

Abstract
Hepatitis C virus (HCV) induces autophagosome formation in infected human hepatocytes. We have previously reported that HCV exploits autophagic machinery in favor of virus growth and survival in host cells (S. Shrivastava et al., Hepatology 53:406–414, 2011); however, the mechanisms for autophagy induction is poorly understood. In the present study, we observed that HCV infection transcriptionally upregulates Beclin1, which forms complex with Vps34, the class III phosphatidylinositol 3-kinase, as a first step for autophagy initiation. Although Bcl-2 has an anti-autophagy effect by its association with Beclin1 in nutrient-deprived cells, our studies revealed that HCV-mediated autophagy occurs independent of Beclin1–Bcl-2 dissociation. Mammalian target of rapamycin (mTOR) is a positive regulator of cell growth and is recognized as an inhibitor of autophagy induction. Our results demonstrated that HCV infection enhances phospho-mTOR expression and its downstream target 4EBP1 activation, suggesting that mTOR is not a negative regulator of HCV-induced autophagy. On the other hand, HCV infection in autophagy-impaired cells reduced phospho-mTOR, mTOR, and phospho-4EBP1 expression. Together, these results suggested that HCV induces autophagy by upregulating Beclin1 and activates mTOR signaling pathway, which in turn may promote hepatocyte growth.
INTRODUCTION
Hepatitis C virus (HCV) infection is a major health problem, and nearly 200 million people are infected with this virus globally (2). HCV chronically infects ∼80% of the infected humans, and among them ∼20% eventually develop liver cirrhosis and hepatocellular carcinoma. HCV is a member of the Flaviviridae family, and its genome contains a positive-strand RNA ∼9.6 kb in length. HCV genome encodes a polyprotein precursor of ∼3,000 amino acids, which is cleaved by both viral and host proteases into structural (core, E1, E2, and p7) and nonstructural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins (36).
Autophagy is an evolutionarily conserved intracellular process that involves the formation of a double membrane structure, called the autophagosome, which engulfs long-lived cytoplasmic macromolecules and damaged organelles and delivers them to lysosomes for degradation and recycling (27). Autophagy is a constitutive process which generally occurs at the basal level but is upregulated in response to extracellular or intracellular stress and signals, such as starvation, growth factor deprivation, endoplasmic reticulum stress, and pathogen infection (50). Viruses are obligate intracellular parasites and their survival is linked to their ability to subvert cellular antiviral defenses and to regulate cellular processes necessary for their own replication. Some of the viruses, such as cytomegalovirus, Kaposi's sarcoma associated herpesvirus, and human herpes simplex virus 1, have evolved strategies to suppress autophagy for their own survival (7, 29). Autophagosome formation occurring during dengue virus, poliovirus, influenza virus A, and coxsackievirus B3 virus infections is associated with enhanced viral replication and an increase in the viral yield (10, 29). We and others have shown that HCV infection induces autophagy in hepatocytes (1, 11, 31, 42, 46). It has also been shown that knockdown of autophagy inhibits production of infectious virus particles (40). We have subsequently shown that disruption of autophagy machinery in HCV-infected hepatocytes activates the interferon (IFN) signaling pathway, resulting in enhancement of the innate immune response (40). HCV-mediated autophagy also controls lipid production (48). However, how HCV induces autophagy remains poorly understood.
There are three major pathways that regulate autophagy induction. The first pathway involves the mammalian target of rapamycin (mTOR), which negatively regulates autophagy. mTOR inhibits ULK1 from recruiting its partners Atg13 and FIP200. ULK–Atg13–FIP200 complex recruits and organizes other proteins for developing autophagosome. In response to insulin or growth factors, class I phosphatidylinositol 3-kinase (PI3K)-induced phosphorylation of Akt activates mTOR that results in inhibition of autophagy (24). During nutrient starvation, activation of AMP protein-activated kinase (AMPK) conversely inhibits mTOR and induces autophagy. The second pathway that regulates autophagy is mediated by Atg6/Beclin1, the first mammalian autophagy protein, which forms a complex with Vps34, the class III PI3K (17, 53). Vps34 produces phosphatidylinositol 3-phosphate, which can recruit other proteins to the complex. On the other hand, binding of Beclin1 by the antiapoptotic protein Bcl-2 inhibits autophagy (34). The third pathway that mediates autophagosome formation and elongation involves 2 ubiquitin-like conjugation processes that generate membrane-bound protein complexes (14, 19). Atg7 and Atg10 mediate the conjugation of Atg12 to Atg5, which subsequently interact with Atg16. The Atg12–Atg5-Atg16 complex binds to the outer membrane and then dissociates upon completion of the autophagosome. The second conjugation reaction involves Atg8 or microtubule-associated protein 1 light chain 3 (LC3). LC3 is constitutively cleaved by Atg4 to produce LC3-I. With a signal to induce autophagy, Atg7 and Atg3 mediate the conjugation of LC3-I to the membrane lipid phosphatidylethanolamine to form LC3-II. LC3-II integrates to both the outer and inner membranes of the autophagosome and helps in membrane elongation and closure. HCV infection in hepatocytes induces LC3-II expression (1, 11, 42), suggesting that this pathway is active.
