

Abstract
Despite the ability of primate lentiviruses to prevent infected cells from being reinfected, cell coinfection has occurred in the past and has shaped virus evolution by promoting the biogenesis of heterozygous virions and recombination during reverse transcription. In vitro experiments have shown that cell coinfection with HIV is more frequent than would be expected if coinfection were a random process. A possible explanation for this bias is the heterogeneity of target cells and the preferred infection of a subpopulation. To address this question, we compared the frequency of double-positive cells measured following coincubation with green fluorescent protein (GFP) and DsRed HIV reporter viruses with that of stochastic coinfection calculated as the product of the frequencies of GFP- and DsRed-positive cells upon incubation with either reporter virus. Coinfection was more frequent than would be expected on the grounds of stochastic infection, due to the underestimation of single-infection frequencies, which mathematically decreased the calculated frequency. Indeed, when cells were incubated with either reporter virus, a fraction of the cells were scored as uninfected yet harbored a silent provirus that was reactivated upon coinfection through cross talk between viral elements. When such cross talk was avoided, experimental and calculated coinfection frequencies matched, indicating random coinfection. The proportion of infected cells harboring a silent provirus was estimated from coinfection experiments and was shown to be cell type dependent but independent of the virus entry route.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) reverse transcriptase introduces 10−4 to 10−5 mutations per site and replication cycle (1, 36, 44). These mutations contribute to the diversity of the molecular species isolated from HIV-infected individuals and the emergence of quasispecies. In addition, the phylogenic analysis of primate lentiviruses has revealed that recombination occurred in the course of virus evolution. For instance, strong evidence suggests that simian immunodeficiency virus agm (SIVagm) and SIVcpz have arisen from the recombination of multiple SIV lineages (2; for a review, see reference 12). An increasing number of HIV-1 primary isolates also appear to be recombinant forms of M clade strains and are designated circulating recombinant forms (CRF) (for reviews, see references 7, 45, and 53). Finally, recombination has been shown to contribute to the diversity of viruses found within HIV-1-infected individuals (8) and to promote the emergence of CXCR4-using strains in a nonhuman model of AIDS (40).
Retroviruses package two genomic RNA (gRNA) molecules that recombine during reverse transcription (RT) due to the ability of RT to switch from one template gRNA molecule to another (13, 36; for reviews, see references 16 and 42). Besides recombination between repeated sequences, template switching is not mutagenic and does not alter the final genetic information carried by the virus cDNA when the two gRNA molecules are identical; however, in cases where virions harbor two unique gRNA molecules, recombination reshuffles alleles and generates mosaic proviruses. Although recombination has been suggested to hamper virus evolution under high selection pressure (6), numerous studies have concluded that it favors the emergence of multiresistant species (3–5, 39, 41, 50; for reviews, see references 20 and 45).
The formation of “heterozygous” virions implies that cells become coinfected with at least two genetically distinct viruses and that progeny virions package two gRNA molecules, each from a unique provirus. Despite the ability of primate lentiviruses to limit cell coinfection through the downregulation of cell surface levels of CD4 (55), a phenomenon known as “superinfection immunity” or “receptor interference” (35, 37), the genetic evidence mentioned above confirms that coinfection played a major role in HIV and SIV evolution (for a review, see reference 53). The presence of cells that harbor multiple genetically distinct proviruses in the lymph nodes and spleens of HIV-1-infected individuals has been reported, supporting the possibility of heterozygous virion biogenesis in vivo and subsequent recombination (23, 28). In vitro experiments conducted with replication-competent reporter viruses have also demonstrated coinfection and recombination events in the course of spreading infection (33). As the weight of this alternative to point mutation in HIV-1 evolution increased, several experimental systems have provided a better understanding of the dynamics of HIV-1 diversification by recombination in the course of virus replication. Such systems have focused on the most relevant parameters leading to virus recombination: (i) the choice of the gRNA molecules packaged into virions generated from cells harboring distinct proviruses, (ii) the recombination rate between two distinct gRNA molecules packaged into the same virion, and (iii) the monitoring of integration events per cell upon infection. These studies have shown that when cells harbor two unique HIV-1 proviruses, the packaging of two gRNA molecules per progeny virion is mostly a random process that generates homozygous or heterozygous virions according to a Hardy-Weinberg distribution (10, 33, 38). This is in sharp contrast with gammaretrovirus gRNA packaging, which mostly leads to the biogenesis of homozygous virions even when cells are multiply infected (29, 46). Upon infection with heterozygous virions, the frequency of recombination during RT is ∼10−4 per site per cycle (27, 39, 50; for reviews, see references 16 and 42), and it was shown to occur almost randomly throughout the RNA genome, with only a few hot spots identified in the constant regions of env (21, 33, 47, 49, 57). Finally, in silico simulations and models derived from the data reported by Jung et al. (28) showed that the number of successful integration events of viral DNA per cell is close to a Poisson distribution (17, 20). While there seems to be a consensus on the packaging and recombination models, coinfection models have to be interpreted cautiously, since they are based on the assumption that cell populations are homogeneous. In addition, they are in conflict with data generated from dual-reporter systems that clearly show a bias in favor of coinfection, which is not taken into account in any model described so far. Indeed, when cells are coincubated with two viral populations, each harboring a distinct reporter gene, the frequency of coinfection events is higher than that calculated based on stochastic infection, regardless of the target cell type and the envelope glycoprotein used for virus pseudotyping (9, 14, 22). These results, which also held true in cell-to-cell coinfection systems (14, 15), suggested the heterogeneity of target cells and the presence of a subpopulation more susceptible to HIV-1 infection (9, 14).
