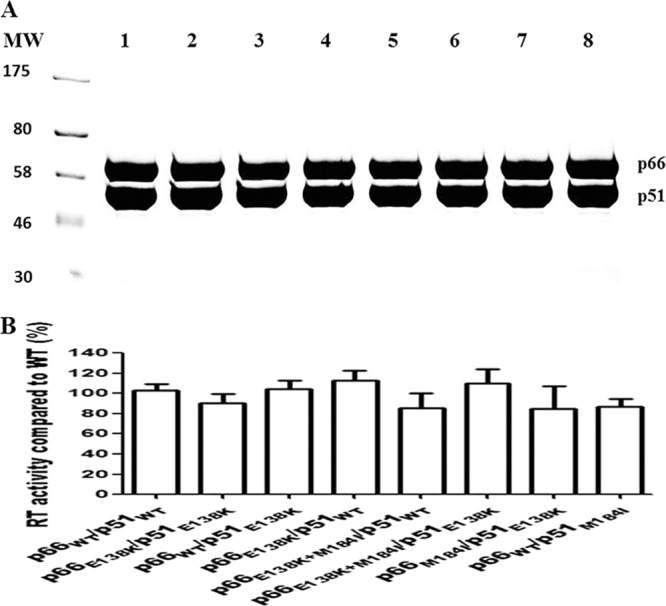
Purification of recombinant HIV-1 RTs. (A) Coomassie brilliant blue staining of purified heterodimer RTs following 8% SDS-PAGE. The purification of heterodimeric RT enzymes was achieved by the attachment of a His6 tag at the N terminus of the p51 subunit through immobilized metal affinity chromatography (IMAC). MW, molecular weight standards (in thousands). Lanes: 1, p66WT/p51WT; 2, p66E138K/p51E138K; 3, p66WT/p51E138K; 4, p66E138K/p51WT; 5, p66E138K+M184I/p51WT; 6, p66E138K+M184I/p51E138K; 7, p66M184I/p51E138K; 8, p66WT/p51M184I. The positions of purified recombinant RT heterodimers are indicated on the right. (B) Comparison of specific activities of recombinant subunit-selective mutant RT enzymes relative to that of the WT enzyme. DNA polymerase activity was assessed as described in Materials and Methods. Data from a representative experiment performed in triplicate are shown as means ± standard deviations.
One concern is that the p66 subunit of RT is highly susceptible to proteolysis, which could yield a p51-like subunit (p51*) that does not contain the His6 tag and thus does not bind to Ni-nitrilotriacetic acid (NTA) resin. However, the attachment of the His6 tag to the C terminus of p66 could result in a situation in which p51*/p66-His6 might be purified and p51* would not contain the desired mutation. With our procedure, it is possible that trace amounts of a His6-p51/p51* homodimer might be purified, but this His6-p51/p51* would not be as enzymatically active as heterodimeric RT enzymes. The intact p66 subunits in our RT heterodimer purifications were verified by SDS-PAGE (Fig. 1).
The E138K mutation in HIV-1 RT confers ETR resistance through the p51 subunit.
To clarify whether the E138K mutation confers resistance to ETR in a subunit-specific fashion, similar to that shown for the TSAO compounds, we performed cell-free RNA-dependent DNA polymerase assays in the presence of various concentrations of ETR using the recombinant RT enzymes p66WT/p51WT, p66E138K/p51WT, p66WT/p51E138K, and p66E138K/p51E138K. The results in Table 2 show that the E138K mutation conferred resistance to ETR (3.6-fold change in the IC50 compared to that of WT RT p66WT/p51WT) when present in both the p66 and p51 subunits of HIV-1 RT p66E138K/p51E138K, in agreement with phenotypic data reported previously (54). In contrast, when the E138K mutation was present only in the p66 subunit (p66E138K/p51WT), the RT susceptibility to ETR was similar (1.1-fold) to that of the WT enzyme. However, when the E138K mutation was present only in p51 (p66WT/p51E138K), HIV-1 RT exhibited 3.5-fold ETR resistance, similar to RT p66E138K/p51E138K. These results confirm that the E138K mutation in HIV-1 RT confers resistance to ETR through p51 and not p66.
Table 2.
Susceptibilities to etravirine of WT and mutant RTs harboring the E138K mutation in different RT subunits
| RT enzyme | Mean IC50 ± SDa (nM) | Fold change in resistanceb |
|---|---|---|
| p66WT/p51WT | 186 ± 15 | |
| p66E138K/p51E138K | 669 ± 29 | 3.6 |
| p66WT/p51E138K | 651 ± 34 | 3.5 |
| p66E138K/p51WT | 204 ± 20 | 1.1 |
The IC50 (50% drug inhibitory concentration) values were determined by recombinant RT assays. Data represent the means ± standard deviations of data from 3 independent experiments.
Values represent the fold changes in IC50 values for mutated RTs compared to WT RT.
The E138K mutation in HIV-1 RT impairs RNase H activity through the p51 subunit.
