

Abstract
In the context of viral infections, autophagy induction can be beneficial or inhibitory. Within the Paramyxoviridae family, only morbilliviruses have been investigated and are reported to induce autophagy. Here we show that morbilliviruses rapidly induce autophagy and require this induction for efficient cell-to-cell spread. Coexpression of both glycoproteins in cells expressing one of the cellular receptors was required for autophagy induction, and LC3 punctum formation, indicative of autophagy, was mainly observed in syncytia. A similar correlation between syncytium formation and autophagy induction was also observed for other paramyxovirus glycoproteins, suggesting that membrane fusion-mediated autophagy may be common among paramyxoviruses and possibly other enveloped viruses.
INTRODUCTION
As obligate intracellular parasites, viruses have evolved to exploit cellular functions, while cells have in turn developed a broad spectrum of antiviral mechanisms. In addition to the well-characterized sensors of pathogen-associated molecular patterns (PAMPs) (18), alteration of autophagy activity is increasingly recognized as an early inducer of innate immune activation (36, 45, 46). Autophagy is a ubiquitous mechanism involved in the maintenance of homeostasis in response to cellular stress by recycling of long-lived proteins and cytoplasmic organelles. Briefly, damaged cellular constituents are sequestered into the autophagosomes, which fuse with lysosomes to transform into autolysosomes (9). Autophagosome formation involves the conjugation of autophagy-related gene 5 (Atg5) and Atg12 by the ubiquitin E1-like enzyme Atg7 and the E2-like enzyme Atg10 (31, 38), followed by the cleavage of the microtubule-associated protein light chain 3 (LC3) in LC3-I and LC3-II (37). LC3 is a major constituent of the autophagosome, and its intracellular localization and conversion from the cytoplasmic form, LC3-I, to its membrane-bound cleavage product, LC3-II, are frequently used to monitor autophagic activity (15, 25). In the context of virus infections, autophagy may be either inhibited to interfere with innate immune activation or induced to promote virus replication (22).
Morbilliviruses constitute a genus in the Paramyxoviridae family within the order Mononegavirales (5). These viruses initially infect a broad range of immune cells and then spread to epithelial cells. Attachment to the target cell is mediated by the hemagglutinin (H) protein, upon which the fusion (F) protein induces fusion of the viral and cellular membranes (19). The signaling lymphocyte activation molecule (SLAM), which is expressed on the surface of activated T and B lymphocytes, macrophages, and dendritic cells (43), serves as the immune cell receptor for all morbilliviruses (39), while CD46, a regulator of complement activation, is used only by attenuated and certain wild-type measles viruses (MeVs) (47). Recent reports demonstrate that both SLAM and CD46 recruit the vps34/beclin1 autophagic complex, suggesting that morbilliviruses might induce autophagy pathways upon receptor binding (2, 14). Furthermore, Scrg1, a newly proposed autophagy marker, was overexpressed in the brain of mice intracerebrally inoculated with a mouse-adapted canine distemper virus (CDV) (7, 8).
The present study aimed at characterizing the role of autophagy in the morbillivirus life cycle in more detail. We found that infection-induced autophagy alone had little impact on the initial replication efficiency but was required for efficient spread. We next investigated the contribution of attachment, fusion, and entry to this process and observed that viral glycoprotein-mediated membrane fusion triggered autophagy. To determine if the observed correlation between membrane fusion and autophagy induction was specific for morbilliviruses or a more generalized phenomenon, the Nipah virus (NiV), Hendra virus (HeV), and mumps virus (MuV) envelope glycoproteins were included in these studies.
MATERIALS AND METHODS
Cells and viruses.
Vero cells (ATCC CCL-81) and Vero cells stably expressing canine SLAM (VerodogSLAMtag) were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented with 5% heat-inactivated fetal bovine serum (FBS; Invitrogen). The CDV wild-type strain 5804P and its enhanced green fluorescent protein (eGFP)-expressing derivative 5804PeH, the eGFP-expressing CDV vaccine strain Onderstepoort (OSeN) (34), the eGFP-expressing MeV wild-type strain IC323 (IC323-EGFP) (11, 35), and the MeV vaccine strain Moraten (MeVvac) (4) and its eGFP-expressing derivative (MeVvacgreen) were used in this study. The CDV neurovirulent strain Snyder Hill carrying a polymerase-mCherry fusion protein (CDV-SHmChL) was generated in the context of an independent study (P. A. Rudd et al., unpublished results) by adding the mCherry open reading frame to the carboxy terminus of the viral polymerase protein. The virus was recovered as described previously (1, 28). All viruses were propagated in VerodogSLAMtag cells, and virus titers were determined by limited dilution method and expressed as 50% tissue culture infectious dose (TCID50).
Transfections.
Cells were seeded at approximately 80% confluence in 12-well plates and transfected using Lipofectamine 2000 (Invitrogen) with 2 μg each of an expression plasmid coding for GFP-microtubule-associated protein light chain 3 (GFP-LC3) and plasmids expressing different viral glycoproteins. For CDV, the wild-type glycoprotein expression plasmids pCG-H5804P and pCG-F5804P as well as the endoplasmic reticulum (ER) retention mutant pCG-F5804Pret (41) were used, while pCG-MeV-Edm/H and pCG-MeV-Edm/F were used to express MeV vaccine strain Edmonston (Edm) glycoproteins (12). The NiV and HeV glycoprotein expression plasmids pCG-NiV/F, pCG-NiV/attachment (G) protein, pCG-HeV/F, and pCG-HeV/G were provided by Bevan Sawatsky, and the MuV glycoprotein expression plasmids pCG-MuV/F and pCG-MuV/hemagglutinin-neuraminidase (HN) protein were a kind gift from Paul Duprex. As an autophagy control, cells were treated with 50 μM rapamycin starting 6 h after transfection. All analyses were performed 20 h later.
siRNA transfections.
