

Abstract
HIV has evolved sophisticated mechanisms to avoid restriction by intracellular innate immune defenses that otherwise serve to control acute viral infection and virus dissemination. Innate defenses are triggered when pattern recognition receptor (PRR) proteins of the host cell engage pathogen-associated molecule patterns (PAMPs) present in viral products. Interferon regulatory factor 3 (IRF3) plays a central role in PRR signaling of innate immunity to drive the expression of type I interferon (IFN) and interferon-stimulated genes (ISGs), including a variety of HIV restriction factors, that serve to limit viral replication directly and/or program adaptive immunity. Productive infection of T cells by HIV is dependent upon the targeted proteolysis of IRF3 that occurs through a virus-directed mechanism that results in suppression of innate immune defenses. However, the mechanisms by which HIV controls innate immune signaling and IRF3 function are not defined. Here, we examined the innate immune response induced by HIV strains identified through their differential control of PRR signaling. We identified viruses that, unlike typical circulating HIV strains, lack the ability to degrade IRF3. Our studies show that IRF3 regulation maps specifically to the HIV accessory protein Vpu. We define a molecular interaction between Vpu and IRF3 that redirects IRF3 to the endolysosome for proteolytic degradation, thus allowing HIV to avoid the innate antiviral immune response. Our studies reveal that Vpu is an important IRF3 regulator that supports acute HIV infection through innate immune suppression. These observations define the Vpu-IRF3 interface as a novel target for therapeutic strategies aimed at enhancing the immune response to HIV.
INTRODUCTION
Virus infection of mammalian cells triggers an intracellular immune response, termed the “innate immune response,” that functions to suppress viral replication and spread (35, 38). Specific motifs within viral products, including genomic RNA, DNA, or nucleic acid replication intermediates, are recognized by the host cell as pathogen-associated molecular patterns (PAMPs) by cellular factors termed pattern recognition receptors (PRRs) (35, 38). Nucleic acid sensors, including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), as well as several classes of DNA sensors, comprise PRRs that engage viral PAMPs to thereby trigger intracellular processes of innate immunity (35, 38). PRR signaling of downstream interferon regulatory factor 3 (IRF3) activation is a central feature of the innate immune response in most cell types, leading to alpha/beta interferon (IFN-α/β) production; the exception is plasmacytoid dendritic cells (pDCs), which utilize IRF7 in this process (17, 38). IFN produced by virus-infected cells and pDCs drives autocrine and paracrine IFN signaling to generate a response in the infected cell and surrounding tissue that induces hundreds of interferon-stimulated genes (ISGs) (38). ISG products have direct antiviral or immune modulator actions that limit virus replication and spread (35, 38). In order to replicate efficiently, many viruses direct strategies to disrupt various aspects of innate immune signaling or response that range from disrupting PRR signaling to inhibiting ISG function (22). Viral control of IRF3 activation presents a central strategy to prevent the onset of the innate immune response, thereby allowing the virus to avoid the limitations on replication imposed by IFN-α/β, proinflammatory cytokines, and other IRF3-responsive gene products (22). Indeed, several studies have linked the direct or indirect regulation of IRF3 to infection outcome and the pathogenesis of human viruses (22).
HIV-1 is a major human pathogen that has evolved sophisticated mechanisms to modulate intracellular innate immune effectors and restriction factors that otherwise serve to control acute retroviral infection and virus dissemination (5, 21, 27). IRF3 plays a central role in induction of innate immunity in T cells and macrophages to drive the expression of IFN and ISGs, including a variety of directly anti-HIV-1 restriction factors, as well as to program downstream adaptive immunity (1, 20, 27, 30, 37). However, a variety of studies have shown that HIV-1-infected peripheral blood mononuclear cells (PBMCs) or T cell lines exhibit only a limited spectrum of ISG expression concomitant with little, if any, IFN production (3, 12, 28, 33), suggesting that during acute HIV-1 infection PRR signaling programs either are not engaged or are counterregulated by virus-directed processes. In support of this notion, we and others have shown that productive infection of T cells by HIV-1 is accompanied by the specific targeted proteolysis of IRF3 that occurs through a virus-directed mechanism resulting in suppression of innate immune defenses (6, 29). These studies revealed that IRF3 activation drives an innate immune response that is highly deleterious to productive HIV-1 infection, suggesting that targeted viral antagonism of IRF3 by HIV-1 may provide an additional level of viral control of the innate immune system. We now demonstrate that the HIV-1 protein Vpu plays an important role in HIV-1 innate immune regulation by targeting and relocalizing IRF3 for proteolysis. Our studies define the IRF3 regulation to be an additional feature of immune control to support HIV-1 infection.