We previously demonstrated that the E138K mutation, when present in both the p66 and p51 subunits of HIV-1 RT, impaired RNase H activity (54). Now, we wished to determine whether the E138K mutation impacted RNase H activity in a subunit-specific manner. We therefore monitored time course RNase H cleavages of a 5′-32P-labeled 41-mer RNA/32-mer DNA substrate using the recombinant enzymes p66WT/p51WT, p66E138K/p51WT, p66WT/p51E138K, and p66E138K/p51E138K (Fig. 2). The cleavage patterns produced by p66WT/p51WT and p66E138K/p51WT were similar to those yielded by recombinant WT RTs purified from an expression construct termed pRT6H-PROT DNA (54, 55). RNA cleavage typically occurred at a position 18 nucleotides (nt) upstream (position −18) of the 3′ end of the primer, and cleavage products were further rapidly cleaved at the −15 position, before RT dissociated from the substrate. The presence of a heparin trap permitted the analysis of RNA cleavage from a single binding event of RT to the substrate. The results (Fig. 2) show that the cleavage activities of the two enzymes containing the E138K mutation in the p51 subunit, p66WT/p51E138K and p66E138K/p51E138K, were impaired compared to the WT p66WT/p51WT enzyme. However, the enzyme containing the E138K mutation solely in the p66 subunit, i.e., p66E138K/p51WT, possessed cleavage activity similar to that of the WT p66WT/p51WT enzyme. Therefore, the E138K mutation impairs RNase H activity via the p51 subunit.
Fig 2.

Subunit-specific analysis of the effect of the E138K mutation in HIV-1 RT on RNase H activity. (A) Graphic representation of the substrate RNA-DNA (kim40R/kim32D) duplex used to monitor the cleavage efficiency of recombinant RT. The 40-mer RNA kim40R was labeled at its 5′ terminus with 32P and annealed to a 32-mer DNA oligonucleotide, kim32D. The positions of cleaved products are indicated by arrows. (B) RNase H activities were analyzed by monitoring substrate cleavage in time course experiments in the presence of a heparin trap. The positions of cleaved products are indicated on the left. The uncleaved substrate indicates the variability in RNase H activity among different RT enzymes. All reactions were resolved by denaturing 6% polyacrylamide gel electrophoresis.
The E138K mutation in RT decreases the rate of polymerization through the p51 subunit.
Previously, we performed RNA-dependent DNA polymerase reactions using recombinant RT enzymes containing E138K mutations and measured the rates of DNA polymerization at both low and high dNTP concentrations. We found that the E138K mutation caused a decrease in the polymerization rate only at high dNTP concentrations (54). Now, we wished to determine whether the E138K mutation in RT impairs polymerization in a subunit-specific manner and therefore performed RNA-dependent DNA polymerase reactions in time course experiments, as described previously (54), for 30 s and 60 s using the recombinant RT enzymes p66WT/p51WT, p66E138K/p51WT, and p66WT/p51E138K (Fig. 3). RT molecules were used at a ∼20-fold excess over the substrate so that any RTs that dissociated from the primer terminus during synthesis would be rapidly replaced and also so that the rate-limiting step would be nucleotide incorporation (18). Polymerase reactions were carried out at high dNTP concentrations (200 μM). The rate of polymerization was calculated as the number of nucleotide additions divided by the reaction time, and the longest extension products generated after 60 s were used to calculate the polymerization rate. When the E138K mutation was present solely in the p66 subunit, the RT enzyme p66E138K/p51WT showed polymerization rates of ∼3.2 nt/s, equivalent to the WT rate (Fig. 3). In contrast, when the E138K mutation was present solely in the p51 subunit, the polymerization rate was diminished to ∼2.0 nt/s, consistent with previously reported maximum polymerization rates for RT containing the E138K mutation in both subunits (54).
Fig 3.
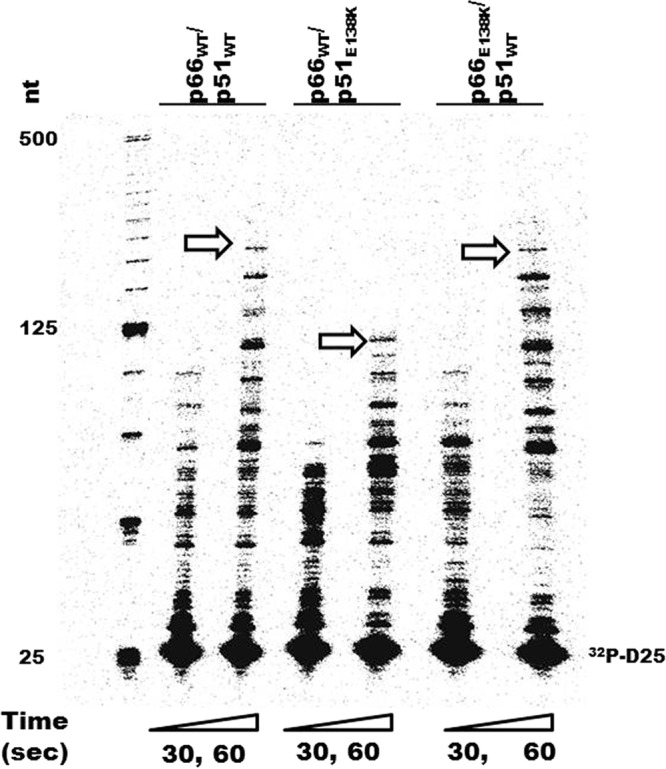
Time course experiments showing the subunit-specific effects of the E138K mutation in HIV-1 RT on rates of DNA polymerization. The 32P-labeled D25 primer (32P-D25) was annealed to the 497-nt RNA template, and extension assays were performed with an excess of recombinant RT enzymes at dNTP concentrations of 200 μM. Reactions were stopped at 30 s (30″) and 60 s (60″). The sizes of some fragments of the 32P-labeled 25-bp DNA ladder (Invitrogen) in nucleotide bases are indicated on the left. The longest extension products generated at 60 s are identified by arrows and indicate differences in polymerization rates.