Small interfering RNA (siRNA) duplexes targeting human Atg7 (116182, 116184, 173705) and negative-control siRNA (12935) were purchased from Invitrogen. VerodogSLAMtag cells were seeded in 12-well plates and transfected with a mix of all Atg7-targeting siRNAs, each at a 25-pmol concentration, or 74 pmol of the control siRNA using Lipofectamine 2000 (Invitrogen). Protein knockdown was evaluated 48 h after siRNA transfection by Western blot analysis.
Replication and dissemination efficacy assessment.
To evaluate the effect of autophagy on morbillivirus infection, VerodogSLAMtag cells were seeded in 12-well plates and transfected with GFP-LC3. The next day, cells were treated with 50 μM chloroquine (Sigma) or 10 mM 3-methyladenine (3-MA; Sigma) to inhibit autophagy or with 50 μM rapamycin (Sigma) to induce autophagy. Alternatively, cells were transfected with control or Atg7 siRNA to inhibit autophagy. Twenty hours after drug treatment or 48 h after siRNA transfection, the cells were infected with the different viruses at a multiplicity of infection (MOI) of 0.01, and the intracellular GFP-LC3 distribution was visualized by confocal microscopy at different times after infection. Cell-associated virus titers were quantified after 20 h.
To assess the impact on spread to noninfected contact cells, VerodogSLAMtag cells were infected at an MOI of 1. After 1 h, the viral inoculum was removed and cells were washed, trypsinized, and mixed at a 1:16 ratio with VerodogSLAMtag cells that had been transfected with control or Atg7 siRNA 48 h previously or pretreated with chloroquine for 20 h. At 3 h after seeding, the medium was removed and cells were overlaid with DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen) and 5% methylcellulose (Sigma). Infectious foci were photographed at different times after mixing.
To determine the importance of viral replication, virus stocks were UV inactivated with three 10-min exposures in a UV box, and inactivation and stock titers were determined by parallel titration of UV and mock-inactivated samples. VerodogSLAMtag cells were seeded in 12-well plates or on chamber slides (Nunc) and infected at an MOI of 2 or 0.05 or the corresponding volume of UV-inactivated virus. At 3 h after infection, chamber slides were fixed for immunofluorescence staining, and whole-cell extracts were harvested at 3 and 20 h after infection from 12-well plates infected and subjected to Western blot analysis.
Western blot analysis.
Cells were lysed in 200 μl of lysis buffer (2% SDS, 10% glycerin, 5% 2-mercaptoethanol, and 0.1% bromophenol blue) and heated for 3 min at 95°C. Protein lysates were separated by SDS-PAGE, transferred to polyvinylidene fluoride membranes (Millipore), and blocked overnight at 4°C in 0.5% blocking reagent (Roche) prepared in Tris-buffered saline (TBS). A polyclonal rabbit anti-LC3 antibody (PD014; MBL Cedarlane), a polyclonal rabbit anti-Atg7 antibody (059K4799; Sigma), a monoclonal mouse anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody (437000; Invitrogen), and anti-MeV H, anti-CDV F, and anti-CDV H rabbit antisera (39) were used as primary antibodies in combination with the appropriate peroxidase-labeled secondary antibodies (Sigma). Bands were visualized using an ECL+ Western blotting detection system (GE Healthcare) with a 4000MM Kodak image station and quantified using Molecular Imaging software (Kodak).
Immunofluorescence.
Cells were visualized either directly or washed twice with cold phosphate-buffered saline (PBS; Invitrogen), fixed with 4% paraformaldehyde in PBS for 20 min at room temperature, and subsequently washed twice with PBS. Cells were permeabilized with 0.2% Triton X-100 (Sigma) in PBS for 10 min, washed twice with PBS, and blocked with 2% bovine serum albumin (Sigma), 0.2% Tween 20 (Sigma) in PBS. Cells were stained with a rabbit anti-LC3 antibody (L10382; Invitrogen), in combination with Alexa Fluor 568 donkey anti-rabbit IgG (A10042; Invitrogen) secondary antibody. The Lysotracker red probe (L7528; Invitrogen) was used to label lysosomes in live cells, as described in the manufacturer's protocol. Fluorescent images were captured using either a standard fluorescence microscope or a confocal laser microscope (MRC 1000; Bio-Rad).
Statistical analyses.
All results shown represent the mean of at least three independent experiments. Analysis of variance (ANOVA) was performed to identify statistically significant differences in LC3-I/LC3-II ratios. P values below 0.05 were considered statistically significant.
RESULTS
Morbilliviruses induce autophagy at early infection stages.
