

Abstract
The vesicular stomatitis virus (VSV) nucleoprotein (N) associates tightly with the viral genomic RNA. This N-RNA complex constitutes the template for the RNA-dependent RNA polymerase L, which engages the nucleocapsid via its phosphoprotein cofactor P. While N and P proteins play important roles in regulating viral gene expression, the molecular basis of this regulation remains incompletely understood. Here we show that mutations in the extreme C terminus of N cause defects in viral gene expression. To determine the underlying cause of such defects, we examined the effects of the mutations separately on encapsidation and RNA synthesis. Expression of N together with P in Escherichia coli results predominantly in the formation of decameric N-RNA rings. In contrast, nucleocapsid complexes containing the substitution NY415A or NK417A were more loosely coiled, as revealed by electron microscopy (EM). In addition, the NEF419/420AA mutant was unable to encapsidate RNA. To further characterize these mutants, we engineered an infectious cDNA clone of VSV and employed N-RNA templates from those viruses to reconstitute RNA synthesis in vitro. The transcription assays revealed specific defects in polymerase utilization of the template that result in overall decreased RNA quantities, including reduced amounts of leader RNA. Passage of the recombinant viruses in cell culture led to the accumulation of compensatory second-site mutations in close proximity to the original mutations, underscoring the critical role of structural features within the C terminus in regulating N function.
INTRODUCTION
Nonsegmented negative-strand (NNS) RNA viruses are enveloped pathogens, which tightly encapsidate their genomes with the nucleoprotein (N) during all stages of replication. N encapsidation is thought to protect the viral RNA from degradation and detection by host defense mechanisms. The ribonucleoprotein (RNP) constitutes the natural template for the viral polymerase machinery and forms long, helical, and flexible nucleocapsids in the cytoplasm of the infected cell (reviewed in reference 21).
Expression of vesicular stomatitis virus (VSV) and rabies virus N proteins in Escherichia coli or Spodoptera frugiperda results in the formation of oligomeric ring complexes comprising 9 to 13 molecules of N that bind cellular RNA (1, 9). The RNA is stored between the C- and N-terminal lobes of the N protein and is accessed by the RNA-dependent RNA polymerase L and its multimeric cofactor P during the transcription process. The RNP is completely resistant to digestion with nucleases during transcription (14), illustrating the tight coordination of the interactions between the transcription apparatus and the viral RNA. Atomic structures of VSV and rabies virus nucleocapsid complexes (2, 8, 11) revealed that N must be displaced for L to copy the RNA. Further structural analysis demonstrated that the C-terminal domain of VSV P (PCTD) binds between adjacent N molecules at an interface formed by a continuous stretch of residues (amino acids [aa] 354 to 386), which exists only in the N-RNA complex (8). Because only minimal structural differences were observed for the PCTD-bound and unbound states, the molecular changes that presumably occur upon binding of the active transcription complex to the nucleocapsid remain elusive. Although the nucleocapsid structures are elongated and flexible in the cytoplasm, they become tightly condensed during the assembly of the bullet-shaped virion.
N is also maintained in the infected cell in a soluble RNA-free state (N0). This is achieved through binding of P protein (N0-P) and is important for preventing the aggregation and premature binding of RNA. A possible strategy by which P may facilitate its chaperone activity has been put forward recently: N protein lacking the 21 N-terminal amino acids was found to bind a peptide of P containing the first 60 amino acids within a region that overlaps with the RNA binding groove (16). This temporary occupation of the RNA binding pocket may prevent nonspecific binding of nucleic acids and N oligomerization.
Both the N and C termini of the VSV nucleoprotein were previously suggested to be essential for encapsidation and RNA synthesis (5, 12, 20, 24). Deletion of the first 22 residues of N (NΔ1-22) prevents the encapsidation of RNA (24). Similarly, deletion of the C-terminal lysine (422K) or of amino acids 418 to 422 (VEFDK) of the N protein of the New Jersey strain of VSV (VSV-NJ) completely abrogates encapsidation (5). In this report, we investigate further the role of the extreme C terminus of N of the Indiana strain of VSV (VSV-IND) in viral replication. We analyze single- and multi-amino-acid substitutions located within the last eight residues of the C terminus of N, some of which are highly conserved across the Rhabdoviridae family. We show that the C terminus of N is an important determinant of encapsidation and RNA synthesis. We also identify several second-site suppressor mutants that compensate for loss of charge or hydrophobicity within this region, providing further support for a structural role of this domain in maintaining N function.
MATERIALS AND METHODS
Plasmids and sequence alignments.
The sequences of the extreme C termini of the N proteins of the following members of the family Rhabdoviridae were aligned using MAFFT software: VSV-IND (VSIV) (NBCI database accession no. NP_041712), VSV-Alagoas (ACB47439), VSV-NJ (NCAP_VSNJO), Chandipura virus (CHPV) (AAU81954), Isfahan virus (ISFV) (Q5K2K7), Cocal virus (CV) (EU373657), Piry virus (PV) (P26037), spring viremia of carp virus (SVCV) (NP_116744), pike fry rhabdovirus (PFV) (ACP27998), Australian bat lyssavirus (ABLV) (NC_003243), Mokola virus (MOKV) (YP_1422350), rabies virus (RABV) (NP_056793), and European bat lyssavirus 1 (EBLV-1) (YP_001285388). Site-directed mutagenesis was performed on plasmids pN (18), pET-N/P (9), pVSV1(+) (22), and pVSV1(+)RFP-P (13) by using standard cloning procedures. A region of the recombinant VSV genome was subcloned to prevent the introduction of inadvertent secondary mutations by the cloning procedure. Individual nucleotide sequences were mutated as follows: Y415A (TAT→GCT), K417A (AAG→GCG), E419A (GAA→GCC), F420A (TTT→GCT), D421A (GAC→GCC), K422A (AAA→GCA), D421N (GAC→AAT), Q405R (CAA→CGC), T361N (ACC→AAT), and Q318L (CAG→CTG). All mutations introduced by site-directed mutagenesis were subsequently verified by sequencing.