It is true that no significant differences were observed among the various purified mutant RT enzymes compared to WT RT in terms of specific activity (Fig. 1B). The activities of all of the RTs were determined by measuring dTTP incorporation using the poly(rA)/p(dT)12–18 template/primer. In the RNA-dependent DNA polymerase reactions that measured rates of polymerization, the 497-nt HIV-1 PBS RNA template was used. In this context, the p66WT/p51E138K RT will pause at multiple sites during the synthesis of longer products, and this will lower the average rate of primer extension. Under conditions of saturating dNTP concentrations, the deficit in the polymerization rate associated with the E138K mutation was more pronounced than for the WT, and this is the rate-limiting step of DNA synthesis. This can be further explained by the M184I RT, which had a deficit in dNTP usage and lower processivity at low dNTP concentrations but exhibited a higher polymerization rate than WT RT at high dNTP concentrations.
We performed our gel-based polymerization rate experiments with different batches of purified enzymes and consistently obtained the same results. Differences in polymerization rates were clearly demonstrated by the maximal sizes of the extended products in the gel-based assays and are in good agreement with our previously reported results (56) and those of others (19); however, it would be difficult to conduct analyses of statistical significance in regard to these results. These data do confirm that the E138K mutation decreases the rate of polymerization through the p51 subunit, and this is the first demonstration that an amino acid residue in the p51 subunit can directly impair the polymerization rate of HIV-1 RT. Crystallization of subunit-specific RT E138K enzymes will help to clarify the mechanisms involved, and such studies are under way.
In our assay, RT molecules were in a ∼20-fold excess over the substrate so that any RTs that dissociated from the primer terminus during synthesis would be rapidly replaced. Under these conditions, the nucleotide addition, instead of RT dissociation, should be rate limiting. We used an RNA template derived from the 5′ end of the HIV-1 RNA genome containing the PBS; thus, the lower polymerization rates measured in our multiple-turnover experiment than the rates of polymerization usually detected in pre-steady-state single-turnover experiments are due to multiple pauses during DNA synthesis, which greatly lower the average rate of primer extension (28). The same rationale justifies the choice of 30-s and 60-s time points rather than earlier time points, since only very little DNA product was detected in the gel-based assay at such earlier times.
The E138K mutation compensates for M184I-mediated deficits in dNTP usage and restores enzyme processivity via both subunits.
The E138K mutation compensates for M184I/V-mediated deficits in dNTP usage and restores enzyme processivity at low dNTP concentrations, thereby restoring a high replication capacity to viruses that possess combinations of the E138K mutation together with the M184I or M184V mutation (54). M184I/V RT is known to have lower enzyme processivity than WT RT, especially at low dNTP concentrations (6, 15, 48), and the M184I mutation is even more impaired than the M184V mutation in regard to processivity (18, 24). Deficits of M184I/V RT in processivity and enzyme activity may be attributable to defective dNTP utilization (15, 18, 24, 48, 51). Now, we wished to investigate whether the compensatory effect of the E138K mutation on the M184I mutation might also be subunit specific, and accordingly, we performed single-cycle processivity assays at low dNTP concentrations with the recombinant RT enzymes p66WT/p51WT, p66M184I/p51WT, p66M184I/p51E138K, p66M184I+E138K/p51E138K, and p66M184I+E138K/p51WT (Fig. 4). The results show that the E138K mutation was unable to restore the impaired processivity associated with the M184I mutation when the former mutation was present solely in the p66 or p51 subunit of HIV-1 RT, i.e., p66M184I+E138K/p51WT and p66M184I/p51E138K. In contrast, enzyme processivity was restored when the E138K mutation was present in both p66 and p51, i.e., p66M184I+E138K/p51E138K. These results indicate that the E138K mutation restores the enzyme processivity of the M184I mutation via both subunits.
Fig 4.

The E138K mutation in HIV-1 RT restores enzyme processivity of the M184I enzyme at low dNTP concentrations via both subunits. The processivity of purified recombinant RT enzymes was analyzed by using a 5′-end-labeled DNA primer (D25) annealed to a 471-nt RNA template as the substrate; the resulting full-length DNA (FL DNA) is 471 nt in size. Processivities were determined by the size distribution of DNA products in fixed-time experiments at low concentrations of dNTPs (0.5 μM) in the presence of a heparin trap. The sizes of some fragments of the 32P-labeled 25-bp DNA ladder (Invitrogen) in nucleotide bases are indicated on the left. All reaction products were resolved by denaturing 6% polyacrylamide gel electrophoresis and visualized by phosphorimaging. Positions of the 32P-labeled D25 primer (32P-D25) and the 471-nt full-length extension DNA (FL DNA) product are indicated on the right.