While autophagy has been observed in the context of both MeV and CDV infections (8, 14), its role in the morbillivirus life cycle remains to be characterized. Toward this, we first investigated the LC3 distribution in GFP-LC3-transfected VerodogSLAMtag cells at different times after infection with the CDV wild-type strain 5804P and the MeV vaccine strain MeVvac at an MOI of 0.01. An increased number of GFP-LC3 puncta, indicative of autophagosome accumulation, was found in rapamycin-treated positive-control and infected samples starting as early as 3 h and reaching a peak at 20 h after infection (Fig. 1A and data not shown). Quantitative analysis of the images revealed GFP-LC3 punctum formation in 75 to 80% of transfected cells in rapamycin-treated or infected samples, while only 25% of the mock-treated samples displayed GFP-LC3 relocalization (Fig. 1B). Since this LC3 punctum accumulation can be the consequence of increased formation or decreased degradation of autophagosomes (16), we next assessed their colocalization with lysosomes using a red-fluorescent lysosome marker. As expected, confocal microscopy imaging revealed a colocalization of GFP-LC3 puncta with lysosomes in rapamycin-treated samples, while GFP-LC3 puncta in cells and syncytia of CDV- or MeV-infected samples were predominantly found in close proximity but separate from lysosomes, indicating that morbillivirus infections induce de novo autophagosome formation but prevent their subsequent fusion with lysosomes (Fig. 1A).
Fig 1.

Morbillivirus infection rapidly induces autophagy. (A) Confocal microscopy analysis of VerodogSLAMtag cells transfected with a GFP-LC3 expression plasmid and infected with CDV 5804P or MeVvac at an MOI of 0.01 or left uninfected. Rapamycin-treated cells were included as an autophagy control. At different times after infection, cells were stained with Lysotracker red, fixed with 4% paraformaldehyde, and imaged by confocal laser microscopy. Results for the 20-h time point are shown. The localization of autophagosomes is indicated by green puncta, and lysosomes are marked in red. Arrows indicate colocalization of GFP-LC3 puncta with lysosomes. (B) Proportion of GFP-expressing cells with punctum formation. For each sample, the total number of GFP-expressing foci and the number of foci with GFP puncta were counted in four separate fields. Data from at least four independent experiments were used for the analysis. Error bars represent the standard deviation, and statistically significant differences (ANOVA, P < 0.05) are indicated by asterisks. (C) Confocal microscopy analysis of GFP-LC3-transfected VerodogSLAMtag cells infected with CDV-SHmChL at an MOI of 0.01. Twenty hours after infection, cells were fixed with 4% paraformaldehyde and visualized by confocal microscopy. The localization of autophagosomes is indicated by green puncta, and that of the CDV polymerase (L)-mCherry fusion protein, indicative of the CDV replication complexes, is marked in red.
Since several viruses use autophagosomes as replication sites (44), we investigated if this is also the case for morbilliviruses. Toward this, we generated a recombinant wild-type CDV strain in which the mCherry fluorescent protein is fused to the carboxy terminus of the polymerase protein (SHmChL), thereby allowing the direct visualization of viral replication complexes. Infection of GFP-LC3-expressing VerodogSLAMtag cells with SHmChL led to GFP-LC3 punctum formation in about 80% of GFP-LC3-expressing cells and syncytia, consistent with the results obtained with other CDV and MeV strains (Fig. 1B). However, subsequent confocal microscopy analysis revealed that GFP-LC3 puncta did not colocalize with CDV replication complexes in infected cells (Fig. 1C), illustrating that morbilliviruses induce autophagy but do not replicate in autophagosomes.
Autophagy induction is not required for efficient replication.
To determine if the observed autophagy induction had an infection-enhancing or -inhibitory effect, VerodogSLAMtag cells were pretreated with chloroquine, which inhibits autophagy by preventing autolysosome formation, or 3-MA, which disrupts autophagosome formation, for 20 h prior to infection with the different CDV and MeV strains at an MOI of 0.01. A 100- to 1,000-fold reduction in cell-associated CDV titers and a more than 10-fold reduction of MeV titers were observed in chloroquine-pretreated cells (P < 0.05; Fig. 2A), even though similar numbers of infected foci were observed in treated and nontreated samples (data not shown). However, siRNA-mediated downregulation of Atg7 (Fig. 2C), one of the key cellular proteins involved in autophagosome formation (38), in VerodogSLAMtag cells yielded similar results for treated and nontreated cells (Fig. 2B), indicating that autophagy alone does not affect viral replication or production of viral progeny.
Fig 2.

Autophagy induction alone is not required for efficient morbillivirus replication. Cell-associated virus production in cells treated with autophagy inhibitors (A) or Atg7-specific siRNAs (B and C). (A) VerodogSLAMtag cells were mock treated or treated with 50 μM chloroquine or with 10 mM 3-methyladenine for 20 h prior to infection with different CDV and MeV strains at an MOI of 0.01. (B) VerodogSLAMtag cells were transfected with control (Ctrl) or Atg7-specific siRNAs for 48 h prior to infection with different CDV and MeV strains at an MOI of 0.01. Twenty hours later, cell-associated virus titers were quantified and expressed as TCID50s/ml. The means of three independent experiments are shown, and error bars indicate the standard deviation. Statistically significant differences (ANOVA, P < 0.05) are indicated by asterisks. (C) VerodogSLAMtag cells were transfected with control or Atg7-specific siRNAs for 48 h. Efficiency of Atg7 downregulation was assessed by Western blot analysis using GAPDH as a loading control.