VSV replicon construction.
A VSV replicon plasmid encoding enhanced green fluorescent protein (eGFP) was prepared with the backbone vector 2.0 (3) by using a previously described cloning strategy to generate an arenavirus replicon analog (15). The noncoding VSV Indiana genomic terminus sequences (GenBank accession no. NC_001560) were cloned and appended to a codon-optimized eGFP open reading frame (ORF) (Clontech Laboratories, Inc., Mountain View, CA). The replicon is flanked by a truncated T7 promoter ending with two guanosine nucleotides on the 5′ end and the hepatitis delta virus ribozyme and T7 terminator sequence on the 3′ end. Intracellular transcription by T7 RNA polymerase produces the following genomic RNA analog: 3′-leader sequence (50 nucleotides [nt]), N gene start signal (10 nt), GAAUUCUCGACCCAUG (the EcoRI restriction site is in boldface, and the eGFP start codon is underlined), eGFP ORF, UGAGUCGAGUGGCC (the eGFP stop codon is underlined, and the nonviral portion of the MscI restriction site is in boldface), gene stop signal (12 nt), trailer sequence (60 nt)-5′.
VSV gene expression replicon assay.
To assess the levels of VSV gene expression supported by mutated pN plasmids, BsrT7 cells were seeded onto 60-mm dishes, grown to 80% confluency, and infected with a recombinant vaccinia virus (vTF7-3) producing T7 RNA polymerase (6). Vectors pL (1.2 μg), pP (2.3 μg), and wild-type or mutagenized pN (5.25 μg) expressing VSV replication proteins (18) were transfected into cells 1 h postinfection by using Lipofectamine 2000 (Life Technologies, Grand Island, NY) along with a negative-sense replicon construct encoding eGFP (6 μg). A construct encoding a catalytically inactive L protein (G713A) was used as a negative control to assess background fluorescence. Expression of eGFP was detected 48 h posttransfection by measuring fluorescence at 488 nm using a Typhoon 9400 phosphorimager (GE Healthcare, United Kingdom). Quantitative analyses were performed using ImageQuant software (GE Healthcare).
Rescue and growth curves of infectious viruses.
As in the replicon assay, BsrT7 cells were seeded onto 60-mm dishes and were infected the next day with vTF7-3. At 1 h postinfection, cells were transfected with plasmids expressing viral replication proteins (pL, pP, and pN) as well as with 6 μg of vector pVSV1(+) (22) or vector pVSV1(+)RFP-P (13). These full-length genomic VSV-IND constructs contained either wild-type or mutagenized N genes. Supernatants containing rescued virions were harvested 36 h posttransfection (passage P0). To confirm the presence of the original mutations and to test for the emergence of second-site mutations, recovered viruses were plaque purified, and viruses from passages P1 to P10 were sequenced by isolation of viral RNA from infected cells using an RNeasy kit (Qiagen, Valencia, CA). For growth curve measurements, BsrT7 cells were infected at a multiplicity of infection (MOI) of 0.1 for 1 h and were washed 3 times with Dulbecco's modified Eagle medium (DMEM) containing 2% fetal bovine serum (FBS), and viral titers were determined by plaque assay analysis at the indicated times (see Fig. 4B).
Fig 4.
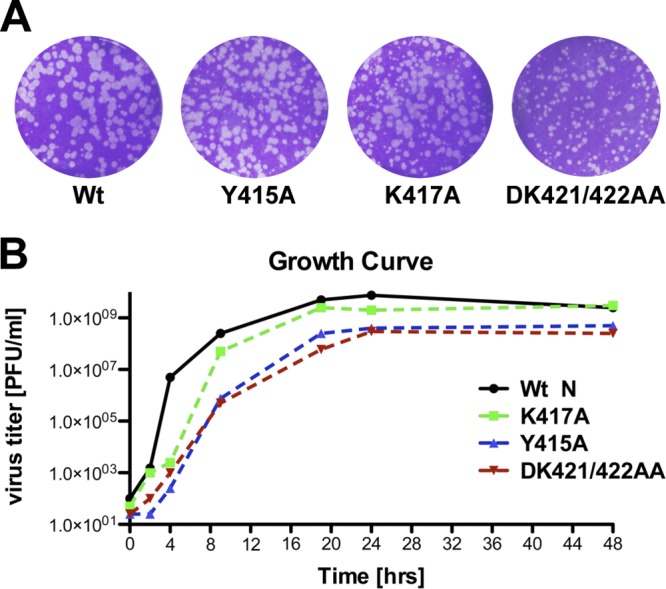
Rescue of recombinant viruses. (A) Plaque assays of rescued mutant viruses after 24 h of incubation at 37°C. Infectious viruses carrying the mutation Y415A, K417A, or DK421/422AA were rescued. Rescue was unsuccessful for all constructs with an alanine substitution at position 420. (B) Growth curves of rescued mutant viruses.