MATERIALS AND METHODS
Cell culture, transfections, and treatments.
All cells were grown under standard conditions as described previously (6). SupT1 cells were cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS), l-glutamine, and antibiotics. HEK293, 293, 293T, and Tzm-bl cells were cultured in Dulbecco modified Eagle medium supplemented with 10% FBS, l-glutamine, and antibiotics. Transfection of cells was performed using the calcium phosphate method or using Fugene 6 transfection reagent (Roche) according to the manufacturer's suggested protocol. Plasmids pcDNA3.1 (Invitrogen), green fluorescent protein (GFP), GFP-labeled IRF3, and Flag-labeled IRF3 were used where indicated and have been described previously (36). All other expression plasmids have been described previously and were obtained through the AIDS Research and Reference Reagent Program, as described below. The protease inhibitors MG115 (50 μM) and MG132 (5 or 10 μM), as well as the endolysosomal inhibitor chloroquine (2, 5, 10, or 50 μM), were used in the indicated experiments, with 10 μM used in single-dose experiments.
Viral stocks and infection.
HIV-1LAI was propagated using standard procedures as described previously (6), and infections utilized a multiplicity of infection of 1. Vpu point mutant proviral constructs have been described previously (YU2, AD8) (32) or were generated via site-directed mutagenesis (NL4-3, JR-CSF). Generation of point mutant proviral clones was accomplished using a QuikChange mutagenesis kit (Stratagene) following the manufacturer's recommended protocol, optimizing for the large size of the plasmids. The following primers were used: JR-CSF A/C Forward (5′-GCAGTAAGTAGTGCATGTACTGCAACCTTTACAAATATTAGCAATAGTAGC-3′) and Reverse (5′-GCTACTATTGCTAATATTTGTAAAGGTTGCAGTACATGCACTACTTACTGC-3′) and NL4-3 A/C Forward (5′-GCAGTAAGTAGTACATGTAGGGCAACCTATAATAGTAGC-3′) and Reverse (5′-GCTACTATTATAGGTTGCCCTACATGTACTACTTACTGC-3′). HIV-1 strains NL4-3, JR-CSF, and YU2 and Vpu-deficient proviral clones were transfected into 293T cells as described previously to generate infectious virus (15, 16). Mock infections represent treatment with conditioned medium. The titers of all HIV-1 strains were determined on Tzm-bl cells to determine the concentration of infectious virus. Sendai virus (SeV) strain Cantell was obtained from Charles River Laboratories.
Immunoblot analysis, coimmunoprecipitation, and immunofluorescence imaging.
SDS-PAGE, immunoblot analysis, and immunofluorescence were performed using standard procedures as described previously (6). The following antibodies were used in the study: mouse (M) anti-p24, goat (Gt) anti-beta-actin (Santa Cruz), rabbit (Rb) total anti-IRF3 (a gift from Michael David), Rb anti-IRF3-p (Cell Signaling), M total anti-IRF3 (13), Rb anti-HIV-1NL4-3 Vpu, Rb anti-HIV-1 Vpr, M anti-HIV-1 Vif (Santa Cruz), and M anti-human LAMP2 (Abcam). For immunoblot detection, the appropriate horseradish peroxidase-conjugated secondary antibody was used (Jackson ImmunoResearch Laboratories), followed by treatment of the membrane with ECL-plus reagent (Roche) and imaging on X-ray film. Densitometry was performed using ImageJ software (NIH) on unsaturated blots. Coimmunoprecipitation assays were performed utilizing standard procedures and anti-Flag M2 agarose (Sigma) to pull down Flag-labeled IRF3. For immunofluorescence imaging, appropriate Alexa Flour secondary antibodies (Invitrogen) were used along with DAPI (4′,6-diamidino-2-phenylindole) during secondary staining for each slide. All images were photographed on a Nikon TE2000-E microscope and processed with Nikon EIS-Elements software.
Dual luciferase assays.
Dual luciferase assays (Promega) were performed according to the manufacturer's specifications and as described previously. The IFN-β promoter plasmid has been described previously (10, 11).
Statistical analysis.