Efficient morbillivirus cell-to-cell spread requires contact-mediated autophagy induction.
The detection of first GFP-LC3 puncta after 3 h indicates that autophagy is induced during early stages of the viral life cycle, leading us to investigate its importance for spread from infected to surrounding cells. Toward this, VerodogSLAMtag cells were infected with wild-type and vaccine MeV and CDV strains for 1 h at an MOI of 1, trypsinized, and mixed with control or Atg7 siRNA- or chloroquine-pretreated cells at a ratio of infected/uninfected cells of 1:16. Once attached, cells were overlaid with medium containing 5% methylcellulose and syncytia were counted 20 h later. A marked reduction in the number of infected foci and smaller syncytia were observed for Atg7 siRNA- or chloroquine-pretreated cocultures (Fig. 3A), demonstrating that autophagy induction in neighboring noninfected cells contributes to efficient cell-to-cell spread. To determine if virus attachment is sufficient to trigger autophagy or if replication is required, we compared the distribution of endogenous LC3 in control and Atg7 siRNA-pretreated VerodogSLAMtag cells exposed to infectious or UV-inactivated viruses. Within 3 h, the LC3 distribution in control siRNA-pretreated cells changed from a diffuse to a punctate cytoplasmic pattern, regardless of the viability of the virus, while no relocalization was observed in Atg7 siRNA-pretreated cells (Fig. 3B). This induction of punctum formation correlated with a small but statistically significant increase (P < 0.05) in LC3-II levels at 3 and 20 h after inoculation, with levels in samples with replicating viruses increasing over time (Fig. 3C and D), suggesting that virus attachment to the target cell is sufficient to induce autophagy through an Atg7-dependent pathway.
Fig 3.

Autophagy is induced at early stages of the morbillivirus life cycle. (A) Autophagy induction contributes to efficient dissemination. VerodogSLAMtag cells were treated with control or Atg7-specific siRNAs or chloroquine. After 48 or 20 h, respectively, these cells were mixed at a 16:1 ratio with VerodogSLAMtag cells previously infected with CDV 5804PeH, OSeN, IC323-EGFP, or MeVvacgreen at an MOI of 1 for 1 h. Once attached, the cells were then overlaid with methylcellulose, and infectious foci were visualized by fluorescence microscopy at a 400-fold magnification after 20 h of incubation. (B) LC3 punctum formation in control or Atg7 siRNA-treated VerodogSLAMtag cells after inoculation with infectious or UV-treated CDV or MeV strains at an MOI of 2. Three hours after infection, cells were fixed with 4% paraformaldehyde and stained for endogenous LC3, and LC3 puncta were visualized using fluorescence microscopy at a 1,000-fold magnification. (C and D) LC3-I and -II distribution in whole-cell extracts from mock- and UV-inactivated cells harvested at 3 h (C) and 20 h (D) after inoculation. Cells harvested at 3 h and 20 h were infected at MOIs of 2 and 0.05, respectively. Rapamycin-treated cells were included as an autophagy control. GAPDH was used as a loading control, and H proteins were detected using cytoplasmic tail-specific rabbit antipeptide antisera. LC3-I and -II band density ratios of noninfected cells were set to 1 to calculate the relative change in LC3-I/LC3-II ratios in infected cells. The mean values of at least three independent experiments are shown, and error bars indicate the standard deviation. Samples that differ statistically significantly (ANOVA, P < 0.05) from untreated controls are indicated by asterisks.
Both envelope glycoproteins are required for autophagy induction.
To further characterize this attachment-mediated autophagy induction, VerodogSLAMtag cells were transiently transfected with GFP-LC3 and CDV 5804P or MeV Edm F- and H-protein expression plasmids. Coexpression of F and H proteins resulted in the same punctate LC3 distribution pattern observed in rapamycin-treated cells, while the LC3 distribution in cells expressing only one of the glycoproteins remained unchanged (Fig. 4A). GFP-LC3 was relocalized in puncta in about 75% of GFP-positive cells in rapamycin-treated samples and 80% in cells transfected with both glycoproteins, whereas only 30% of GFP-positive cells displayed a punctum in mock-transfected samples and cells expressing only one of the viral glycoproteins (Fig. 4B). Western blot analysis revealed ratios of LC3-I to LC3-II consistent with the observed intracellular distribution pattern in cells expressing both glycoproteins or treated with rapamycin, while expression of the F or H protein alone had no effect (Fig. 4C). As observed previously in the absence of the H protein or for F proteins carrying an ER retention sequence (41), F-protein processing was less efficient, resulting in smaller amounts of the mature F1 subunit and the detection of an additional band that likely represents partially glycosylated F0 precursor proteins. Coexpression of CDV H with the ER-retained F protein, which was no longer expressed at the cell surface, also failed to induce autophagy (Fig. 4C), further confirming that expression of both glycoproteins at the surface membrane is required and sufficient to induce autophagy.
Fig 4.