Previous steady-state kinetic studies demonstrated that the E138K mutation resulted in decreased Km values for dTTP (54). Structural modeling also showed that the addition of the E138K mutation to the M184I/V mutations promoted tighter dNTP binding (54). In the present study, we also performed steady-state kinetic assays using subunit-selective mutant RT enzymes to investigate the subunit-specific effects of the E138K mutation on dNTP usage (Table 3). When the E138K and M184I mutations were jointly present in p66, RT (p66E138K+M184I/p51WT) had higher Km values (1.8-fold) for dTTP than WT RT (p66WT/p51WT) but behaved similarly to M184I RT (p66M184I/p51WT) (2.2-fold). RT p66M184I/p51E138K behaved similarly to p66M184I/p51WT. In contrast, the presence of the E138K mutation in both subunits led to RT (p66E138K+M184I/p51E138K) having a Km value (1.1-fold) for dTTP similar to that of WT RT (p66WT/p51WT). These results are in agreement with processivity results indicating that the E138K mutation enhances the dNTP affinity through both subunits (16).
Table 3.
Kinetic parameters of recombinant RT enzymes as determined by analysis of steady-state kinetics
| RT | Avg kcat (min−1) ± SD | Avg Kma (μM) ± SD (fold changeb) |
|---|---|---|
| p66WT/p51WT | 14.2 ± 1.3 | 4.89 ± 0.3 (1) |
| p66M184I/p51WT | 18.4 ± 1.2 | 10.8 ± 0.7 (2.2) |
| p66M184I/p51E138K | 17.0 ± 1.6 | 9.8 ± 0.9 (2.0) |
| p66E138K+M184I/p51WT | 17.0 ± 1.4 | 8.7 ± 0.5 (1.8) |
| p66E138K+M184I/p51E138K | 16.6 ± 1.6 | 5.22 ± 0.4 (1.1) |
The steady-state kinetic parameters kcat and Km for dTTP of WT HIV-1 RT and its mutant derivatives were measured by using poly(rA)/p(dT)12–18 template/primers. The recombinant RT enzymes were purified in a heterodimeric form, and mutations were introduced into specific subunits. Values are averages ± standard deviations from representative experiments performed in triplicate.
Fold changes of the Km values of mutant RT variants compared to the WT.
DISCUSSION
HIV-1 drug resistance mutations are associated with anti-HIV therapy and can be the cause of treatment failure. However, drug-resistant viruses are usually less fit than WT viruses, which can affect the detectability of drug resistance mutations in transmitted resistance. The M184I/V mutations are associated with high-level resistance to 3TC and FTC and are common in treatment failures. The M184I/V mutations are also associated with decreased dNTP usage and have a strong negative impact on viral replication fitness (5, 6, 14, 15, 18, 24, 50). Under selection pressure with 3TC or FTC, both in vitro and in vivo, the M184I mutation usually emerges first and is rapidly replaced by the M184V mutation due to the relative fitness advantage of the latter over the former mutation (13, 15, 29, 48). However, M184I/V mutations are rarely found in cases of transmitted drug resistance (TDR), although the M184V mutation can be detected in newly infected individuals with detection methods that are more sensitive than standard genotyping (12, 47, 52). As a consequence of low viral fitness, the prevalence of the M184V mutation as a single mutation in newly infected individuals wanes over time (52). However, the E138K mutation, which is an important mutation for the second-generation NNRTIs ETR and RPV, can compensate for this deficit in dNTP usage and restore enzyme processivity and viral fitness (54). Although the E138K mutation alone is associated with an impaired polymerization rate and low viral fitness, the M184I/V mutations can also compensate for this deficit (54). We believe that these mutual compensatory interactions between the E138K and M184I/V mutations are clinically relevant in treatment failures involving 3TC-FTC and ETR-RPV and may also be of importance for the potential transmission of drug-resistant HIV-1 variants. Our current findings demonstrate that the M184I/V and E138K mutations can be selected in viruses containing either of these mutations individually under appropriate drug selective pressure in cell types containing high or low dNTP pools.
The transmission of HIV-1 resistance mutations is driven by the treatment rate, the rate of development of acquired resistance, and the fitness of the drug-resistant viruses that emerge (9, 10). Intracellular dNTP pools play an important role in impacting HIV-1 replication kinetics and mutation rates (6, 7, 24, 27, 36, 37). The cellular dNTP concentrations of two different types of HIV-1 target cells, activated/dividing CD4+ T cells and terminally differentiated/nondividing macrophages, were recently determined, and the latter cells contain very low dNTP concentrations (16). Among cells frequently used to cultivate HIV-1 in vitro, the dNTP levels are ∼20-fold higher in established T cell lines than in primary peripheral blood mononuclear cells (6). The replication deficit of the M184I/V mutations is detectable only in cells with low dNTP pools (6, 24); in cells with high dNTP pools, the replication rates of these mutated viruses are comparable to those of the wild-type virus. Viruses containing the M184I mutation are more impacted than those containing the M184V mutation, due to a more severe impairment in dNTP usage by RT. However, the M184I mutation is the result of a G→A hypermutation, in comparison to A→G, which yields the M184V mutation, and therefore, the M184I mutation is usually present before the M184V mutation. The M184V mutation, although clearly less fit than the wild type, is more fit than the M184I mutation and rapidly outgrows the former in vivo (29). Our selection experiments using WT viruses show that the M184I mutation was consistently selected under 3TC-FTC pressure in MT2 cells, which have high dNTP concentrations, whereas the M184V mutation was more often selected in CBMCs, which have lower dNTP concentrations, confirming that differences in dNTP usage can contribute to the differential selection of these two mutations (5, 6, 24, 54). This notwithstanding, it cannot be excluded that the metabolism of 3TC and FTC in the different cells used may have influenced mutational selection. Although we did not measure dNTP concentrations in the cells employed in our study, others have shown previously that dNTP levels are ∼20-fold higher in established T cell lines than in primary peripheral blood mononuclear cells (6). The presence of the E138K mutation does not change the evolutionary pattern in either cell type, indicating that the E138K mutation cannot prevent the emergence of the M184I/V mutations under 3TC-FTC pressure.