Protein expression in mammalian cells.
vTF7-3-infected BsrT7 cells were transfected with 5.25 μg of wild-type plasmid pN or mutagenized plasmid pN containing the alterations NY415A, NK417A, NE419A, NF420A, NEF419/420AA, NDK421/422AA, or NEFDK419-422A4. At 5 h posttransfection, cells were starved of l-methionine and l-cysteine for 1 h in the presence of 10 μg ml−1 actinomycin D (Sigma, St. Louis, MO). This was followed by metabolic incorporation of [35S]Met from 10 to 16 h posttransfection and subsequent analysis of total cytoplasmic protein by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% SDS-PAGE). The radioactive signal was detected using a Typhoon 9400 phosphorimager (GE Healthcare).
Purification of N-P-RNA complexes and electron microscopy.
Wild-type and mutant (NY415A, NK417A, NEF419/420AA, and NDK421/422AA) pET-N/P (9) constructs were expressed in E. coli. N-P protein complexes were purified with Ni-nitrilotriacetic acid (NTA) agarose via a 10× His tag at the N terminus of P, followed by gel filtration (Superdex 200 HR 10/30 column; GE Healthcare). Eluted peak fractions were analyzed for the presence of N and P proteins by SDS-PAGE and Coomassie blue staining. For electron microscopy (EM), purified N-P complexes were transferred to copper grids, stained for 30 s with 2% phosphotungstic acid (PTA) in H2O (pH 7.5), and washed twice for 30 s with H2O. The grids were then examined in a TecnaiG2 Spirit BioTWIN transmission electron microscope (FEI, Hillsboro, OR). Images were recorded with an AMT 2k charge-coupled device (CCD) camera.
Nuclease digestion of encapsidated templates.
Purified N-P-RNA complexes (5 mg) were subjected to treatment with 100 U micrococcal nuclease (New England BioLabs, Ipswich, MA) for 30 min at 37°C. The reaction was stopped by the addition of 20 mM EGTA and 10% SDS (final concentrations), and the RNA was phenol-chloroform extracted. For visualization, the RNA was 5′ end labeled with [γ-32P]ATP (Perkin-Elmer, Waltham, MA) and was electrophoresed on a 6% polyacrylamide gel.
Transcription of viral mRNAs in vitro.
Viral RNAs were transcribed in vitro as described previously (4) with minor modifications (23). For RNA synthesis reactions using detergent-activated virus, wild-type and mutant (NY415A, NK417A, and NDK421/422AA) recombinant VSVs (rVSV–red fluorescent protein [RFP]–P) were purified and incubated (10 μg) in the presence of nucleoside triphosphates (1 mM ATP and 0.5 mM each CTP, GTP, and UTP), 10 μg ml−1 actinomycin D (Sigma), rabbit reticulocyte lysate (Life Technologies), and 15 μCi of [α-32P]GTP (Perkin-Elmer). For transcription from purified nucleocapsids (5 μg), genomic N-RNA templates from infectious viruses (rVSV) were prepared as described previously (19), and reaction mixtures were supplemented with recombinant L (1 μg) and P (0.5 μg) proteins. Briefly, 6× His-tagged L and 10× His-tagged P proteins were expressed in Spodoptera frugiperda 21 cells and were affinity purified with Ni-NTA agarose (Qiagen) as described previously (19). This was followed by ion-exchange chromatography (L protein) or gel filtration (P protein). Leader RNA was transcribed as described previously (17). Portions (155 ng) of the template were mixed with 0.2 μM VSV L and 0.3 μM VSV P in a reaction mixture containing 20 mM Tris base (pH 8), 50 mM NaCl, 6 mM MgCl2, 200 μM UTP, 1.5 mM ATP, 1.5 mM CTP, 250 μM GTP, and 330 nM [α-32P]GTP (3,000 Ci/mmol). Reaction mixtures were incubated at 30°C for 3 h, and reactions were stopped by the addition of EDTA-formamide. Reaction products were resolved using denaturing polyacrylamide gel electrophoresis (20% polyacrylamide, 7 M urea) in Tris-borate-EDTA (TBE) buffer and were analyzed by autoradiography. Radioisotopes were purchased from Perkin-Elmer.
RESULTS
Conservation of amino acids at the C termini of rhabdovirus N proteins.
The C-terminal five amino acids of the VSV-NJ N protein were previously implicated in the encapsidation and replication of the genome, with K422 playing a critical role (5). An alignment of the C termini of N proteins within the family Rhabdoviridae, however, shows that K422 of VSV-NJ is not completely conserved within the genus Vesiculovirus (Fig. 1A). This alignment also shows that the physiochemical properties of several other amino acids within this region, including F420 and Y415, are highly conserved. We also noted that position 417 maintains a positive charge (K/R) for the vesiculoviruses but is an N for the lyssaviruses. As a first step in mechanistically probing the role of the extreme C terminus of the VSV-IND N protein in encapsidation and RNA synthesis, we performed site-directed mutagenesis to introduce the following changes: Y415A, K417A, E419A, F420A, EF419/420AA, DK421/422AA, and EFDK419-422A4 (Fig. 1B).
Fig 1.

Sequence alignment of the extreme C terminus of the N protein in the vesiculoviruses and lyssaviruses of the Rhabdoviridae family. (A) Alignments shown are from VSV-IND (VSIV), VSV-Alagoas, VSV–NJ, Chandipura virus (CHPV), Isfahan virus (ISFV), Cocal virus (CV), Piry virus (PV), spring viremia of carp virus (SVCV), pike fry rhabdovirus (PFV), Australian bat lyssavirus (ABLV), Mokola virus (MOKV), rabies virus (RABV), and European bat lyssavirus 1 (EBLV-1). Residues are colored according to their physiochemical properties; shading represents conservation. Numbers at the top correspond to amino acid positions of VSV-IND N. The vertical black line on the left indicates members of the genus Vesiculovirus; the gray line, members of the genus Lyssavirus. (B) Amino acid changes to the C terminus of VSV-IND N engineered in this study.