In the present study, we show that HCV upregulates Beclin1 expression and augments autophagy initiation. Further studies suggest that HCV induced autophagy acts upstream of mTOR signaling pathways, which may in turn promote cell growth.
MATERIALS AND METHODS
Cell culture, transfection and HCV infection.
Immortalized human hepatocytes (IHH) and human hepatoma (Huh 7.5) cells were maintained in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum, 100 U of penicillin G/ml, and 100 μg of streptomycin/ml at 37°C in a 5% CO2 atmosphere. HCV genotype 2a was grown in Huh 7.5 cells or IHH. For infection, cells were incubated with HCV genotype 2a (multiplicity of infection of 0.1) in a minimum volume of medium. After 8 h of virus adsorption on hepatocytes, DMEM supplemented with 5% heat-inactivated fetal bovine serum was added. IHH, transfected with control (scrambled) or Beclin1 (BCN1) small interfering RNA (siRNA) using Lipofectamine 2000 (Invitrogen), were similarly infected with cell culture grown HCV. The siRNAs used here were mixture of three siRNAs and were purchased from Santa Cruz (the sequences are not shown).
For autophagosome-lysosome fusion, the control and Flag-NS5A expressing IHH were transfected with LC3-GFP and treated with 1 μM LysoTracker Red DND-99 (Invitrogen) at 37°C for 30 min as described previously (40). Two-channel optical images (green fluorescent protein [GFP] and LysoTracker red) were collected using the sequential scanning mode (488- and 543-nm excitation and 522- and 595-nm emission, respectively) of the Olympus FV1000 confocal system. NS5A was stained with a mouse anti-Flag antibody and anti-mouse immunoglobulin conjugated to Alexa Fluor 647 (Molecular Probes). Images were superimposed digitally for fine comparisons.
Immunoblot analysis.
Mock- or HCV-infected cells were lysed using an SDS-PAGE sample loading buffer. The proteins were subjected to electrophoresis on polyacrylamide gel and transferred onto nitrocellulose membrane. The membrane was probed with antibodies to phospho-mTOR, mTOR, phospho-4EBP1, phospho-ERK1/2, phospho-MEK1/2, MEK1/2, Vps34, p110β, Beclin1, or Bcl-2 (Cell Signaling or Santa Cruz). Proteins were detected using an enhanced chemiluminescent ECL Western blot substrate (Pierce, IL). The membrane was reprobed for actin or tubulin as an internal control. All immunoblot experiments were performed three to five times for reproducibility.
Immunoprecipitation assay.
Mock-treated or virus-infected cells were lysed with radioimmunoprecipitation assay buffer supplemented with protease inhibitor complex (Roche). Cell lysates were centrifuged at 14,000 rpm for 15 min, and soluble protein was incubated with primary antibody (Beclin1, Bcl-2, or p110β) overnight at 4°C. Protein G-agarose beads were added, followed by incubation at 4°C for 1 h. The precipitates were then washed four to five times with lysis buffer, boiled with 1× protein loading dye, and subjected to Western blotting. Flag-Beclin1 was precipitated with anti-flag antibody, and the samples were processed as described above.
RNA quantitation.
IHH were infected with HCV and incubated for 72 h. Total RNA was isolated using a Qiagen RNeasy kit (Qiagen, CA). cDNA was synthesized using random hexamer and Superscript III reverse transcriptase kit (Invitrogen, CA). Real time-PCR was performed with the cDNA for RNA quantitation using TaqMan gene expression PCR master mix (Applied Biosystems) and FAM-MGB probe for BCN1 (Hs00186838_ml). FAM-MGB probe for GAPDH (Hs99999905_m1) was used as an endogenous control.
Luciferase assay.
BCN1 promoter sequence (from −644 to +197) was PCR amplified and cloned into pGL3-Basic vector encoding the firefly luciferase gene (BCN1-luc). Cells were transfected with BCN1-luc and full-length HCV cDNA (FL-HCV) plasmid DNAs or vector alone. The cells were lysed at 48 h posttransfection with reporter lysis buffer (Promega, WI), and the luciferase activity was determined using a luminometer (Optocomp II; MGM Instruments, Hamden, CT). Luciferase activity was normalized with respect to the protein concentration of cell lysates.