We also investigated whether the M184I/V mutations can prevent the emergence of the E138K mutation under ETR selection. In CBMCs, the E138K mutation was consistently selected under ETR pressure using both M184I and M184V clonal variants. In contrast, the E138G/Q mutations were selected in MT2 cells under similar circumstances, instead of the E138K mutation. It is not known whether such differences may be attributable to enhanced dNTP usage and a higher processivity associated with the E138K mutation. This is even true in comparison with WT RT under conditions of low dNTP concentrations (54), which makes the HIV-1 E138K mutant more replication competent in cells with low dNTP pools.
Genotyping at various time points after the application of FTC pressure showed that there were no mutations at week 10 and a mixture of M184I/M/V mutations at week 13 and that the M184V mutation was fully represented by week 19. With 3TC pressure, there were no mutations at week 7, a mixture of M184I/M/V mutations at week 10, and the M184V mutation alone at week 13. Previous experiments have indicated that the M184V mutation prevails in MT2 cell culture selections, if drug pressure is continued with either 3TC or FTC (data not shown).
The compensatory effect between the E138K and M184I mutations in enhancing dNTP usage may also stabilize the M184I mutation in CBMCs. The WT virus did not generate the E138K mutation in MT2 cells, which may also be related to the dNTP pool imbalance, since changing the intracellular dCTP/dTTP ratio can alter the evolutionary pattern at E138 under TSAO pressure (7). Possibly, the different sizes of intracellular dNTP pools in MT2 cells and CBMCs also impact the dNTP imbalance bias. The differences among the cell types studied here in regard to dNTP levels may have contributed to differences in results obtained by our group and other groups in regard to considerations of viral fitness among viruses harboring both the M184I and E138K mutations when grown in different cell types (31, 54). Our selection data clearly show that the E138K and M184I/V mutations can be mutually favored in evolution under appropriate selection pressure. It may be anticipated that the increased use of second-generation NNRTIs such as ETR and RPV will lead to a greater selection of E138K mutations in the future. It is therefore relevant to ask whether the E138K mutation may enhance the transmission rates of detectable M184I/V mutations, and methods more sensitive than classical genotyping may be necessary to provide an answer. To try to shed further light on these topics, we have also performed biochemical analyses of RT molecules containing the above-described mutations.
Previous studies have shown that the β7-β8 loop spanning amino acids 133 to 140 of the HIV-1 RT p51 subunit participates in forming the floor of the NNRTI binding platform in the RT crystal structure (17) and helps to maintain the stable, functional heterodimeric enzyme (39, 40, 49). Subunit-selective mutagenesis also showed that the E138K mutation confers resistance to TSAO compounds through the p51 subunit (8, 11); however, no experimental data have been available until now in regard to second-generation NNRTIs. The present study on the use of subunit-selective E138K mutants provides the first enzymatic analysis to confirm that the E138K mutation confers resistance to ETR through the p51 subunit and also shows that E138 in the p51 subunit is directly involved in regulating RNase H processing activity and RT polymerization. We also provide evidence that the E138K mutation compensates for M184I-mediated deficits in dNTP usage through both RT subunits. Our claim that the E138K mutation compensates for M184I-mediated deficits in dNTP usage and restores enzyme processivity via both subunits is based on both Km changes and data from the gel-based processivity assay.
The p51 subunit has been shown to have a critical role in the fine-tuning of HIV-1 RT RNase H activity. Recently, subunit-specific analyses showed that the decreased RNase H activity conferred by the N348I mutation mapped to the p51 subunit (44, 57). We have now extended our previous results (54) to show that the E138 residue in the β7-β8 loop of the p51 subunit also plays a key role in regulating the RNase H processing activity. The decreased RNase H activity associated with the E138K mutation together with a lower rate of polymerization help to explain the lower replication capacity of the E138K virus. Our data on RNase H activity were generated under processive conditions so as to demonstrate the effect of subunit-specific mutations on RNase H cleavage activity alone and not on enzyme-substrate reassociations. In the virion, more RT molecules than RNA genomes are present, thus ensuring that RNase H cleavage can proceed as long as the virus is still replication competent, even though a mutation of interest may diminish RNase H activity. Our finding that the E138K mutation also diminishes the RT polymerization rate is the first evidence that p51 is also involved in the regulation of polymerase activity. It is interesting that the E138K mutation compensates for the M184I-mediated deficit in dNTP usage via both subunits.