Amino acid substitutions in the C terminus of N diminish viral gene expression.
To examine the effects of mutations in the N gene on viral gene expression, we employed a cell-based reporter gene assay (Fig. 2A). Transcription from the viral replicon RNA requires coexpression of the VSV N, P, and L proteins, and eGFP reporter protein expression occurs only in the presence of a functional VSV RNA synthesis complex. Each of the altered N proteins was expressed at levels comparable to wild-type levels, as determined by metabolic incorporation of [35S]Met from 10 to 16 h posttransfection and subsequent analysis of total cytoplasmic protein by SDS-PAGE (Fig. 2B). To measure VSV gene expression, we compared eGFP expression at 48 h posttransfection (Fig. 2C). Visual inspection of the transfected cells revealed that expression of eGFP was L dependent and was reduced by each of the N gene mutations (Fig. 2C). Quantification of the fluorescence intensities of the cells using a fluorescence imager (Typhoon 9400 phosphorimager) and ImageQuant software demonstrated that all substitutions involving F420A showed a reduction in gene expression to about 5%, and substitutions Y415A and K417A showed reductions to about 10%, of wild-type N expression (Fig. 2D). The effects of these substitutions on gene expression are consistent with the fact that hydrophobic side chains are retained across all of the rhabdoviruses at positions 415 and 420, while a positive charge at position 417 is conserved within vesiculoviruses. Although the two terminal residues D421 and K422 (DK421/422) differ across vesiculoviruses, replacement with alanine at these positions reduced gene expression >80% (Fig. 2D). In contrast, alanine replacement of E419, which is also not a highly conserved residue, decreased gene expression only modestly (Fig. 2D). These data demonstrate that amino acid substitutions within the extreme C terminus of the VSV-IND N protein inhibit viral gene expression, and they define a set of residues hypersensitive to substitution. Since each of the proteins was expressed in transfected cells (Fig. 2B), this block to gene expression appears to be downstream of protein synthesis.
Fig 2.

Substitutions within the extreme C terminus of N result in a loss of gene expression. (A) Schematic of the VSV gene expression assay. BsrT7 cells expressing T7 RNA polymerase from vaccinia virus vTF7-3 were transfected with plasmids encoding the VSV replication proteins N (wild type or mutant), P, and L, as well as with a minireplicon construct that serves as a template for the VSV polymerase and results in the expression of eGFP. (B) Protein expression of T7-driven constructs in transfected BsrT7 cells. N protein expression was assessed by metabolic incorporation of [35S]methionine and SDS-PAGE analysis. Untransfected cells (−) served as a negative control. (C) Images of entire wells showing eGFP expression of cells transfected with wild-type (Wt) or mutant nucleoprotein. Linact. encodes a catalytically inactive mutant (G713A) of the L protein, which was coexpressed with Wt N and served as a negative control. (D) Quantification of fluorescence intensities shown in panel B. The gene expression level of Wt N was set to 100%, while that from the catalytically inactive L protein was used to determine background fluorescence, which was subtracted from all conditions. Error bars represent standard errors of the means from three independent experiments.
C-terminal amino acid substitutions in N affect the assembly of nucleocapsid complexes.
To further analyze the molecular basis of the gene expression defects described above, we engineered a subset of those mutations into a vector coexpressing the VSV N and P genes in E. coli (9). Since we detected only modest effects of the E419A substitution on gene expression, we excluded this substitution from future analysis. N-P complexes were purified via an N-terminal 10× His tag on the P protein; their protein contents were examined by SDS-PAGE; and their morphologies were analyzed by electron microscopy. None of the substitutions impacted the ability of the N protein to form a complex with P (Fig. 3A to E). Purified N-P complexes containing wild-type N protein formed characteristic decameric rings of 160 Å in diameter (Fig. 3A) (11). N-P complexes assembled from NDK421/422AA also formed similarly sized rings (Fig. 3B). However, each of the substitutions NK417A and NY415A resulted in distinct N-P complex formation, displaying a heterogeneous mixture of incomplete and closed ring structures. Closed rings were irregular in size; some complexes measured as much as 270 Å in diameter (Fig. 3C and D). N proteins harboring alterations at position 420 completely failed to arrange themselves into ring structures (Fig. 3E). These data show that substitutions within the extreme C terminus affect nucleocapsid complex assembly.
Fig 3.

Mutations in the extreme C terminus of the N gene lead to defects in N-RNA complex assembly and RNA encapsidation. (A to E) E. coli-expressed, purified N-P complexes carrying substitutions at the extreme C terminus of the N protein were purified by gel chromatography, run on SDS-PAGE gels to confirm the presence of N and P proteins by Coomassie staining, and analyzed for abnormal complex formation by electron microscopy. Ring formation was observed in all samples (A to D, arrows) except for N carrying the substitution EF419/420AA (E). Bars, 100 nm in figures and 50 nm in insets. Wt, wild type. (F) (Left panel) E. coli-expressed, purified N-P complexes were tested for their abilities to encapsidate RNA and to protect this RNA from nuclease digestion. Samples were treated with micrococcal nuclease (MN) for 30 min, after which the reaction was terminated by treatment with 10% SDS and 20 mM EGTA (final concentrations). The RNA was phenol-chloroform extracted, radioactively end labeled, and run on a 6% polyacrylamide gel. Naked RNA was additionally subjected to RNase treatment to control for enzyme activity. All mutant N proteins were competent for RNA encapsidation, except for the EF419/420AA mutant, which demonstrated a lack of RNA presence even before digestion with nuclease. (Right panel) The A260/280 ratios of peak fractions eluted from the gel filtration column confirm the absence of nucleic acid in complexes carrying the substitution EF419/420AA.