Generation of lenti-NS5A and transduction of hepatocytes.
HCV NS5A region was amplified using forward primer 5′-ACCTGGCTGAATTCCAAGCTCATGCCA-3′ and reverse primer 5′- CAGGAATCTAGACATTCAGCAGCA-3′ from full-length HCV cDNA clone by PCR. The amplified product was digested with EcoRI and XbaI and inserted into the lentivirus vector pLVX-puro (Clontech). HEK293T cells were transfected with pLVX-NS5A plasmid along with lentivirus packaging mix (Clontech). Cell culture supernatants containing lentivirus particles expressing HCV NS5A were collected at 48 h after transfection, filtered, and stored at −70°C for further use. Titration of lenti-NS5A was performed, as suggested by the manufacturer. IHH were transduced with lenti-NS5A or lenti-puro (as a control), and cell lysates were prepared after 72 h for subsequent analyses.
Flow cytometric analyses.
IHH treated with control siRNA or BCN1 siRNA were infected with cell culture grown HCV or transduced with lenti-NS5A. Mitochondrion-associated reactive oxygen species (ROS) levels were measured after 72 h infection by staining cells with MitoSOX (Molecular Probes/Invitrogen) at 5 μM for 40 min at 37°C. The cells were washed with phosphate-buffered saline (PBS), treated with trypsin, and resuspended in PBS containing 1% fetal bovine serum for fluorescence-activated cell sorting analysis.
RESULTS AND DISCUSSION
HCV upregulates Beclin1 at the transcriptional level.
Beclin1 is an interacting partner for the mammalian class III PI3K Vps34 (15, 17, 23, 26, 30). Beclin1 forms complex with Vps34, Vps15, and Atg14L to initiate autophagosome formation (17, 53). We observed an increased expression of Beclin1 protein at the early stage of HCV infection of hepatocytes (1). To further investigate how HCV enhances Beclin1 expression, we examined its regulation at the mRNA level. An increased expression of Beclin1 at the mRNA level in virus-infected hepatocytes was observed up to 6 days (Fig. 1A). The Beclin1 promoter region (−644 to +197) was cloned into pGL3-Basic vector to examine its modulation at the transcriptional level. Our data suggested that exogenous expression of HCV full-length cDNA enhances Beclin1 promoter activity to ∼8-fold compared to vector-transfected control (Fig. 1B). We further examined whether individual HCV proteins regulate Beclin1 gene expression and observed that HCV NS5A significantly enhances the Beclin1 promoter activity compared to other HCV proteins (Fig. 1C). Overexpression of HCV NS5A also increased endogenous Beclin1 protein expression similar to HCV-infected cells (Fig. 1D). HCV protein expression from virus infection or lentivirus transduction is also shown (Fig. 1D). Together, these results suggest that HCV mediated upregulation of Beclin1 expression occurs at the transcriptional level, which in turn, may initiate autophagy induction.
Fig 1.

HCV infection upregulates Beclin1 expression. (A) Total RNA was isolated from mock or HCV-infected IHH at different days postinfection. BCN1 mRNA expression was examined by quantitative reverse transcription-PCR, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as an internal control. The fold changes of mRNA are presented after normalizing with internal control and was arbitrarily set as 1. The results are presented as means from three independent experiments with standard errors. (B and C) IHH were cotransfected with the reporter plasmid containing −644/+197 region of BCN1 promoter with luciferase reporter gene and plasmid DNAs containing FL-HCV or individual HCV genomic regions. Luciferase activity was measured at 48 h posttransfection. The results are representative of three independent experiments. *, P < 0.038; **, P < 0.001. (D) IHH were infected with HCV or transduced with lentivirus expressing HCV NS5A. Cell lysates were analyzed for Beclin1, HCV core, or NS5A expression by Western blotting with a specific antibody. The blot was reprobed with an antibody to actin for comparison of protein load.
HCV induces autophagy independent of Beclin1–Bcl-2 interaction.