Hydrogen exchange mass spectrometry of the p66/p51 RT heterodimer showed that the binding of EFV to the p66 subunit could cause an extensive allosteric change in the p51 subunit as well (19). This example of allosteric subunit cross talk complements our in silico analysis, which suggests that the E138K mutation in p51 causes allosteric changes in the p66 subunit. This also helps to elucidate how the E138K mutation, when located within p51, can affect RNase H activity, even though the E138K mutation is remote from the active site of RNase H. The impact of the E138K mutation on dNTP usage might also occur, in part, through allosteric changes in the p66/p51 heterodimer. The position of the E138K mutation within p51 can also impact the functions of other drug resistance mutations located within p66. Although certain mutational interactions are difficult to predict and have come to light only as a result of the ECHO and THRIVE clinical trials, it now seems likely that future drug development may need to consider the possibility of unforeseen mutational interactions that may have adverse consequences. As an example, the potential sexual transmission of replication-competent multiply drug-resistant viruses containing both the E138K and M184I mutations could have adverse implications for public health. It will also be important to develop new NNRTIs that retain activity against E138K-containing viruses.
One question that remains is why patients who developed the M184I/V mutations have not also spontaneously generated the E138K mutation, if, indeed, the simultaneous presence of the latter is associated with a restoration of replication competence. A search of multiple clinical databases has failed to reveal evidence of the presence of the E138K mutation in individuals who developed M184I/V mutations (not shown), and our results clearly show that viruses that contain the M184I/V mutations can be selected with ETR to yield the E138K substitution. Perhaps other mutations that are selected by antiretroviral drugs (ARVs) in the clinic, including some in the connection and RNase H domains, may be able to prevent the generation of the E138K mutation. In addition, we have never witnessed the spontaneous emergence of the E138K mutation in M184I/V clonal viruses that have been grown over periods of many months in either the presence or the absence of 3TC. A more common occurrence is a reversion of the M184I/V mutation to WT M184, followed by the more rapid growth of the WT M184-containing virus. The basis for the bias against the spontaneous selection of the E138K mutation by M184I/V-containing viruses in the clinic will require further investigation.
In summary, we have determined that the activities of the E138K mutation to confer ETR resistance, impair RNase H activity, and decrease the RT polymerization rate all occur through the p51 subunit, while the enhancement of dNTP usage in the context of the M184I mutation results from the involvement of both subunits. In addition to providing structural integrity for the p51/p66 heterodimer, p51 is also important for determining rates of polymerization, RNase H processing, and drug resistance. The compensatory effect between the E138K and M184I/V mutations may have clinical significance in regard to treatment failures involving ETR-RPV as well as the detectability of these mutations in transmitted resistance.
ACKNOWLEDGMENTS
This work was supported by research grants from the Canadian Institutes of Health Research (CIHR).
We thank Stuart F. J. Le Grice for providing HIV-1 RT expression DNAs pRT6H-PROT, pRT, and p6H51 and Tomozumi Imamichi for pNL4.3PFB plasmid DNA.
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 23 May 2012
REFERENCES
- 1. Andries K, et al. 2004. TMC125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 48:4680–4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arts EJ, et al. 1994. Comparison of deoxyoligonucleotide and tRNA(Lys-3) as primers in an endogenous human immunodeficiency virus-1 in vitro reverse transcription/template-switching reaction. J. Biol. Chem. 269:14672–14680 [PubMed] [Google Scholar]
- 3. Asahchop EL, et al. 2011. Characterization of the E138K resistance mutation in HIV-1 reverse transcriptase conferring susceptibility to etravirine in B and non-B HIV-1 subtypes. Antimicrob. Agents Chemother. 55:600–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Azijn H, et al. 2010. TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTI-resistant HIV-1. Antimicrob. Agents Chemother. 54:718–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Back NK, Berkhout B. 1997. Limiting deoxynucleoside triphosphate concentrations emphasize the processivity defect of lamivudine-resistant variants of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 41:2484–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Back NK, et al. 1996. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase enzyme. EMBO J. 15:4040–4049 [PMC free article] [PubMed] [Google Scholar]
- 7. Balzarini J, et al. 2001. Exploitation of the low fidelity of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase and the nucleotide composition bias in the HIV-1 genome to alter the drug resistance development of HIV. J. Virol. 75:5772–5777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Balzarini J, et al. 1994. Sensitivity of (138 Glu→Lys) mutated human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) to HIV-1-specific RT inhibitors. Biochem. Biophys. Res. Commun. 201:1305–1312 [DOI] [PubMed] [Google Scholar]