Differences between groups were analyzed for statistical significance by the Student t test.
The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: Tzm-bl cells were from John C. Kappes, Xiaoyun Wu, and Tranzyme Inc., monoclonal antibody to HIV-1 p24 (AG3.0) was from Jonathan Allan, pNL4-3 was from Malcolm Martin, pcDNA-HVif and pcDNA-Vphu were from Stephan Bour and Klaus Strebel, pEGFP-Vpr was from Warner C. Greene, HIV-1NL4-3 Vpu antiserum was from Klaus Strebel, and HIV-1 Vpr (1-50) antibody was from Jeffrey Kopp.
RESULTS
Role for Vpu in disruption of IRF3-dependent signaling.
To identify viral determinants that direct IRF3 regulation during acute HIV-1 infection, we screened a series of HIV-1 proviral constructs for their ability to regulate IRF3-dependent IFN-β promoter induction in response to SeV infection, a potent inducer of IRF3 activation. In contrast to lab strains of HIV-1 such as NL4-3 and JR-CSF, which mediate the degradation of IRF-3 to block its induction of IFN-β (6), we found that expression of the HIV-1YU2 strain failed to inhibit virus-induced signaling (Fig. 1A). Interestingly, YU2 is one of only ∼1% of all sequenced HIV-1 primary isolates carrying a mutation in the accessory gene vpu (4), harboring a single A-to-C transversion in the start codon of the Vpu open reading frame (ORF) that fails to produce a functional protein (Fig. 1B and 2A). To test the hypothesis that Vpu may be responsible for the ability of HIV to downmodulate IRF3-dependent signaling, we examined the effect of Vpu overexpression signaling of the IFN-β promoter. We found that ectopic expression of Vpu disrupted IFN-β promoter signaling in a dose-dependent manner and occurred in a manner similar to that observed with cognate HIV-1 provirus (Fig. 1C). As we have reported previously (6), expression of similar levels of Vif did not alter signaling to the IFN-β promoter (Fig. 1C). Vpu is a late HIV-1 gene product, being produced off transcripts along with Env. To determine if Vpu is expressed with kinetics similar to that of the depletion of IRF3 protein, we infected SupT1 cells and probed for Vpu expression. We observed low levels of Vpu within 8 h postinfection of cells with HIV-1LAI (Fig. 1D), with peak expression levels coinciding with the strongest depletion of IRF3 at 24 h postinfection.
Fig 1.

Role for Vpu in disruption of IRF3-dependent signaling. (A) Proviral constructs for HIV strains NL4-3, JR-CSF, and YU2 or control plasmid were transfected in 293 cells along with the IFN-β promoter luciferase construct and challenged with SeV to drive IRF3-dependent signaling. (B) (Top) Alignment of LAI, NL4-3, JR-CSF, and YU2 highlighting the start codon (arrow) for Vpu and the A-to-C transversion mutation found in the YU2 strain. *, upstream, nonproductive ATG. (Bottom) Amino acid alignment of Vpu from the same strains. (C) Vpu or Vif overexpression constructs tested as described for panel A. Vpu expression was titrated by expressing 50 ng to 375 ng of expression plasmid within equal DNA dosage transfections using control plasmid as filler; Vif expression from 375 ng of expression plasmid is shown. (D) Immunoblot analysis of either mock-infected HIV-1LAI-infected SupT1 cells or HIV-1LAI-infected SupT1 cells at 8, 24, or 48 h postinfection. Cell lysates were probed for IRF3, Vpu, HIV-1 Gag, or actin as a loading control. Luciferase reporter gene experiments were repeated 3 or more times, and representative immunoblot analyses are shown.
Fig 2.

Vpu is necessary and sufficient for disruption of IRF3-dependent signaling and IRF3 depletion. (A) Wild-type and Vpu-deficient proviral mutants of JR-CSF, pNL4-3, AD8, and YU2 were transfected and tested for signaling in response to SeV as described for Fig. 1A; constructs were tested for expression of Vpu, Vpr, Vif, HIV-1 Gag, or actin by immunoblotting. *, P < 0.05; **, P < 0.01. (B) Cells were transfected as described for Fig. 1A and immunoblotted for IRF3, Vpu, and actin. (C) Cells were treated as described for panel A, with Vpu added in trans in increasing doses along with the Vpu-deficient pNL4-3 proviral construct. Luciferase reporter gene experiments were repeated 3 or more times, and representative immunoblot analyses are shown.