In the present work, we investigate coinfection with homozygous green fluorescent protein (GFP)- or DsRed-encoding proviruses derived from HIV-1 and Moloney murine leukemia virus (MLV). We show that when cells are coincubated with two HIV-1 populations, each harboring a distinct reporter gene, the percentage of coinfected cells is ∼5 to 20 times that calculated based on stochastic infection. We also analyze the specificity of this phenotype and demonstrate that viral elements, including the transactivation property of Tat, account for apparently biased coinfection.
MATERIALS AND METHODS
Cell culture.
All tissue culture media and antibiotics were purchased from Invitrogen (Cergy-Pontoise, France), except fetal calf serum, purchased from PAA (Austria). 293T cells and HeLa cells were obtained through the ATCC and the AIDS Research and Reference Reagents Program, Division of AIDS, NIAID, NIH, respectively, and grown in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum, 100 IU of penicillin/ml, and 100 μg of streptomycin/ml (complete DMEM). HPB-ALL cells were a kind gift from G. Bismuth (Institut Cochin, Paris, France) and were cultured in RPMI 1640 supplemented with 10% fetal calf serum, 100 IU of penicillin/ml, and 100 μg of streptomycin/ml (complete RPMI). Primary CD3+ T cells were purified from whole-blood samples with the RosetteSepHLA human lymphoid cell enrichment kit according to the instructions of the manufacturer (Stemcell Technologies, Grenoble, France), aliquoted, and frozen until further use. The cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
Virus production.
All viruses were made in 293T cells, as described elsewhere (30). Briefly, cells (∼2.5 × 106) were seeded in T75 flasks and transfected the next day by the calcium phosphate precipitation technique with 3 μg of HIV-1 Gag/Pol-encoding pCMVΔP1ΔenvpA, 3 μg of HIV-1-derived reporter provirus encoding DsRed (pHIvec2.DsRed) or GFP (pHIvec2.GFP), and 1 μg of HIV-1 Env-encoding pSVIIIEnv, all of which (except pHIvec2.DsRed) are described elsewhere (26, 43, 52), and carrier DNA up to 15 μg. pHIvec2.DsRed was generated by exchanging the GFP open reading frame (ORF) in pHIvec2.GFP (BamHI/XbaI) with that of DsRed from pLVX-DsRed-Monomer (Clontech, Saint-Germain-en-Laye, France). The pRRLSINcPPT PGK GFP WPRE plasmid described elsewhere (19, 58, 59) was used to generate Tat-independet GFP reporter viruses (HIV-GFP). In this construct, the 5′ long terminal repeat (LTR) U3 sequence has been replaced with that of Rous sarcoma virus (RSV), the 3′ LTR U3 sequence has been mutated, and enhanced green fluorescent protein (EGFP) expression is driven by the murine phosphoglycerokinase 1 gene (PGK1) promoter inserted 5′ to the EGFP cDNA. This ensures Tat-independent DNA transcription in the context of the plasmid and integrated proviruses. When viruses were pseudotyped with the vesicular stomatitis virus G (VSV-G) glycoprotein, 0.5 μg of each Rev-encoding plasmid (54) and a VSV-G-encoding plasmid (phCMV; obtained from F. L. Cosset, Université de Lyon, Lyon, France) were added to the transfection mixture. The cells were washed 6 and 24 h posttransfection and cultured for 24 h, after which the supernatant was collected, spun, filtered through a 0.45-μm-pore-size filter, and stored at −80°C. A similar protocol was used to make MLV virions, in which MLV Gag/Pol-encoding pTG5349 and pTG13077, an MLV-based GFP reporter provirus (both obtained from F. L. Cosset), were substituted for pCMVΔP1ΔenvpA and pHIvec2.GFP, respectively. The MLV-based DsRed reporter provirus was generated by exchanging the AgeI/SalI fragment encompassing the GFP ORF in pTG13077 with the DsRed ORF of pLVX-DsRed-Monomer (Clontech), amplified by PCR with primers containing the appropriate restriction sites.