Beclin1 was initially identified as Bcl-2 interacting protein (28). During nutrient starvation, the dissociation between Beclin1 and Bcl-2 occurs either by JNK mediated phosphorylation of Bcl-2 or the interaction between HMGB1 and Beclin1 (35, 45, 52). We next examined whether dissociation of Beclin1 and Bcl-2 occurs in hepatocytes infected with HCV. Our results suggested that endogenous Bcl-2 or Beclin1 is coimmunoprecipitated by Beclin1 or Bcl-2 in HCV-infected hepatocytes (Fig. 2A and B), suggesting that HCV does not dissociate Beclin1 and Bcl-2 complex for autophagy induction. For further verification, IHH were transfected with Bcl-2 and Beclin1 in cells expressing full-length HCV (FL-HCV). The results suggested that association between Beclin1 and Bcl-2 is not inhibited by FL-HCV (Fig. 2C). However, in nutrient starved condition, very little association of Beclin1 and Bcl-2 is observed compared to cells with normal growth media (Fig. 2D). Therefore, it appears that Beclin1–Bcl-2 dissociation is not important for HCV-mediated autophagy induction. In Bcl-2-overexpressing HCV-infected cells, we observed LC3 puncta formation, suggesting that increased Bcl-2 expression does not inhibit autophagy in HCV-infected cells (data not shown). Shimizu et al. (39) reported that Bcl-2 or Bcl-XL overexpression increases autophagy in transformed murine embryonic fibroblasts treated with etoposide. We and others have shown earlier that knockdown of Beclin1 in hepatocytes impairs HCV-induced autophagy (11, 40), suggesting an involvement of Beclin1 in the processes. Our present finding is in agreement with our observations, suggesting that Bcl-2 may function as a positive regulator of autophagy.
Fig 2.

Bcl-2 and Beclin1 associate in HCV-infected hepatocytes. (A and B) IHH were infected with HCV, and cell lysates prepared after 3 days of infection were immunoprecipitated with mouse monoclonal Bcl-2 or rabbit anti-Beclin1 antibody. Mock-infected IHH were treated similarly as control. Interaction of Beclin1, Vps34, or Bcl-2 was detected by immunoblotting with specific antibody. (C) Cells were transfected with Bcl-2 and flag-tagged Beclin1 plasmid DNAs in the presence or absence of FL-HCV. Cell lysates prepared after 2 days of transfection were immunoprecipitated with a mouse monoclonal antibody to Bcl-2. The interaction of Beclin1 or Bcl-2 was detected by immunoblotting with specific antibody. (D) Control and nutrient-starved (for 4 h) cell lysates were prepared for immunoprecipitation with a mouse monoclonal antibody to Bcl-2. The interaction of Beclin1 or Bcl-2 was detected by immunoblotting with specific antibody.
HCV infection induces p110β, but does not associate with Vps34.
Vps34 is conserved from lower eukaryotes to plants and mammals (47). Beclin1 and Vps34 are known to be positive regulators of autophagy, through initial nucleation and assembly of the primary autophagosome membrane (41). The assembly of the phagophore requires the Beclin1 complex with Vps34, Vps15, and Atg14L (3). We observed an association of Beclin1-Vps34 in HCV-infected hepatocytes, as expected (Fig. 2B). The PI3Ks are lipid kinases and are critical signaling molecules in cell growth regulation. PI3Ks are grouped into three classes: class I, class II, and class III based on substrate specificity and sequence homology (8). Class I PI3Ks are divided into two groups: class IA and class IB. Class IA PI3Ks are heterodimeric proteins consisting of an 85-kDa regulatory subunit and one of the three 110-kDa catalytic subunits: p110-α, p110-β, and p110-δ (6, 12, 13, 21). Dou et al. (9) have recently suggested that p110β associates with Vps34-Vps15-Beclin1-Atg14L complex for autophagosome initiation.
Since dissociation of Bcl2 and Beclin1 was not observed, we examined the status of p110β in HCV-infected hepatocytes. An increased expression of p110β in HCV-infected hepatocytes was observed (Fig. 3A). We therefore investigated whether p110β forms complex with Beclin1 in HCV-infected cells. Our results suggested that p110β does not coprecipitate with Beclin1 or Vps34 (Fig. 3B). Cellular transformation via p110γ and p110β is activated by MEK1/2 and ERK1/2 signaling. MEK/ERK molecules have been implicated in enhancing autophagy by regulating Beclin1 for inhibiting mTOR pathway (51). An increased levels of phosphorylated MEK1/2 and ERK1/2 was observed in HCV-infected cells compared to mock-infected controls (Fig. 3C). However, treatment of HCV-infected cells with phospho-ERK inhibitor did not impair autophagy formation. Overexpression of wild-type p110β, p110γ, or p110δ is sufficient to induce cellular transformation in chicken embryo fibroblasts (25). Knockouts of p110α and p110β are embryonically lethal (4, 5). Therefore, it is possible that HCV mediated increased p110β expression occurs through MEK/ERK activation and plays an important role in HCV-infected hepatocyte survival.
Fig 3.