Vpu is necessary and sufficient for IRF3 depletion and disruption of IRF3-dependent signaling.
To determine how the HIV-1YU2 vpu mutation impacts IRF3-dependent signaling, we characterized the same mutation engineered into the same context within three otherwise Vpu-positive proviruses (JR-CSF, NL4-3, and AD8). When examined for Vpu protein expression, we found that each of the resulting viruses failed to express Vpu protein, thus releasing the HIV-1-mediated blockade of innate immune signaling (Fig. 2A). Importantly, placement of a functional start codon into the vpu ORF of HIV-1YU2 restored Vpu expression, resulting in a gain of function to disrupt virus signaling of IFN-β induction (Fig. 2A). Moreover, this gain of function associated with a reduction in IRF3 abundance within cells expressing HIV-1 provirus (see Fig. 5C) or Vpu alone (Fig. 2B). In these experiments, all proviruses expressed similar levels of Vpr, Vif, and Gag proteins, demonstrating that Vpu deficiency does not impact global HIV-1 protein production or processing (Fig. 2A). Additionally, when coexpressed in trans along with a vpu mutant provirus, wild-type (wt) Vpu was able to restore disruption of virus signaling of IFN-β induction in a manner similar to that for wt HIV-1 provirus (Fig. 2C). These results identify Vpu as an HIV-1 antagonist of IRF3-dependent signaling that may function in part to suppress IRF3 protein levels in the host cell.
Fig 5.

Vpu promotes the endolysosomal degradation of IRF3. (A) SupT1 cells were infected with HIV-1LAI or mock treated. At 24 h after infection, HIV-1-infected samples were treated with the proteasome inhibitors MG115 and MG132 or mock treated (HIV-1) for an additional 8 h. Cells were harvested and probed for IRF3 and HIV-1 p24 levels. IRF3 levels are quantified and displayed as a percentage of the IRF3 of the mock-infected sample. (B) HIV-1LAI-infected SupT1 cells or mock-infected cells were treated with increasing doses of chloroquine. *, noticeable cell toxicity was apparent in culture. IRF3 levels were determined by immunoblotting and quantified as described for panel A. (C) 293 cells were transfected with HIVYU2 or HIVYU2 C/A provirus in the presence or absence of chloroquine. Lysates were immunoblotted for IRF3, p24 (HIV-1), and actin as a loading control. (D) IFN-β signaling determined in the presence of control, wt Vpu, or Vpu (S52, 56N) mutation plasmids, with cells treated as described in the legend to Fig. 1A. Immunoblot for Vpu and actin as controls for expression and loading. RLU, relative light units.
Vpu complexes with IRF3, which is strengthened upon virus-induced IRF3 activation.
To determine how Vpu impacts the abundance and metabolism of IRF3, we first performed coimmunoprecipitation analysis to assess a possible Vpu-IRF3 interaction. We transfected HIVJR-CSF or HIVJR-CSF A/C proviruses with Flag-tagged IRF-3 in 293 cells and performed immunoprecipitation of IRF3 using anti-Flag beads. The recovered complexes were then assessed for the presence of both IRF3 and HIV proteins. In cells expressing HIV-1JR-CSF provirus, Vpu formed a stable complex with IRF3 but not with the viral Vpr protein (Fig. 3A). Neither Vpu nor Vpr was found to be associated in the mutant HIVJR-CSF A/C, which fails to express Vpu but expresses normal levels of Vpr. Moreover, when expressed in the absence of other viral products, Vpu but not Vpr associated with IRF3 in transfected cells (Fig. 3B). In addition to Vpu, Vpr altered IRF3 levels, possibly explaining previous observations that implicated Vpr in IRF3 regulation (6, 29). However, we note that differences in the expression constructs used here and in other studies preclude a direct comparison of Vpu and Vpr (6). Interestingly, the Vpu-IRF3 interaction was strengthened if IRF3 was activated by SeV infection, resulting in a 5-fold or more increase in Vpu associated with IRF3 compared to that for cells not infected with SeV, despite lower levels of recoverable IRF3 in the presence of Vpu (Fig. 3B and C). These data together suggest that Vpu complexes with IRF3 and this interaction may be more robust if IRF3 is activated/phosphorylated.
Fig 3.