Infection assay.
HeLa cells were seeded in 24-well plates at a density of 3 × 104 cells/well. After an overnight incubation, the cells were incubated with viruses as described in the figure legends, washed 24 h postinfection, incubated for 48 h, harvested, fixed in phosphate-buffered saline (PBS) supplemented with 3.7% formaldhehyde (Sigma), and analyzed by flow cytometry. When required, tumor necrosis factor alpha (TNF-α) (R&D Systems Europe, Lille, France) was added to the culture medium 24 h postinfection. The cells were incubated for 24 h, washed, and incubated in complete DMEM for an additional 24 h. When indicated, Lipofectamine 2000 (Invitrogen) was used to transfect cells with pHIvec2.dsRed or pcDNA3.DsRed, together with pUC or a Tat-encoding plasmid, and infected 24 h later. HPB-ALL cell infection was carried out as follows. Cells were seeded in 96-well plates at a density of 3 × 105 cells/well and incubated with viruses as described in the figure legends. Complete medium was added the next day, and the cells were cultured for an additional 48-h incubation time, after which cells were harvested, fixed, and analyzed as described above. Primary cells were activated prior to infection as follows. Non-tissue-culture-treated 24-well plates were coated overnight at 4°C with anti-CD3 and anti-CD28 monoclonal antibodies (MAbs) (3 μg/ml each in PBS; 500 μl/well), after which unbound MAbs were washed off and the cells were seeded (∼1 × 106 cells/well in 1 ml) in complete RPMI. Cells were harvested after a 48-h incubation time, resuspended in complete RPMI supplemented with 10 U/ml interleukin 2 (IL-2) (R&D Systems Europe), and dispensed in non-tissue-culture-treated 48-well plates (∼1 × 105 cells/well in 300 μl). GFP and DsRed reporter viruses, either separately or combined, were then added to the cells. Complete medium was added the next day, and the cells were cultured for an additional 48-h incubation time, after which cells were harvested, fixed, and analyzed as described above. Samples were analyzed on a FACSCalibur (Becton, Dickinson) with CellQuest Pro software.
Cell synchronization and cell cycle analysis.
HeLa-CD4 cells were seeded at a density of 2.5 × 106 cells/T75 flask or 105 cells/well in 24-well plates. Upon adherence, the cell culture medium was replaced with complete medium supplemented with 2.5 mM thymidine (Sigma) and the cells were incubated for 16 h. The cells were then washed with PBS and incubated in complete medium for 8 h, after which the medium was again replaced with complete medium supplemented with thymidine and the cells were incubated for 16 h. Cells arrested in G0/G1 were allowed to cycle again by washing off the excess thymidine. Cells were harvested at the indicated time points after a 1-h pulse with 10 μM bromodeoxyuridine (BrdU) (Sigma) and fixed overnight at 4°C in 70% ethanol. The fixed cells were washed in PBS and incubated in 2 M HCl supplemented with 0.5% Triton at room temperature for 30 min to ensure double-stranded DNA (dsDNA) denaturation. Samples were treated with 0.1 M Na2B4O7, and the cells were incubated for 1 h at room temperature with an anti-BrdU antibody (1:500; AbD Serotec), washed, and incubated for 30 min at room temperature with Alexa 488-conjugated immunoglobulin (1:400; Invitrogen). The cells were then stained with 20 μg/ml of propidium iodide (Sigma-Aldrich) following treatment with RNase and analyzed by flow cytometry.
Model and statistics.