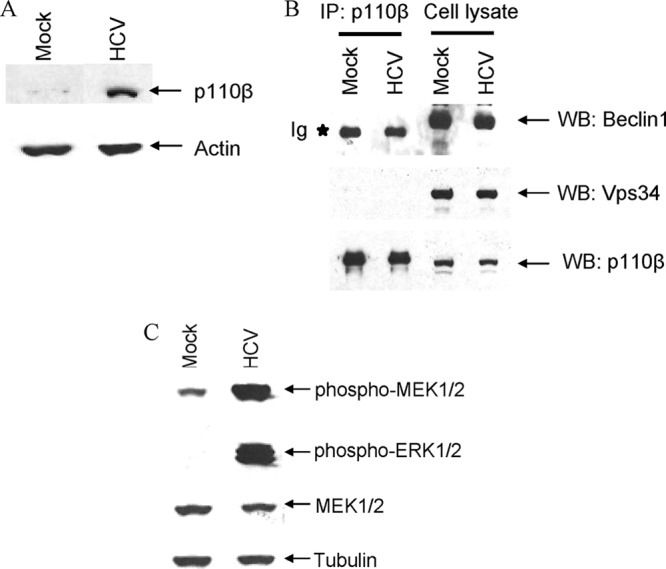
HCV infection induces p110β but does not associate with Beclin1 or Vps34. (A) IHH were infected with HCV, and infected cell lysates were prepared 3 days postinfection. Cell lysates were analyzed for expression of p110β by Western blotting with specific antibody. Mock-infected IHH were treated similarly to the control. The blot was reprobed with an antibody to actin for a comparison of the protein load. (B) IHH were infected with HCV, and cell lysates were immunoprecipitated with anti-p110β antibody. Mock-treated IHH were used as a control in parallel. The interaction of p110β with Beclin1 and Vps34 was examined by immunoblotting with anti-Beclin1 or anti-Vps34 antibody. (C) HCV-infected cell lysates were analyzed for activation of MEK/ERK by Western blot analysis. Mock-treated IHH were used as a control in parallel. The blot was reprobed with an antibody to total MEK1/2. The blot was also reprobed with an antibody to tubulin for comparison of the protein load in each lane.
HCV activates mTOR signaling pathway.
mTOR is an important signaling molecule that regulates diverse cellular functions, such as initiation of mRNA translation, cell growth and proliferation, ribosome biogenesis, transcription, cytoskeletal reorganization, long-term potentiation, and autophagy. Activation of mTOR in nutrient-proficient cells acts as a negative regulator of autophagy (20, 22, 33, 37). However, the cross talk between mTOR pathway and autophagy induction during HCV infection remains unknown. We observed an increased expression of total mTOR and phospho-mTOR in HCV-infected hepatocytes (Fig. 4A). Subsequently, a significant enhancement of phospho-4EBP1 (Thr37/46), a downstream substrate of mTOR, was noted in HCV-infected cells (Fig. 4B). This unexpected result prompted us to assume that HCV-mediated autophagy induction may not be regulated by mTOR signaling. Recent reports suggested that autophagy acts as an upstream positive regulator of mTORC1 (32, 38). We therefore examined the expression level of phospho-mTOR, mTOR, and phospho-4EBP1 in autophagy-knockdown HCV-infected cells. Our results demonstrated a decreased expression of phospho-mTOR and total mTOR (Fig. 4C) and a significant decrease of phospho-4EBP1 in autophagy-knockdown HCV-infected cells (Fig. 4D). Cells were transfected with Beclin1 siRNA or control siRNA (scrambled), and expression of Beclin1 was examined at the protein level. A significant inhibition of Beclin1 at protein levels (>90%) was observed after siRNA treatment (Fig. 4E). Together, these data suggest that HCV-mediated autophagy may act on upstream of mTOR signaling pathway. Further, HCV-mediated autophagy may regulate phospho-4EBP1 (Thr37/46) expression for promotion of cell growth. Indeed, further work is necessary to unravel this mechanism.
Fig 4.

HCV activates mTOR signaling. (A and B) Mock-treated or HCV-infected IHH lysates were analyzed for phospho-mTOR (Ser2448), total mTOR, and phospho-4EBP1 (Thr37/46) by Western blotting with specific antibodies. The blot was reprobed with an antibody to actin for comparison of protein load. (C and D) HCV infection inhibits phospho-mTOR, mTOR, and phospho-4EBP1 in Beclin1-knockdown hepatocytes. IHH treated with BCN1 siRNA were infected with HCV or mock treated. Cell lysates were analyzed for phospho-mTOR, mTOR, and phospho-4EBP1 by Western blotting. The blot was also reprobed with an antibody to actin for comparison of the protein load. (E) IHH were transfected with Beclin1 siRNA or control (scrambled) siRNA. Cell lysates were subjected to Western blot analysis with specific antibody. The blot was reprobed with an antibody to actin for comparison of protein load in each lane.
HCV NS5A protein is sufficient for autophagy induction.