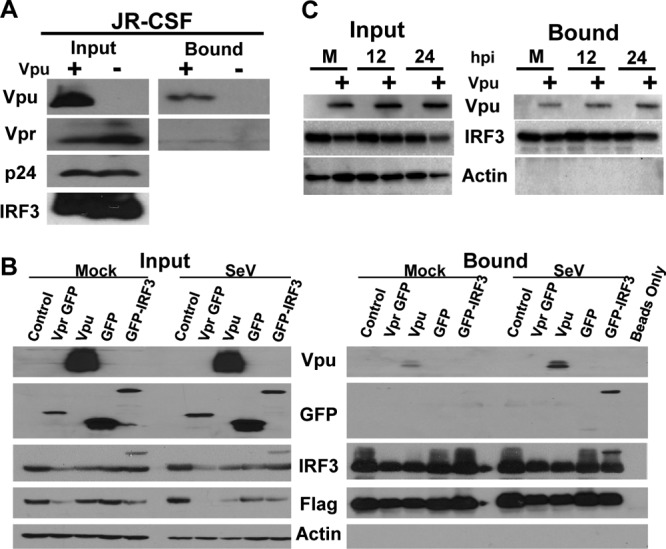
Vpu coimmunoprecipitates with IRF3, and the interaction is strengthened upon IRF3 activation. (A) 293 cells were transfected with Flag-tagged IRF3 as well as proviral constructs for JR-CSF and JR-CSF A/C (Vpu + and −, respectively). Cell lysates were exposed to anti-Flag beads for binding and then washed extensively. Input and bound fractions were immunoblotted for the presence of Vpu, Vpr, p24, and IRF3. (B) Vpu, Vpr, and control plasmids (vector control, GFP alone, GFP-IRF3) were transfected along with Flag-tagged IRF3 in 293 cells. At 24 h posttransfection, cells were either mock or SeV treated to activate IRF3. After 12 h, cells were harvested, IRF3 was immunoprecipitated, and samples were probed for Vpu, GFP, IRF3, Flag, and beta-actin. A portion of the total cell lysate was saved and probed as input. (C) Overexpressed Vpu was isolated with Flag-tagged IRF3 during activation of IRF3 with SeV for increasing times (mock [M], 12 h, or 24 h) posttreatment (hpi), as described for panel B.
IRF3 and Vpu colocalize with lysosomal markers during HIV protein expression.
IRF3 normally displays a cytoplasmic localization in resting cells but translocates to the nucleus upon its activation by virus infection, as demonstrated by SeV stimulation (Fig. 4A). However, immunofluorescence microscopy analysis revealed that during HIV-1 provirus expression but prior to maximal depletion, IRF3 becomes redistributed to colocalize with Vpu into punctate bodies (Fig. 4B) that codistribute with LAMP2, a protein marking the endolysosome (Fig. 4C and E), as previously described for Vpu localization (19). In contrast, within neighboring cells that lack detectable levels of Vpu, IRF3 remained normally distributed and highly abundant throughout the cytoplasm. Image quantification of micrographs from these experiments revealed that over 80% of Vpu-expressing cells exhibited IRF3 localized into punctate structures (Fig. 4D). In contrast, this pattern of IRF3 distribution was not observed in Vpu-deficient cells from the same fields (Fig. 4E). Moreover, IRF3 staining also colocalized with endolysosomal markers in cells expressing HIV-1JR-CSF (Fig. 4E). These results demonstrate that Vpu forms a stable and specific complex with IRF3 in HIV-1-infected cells, resulting in its specific sequestration within the endolysosomal compartment. Therefore, the Vpu-IRF3 interaction may alter the cellular metabolism of IRF3 in HIV-1-infected cells (Fig. 1D) (6).
Fig 4.

IRF3 and Vpu colocalize with lysosomal markers during HIV protein expression. (A) Tzm-bl cells were mock or SeV infected overnight, stained with anti-IRF3 (green) and DAPI (blue), and visualized by immunofluorescence microscopy. (B) Tzm-bl cells were transfected with JR-CSF provirus for 24 h and stained with anti-Vpu (red), anti-IRF3 (green), and DAPI (blue). Cells were visualized with two fields presented for IRF3/Vpu staining. Arrows, areas of strong colocalization (C) Cells treated as described for panel B but stained with anti-Vpu (red), anti-LAMP2 (green), and DAPI (blue). (D) Quantification of the images in panel B with a total of 20 fields from 3 experiments of control or Vpu-positive cells is displayed. (E) Additional cells treated as described for panels B and C but stained with anti-LAMP2 (red), anti-IRF3 (green), and DAPI (blue). For all panels, representative cells are shown, with images of individual channels and a merged image of all three signals shown. Control cells were transfected with vector alone and treated as with their JR-CSF-matched staining panels.