When cells are incubated separately with a GFP or a DsRed reporter virus and the percentages of GFP- or DsRed-positive cells are measured by flow cytometry (a and b, respectively), the following equations allow the estimation of the percentages of GFP−/DsRed−, GFP+/DsRed−, GFP+/DsRed+, and GFP−/DsRed+ cells (A, B, C, and D, respectively) when cells are coincubated with both viruses and coinfection is random.
| (1) |
| (2) |
| (3) |
| (4) |
When the experimental percentage of coinfected cells (C′) does not match that predicted from equation 3, it is possible to estimate theoretical single-infection percentages (a′ and b′) from equations 5 and 6 that would lead to C′ if coinfection was not biased, assuming (i) a/b and a′/b′ are equal and (ii) reactivation of all silent proviruses upon coinfection. The percentage of infected yet reporter-negative cells is then calculated by subtracting a from a′.
| (5) |
| (6) |
The experimental and calculated coinfection frequencies were compared using a two-proportion z test. With pco.2 and nco.2 being the experimental frequency and the number of coinfection events, respectively, the number of events acquired was adjusted to satisfy values for pco.2 · nco.2 of >5 and for (1 − pco.2) · nco.2 of >5, allowing normal approximation. The theoretical infection frequency according to equation 3 was as follows: pco.1 = C/100. Arbitrarily setting nco.1 to satisfy the equations pco.1 · nco.1 > 5 and (1 − pco.1) · nco.1 > 5 was a legitimate approximation, given that the number of positive events corresponding to a and b represent more than 1,000 events and a/100 · 1,000 is more than 5, (100 − a)/100 · 1,000 is more than 5, b/100 · 1,000 is more than 5, and (100 − b)/100 · 1,000 is more than 5. The z score was calculated and the P value was determined as indicated in the figure legends.
RESULTS
Cellular and viral specificities of biased coinfection.
In order to avoid complex events arising from multiple rounds of replication, cell coinfection was analyzed with single-round entry competent viruses. A dual-color assay was used that consisted of incubating cells in the presence of reporter viruses that encode DsRed or GFP. Coinfection was first investigated in primary T cells incubated with VSV-G-pseudotyped viruses, an experimental system in which biased coinfection has been reported previously (9). Incubation of CD3+ T cells with a fixed amount of DsRed or GFP reporter virus resulted in 7.37% and 7.40% reporter-positive cells, respectively, considered productive infection (Fig. 1A, b and c). Using similar virus amounts and concentrations, coincubation of cells with both viruses resulted in 2.30% coinfection events (Fig. 1A, d). As shown in Fig. 1B, and in agreement with published results, this number was significantly higher than what would be expected if coinfection was occurring randomly and calculated as 7.37 × 7.40/100, equal to 0.55% (9, 14). Similar discrepancies between experimental and calculated percentages of coinfection were noticed with cells from another donor (Fig. 1A, e to g, and B, right). We then analyzed coinfection in cell lines frequently used in virus infectivity assays. Figure 1C shows that under similar settings, HPB-ALL T cells were more frequently coinfected than would be expected based on stochastic infection, whether the viruses were pseudotyped with HIV-1HXBc2 Env or VSV-G (Fig. 1C, left and right, respectively). A similar phenotype was observed with HeLa-CD4 cells of epithelial lineage (Fig. 1D). These results confirm that, regardless of the target cell type and the route of entry, coinfection with HIV-1 occurs more frequently than would be predicted on the basis of stochastic infection (see Fig. 6, top and middle, for a dose-response experiment plotted as described in reference 15). We thus chose to investigate the mechanisms responsible for this phenotype in HeLa cells with VSV-G-mediated virus entry.
Fig 1.

Biased productive coinfection of cells with GFP and DsRed reporter HIV-1-derived viruses. (A) CD3+ T cells purified from two healthy individuals were mock infected (a and e) and incubated with VSV-G-pseudotyped DsRed reporter virus (b and f) or GFP reporter virus (c and g) or coincubated with both viruses (d and h) and analyzed by flow cytometry (∼2 ng of p24 of each virus). The percentages of single- and double-positive cells are indicated on each dot plot. (B) The percentages of infected cells measured upon separate incubations with the DsRed or the GFP reporter virus were plotted, together with the experimental percentage of coinfection measured when the cells were coincubated with both viruses. The percentages of coinfection in case of stochastic infection were also calculated as the product of the percentages of DsRed-positive cells and GFP-positive cells measured (b or f and c or g) divided by 100. (C) HPB-ALL cells were treated and analyzed as for panel A with ∼2 ng or ∼ 0.1 ng of p24 of HIV-1HXBc2 envelope glycoprotein- or VSV-G-pseudotyepd viruses, respectively. The results are presented as in panel B. (D) HeLa CD4 cells were treated and analyzed as for panel A with ∼20 ng or ∼1 ng of p24 of HIV-1HXBc2 envelope glycoprotein- or VSV-G-pseudotyped viruses, respectively. The results are presented as in panel B. Representative results of experiments performed 3 times in duplicate are shown in panels C and D. Two-proportion z test; ***, z > 3.29, P < 0.001; **, z > 2.57, P < 0.01. The error bars indicate standard deviations.