To delineate the role of individual viral proteins in autophagy induction, we transfected the cells with DNA constructs encoding individual HCV proteins and examined the autophagy induction analyzing LC3 punctate dot formation using immunofluorescence. Transduction with lenti-NS5A displayed enlarged punctate dot formation compared to lenti-puro-transduced cells (Fig. 5A and B). A deficiency of autophagosome-lysosome fusion would predict the accumulation of autophagosome. To examine this, we transfected IHH with LC3-GFP and HCV NS5A. After 44 h of transfection, LysoTracker Red was added, and LC3 was found to be colocalized with LysoTracker red, suggesting the formation of the autolysosomes in NS5A-expressing cells (Fig. 5C). We observed a significant increase in LC3-II protein in NS5A-transduced cells, and the ratio of LC3-II to LC3-I was much higher in NS5A-transduced cells compared to the control (Fig. 5D). Thus, the results suggested that NS5A-mediated autophagy cascade proceeds by fusion of autophagosome with lysosome.
Fig 5.

HCV NS5A induces autophagy. (A) IHH were transduced with lenti-puro (mock) or lenti-NS5A for 48 h. Confocal microscopy images displaying subcellular localization of endogenous LC3 (red) and HCV NS5A protein (green) in merged image panels are shown. The nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole) (blue). Punctate localization of LC3 on autophagic vacuoles (shown by arrows) was clearly visible in NS5A-transduced IHH but not in the mock-treated control. (B) HCV NS5A-induced LC3 punctate dots were counted and are presented as a bar graph. (C) Confocal microscopy of autolysosome formation by HCV NS5A. IHH were transfected with GFP-LC3 and HCV NS5A. Lysosomes were stained by LysoTracker dye (red), autophagosomes were stained by GFP-LC3 (green), and NS5A was stained with specific antibody, followed by Alexa Fluor 647 (blue). Autolysosomes stained by LysoTracker red and GFP-LC3 displayed a yellow signal (merged panel). (D) Cells transduced with lenti-NS5A displayed increased LC3 lipidation compared to lenti-puro-transduced cells. NS5A expression is shown in the bottom panel. The blot was reprobed with an antibody to actin for comparison of equal protein load.
To further examine whether HCV NS5A expression in autophagy-impaired cells modulates IFN signaling and cell growth, we transduced lenti-NS5A in BCN1-knockdown IHH. RNA isolated from these cells was analyzed for IFN signaling molecules. Our results demonstrated that HCV NS5A alone upregulates IFNA1 and IFN-β mRNA expression in autophagy-impaired cells (Fig. 6A) and induces PARP cleavage (Fig. 6B). Mitochondrial ROS has been implicated for induction of IFN signaling in autophagy-impaired cells (44). The absence of autophagy resulted into ROS accumulation in mitochondria. We therefore examined mitochondrial ROS production in HCV-infected or NS5A-transduced Beclin1-knockdown cells. An enhancement of mitochondrial ROS production was observed in Beclin1-knockdown cells infected with HCV (Fig. 6C) or transduced with NS5A (Fig. 6D) compared to control cells treated similarly. Mitochondrial ROS production remained unchanged in Beclin1-knockdown cells compared to control cells (data not shown). We also observed that NS5A enhances Beclin1 promoter activity and protein expression (Fig. 1). Together, these results suggest that HCV NS5A alone can induce autophagy. It is possible that more than one HCV protein is involved in the induction of autophagy, as suggested by other investigators (18, 43). HCV NS4B has recently been reported to form complex with Rab5 and Vps34 and induces autophagy (43).
Fig 6.

HCV NS5A upregulates IFN signaling molecules in Beclin1-knockdown IHH. (A) Transduction of lenti-NS5A in Beclin1-knockdown cells enhances IFNA1 and IFN-β mRNA expression. Total cellular RNA was extracted from HCV NS5A-transduced siBCN1-IHH or control IHH after 3 days. Intracellular mRNA expression of IFNA1 or IFN-β was measured by quantitative reverse transcription-PCR. GAPDH was used as an internal control, and the fold changes of mRNA are presented after normalization with internal control. The results are presented as means from three independent experiments. (B) Transduction of lenti-NS5A in Beclin1-knockdown cells induces PARP cleavage. Cell lysates were subjected to Western blot analysis with a specific antibody for the detection of PARP. PARP was significantly cleaved to an ∼86-kDa signature peptide in HCV NS5A-transduced Beclin1-knockdown cells compared to control siRNA-treated IHH. The blot was reprobed with an antibody to actin for comparison of equal protein load. (C and D) Autophagy-impaired HCV-infected or NS5A-transduced IHH have increased levels of mitochondrial ROS. IHH treated with control siRNA or BCN1 siRNA were infected with HCV or transduced with lenti-NS5A. The levels of mitochondrion-associated ROS in cells were analyzed by MitoSOX labeling.