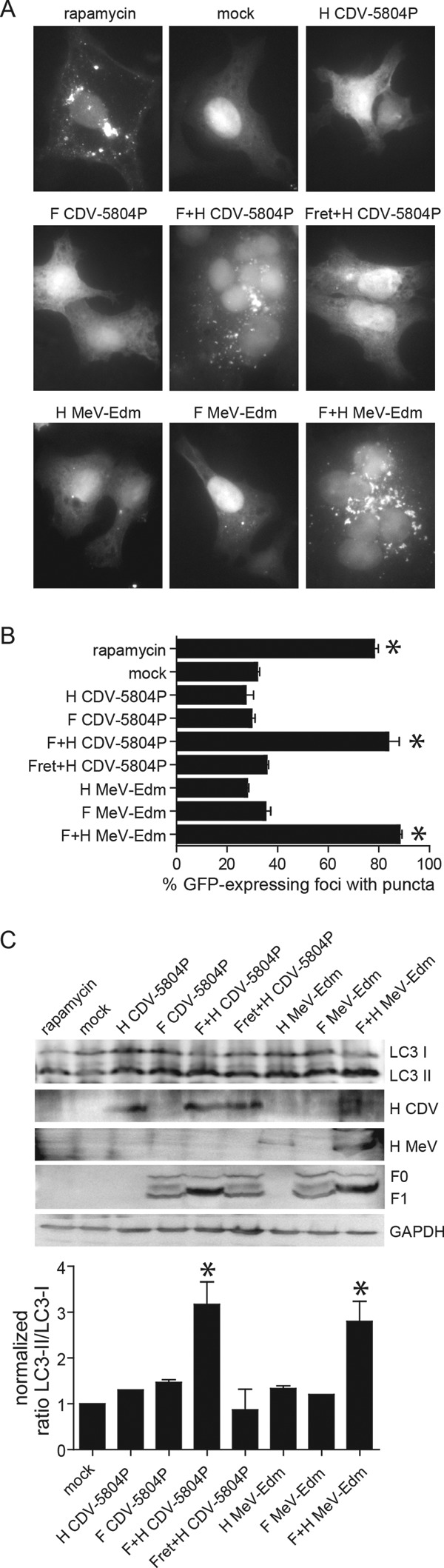
Expression of both morbillivirus glycoproteins is required for autophagy induction. (A) LC3 punctum formation in VerodogSLAMtag cells cotransfected with GFP-LC3 and CDV 5804P and MeV Edm F and H expression plasmids. Samples were analyzed 20 h after transfection, and pictures were taken at a 1,000-fold magnification. (B) Proportion of GFP-expressing cells with punctum formation. For each sample, the total number of GFP-expressing foci and the number of foci with GFP puncta were counted in four separate fields. Data from at least four independent experiments were used for the analysis. Error bars represent the standard deviation, and statistically significant differences (ANOVA, P < 0.05) are indicated by asterisks. (C) LC3-I and -II distribution in VerodogSLAMtag cells transfected with MeV Edm or CDV 5804P glycoprotein expression plasmids. Rapamycin-treated cells were included as an autophagy control for each experiment, and a CDV F-protein expression plasmid carrying an ER retention signal (Fret) was used to assess the importance of F-protein surface expression. GAPDH was used as a loading control, and viral glycoproteins were detected using cytoplasmic tail-specific rabbit antipeptide antisera. The mean values of LC3-I/LC3-II ratios of at least three independent experiments are shown, and error bars indicate the standard deviation. Samples that differ statistically significantly (ANOVA, P < 0.05) from untreated controls are indicated by asterisks.
Contact-mediated autophagy induction is fusion dependent.
Coexpression of morbillivirus F and H proteins in receptor-expressing cells results in extensive syncytium formation (40, 42). To determine the importance of fusion for autophagy induction, parental Vero cells, which are efficiently infected with vaccine but lack the receptors used by wild-type viruses, and VerodogSLAMtag cells, which express the morbillivirus immune cell receptor SLAM, were transiently transfected with GFP-LC3 and either wild-type CDV 5804P or vaccine MeV Edm F- and H-protein expression plasmids. Consistent with previous reports (40), vaccine MeV Edm glycoproteins resulted in syncytium formation in Vero cells, while wild-type CDV 5804P glycoprotein-mediated fusion was SLAM dependent (Fig. 5A). A GFP-LC3 relocalization as puncta was observed in up to 85% of cells treated with rapamycin or transfected with the MeV Edm glycoproteins, whereas all other samples were similar to mock-transfected cells (Fig. 5A and B). Furthermore, a statistically significant increase (ANOVA, P < 0.05) in LC3-II level was observed only in the presence of syncytia (Fig. 5A and C), indicating that glycoprotein-receptor interactions are essential for autophagy induction. To investigate if this interaction is also sufficient for autophagy induction, we treated Vero cells expressing both MeV Edm glycoproteins with fusion-inhibiting peptide (FIP), thus allowing receptor binding but preventing subsequent membrane fusion. Neither syncytia nor increased LC3-II levels were detected in FIP-treated cells (Fig. 5A and C), illustrating the important role of membrane fusion in morbillivirus autophagy induction.
Fig 5.