Lysosomal disruption specifically rescues IRF3 depletion during HIV infection.
To maintain immune homeostasis, active IRF3 is typically metabolized through autoregulatory processes of proteasomal degradation after being marked by ubiquitin at late times during the innate immune response (5, 6). We therefore assessed the role of proteasomal degradation in regulating IRF3 levels during HIV-1 infection. While HIV-1 infection significantly reduces IRF3 levels, treatment of HIV-1-infected cells with the proteasomal inhibitor MG132 resulted in only a slight rescue of IRF3, indicating that enhanced proteasomal targeting is not the major process of HIV-1-mediated IRF3 suppression (Fig. 5A).
Considering that Vpu can direct the relocalization of cellular factors, including the HIV-1 restriction factor tetherin/BST2, to the endolysosomal compartment for degradation (9, 19, 26), we assessed if Vpu could similarly target IRF3 to the lysosome for proteolysis. Treatment of cells to disrupt endolysosomal acidification prevented the degradation of IRF3 that otherwise occurred during HIV-1LAI infection (Fig. 5B). Treatment with increasing concentrations of the inhibitor resulted in rescue of IRF3 levels in a dose-dependent manner. This recovery of IRF3 peaked at ∼65% rescue, after which cell toxicity became apparent in the cultures, precluding full recovery of IRF3 levels. Importantly, HIV-1YU2 C/A containing a functional Vpu ORF restored the ability of this HIV-1 strain to suppress IRF3 levels, but treatment of these cells with lysosome inhibitor rescued IRF3 from degradation in the presence of Vpu (Fig. 5C). Two key serine residues in Vpu (S52, S56) have been described to be essential for its ability to downmodulate both CD4 and tetherin/BST2 in β-TrCP and lysosome-dependent mechanisms (8, 24). We therefore tested these mutants to determine if the same residues were also important for Vpu to disrupt IRF3-dependent signaling. We found that the S52 and 56N double mutation of Vpu completely ablated its ability to block innate immune signaling (Fig. 5D). Taken together, these results indicate that Vpu complexes with IRF3 and targets it to the lysosome for proteolytic degradation in a manner mechanistically similar to antagonism of CD4 and tetherin/BST2.
DISCUSSION
The degradation of IRF3 in HIV-1-infected cells is specific and occurs rapidly during acute infection of T cells and myeloid cells in vitro and within mucosal T cells ex vivo (6, 29). Our study defines Vpu as an IRF3 antagonist encoded by HIV-1 and uncovers a novel role for Vpu in directing IRF3 degradation and control of innate antiviral immunity in HIV-1-infected cells. Previous work suggested possible roles for the viral protein Vif or Vpr in targeting IRF3 (29); however, we have not observed an effect of Vif on IRF3 binding or suppression of IRF-3 levels (Fig. 1C) (6), and the effects of Vpr on IRF3 regulation have been only modest in cell models of Vpr expression (6). Here we report a stable interaction between IRF3 and Vpu that appears to not involve either Vif or Vpr (Fig. 3A and B). Indeed, our genetic and biochemical evidence indicates that Vpu is necessary and sufficient for IRF3 degradation by HIV-1. Of additional note, however, is that by providing Vpu in trans to otherwise Vpu-deficient proviruses (Fig. 2C), we were able to impose a reduction of IRF3 more potent than that when ectopic Vpu was expressed alone in the absence of provirus. This additional boost in inhibiting the innate immune response within the context of full HIV-1 protein expression could suggest a secondary role in innate immune regulation facilitated by additional viral proteins (such as Vpr) or possibly reflect other known HIV-1 strategies to suppress the host cell response to infection (21, 27, 34, 39).