We and others have shown earlier that HCV induces autophagy formation. Here, we demonstrated that HCV infection upregulates Beclin1 at the transcriptional level and that virus-mediated autophagy induction occurs independent of Beclin1–Bcl-2 dissociation. HCV infection also enhances MEK/ERK phosphorylation but does not inhibit mTOR signaling. Beclin1 acts as a nexus point in autophagy induction pathway by forming a complex with Vps34. The activation of MEK/ERK with moderate increase in Beclin1 expression resulted in cytoprotective autophagy (51). We and others have shown earlier that Beclin1 knockdown impairs HCV-mediated autophagy induction (11, 40). Together, our results indicate that HCV-mediated autophagy induction is Beclin1 dependent.
Both mTORC1 activation and eIF4E phosphorylation are involved in tumorigenesis (16, 49). The canonical pathway involves mTORC1-mediated negative regulation of autophagy, especially in the case of starvation. We observed that HCV induces phospho-mTOR and its substrate phospho-4EBP1 expression in IHH. On the other hand, Su et al. (43) did not observe a change in phospho-4EBP1 expression in HCV-infected Huh 7.5 cells compared to mock-infected cells, and this difference may be due to the nature of the experiments and different cell lines used. Paradoxically, we observed inhibition of mTOR and 4EBP1 phosphorylation in autophagy-knockdown HCV-infected cells. Further, Bcl-2 is known to have an anti-autophagy effect via its association with Beclin1 (28). However, our results suggested that this association do not have an effect on HCV-induced autophagy. We conclude from these data that HCV-mediated autophagy and mTOR signaling act concurrently or that induction of autophagy occurs upstream of mTOR signaling pathway in HCV-infected hepatocytes (Fig. 7). A potential explanation of this paradox is likely that HCV infection is inducing autophagy for establishment of infection, while activating mTOR signaling for hepatocyte growth.
Fig 7.
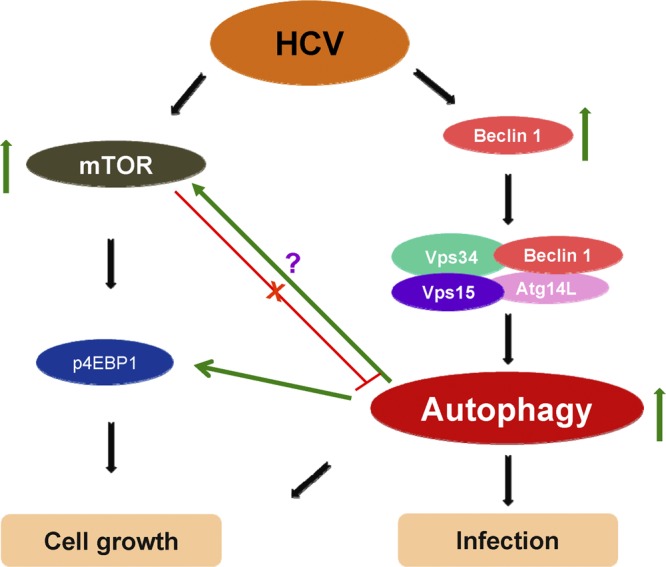
Cross talk between autophagy and mTOR signaling in HCV-infected hepatocytes.
ACKNOWLEDGMENTS
We thank Beth Levine for providing the Flag-Beclin1 plasmid DNA.
This study was supported by research grants DK081817 and AI065535 from the National Institutes of Health.