To test directly whether the nucleocapsid complexes were capable of encapsidating RNA and protecting this RNA from nuclease digestion, we subjected the purified N-P complexes to RNase treatment prior to RNA isolation. Except for NEF419/420AA, all of the N proteins encapsidate RNA and protect it from nuclease digestion (Fig. 3F). To further substantiate the RNA binding, we measured the ratio of UV absorbance at 260 nm to that at 280 nm. Purified N-P complexes containing the mutant NY415A, NK417A, or NDK421/422AA exhibited absorbance ratios around 0.9. However, the protein complex formed by NEF419/420AA exhibited an A260/280 value of only about 0.5, verifying that no nucleic acid is associated with the latter complex. Taking these results together, we discovered defects in RNP complex formation and RNA encapsidation for the NY415A, NK417A, and NEF419/420AA mutants. NY415A and NK417A formed oligomeric complexes distinct from those of wild-type N, whereas NEF419/420AA failed to oligomerize and associate with RNA.
Recombinant viruses containing N gene mutations display defects in growth.
In the analysis described above, we observed a range of defects in N oligomerization and N-P-RNA complex assembly associated with individual C-terminal substitutions. However, all of the mutants, except for NEF419/420AA, were able to encapsidate RNA. For further definition of the postencapsidation block, we generated a panel of recombinant viruses to test the role of the extreme C terminus of N in RNA synthesis. For ease of analysis, we elected to use an infectious clone of VSV in which the P gene was fused to RFP, a fusion that we previously demonstrated was inert with regard to viral replication (13). Recombinant viruses were recovered from NY415A, NK417A, and NDK421/422AA (Fig. 4A), but despite repeated transfections, we were unable to recover infectious virus from NEFDK419-422A4, NEF419/420AA, or NF420A. These results are consistent with the levels of eGFP expression observed with each of the substitutions in our cell-based assay (Fig. 2C and D) and the inability of NEF419/420AA to associate with RNA (Fig. 3F). Each of the recombinants showed a reduction in plaque size from that of the rVSV-RFP-P parental clone. Growth kinetics and endpoint titers of NK417A yielded titers comparable to wild-type levels of 2 × 109 to 3 × 109 PFU ml−1 (Fig. 4B). In contrast, NY415A and NDK421/422AA showed slower growth kinetics and reached endpoint titers of 2.5 × 108 and 5 × 108 PFU ml−1, respectively (Fig. 4B).
The C terminus of the N protein is required for efficient transcription by enabling full engagement of the polymerase complex.
We then tested the infectious mutant viruses for their abilities to transcribe RNA in an in vitro transcription assay. Using detergent-activated virus, we found that mutants NY415A and NK417A consistently transcribed significantly smaller amounts of mRNA (not shown) than wild-type viruses. To ensure that the effect on transcription efficiency was entirely dependent on the template-associated N protein, we isolated N-RNA templates from infectious virus and reconstituted RNA synthesis in vitro with purified recombinant wild-type L and P proteins. For this purpose, we used templates purified from an engineered version of VSV containing a wild-type P gene. In these assays, mRNA transcription was diminished significantly, to about 55% for the NK417A mutant and to 25% for the NY415A mutant, from wild-type N levels (Fig. 5A and C). The NDK421/422AA mutant N-RNA template supported transcription of mRNAs to approximately wild type levels (Fig. 5C). Quantification of the amounts of leader RNA produced (Fig. 5B and D) gave comparable results, in which the mutant NY415A produced less leader RNA (45%) than the mutant NK417A (79%). The nucleotide pattern of <50 bp, however, appeared unchanged from that of wild-type templates, suggesting that the substitutions did not affect transcription initiation. Transcription of leader RNA with the mutant NDK421/422AA was only minimally affected (Fig. 5D). These data show that mutations in the extreme C terminus of N adversely affect overall utilization of the N-RNA template for transcription but do not inhibit the processivity of the polymerase complex. This defect of the template in efficiently engaging the polymerase leads, consequently, to decreased RNA quantities, including reduced quantities of the leader RNA. Our data thus point to a subset of specific amino acid residues within the C terminus of N that, even though they do not appear to form direct contacts with the encapsidated RNA, facilitate efficient RNA synthesis. Whether the C terminus of N makes direct contact with a region of P or L, or both, in the L-P complex remains unknown.
Fig 5.

C-terminal substitutions in the N protein decrease mRNA transcription from purified templates. (A) N-RNA templates were purified from infectious mutant viruses (rVSV) and were tested in in vitro transcription reactions by supplementing the assay mixture with purified P and L proteins. L, G, N, M, and P indicate the five viral mRNA products. Wt, wild type. (B) Mutant viruses (rVSV) were tested for their abilities to initiate transcription by analyzing levels of leader transcripts (Le). (C) Quantification of the gel in panel A. Error bars represent standard errors of the means from three independent experiments. (D) Quantification of the gel in panel B. Error bars represent standard errors of the means from three independent experiments.
Second-site mutations that restore viral gene expression arise during the passage of mutant viruses.