It is surprising that Vpu, a late HIV-1 gene product and membrane protein, would be responsible for promoting modulation of the cellular environment so that it is conducive to viral growth by preventing the innate sensing of infection. However, we found that Vpu expression coincides with IRF3 suppression, suggesting that the Vpu-IRF3 interaction represents a point in the HIV-1 infection cycle where viral products are sensed by the host cell to otherwise trigger innate immune signaling and IRF3 activation. Our working model of IRF3 regulation by HIV-1 therefore suggests that Vpu antagonizes IRF3 at a key point later in the viral life cycle, perhaps as replication intermediates accumulate and trigger innate immune signaling that would otherwise suppress HIV-1 infection. This model is consistent with recent reports describing innate immune activation in other contexts, including IRF3 activation during infection in TREX-deficient cells (7, 40). HIV-1 PAMP recognition and signaling by specific but yet to be defined PRRs would thereby activate IRF3, further promoting its interaction with Vpu. This process might also explain the ability of Vpu to interact with a cytoplasmic IRF3, which might move in closer proximity to membranes during innate immune synapse formation on a variety of cellular membranes, as has been recently described (18). This triggering of innate immune signaling likely occurs independently of host cell sensing of HIV-1 capsids, where incoming virions are sensed and then trigger innate immune programs in an AP-1- and NF-κB-dependent mechanism (31), and is likely specific to distinct cell types. More studies are necessary to delineate the full breadth of these various sensing pathways and the full context of HIV antagonism of these programs.
By interacting with IRF3 and targeting it to the lysosome (and, to a lesser extent, the proteasome), Vpu acts to facilitate IRF3 proteolysis and prevent the expression of genes involved in innate immune defenses against HIV-1, including type I IFN, ISGs, and direct IRF3-target genes that mediate antiviral actions. Included among the genes responsive to IRF3 are known HIV-1 restriction factors, including APOBEC3G, tetherin, ISG15, and others. Indeed, HIV-1 suppression of IRF3 may serve to enhance cell permissiveness for infection by relieving innate immune restriction of virus replication and cell spread during the critical stage of acute infection (see the accompanying paper [4a]).
These conclusions are supported by the observations that many pathogenic viruses have evolved means to suppress IRF3 function (2, 14, 22). Most relevant is that hepatitis C virus, which, like HIV-1, typically mediates chronic infection, encodes its own protease to ablate signaling of IRF3 activation to suppress innate immunity and support viral persistence (11, 23). Our data underscore a major feature of Vpu to target key host proteins of the immune response, including tetherin/BST2 and CD4, for lysosomal destruction, while revealing that IRF3 is a new target of Vpu control. IRF3 signaling is essential to promote cell expression of proinflammatory and immunomodulatory cytokines and chemokines from the site of infection that are required for effective adaptive immune responses (5). IRF3 regulation by HIV-1 is therefore expected to contribute to early immune dysfunction in HIV-1-infected patients, thus serving to establish a permissive environment for seeding the initial infection. Once HIV-1 infection is established, IRF3 suppression by Vpu may also serve to promote the systemic dissemination of the virus, likely promoting the innate immune dysfunction that is linked to the end stages of the viral eclipse period (25). Strategies to disrupt the Vpu-IRF3 interaction and/or block Vpu lysosomal targeting actions could serve as a new therapeutic avenue against HIV-1.
ACKNOWLEDGMENTS
We thank Hilario Ramos and Mehul Suthar for helpful conversation and review.
This work was supported by grants from the U.S. Public Health Service, including BMGF CAVD 38645 and NIH/NIAID U01 AI068618. This work was also supported by funds from the state of Washington, NIH grants DA024563 (to M.G.), AI078768 (to B.P.D.), 5T32GM007266-34 (to A.R.), and AI07140 (to A.R.), and the Burroughs-Wellcome Fund (to M.G.).