Footnotes
Published ahead of print 6 June 2012
REFERENCES
- 1. Ait-Goughoulte M, et al. 2008. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 82:2241–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Armstrong GL, et al. 2006. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann. Intern. Med. 144:705–714 [DOI] [PubMed] [Google Scholar]
- 3. Axe EL, et al. 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182:685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. 1999. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110 subunit of phosphoinositide 3-kinase. J. Biol. Chem. 274:10963–10968 [DOI] [PubMed] [Google Scholar]
- 5. Bi L, Okabe I, Bernard DJ, Nussbaum RL. 2002. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm. Genome. 13:169–172 [DOI] [PubMed] [Google Scholar]
- 6. Cantley LC. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655–1657 [DOI] [PubMed] [Google Scholar]
- 7. Chaumorcel M, et al. 2012. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin1. J. Virol. 86:2571–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Domin J, Waterfield MD. 1997. Using structure to define the function of phosphoinositide 3-kinase family members. FEBS Lett. 410:91–95 [DOI] [PubMed] [Google Scholar]
- 9. Dou Z, et al. 2010. The class IA phosphatidylinositol 3-kinase p110-beta subunit is a positive regulator of autophagy. J. Cell Biol. 191:827–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dreux M, Chisari FV. 2010. Viruses and the autophagy machinery. Cell Cycle 9:1295–1307 [DOI] [PubMed] [Google Scholar]
- 11. Dreux M, Gastaminza P, Wieland SF, Chisari FV. 2009. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:14046–14051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Engelman JA, Luo J, Cantley LC. 2006. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7:606–619 [DOI] [PubMed] [Google Scholar]
- 13. Fruman DA, Meyers RE, Cantley LA. 1998. Phosphoinositide kinases. Annu. Rev. Biochem. 67:481–507 [DOI] [PubMed] [Google Scholar]
- 14. Fujita N, et al. 2008. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19:2092–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Funderburk SF, Wang QJ, Yue Z. 2010. The Beclin1-VPS34 complex: at the crossroads of autophagy and beyond. Trends Cell Biol. 20:355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Furic L, et al. 2010. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. U. S. A. 107:14134–14139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Furuya N, Yu J, Byfield M, Pattingre S, Levine B. 2005. The evolutionarily conserved domain of Beclin1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 1:46–52 [DOI] [PubMed] [Google Scholar]
- 18. Guevin C, et al. 2010. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 405:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hanada T, et al. 2007. The Atg12–Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 282:37298–37302 [DOI] [PubMed] [Google Scholar]
- 20. Hara K, et al. 1998. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 273:14484–14494 [DOI] [PubMed] [Google Scholar]
- 21. Hawkins PT, Anderson KE, Davidson K, Stephens LR. 2006. Signaling through class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 34:647–662 [DOI] [PubMed] [Google Scholar]
- 22. Inoki K, Zhu T, Guan KL. 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590 [DOI] [PubMed] [Google Scholar]
- 23. Itakura E, Kishi C, Inoue K, Mizushima N. 2008. Beclin1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 19:5360–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. 2010. mTOR regulation of autophagy. FEBS Lett. 584:1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kang S, Denley A, Vanhaesebroeck B, Vogt PK. 2006. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. U. S. A. 103:1289–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kihara A, Noda T, Ishihara N, Ohsumi Y. 2001. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 152:519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klionsky DJ. 2007. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell. Biol. 8:931–937 [DOI] [PubMed] [Google Scholar]
- 28. Liang XH, et al. 1998. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 72:8586–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin LT, Dawson PW, Richardson CD. 2010. Viral interactions with macroautophagy: a double-edged sword. Virology 402:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsunaga K, et al. 2009. Two Beclin1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11:385–396 [DOI] [PubMed] [Google Scholar]
- 31. Mizui T, et al. 2010. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J. Gastroenterol. 45:195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Narita M, et al. 2011. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332:966–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Noda T, Ohsumi Y. 1998. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 273:3963–3966 [DOI] [PubMed] [Google Scholar]
- 34. Pattingre S, et al. 2005. Bcl-2 antiapoptotic proteins inhibit Beclin1-dependent autophagy. Cell 122:927–939 [DOI] [PubMed] [Google Scholar]
- 35. Pattingre S, et al. 2009. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 284:2719–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Poenisch M, Bartenschlager R. 2010. New insights into structure and replication of the hepatitis C virus and clinical implications. Semin. Liver Dis. 30:333–347 [DOI] [PubMed] [Google Scholar]
- 37. Ravikumar B, et al. 2010. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90:1383–1435 [DOI] [PubMed] [Google Scholar]
- 38. Settembre C, et al. 2011. TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shimizu S, et al. 2004. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 6:1221–1228 [DOI] [PubMed] [Google Scholar]
- 40. Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. 2011. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 53:406–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simonsen A, Tooze SA. 2009. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 186:773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sir D, et al. 2008. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Su WC, et al. 2011. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 85:10561–10571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tal MC, et al. 2009. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U. S. A. 106:2770–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang D, et al. 2010. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 190:881–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tanida I, et al. 2009. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 5:937–945 [DOI] [PubMed] [Google Scholar]
- 47. Vanhaesebroeck B, et al. 2001. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 70:535–602 [DOI] [PubMed] [Google Scholar]
- 48. Vescovo T, et al. 2011. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gastroenterology 142:644–653 [DOI] [PubMed] [Google Scholar]
- 49. Villanueva A, et al. 2008. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 135:1972–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Virgin HW, Levine B. 2009. Autophagy genes in immunity. Nat. Immunol. 10:461–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang J, et al. 2009. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin1. J. Biol. Chem. 284:21412–21424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. 2008. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30:678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zeng X, Overmeyer JH, Maltese WA. 2006. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J. Cell Sci. 119:259–270 [DOI] [PubMed] [Google Scholar]