To test whether second-site mutations arose during recovery or passage of the recombinant viruses, we performed sequence analysis. For this purpose, we elected to sequence the entire N and P genes of the original rescued viruses, and those viruses from every other passage for a total of 10 passages. For each virus, the recombinants initially rescued contained only the engineered mutation within the N gene (Fig. 6A, top). Following passage, each of the recombinant viruses displayed one or more second-site mutations within N (Fig. 6A, bottom), but no mutations were detected within the P gene. Recombinant NY415A contained the substitution Q318L following three passages, while recombinant NK417A acquired two independent substitutions, Q405R and D421N, following the ninth passage. NDK421/422AA acquired the T361N substitution after passage 7 (Fig. 6A, bottom). Growth curves demonstrated a gain in fitness for some of these viruses, in particular for the mutant viruses NY415A/Q318L and NK417A/D421N, which reached titers as high as 5 × 109 PFU ml−1 and 1.4 × 1010 PFU ml−1, respectively (Fig. 6B). This suggests that the uncovered second-site substitutions arose to compensate for the original substitutions. We therefore mapped the locations of the second-site substitutions onto an N–PCTD–RNA atomic structure (8). The C-terminal 14 amino acids (aa 409 to 422) of the VSV-IND N protein form an α-helical domain (α-helix 15) (Fig. 6C, top), which does not make direct contact with the encapsidated genomic RNA or with the PCTD. With the exception of T361N, the compensatory substitutions lie within (D421N) or in close proximity (Q318 and Q405) to the mutagenized α-helix (Fig. 6C, center and bottom). All the second-site substitutions compensate for the loss of charge or the change in polarity/hydrophobicity introduced by the original mutation. For example, upon loss of the extremely hydrophobic tyrosine Y415, a polar residue was substituted for an amino acid containing a more hydrophobic side chain (Q318L), and loss of a basic residue (K417) resulted in the replacement of an acidic side chain for a polar residue (D421N). The Q405R substitution was the most compelling exchange, reintroducing a basic amino acid residue in response to the loss of K417.
Fig 6.

Sequence analysis of passaged mutant viruses uncovers second-site mutations. (A) Sequences of mutant viruses (top) and their respective second-site mutations (bottom). Stars indicate the positions of the nucleotide changes of individual second-site mutations. (B) Maximal viral titers of recombinant viruses carrying second-site mutations in comparison to titers obtained with infectious viruses carrying the original mutation alone. (C) (Top) The final 14 amino acids of the VSV N protein form an α-helix (bright pink) within the N-P-RNA crystal structure (Protein Data Bank [PDB] code 3HHZ). The location of the α-helix is shown in relation to five N molecules (alternating green and blue) in complex with RNA (gray) and the C-terminal N protein-binding domain of the P protein (PCTD) (yellow). (Center and bottom) The location of each second-site substitution (purple) within the N-P-RNA structure is shown in relation to the original mutation (orange).
To examine directly whether the second-site mutations rescue the defects in gene expression, we introduced these into plasmid pN and tested them in our cell-based replicon assay (Fig. 7A). The Q318L substitution, which arose in conjunction with the Y415A modification, restored gene expression to about 90% of wild-type N levels (Fig. 7B). Similarly, D421N and Q405R, which were both discovered as independent second-site alterations of the NK417A mutant virus, restored gene expression to 57% and 100% of wild-type N levels, respectively. This indicates that the major compensating mutation was Q405R, even though this is not reflected in the viral growth curve measurements. The second-site substitution T361N was the least efficient, increasing gene expression to 30% from the 20% base level of NDK421/422AA (Fig. 7B). Collectively, these data show that the second-site mutations function as suppressors for defects induced by the original mutations. We conclude that the extreme C terminus of N underlies some degree of plasticity in which specific physiochemical properties within this area, including polarity and hydrophobicity distributions, are key for efficient N-RNA complex assembly and RNA synthesis, and consequently for viral gene expression.
Fig 7.
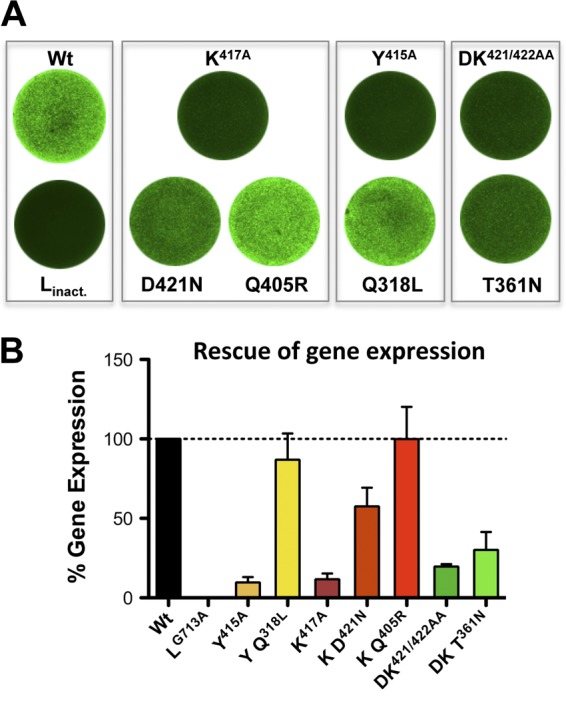
Second-site suppressor substitutions compensate for the loss of viral gene expression. (A) Representative images of VSV gene expression assessed by fluorescence (eGFP) intensities. Plasmids encoding N protein either contained the original alterations alone or, in addition, their respective second-site mutations. Wt, wild type. (B) Quantification of eGFP fluorescence from panel A to assess the extent of the compensatory effects of second-site mutations on viral gene expression. Error bars represent standard errors of the means for three independent experiments.
DISCUSSION
In the current study, we determined that amino acid substitutions to the C-terminal region of VSV N inhibit viral gene expression. By analysis of the mechanism by which such substitutions impede viral gene expression, we define residues critical for N-RNA assembly, encapsidation, and RNA transcription, and through the use of recombinant viruses, we uncover second-site suppressors of such deleterious mutations. This work indicates that structural features within the C terminus of N, and not specific amino acid residues, dictate efficient viral gene expression.