Footnotes
Published ahead of print 16 May 2012
REFERENCES
- 1. Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801 [DOI] [PubMed] [Google Scholar]
- 2. Barro M, Patton JT. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. U. S. A. 102:4114–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bosinger SE, et al. 2004. Gene expression profiling of host response in models of acute HIV infection. J. Immunol. 173:6858–6863 [DOI] [PubMed] [Google Scholar]
- 4. Dejucq N, Simmons G, Clapham PR. 2000. T-cell line adaptation of human immunodeficiency virus type 1 strain SF162: effects on envelope, Vpu and macrophage-tropism. J. Gen. Virol. 81:2899–2904 [DOI] [PubMed] [Google Scholar]
- 4a. Doehle BP, et al. 2012. Vpu-deficient HIV strains stimulate innate immune signaling responses in target cells. J. Virol. 8499–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doehle BP, Gale M., Jr 2012. Innate immune evasion strategies of HCV and HIV: common themes for chronic viral infection. In Sambhara S, Fujita T. (ed), Nucleic acid sensors and antiviral immunity. Landes Bioscience, Austin, TX [Google Scholar]
- 6. Doehle BP, Hladik F, McNevin JP, McElrath MJ, Gale M., Jr 2009. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 83:10395–10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Doitsh G, et al. 2010. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143:789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Douglas JL, et al. 2010. The great escape: viral strategies to counter BST-2/tetherin. PLoS Pathog. 6:e1000913 doi:10.1371/journal.ppat.1000913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Douglas JL, et al. 2009. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a βTrCP-dependent mechanism. J. Virol. 83:7931–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Foy E, et al. 2005. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. U. S. A. 102:2986–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Foy E, et al. 2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 300:1145–1148 [DOI] [PubMed] [Google Scholar]
- 12. Geiss GK, et al. 2000. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology 266:8–16 [DOI] [PubMed] [Google Scholar]
- 13. Harman AN, et al. 2011. HIV infection of dendritic cells subverts the IFN induction pathway via IRF-1 and inhibits type 1 IFN production. Blood 118:298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hilton L, et al. 2006. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J. Virol. 80:11723–11732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hladik F, et al. 1999. Dendritic cell–T-cell interactions support coreceptor-independent human immunodeficiency virus type 1 transmission in the human genital tract. J. Virol. 73:5833–5842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hladik F, Lentz G, Delpit E, McElroy A, McElrath MJ. 1999. Coexpression of CCR5 and IL-2 in human genital but not blood T cells: implications for the ontogeny of the CCR5(+) Th1 phenotype. J. Immunol. 163:2306–2313 [PubMed] [Google Scholar]
- 17. Honda K, et al. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777 [DOI] [PubMed] [Google Scholar]
- 18. Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr 2011. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 108:14590–14595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iwabu Y, et al. 2009. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 284:35060–35072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384 [DOI] [PubMed] [Google Scholar]
- 21. Kirchhoff F. 2010. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8:55–67 [DOI] [PubMed] [Google Scholar]
- 22. Loo YM, Gale M., Jr 2007. Viral regulation and evasion of the host response. Curr. Top. Microbiol. Immunol. 316:295–313 [DOI] [PubMed] [Google Scholar]
- 23. Loo YM, et al. 2006. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 103:6001–6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Margottin F, et al. 1998. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1:565–574 [DOI] [PubMed] [Google Scholar]
- 25. McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. 2010. The immune response during acute HIV-1 infection: clues for vaccine development. Nat. Rev. Immunol. 10:11–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitchell RS, et al. 2009. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 5:e1000450 doi:10.1371/journal.ppat.1000450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neil S, Bieniasz P. 2009. Human immunodeficiency virus, restriction factors, and interferon. J. Interferon Cytokine Res. 29:569–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ockenhouse CF, Bernstein WB, Wang ZN, Vahey MT. 2005. Functional genomic relationships in HIV-1 disease revealed by gene-expression profiling of primary human peripheral blood mononuclear cells. J. Infect. Dis. 191:2064–2074 [DOI] [PubMed] [Google Scholar]
- 29. Okumura A, et al. 2008. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology 373:85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pasare C, Medzhitov R. 2004. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 6:1382–1387 [DOI] [PubMed] [Google Scholar]
- 31. Pertel T, et al. 2011. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472:361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Richards KH, Clapham PR. 2007. Effects of Vpu start-codon mutations on human immunodeficiency virus type 1 replication in macrophages. J. Gen. Virol. 88:2780–2792 [DOI] [PubMed] [Google Scholar]
- 33. Schaefer TM, et al. 2006. Increased expression of interferon-inducible genes in macaque lung tissues during simian immunodeficiency virus infection. Microbes Infect. 8:1839–1850 [DOI] [PubMed] [Google Scholar]
- 34. Sgarbanti M, et al. 2002. Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors. J. Exp. Med. 195:1359–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381 [DOI] [PubMed] [Google Scholar]
- 36. Sumpter R, et al. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Suthar MS, et al. 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 6:e1000757 doi:10.1371/journal.ppat.1000757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilkins C, Gale M., Jr 2010. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 22:41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yan N, Lieberman J. 2011. Gaining a foothold: how HIV avoids innate immune recognition. Curr. Opin. Immunol. 23:21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. 2010. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11:1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]