Induction of autophagy requires viral glycoprotein-mediated membrane fusion. (A) Syncytium formation and intracellular distribution of GFP-LC3 puncta in Vero cells cotransfected with GFP-LC3 and CDV 5804P and MeV Edm glycoprotein expression plasmids. Pictures were taken 20 h after transfection. Phase-contrast images are shown at a 400-fold magnification, and fluorescence images are shown at a 1,000-fold magnification. (B) Proportion of GFP-expressing cells with punctum formation. For each sample, the total number of GFP-expressing foci and the number of foci with GFP puncta were counted in four separate fields. Data from at least four independent experiments were used for the analysis. Error bars represent the standard deviation, and statistically significant differences (ANOVA, P < 0.05) are indicated by asterisks. (C) LC3-I/LC3-II ratios in Vero cells transfected with the CDV and MeV glycoprotein expression plasmids. Fusion-inhibiting peptide (FIP) was used to prevent membrane fusion in MeV glycoprotein-expressing cells. GAPDH was used as a loading control, and viral glycoproteins were detected using rabbit antipeptide antisera against the F- and H-protein cytoplasmic tails, respectively. Mean values of LC3-I/LC3-II ratios of at least three independent experiments are shown, and error bars indicate the standard deviation. Statistically significant (ANOVA, P < 0.05) differences between samples and the control are indicated by asterisks.
Membrane fusion-mediated autophagy induction is common among paramyxoviruses.
All paramyxoviruses enter cells by membrane fusion, regardless of the specificity of their attachment proteins. To investigate if the membrane fusion-mediated autophagy induction observed for morbilliviruses also occurs with other paramyxovirus glycoproteins, we coexpressed NiV, HeV, and MuV G/HN and F proteins with GFP-LC3 in VerodogSLAMtag cells. All paramyxovirus glycoproteins induced syncytium formation and resulted in a punctate distribution of GFP-LC3, similar to the localization observed for morbillivirus glycoproteins (Fig. 6A). Quantification of the cells with a punctate GFP-LC3 distribution and analysis of LC3-I/LC3-II ratios confirmed these observations, with all paramyxovirus glycoproteins resulting in a statistically significant increase (ANOVA, P < 0.05) in LC3-II (Fig. 6B and C). Taken together, these results indicate that the observed membrane fusion-mediated autophagy induction may be common among viruses with fusogenic viral glycoproteins.
Fig 6.

Fusogenic viral glycoproteins from different paramyxoviruses induce autophagy. (A) Syncytium formation and localization of GFP-LC3 puncta in VerodogSLAMtag cells cotransfected with plasmids encoding GFP-LC3 and CDV, MeV, NiV, HeV, and MuV glycoproteins. Cells were stained 20 h after transfection with Lysotracker red and fixed with 4% paraformaldehyde. The intracellular localization of lysosomes (red) and autophagosomes (GFP-LC3 puncta) was visualized using a confocal laser microscope. Rapamycin-treated cells were included as an autophagy control for each experiment. Arrows indicate colocalization of GFP-LC3 puncta with lysosomes. (B) Proportion of GFP-expressing cells with punctum formation. For each sample, the total number of GFP-expressing foci and the number of foci with GFP puncta were counted in four separate fields. Data from at least four independent experiments were used for the analysis. Error bars represent the standard deviation, and statistically significant differences (ANOVA, P < 0.05) are indicated by asterisks. (C) Cell lysates were harvested 20 h after transfection, and LC3-I/LC3-II ratios were analyzed by Western blotting. Mean values of at least three independent experiments are shown, and error bars indicate the standard deviation. Statistically significant (ANOVA, P < 0.05) differences between samples and the control are indicated by asterisks.
DISCUSSION
As a cellular lysosomal degradation pathway, autophagy plays an important role in cellular homeostasis, cell survival under stress conditions, and cellular defense against pathogens (21, 29). Thus, the survival of viruses is tightly linked to their ability to counteract autophagy-associated antiviral defenses (32). However, an increasing number of viruses have been recognized to use autophagy pathways to their own benefit (6, 49). Little is known regarding the role of autophagy in the life cycle of paramyxoviruses, but there are recent reports that the morbillivirus immune cell receptor SLAM as well as the MeV vaccine strain receptor CD46 recruits autophagic complexes (2, 14). Here we investigated the role of autophagy in the morbillivirus life cycle in more detail. We initially confirmed that CDV and MeV infection results in autophagy induction and showed that this induction occurs at early infection stages and contributes to efficient viral spread. Further characterization revealed that viral glycoprotein-mediated membrane fusion triggers autophagy and that this process is not limited to morbilliviruses but is common to paramyxovirus glycoproteins and possibly even other virus-mediated membrane fusion processes.
Early autophagy induction may play an important role in spreading of the virus through the host.
Several positive-stranded RNA viruses, including poliovirus, coronavirus, dengue virus, and hepatitis C virus, require autophagy induction for efficient replication (13, 26, 27, 33). Here we provide evidence that morbilliviruses, as negative-stranded nonsegmented RNA viruses, exploit autophagy pathways rather than replication for spread to neighboring cells. Upon contact with either CDV or MeV virions or envelope glycoproteins, we observed a rapid redistribution of LC3 in a punctate cytoplasmic pattern and a corresponding increase in LC3-II, indicating that autophagic processes are triggered by virus-cell or cell-cell fusion. However, in contrast to positive-stranded RNA viruses, where the accumulated autophagic vacuoles serve as replication sites (44), the morbillivirus replication complex did not colocalize with autophagosomes. Furthermore, autophagy induction alone was not required for efficient replication, as illustrated by similar cell-associated virus titers obtained in control and Atg7-knockdown cells. Instead, inhibition of autophagy induction greatly reduced cell-cell fusion and spread of the virus from the initially infected cell to neighboring noninfected cells. There is increasing evidence that the primary mode of dissemination throughout the organism for morbilliviruses and probably most paramyxoviruses is via contact-mediated cell-to-cell spread (20, 23, 24), and our data suggest that autophagy induction may play an important role in this process.