- 9. Blower S, Bodine E, Kahn J, McFarland W. 2005. The antiretroviral rollout and drug-resistant HIV in Africa: insights from empirical data and theoretical models. AIDS 19:1–14 [DOI] [PubMed] [Google Scholar]
- 10. Blower SM, Aschenbach AN, Gershengorn HB, Kahn JO. 2001. Predicting the unpredictable: transmission of drug-resistant HIV. Nat. Med. 7:1016–1020 [DOI] [PubMed] [Google Scholar]
- 11. Boyer PL, Ding J, Arnold E, Hughes SH. 1994. Subunit specificity of mutations that confer resistance to nonnucleoside inhibitors in human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 38:1909–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buckton AJ, et al. 2011. Increased detection of the HIV-1 reverse transcriptase M184V mutation using mutation-specific minority assays in a UK surveillance study suggests evidence of unrecognized transmitted drug resistance. HIV Med. 12:250–254 [DOI] [PubMed] [Google Scholar]
- 13. Cheynier R, Gratton S, Vartanian JP, Meyerhans A, Wain-Hobson S. 1997. G → A hypermutation does not result from polymerase chain reaction. AIDS Res. Hum. Retroviruses 13:985–986 [DOI] [PubMed] [Google Scholar]
- 14. Diallo K, et al. 2003. The M184V substitution in human immunodeficiency virus type 1 reverse transcriptase delays the development of resistance to amprenavir and efavirenz in subtype B and C clinical isolates. Antimicrob. Agents Chemother. 47:2376–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diallo K, Gotte M, Wainberg MA. 2003. Molecular impact of the M184V mutation in human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 47:3377–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diamond TL, et al. 2004. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 279:51545–51553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Esnouf RM, et al. 1997. Unique features in the structure of the complex between HIV-1 reverse transcriptase and the bis(heteroaryl)piperazine (BHAP) U-90152 explain resistance mutations for this nonnucleoside inhibitor. Proc. Natl. Acad. Sci. U. S. A. 94:3984–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao L, et al. 2008. Apparent defects in processive DNA synthesis, strand transfer, and primer elongation of Met-184 mutants of HIV-1 reverse transcriptase derive solely from a dNTP utilization defect. J. Biol. Chem. 283:9196–9205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goff SP. 1990. Retroviral reverse transcriptase: synthesis, structure, and function. J. Acquir. Immune Defic. Syndr. 3:817–831 [PubMed] [Google Scholar]
- 20. Howard KJ, Frank KB, Sim IS, Le Grice SF. 1991. Reconstitution and properties of homologous and chimeric HIV-1.HIV-2 p66.p51 reverse transcriptase.J. Biol. Chem. 266:23003–23009 [PubMed] [Google Scholar]
- 21. Hu Z, Kuritzkes DR. 2011. Interaction of reverse transcriptase (RT) mutations conferring resistance to lamivudine and etravirine: effects on fitness and RT activity of human immunodeficiency virus type 1. J. Virol. 85:11309–11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang W, Gamarnik A, Limoli K, Petropoulos CJ, Whitcomb JM. 2003. Amino acid substitutions at position 190 of human immunodeficiency virus type 1 reverse transcriptase increase susceptibility to delavirdine and impair virus replication. J. Virol. 77:1512–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Imamichi T, et al. 2000. Relative replication fitness of a high-level 3′-azido-3′-deoxythymidine-resistant variant of human immunodeficiency virus type 1 possessing an amino acid deletion at codon 67 and a novel substitution (Thr→Gly) at codon 69. J. Virol. 74:10958–10964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jamburuthugoda VK, et al. 2008. Reduced dNTP binding affinity of 3TC-resistant M184I HIV-1 reverse transcriptase variants responsible for viral infection failure in macrophage. J. Biol. Chem. 283:9206–9216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Janssen PA, et al. 2005. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (R278474, rilpivirine). J. Med. Chem. 48:1901–1909 [DOI] [PubMed] [Google Scholar]
- 26. Johnson VA, et al. 2011. 2011 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 19:156–164 [PMC free article] [PubMed] [Google Scholar]
- 27. Julias JG, Pathak VK. 1998. Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate. J. Virol. 72:7941–7949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kati WM, Johnson KA, Jerva LF, Anderson KS. 1992. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267:25988–25997 [PubMed] [Google Scholar]
- 29. Keulen W, Back NK, van Wijk A, Boucher CA, Berkhout B. 1997. Initial appearance of the 184Ile variant in lamivudine-treated patients is caused by the mutational bias of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 71:3346–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. 1992. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256:1783–1790 [DOI] [PubMed] [Google Scholar]
- 31. Kulkarni R, et al. 2012. The HIV-1 reverse transcriptase M184I mutation enhances the E138K-associated resistance to rilpivirine and decreases viral fitness. J. Acquir. Immune Defic. Syndr. 59:47–54 [DOI] [PubMed] [Google Scholar]
- 32. Kulkarni SS, et al. 2009. Highly complex neutralization determinants on a monophyletic lineage of newly transmitted subtype C HIV-1 Env clones from India. Virology 385:505–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Le Grice SF, Cameron CE, Benkovic SJ. 1995. Purification and characterization of human immunodeficiency virus type 1 reverse transcriptase. Methods Enzymol. 262:130–144 [DOI] [PubMed] [Google Scholar]