Structural properties of the C terminus of N impact encapsidation and RNA synthesis.
Our work shows that the C terminus of the N protein underlies some degree of plasticity in which the physiochemical properties of structurally neighboring residues are capable of influencing and maintaining the function of this region. This conclusion is apparent from second-site suppressor mutations that retain a charge and/or hydrophobic properties within the C terminus of N. Loss of Y415, for instance, resulted in the replacement of the polar, uncharged Q318 residue with the hydrophobic residue L. Similarly, loss of the positively charged K417 residue led to two independent second-site mutations, Q405R and D421N, both of which compensate for the loss of charge. Each of these amino acid substitutions was beneficial overall, since NY415A/Q318L, NK417A/D421N, and NK417A/Q405R unambiguously restored gene expression (Fig. 7), and NY415A/Q318L and NK417A/D421N clearly improved the fitness of infectious viruses (Fig. 6). The NK417A/D421N mutant attained a gain in fitness: it surpassed standard wild-type titers, growing to 1.4 × 1010 PFU ml−1. Correspondingly, analysis of the second-site mutations using our cell-based replicon assay further showed that restoring physiochemical amino acid properties within, or near, the C-terminal helical domain allows for the restoration of functions that are important for gene expression. Consequently, it is not a defined amino acid sequence that determines the ability of the C-terminal helix to mediate protein-protein or RNA-protein interactions, but the overall structural architecture of this region.
As an exception to all other second site-substitutions, the alteration T361N, which was found in combination with the NDK421/422AA mutant virus, is positioned close to the PCTD binding site. This second-site mutation only minimally rescued gene expression and viral fitness. Prior work had implicated T361 in RNA replication, though not in transcription (12), but this substitution does not appear to act as a second-site suppressor for the DK421/422AA substitution. In contrast to our findings, prior work had shown that alanine replacement of the two terminal residues of VSV N disrupts encapsidation (5). Although we observed a reduction in gene expression for this substitution, we did not observe specific defects in either encapsidation or nucleocapsid assembly. The assays employed in the prior study were quite distinct from those utilized here in that the prior work examined the ability of N to encapsidate a radiolabeled leader RNA in vitro (5). In contrast, we coexpressed N and P in E. coli, followed by isolation of the resulting nucleocapsid complexes. Moreover, we successfully rescued infectious virions for this mutant, indicating that the effects of this substitution on virus replication are not lethal, a phenotype that would be expected from a mutant that is incompetent for RNA encapsidation.
Role of the extreme C terminus in nucleocapsid assembly.
Coexpression of the N and P proteins in E. coli results in the predominant formation of a ring structure comprising 10 molecules of N that encapsidates 90 nt of RNA (11). Here we found that nucleocapsid complexes containing NY415A or NK417A formed heterogeneous mixtures of larger, more loosely coiled N-RNA, indicating that some aspect of nucleocapsid assembly was impacted (Fig. 3). Within those complexes, however, the RNA remained protected from nuclease digestion, consistent with correct encapsidation.
We considered the possibility that such substitutions, in addition to impacting the assembly of N-RNA, may also impact virus particle assembly, since this requires compaction of the nucleocapsid. Cryo-EM analysis of VSV particles reveals that the nucleocapsid is constrained such that there are approximately 10 molecules of N per turn at the very tip and that this expands to 37.5 units per turn in the body region of the bullet-shaped particle (7). The constraints on N in the particle tip, therefore, resemble closely the arrangement of N in the decameric rings obtained following expression in E. coli. The change in N residues per turn in the different parts of the virion results in changes within interactions between adjacent N molecules. Among the most significant adjustments are hydrogen bond interactions between residues R309 and E419 and the hydrophobic interaction I237-Y324 along the interface between adjacent C lobes in the decamer (10 subunits/turn), which show an increased distance of 9 Å over the distance between respective partners in the trunk region. (37.5 subunits/turn). Consistent with the importance of such interactions, the R309A substitution results in the formation of larger ring structures in the E. coli encapsidation assay (7). We therefore reasoned that disrupting such N-N interactions may result in an altered N-RNA structure that could impact the packaging of N-RNA into particles. Despite this possibility, mutations in the C terminus of N that produced nucleocapsid structures showing alterations in morphology in our E. coli encapsidation assay did not affect the morphology of the virus particles (not shown).
Previous reports have also shown that while intermolecular contacts among N molecules are necessary for encapsidation, RNA binding is not a prerequisite for the assembly of stable ring-like N oligomers. The NS290W mutant, for example, forms decameric ring structures devoid of RNA (24). The RNA can also be removed from fully assembled complexes without disrupting the assembly of the N protein (10). It is interesting that even though amino acids F420, Y415, and K417 are not directly involved in mediating N-N interactions, these residues appear to influence N-RNA assembly. Our data thus provide additional evidence for a role of the C terminus in stabilizing intermolecular interactions between N molecules and the nucleocapsid complex as a whole.
Function of the C terminus in RNA synthesis.