Virus glycoprotein-mediated membrane fusion triggers autophagy.
For MeV, an association between receptor binding and autophagy induction has been previously reported (14), and the receptor SLAM also recruits the autophagy complex vps34/beclin1 (2). Our study confirms the importance of attachment to the target cells for morbillivirus infection-associated autophagy. However, further investigation revealed that autophagy induction also requires fusion between viral glycoprotein-expressing and target cell membranes. A similar effect has been described for the fusogenic HIV envelope glycoprotein gp41, which induces autophagy in bystander CD4+ T lymphocytes through contact with CXCR4 and subsequent fusion (3). In HIV, this contact-mediated autophagy induction facilitates efficient infection of or initiates apoptosis in these bystander cells, depending on their activation status, thereby contributing at the same time to viral spread and to the severe immunosuppression and loss of CD4+ T lymphocytes associated with the disease (10). A similar mechanism may also be involved in morbillivirus leukopenia and immunosuppression, since contact-mediated apoptosis and alteration of immune cell reactivity are well documented (17, 30). Further investigations in immune cells will be needed to confirm this dual role of autophagy in morbillivirus pathogenesis.
Fusion-mediated autophagy induction may be a widespread phenomenon.
The autophagy induction that we observed after coexpression of glycoproteins from different members of the Paramyxoviridae family, together with the reported similar activity of HIV gp41 (3), strongly suggests that there is a common link between membrane fusion and autophagy. While it is still possible that binding to the cellular receptor activates autophagy via a specific intracellular signaling cascade, the lack of autophagy induction in cells expressing only the CDV or MeV H protein and the gp41-mediated autophagy in cells expressing intracellular signaling-deficient CXCR4 (3) point toward a more direct activation mechanism in response to plasma membrane disruption, possibly similar to the response observed in neurons after traumatic injury (48). Since fusion between viral and cellular membranes is the first step in the life cycle of all enveloped viruses and fusion between infected and noninfected neighboring cells is one of the main mechanisms of dissemination within the infected host, future studies will likely reveal how viruses have evolved to exploit or at least prevent detrimental effects of the resulting autophagy induction.
ACKNOWLEDGMENTS
We thank Christopher Richardson for helpful comments on the manuscript. The NiV and HeV glycoprotein expression plasmids were kindly provided by Bevan Sawatsky, and MuV glycoprotein expression plasmids were provided by Paul Duprex. We are grateful to Yusuke Yanagi for the IC323-EGFP virus. We thank all laboratory members for continuing support and constructive discussion and Chantal Thibault for technical support.
This work was supported by grants from CIHR (MOP-66989), NSERC (299385-04), and CFI (9488) to V.V.M. P.L. was supported by a CIHR grant (MOP-93792) and received salary support from the Fonds de recherche en Santé du Québec (FRSQ).
Footnotes
Published ahead of print 30 May 2012
REFERENCES
- 1. Anderson DE, von Messling V. 2008. Region between the canine distemper virus M and F genes modulates virulence by controlling fusion protein expression. J. Virol. 82:10510–10518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berger SB, et al. 2010. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nat. Immunol. 11:920–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Denizot M, et al. 2008. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 4:998–1008 [DOI] [PubMed] [Google Scholar]
- 4. Devaux P, von Messling V, Songsungthong W, Springfeld C, Cattaneo R. 2007. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology 360:72–83 [DOI] [PubMed] [Google Scholar]
- 5. Diallo A. 1990. Morbillivirus group: genome organisation and proteins. Vet. Microbiol. 23:155–163 [DOI] [PubMed] [Google Scholar]
- 6. Dreux M, Chisari FV. 2009. Autophagy proteins promote hepatitis C virus replication. Autophagy 5:1224–1225 [DOI] [PubMed] [Google Scholar]
- 7. Dron M, et al. 2005. Scrg1 is induced in TSE and brain injuries, and associated with autophagy. Eur. J. Neurosci. 22:133–146 [DOI] [PubMed] [Google Scholar]
- 8. Dron M, et al. 2006. SCRG1, a potential marker of autophagy in transmissible spongiform encephalopathies. Autophagy 2:58–60 [DOI] [PubMed] [Google Scholar]
- 9. Eskelinen EL. 2005. Maturation of autophagic vacuoles in mammalian cells. Autophagy 1:1–10 [DOI] [PubMed] [Google Scholar]
- 10. Espert L, et al. 2006. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 116:2161–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hashimoto K, et al. 2002. SLAM (CD150)-independent measles virus entry as revealed by recombinant virus expressing green fluorescent protein. J. Virol. 76:6743–6749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu A, Cathomen T, Cattaneo R, Norrby E. 1995. Influence of N-linked oligosaccharide chains on the processing, cell surface expression and function of the measles virus fusion protein. J. Gen. Virol. 76(Pt 3):705–710 [DOI] [PubMed] [Google Scholar]
- 13. Jackson WT, et al. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156 doi:10.1371/journal.pbio.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joubert PE, et al. 2009. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 6:354–366 [DOI] [PubMed] [Google Scholar]