- 34. Macarthur RD. 2011. Clinical trial report: TMC278 (rilpivirine) versus efavirenz as initial therapy in treatment-naive, HIV-1-infected patients. Curr. Infect. Dis. Rep. 13:1–3 [DOI] [PubMed] [Google Scholar]
- 35. Menendez-Arias L, Betancor G, Matamoros T. 2011. HIV-1 reverse transcriptase connection subdomain mutations involved in resistance to approved non-nucleoside inhibitors. Antiviral Res. 92:139–149 [DOI] [PubMed] [Google Scholar]
- 36. Meyerhans A, et al. 1994. Restriction and enhancement of human immunodeficiency virus type 1 replication by modulation of intracellular deoxynucleoside triphosphate pools. J. Virol. 68:535–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nikolenko GN, Svarovskaia ES, Delviks KA, Pathak VK. 2004. Antiretroviral drug resistance mutations in human immunodeficiency virus type 1 reverse transcriptase increase template-switching frequency. J. Virol. 78:8761–8770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oliveira M, Brenner BG, Wainberg MA. 2009. Isolation of drug-resistant mutant HIV variants using tissue culture drug selection. Methods Mol. Biol. 485:427–433 [DOI] [PubMed] [Google Scholar]
- 39. Pandey PK, et al. 2002. Insertion of a small peptide of six amino acids into the beta7-beta8 loop of the p51 subunit of HIV-1 reverse transcriptase perturbs the heterodimer and affects its activities. BMC Biochem. 3:18 doi:10.1186/1471-2091-3-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pandey PK, Kaushik N, Talele TT, Yadav PN, Pandey VN. 2001. The beta7-beta8 loop of the p51 subunit in the heterodimeric (p66/p51) human immunodeficiency virus type 1 reverse transcriptase is essential for the catalytic function of the p66 subunit. Biochemistry 40:9505–9512 [DOI] [PubMed] [Google Scholar]
- 41. Quan Y, et al. 2003. Drug resistance profiles of recombinant reverse transcriptases from human immunodeficiency virus type 1 subtypes A/E, B, and C. AIDS Res. Hum. Retroviruses 19:743–753 [DOI] [PubMed] [Google Scholar]
- 42. Reutrakul V, et al. 2010. Anti-HIV-1 and anti-inflammatory lupanes from the leaves, twigs, and resin of Garcinia hanburyi. Planta Med. 76:368–371 [DOI] [PubMed] [Google Scholar]
- 43. Richmond NJ, et al. 2006. GALAHAD. 1. Pharmacophore identification by hypermolecular alignment of ligands in 3D. J. Comput. Aided Mol. Des. 20:567–587 [DOI] [PubMed] [Google Scholar]
- 44. Schuckmann MM, et al. 2010. The N348I mutation at the connection subdomain of HIV-1 reverse transcriptase decreases binding to nevirapine. J. Biol. Chem. 285:38700–38709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smerdon SJ, et al. 1994. Structure of the binding site for nonnucleoside inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 91:3911–3915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tambuyzer L, et al. 2010. Characterization of genotypic and phenotypic changes in HIV-1-infected patients with virologic failure on an etravirine-containing regimen in the DUET-1 and DUET-2 clinical studies. AIDS Res. Hum. Retroviruses 26:1197–1205 [DOI] [PubMed] [Google Scholar]
- 47. Turner D. 2011. Does the M184V resistance mutation in reverse transcriptase reduce HIV transmission? HIV Med. 12:193–194 [DOI] [PubMed] [Google Scholar]
- 48. Turner D, Brenner B, Wainberg MA. 2003. Multiple effects of the M184V resistance mutation in the reverse transcriptase of human immunodeficiency virus type 1. Clin. Diagn. Lab. Immunol. 10:979–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Upadhyay A, Pandey N, Mishra CA, Talele TT, Pandey VN. 2010. A single deletion at position 134, 135, or 136 in the beta 7-beta 8 loop of the p51 subunit of HIV-1 RT disrupts the formation of heterodimeric enzyme. J. Cell. Biochem. 109:598–605 [DOI] [PubMed] [Google Scholar]
- 50. Van Cor-Hosmer SK, Daddacha W, Kim B. 2010. Mechanistic interplay among the M184I HIV-1 reverse transcriptase mutant, the central polypurine tract, cellular dNTP concentrations and drug sensitivity. Virology 406:253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wainberg MA. 2004. The impact of the M184V substitution on drug resistance and viral fitness. Expert Rev. Anti Infect. Ther. 2:147–151 [DOI] [PubMed] [Google Scholar]
- 52. Wainberg MA, Moisi D, Oliveira M, Toni TD, Brenner BG. 2011. Transmission dynamics of the M184V drug resistance mutation in primary HIV infection. J. Antimicrob. Chemother. 66:2346–2349 [DOI] [PubMed] [Google Scholar]
- 53. Xu H, et al. 2009. Human immunodeficiency virus type 1 recombinant reverse transcriptase enzymes containing the G190A and Y181C resistance mutations remain sensitive to etravirine. Antimicrob. Agents Chemother. 53:4667–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu HT, et al. 2011. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J. Virol. 85:11300–11308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu HT, Oliveira M, Quan Y, Bar-Magen T, Wainberg MA. 2010. Differential impact of the HIV-1 non-nucleoside reverse transcriptase inhibitor mutations K103N and M230L on viral replication and enzyme function. J. Antimicrob. Chemother. 65:2291–2299 [DOI] [PubMed] [Google Scholar]
- 56. Xu HT, et al. 2010. Comparative biochemical analysis of recombinant reverse transcriptase enzymes of HIV-1 subtype B and subtype C. Retrovirology 7:80 doi:10.1186/1742-4690-7-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yap SH, et al. 2007. N348I in the connection domain of HIV-1 reverse transcriptase confers zidovudine and nevirapine resistance. PLoS Med. 4:e335 doi:10.1371/journal.pmed.0040335 [DOI] [PMC free article] [PubMed] [Google Scholar]