The determination of the structure of the N protein has enhanced our understanding of its function. Beyond a structural role of N in RNA encapsidation, recent work has further expanded our view of the role of N in gene expression. The template-associated N stimulates the processivity of the polymerase complex (17). Mutations of residues that are located in close proximity to the RNA binding pocket of N have previously been shown to reduce transcription and RNA replication (20). In addition, mutations in the C-terminal loop differentially affect RNA replication and transcription (12). Our present study adds to these findings by defining the extreme C terminus of the N protein as an important determinant of transcription efficiency. We show that while the L-P complex can recognize and transcribe from templates encapsidated by N proteins carrying C-terminal amino acid substitutions (NY415A and NK417A), the overall amount of mRNA produced from these templates is decreased. There are several possible explanations for this reduction. First, there may simply be a reduction in the ability of the polymerase complex to engage the template. Second, the substitutions could impact the ease with which N can be displaced by the polymerase complex, causing steric hindrance to the forward movement of the polymerase on the RNA. Our work cannot distinguish between these possibilities.
We can, however, infer from our experiments that once the polymerase has initiated the transcription of mRNAs, it is not more likely to disengage, or to fall off from the mutagenized templates, since this would result in an altered transcription gradient. Consequently, our data are more consistent with a defect in which the polymerase complex overall is unable to fully engage the N-RNA template for efficient RNA synthesis rather than with a defect affecting the regulation of viral gene expression. Interactions between the polymerase complex and the first N molecule at the 3′ end may hinder the initial loading of the transcription machinery or, alternatively, slow down the rate of RNA synthesis. In either scenario, the observed defect in transcription efficiency suggests that we have affected residues that are involved in a dynamic part of the RNP. In this regard, it may be of interest to examine whether a C-terminal peptide of N serves as an inhibitor of template copying.
ACKNOWLEDGMENTS
We thank the protein production core of the New England Regional Center for Excellence in Biodefense and Emerging Infectious Disease, in particular Robin Ross and Benjamin Seiler, for assistance with the purification of N-P-RNA complexes. We also acknowledge the Harvard Medical School EM facility and Silvia Piccinotti for help with the electron microscopy studies and Philip Kranzusch for helpful comments on the manuscript.
This study was supported by NIH grant AI059371 to S.P.J.W. S.P.J.W. is a recipient of the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Award.
Footnotes
Published ahead of print 6 June 2012
REFERENCES
- 1. Albertini AA, et al. 2007. Isolation and crystallization of a unique size category of recombinant rabies virus nucleoprotein-RNA rings. J. Struct. Biol. 158:129–133 [DOI] [PubMed] [Google Scholar]
- 2. Albertini AA, et al. 2006. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313:360–363 [DOI] [PubMed] [Google Scholar]
- 3. Ball LA. 1992. Cellular expression of a functional nodavirus RNA replicon from vaccinia virus vectors. J. Virol. 66:2335–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baltimore D. 1970. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 226:1209–1211 [DOI] [PubMed] [Google Scholar]
- 5. Das T, Chakrabarti BK, Chattopadhyay D, Banerjee AK. 1999. Carboxy-terminal five amino acids of the nucleocapsid protein of vesicular stomatitis virus are required for encapsidation and replication of genome RNA. Virology 259:219–227 [DOI] [PubMed] [Google Scholar]
- 6. Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 83:8122–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ge P, et al. 2010. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science 327:689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Green TJ, Luo M. 2009. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor, P. Proc. Natl. Acad. Sci. U. S. A. 106:11713–11718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Green TJ, et al. 2000. Study of the assembly of vesicular stomatitis virus N protein: role of the P protein. J. Virol. 74:9515–9524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Green TJ, et al. 2011. Access to RNA encapsidated in the nucleocapsid of vesicular stomatitis virus. J. Virol. 85:2714–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Green TJ, Zhang X, Wertz GW, Luo M. 2006. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313:357–360 [DOI] [PubMed] [Google Scholar]
- 12. Harouaka D, Wertz GW. 2009. Mutations in the C-terminal loop of the nucleocapsid protein affect vesicular stomatitis virus RNA replication and transcription differentially. J. Virol. 83:11429–11439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog. 6:e1000958 doi:10.1371/journal.ppat.1000958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iseni F, Baudin F, Blondel D, Ruigrok RW. 2000. Structure of the RNA inside the vesicular stomatitis virus nucleocapsid. RNA 6:270–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kranzusch PJ, et al. 2010. Assembly of a functional Machupo virus polymerase complex. Proc. Natl. Acad. Sci. U. S. A. 107:20069–20074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leyrat C, et al. 2011. Structure of the vesicular stomatitis virus N-P complex. PLoS Pathog. 7:e1002248 doi:10.1371/journal.ppat.1002248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morin B, Rahmeh AA, Whelan SP. 2012. Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J. 31:1320–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pattnaik AK, Wertz GW. 1990. Replication and amplification of defective interfering particle RNAs of vesicular stomatitis virus in cells expressing viral proteins from vectors containing cloned cDNAs. J. Virol. 64:2948–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rahmeh AA, et al. 2010. Molecular architecture of the vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 107:20075–20080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rainsford EW, Harouaka D, Wertz GW. 2010. Importance of hydrogen bond contacts between the N protein and RNA genome of vesicular stomatitis virus in encapsidation and RNA synthesis. J. Virol. 84:1741–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruigrok RW, Crepin T, Kolakofsky D. 2011. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 14:504–510 [DOI] [PubMed] [Google Scholar]
- 22. Whelan SP, Ball LA, Barr JN, Wertz GT. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. U. S. A. 92:8388–8392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whelan SP, Wertz GW. 2002. Transcription and replication initiate at separate sites on the vesicular stomatitis virus genome. Proc. Natl. Acad. Sci. U. S. A. 99:9178–9183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang X, Green TJ, Tsao J, Qiu S, Luo M. 2008. Role of intermolecular interactions of vesicular stomatitis virus nucleoprotein in RNA encapsidation. J. Virol. 82:674–682 [DOI] [PMC free article] [PubMed] [Google Scholar]