- 15. Kadowaki M, Karim MR. 2009. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 452:199–213 [DOI] [PubMed] [Google Scholar]
- 16. Klionsky DJ, et al. 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumagai K, Yamaguchi R, Uchida K, Tateyama S. 2004. Lymphoid apoptosis in acute canine distemper. J. Vet. Med. Sci. 66:175–181 [DOI] [PubMed] [Google Scholar]
- 18. Kumar H, Kawai T, Akira S. 2011. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30:16–34 [DOI] [PubMed] [Google Scholar]
- 19. Lamb RA. 1993. Paramyxovirus fusion: a hypothesis for changes. Virology 197:1–11 [DOI] [PubMed] [Google Scholar]
- 20. Lemon K, et al. 2011. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog. 7:e1001263 doi:10.1371/journal.ppat.1001263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Levine B, Klionsky DJ. 2004. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6:463–477 [DOI] [PubMed] [Google Scholar]
- 22. Lin LT, Dawson PW, Richardson CD. 2010. Viral interactions with macroautophagy: a double-edged sword. Virology 402:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ludlow M, Allen I, Schneider-Schaulies J. 2009. Systemic spread of measles virus: overcoming the epithelial and endothelial barriers. Thromb. Haemost. 102:1050–1056 [DOI] [PubMed] [Google Scholar]
- 24. Mathieu C, et al. 2011. Nipah virus uses leukocytes for efficient dissemination within a host. J. Virol. 85:7863–7871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mizushima N. 2009. Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol. 452:13–23 [DOI] [PubMed] [Google Scholar]
- 26. Panyasrivanit M, Khakpoor A, Wikan N, Smith DR. 2009. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 90:448–456 [DOI] [PubMed] [Google Scholar]
- 27. Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. 2004. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 279:10136–10141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rudd PA, Bastien-Hamel LE, von Messling V. 2010. Acute canine distemper encephalitis is associated with rapid neuronal loss and local immune activation. J. Gen. Virol. 91:980–989 [DOI] [PubMed] [Google Scholar]
- 29. Schmid D, Munz C. 2007. Innate and adaptive immunity through autophagy. Immunity 27:11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schobesberger M, Summerfield A, Doherr MG, Zurbriggen A, Griot C. 2005. Canine distemper virus-induced depletion of uninfected lymphocytes is associated with apoptosis. Vet. Immunol. Immunopathol. 104:33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shintani T, et al. 1999. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 18:5234–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shoji-Kawata S, Levine B. 2009. Autophagy, antiviral immunity, and viral countermeasures. Biochim. Biophys. Acta 1793:1478–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sir D, et al. 2008. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Suter SE, et al. 2005. In vitro canine distemper virus infection of canine lymphoid cells: a prelude to oncolytic therapy for lymphoma. Clin. Cancer Res. 11:1579–1587 [DOI] [PubMed] [Google Scholar]
- 35. Takeda M, et al. 2000. Recovery of pathogenic measles virus from cloned cDNA. J. Virol. 74:6643–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tal MC, Iwasaki A. 2009. Autophagy and innate recognition systems. Curr. Top. Microbiol. Immunol. 335:107–121 [DOI] [PubMed] [Google Scholar]
- 37. Tanida I. 2011. Autophagy basics. Microbiol. Immunol. 55:1–11 [DOI] [PubMed] [Google Scholar]
- 38. Tanida I, et al. 1999. Apg7p/Cvt2p: a novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell 10:1367–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tatsuo H, Yanagi Y. 2002. The morbillivirus receptor SLAM (CD150). Microbiol. Immunol. 46:135–142 [DOI] [PubMed] [Google Scholar]
- 40. Vongpunsawad S, Oezgun N, Braun W, Cattaneo R. 2004. Selectively receptor-blind measles viruses: identification of residues necessary for SLAM- or CD46-induced fusion and their localization on a new hemagglutinin structural model. J. Virol. 78:302–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. von Messling V, Cattaneo R. 2003. N-linked glycans with similar location in the fusion protein head modulate paramyxovirus fusion. J. Virol. 77:10202–10212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. von Messling V, Zimmer G, Herrler G, Haas L, Cattaneo R. 2001. The hemagglutinin of canine distemper virus determines tropism and cytopathogenicity. J. Virol. 75:6418–6427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang N, et al. 2004. The cell surface receptor SLAM controls T cell and macrophage functions. J. Exp. Med. 199:1255–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wileman T. 2006. Aggresomes and autophagy generate sites for virus replication. Science 312:875–878 [DOI] [PubMed] [Google Scholar]
- 45. Xu Y, Eissa NT. 2010. Autophagy in innate and adaptive immunity. Proc. Am. Thorac. Soc. 7:22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu Y, et al. 2007. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27:135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yanagi Y, Takeda M, Ohno S. 2006. Measles virus: cellular receptors, tropism and pathogenesis. J. Gen. Virol. 87:2767–2779 [DOI] [PubMed] [Google Scholar]
- 48. Zhang H, Song LC, Jia CH, Lu YL. 2008. Effects of ATP sensitive potassium channel opener on the mRNA and protein expressions of caspase-12 after cerebral ischemia-reperfusion in rats. Neurosci. Bull. 24:7–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou Z, et al. 2009. Autophagy is involved in influenza A virus replication. Autophagy 5:321–328 [DOI] [PubMed] [Google Scholar]