Fig 6.
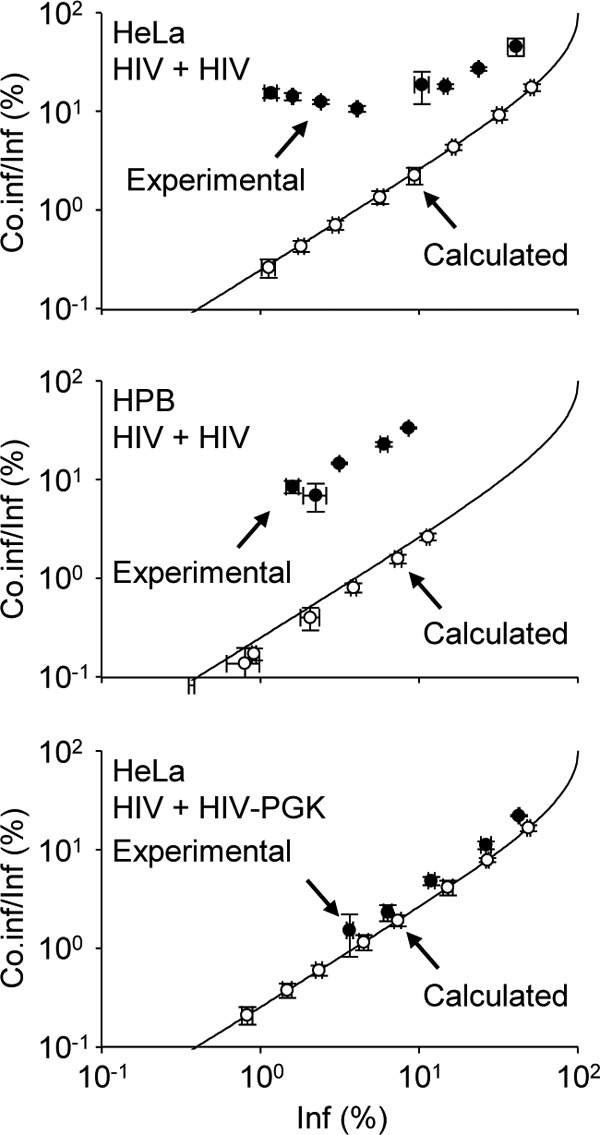
Coinfection with Tat-dependent and Tat-independent virus is random. Coinfection analyses were performed as for Fig. 1. Using a starting input of GFP and DsRed reporter viruses that ensured similar percentages of productive infection, each virus was serially diluted and incubated with cells either separately or in combination. For each virus input, the experimental and calculated percentages of coinfected cells (Co.inf) among infected cells (Inf) were plotted as a function of the percentage of infected cells. Experimental values were plotted only when the number of coinfection events collected by flow cytometry met the requirements for statistical significance. The Poisson distribution of multiple infection was calculated as described previously (15) and plotted (solid line). HPB-ALL or HeLa cells (top and middle, respectively) were incubated with VSV-G-pseudotyped Tat-dependent GFP and/or DsRed reporter HIV-1 (HIV). Coinfection of HeLa cells with a Tat-dependent DsRed reporter virus (HIV) and a Tat-independent GFP reporter virus (HIV PGK) (bottom) was analyzed and plotted as for the top and middle graphs. Representative results of experiments performed 3 times in duplicate are shown. The error bars indicate standard deviations.
The gap between experimental and calculated coinfection events was documented using various virus inputs. Starting from ∼8 ng of p24 for GFP or DsRed reporter viruses, which ensured similar percentages of productive infection, 2-fold serial dilutions of each virus were incubated on cells, either separately or in combination, and the cells were analyzed as for Fig. 1. As expected, the percentage of reporter-positive cells upon separate infection was proportional to the virus input (Fig. 2A). The ratio between the experimental and calculated values, referred to here as the coinfection index, was negatively correlated with the virus input (Fig. 2B). Therefore, when coinfection was analyzed under various experimental conditions, virions were first titrated individually under similar experimental conditions to determine the input leading to comparable percentages of infected cells. These settings were then used with both viruses combined, and the coinfection index was calculated. We then asked whether the infection time course influences the coinfection index. HeLa cells were coincubated with GFP and DsRed reporter viruses or incubated first with a DsRed reporter virus and 24 h later with a GFP reporter virus. Under both conditions, the frequencies of coinfected cells were significantly different from those calculated based on stochastic infection (Fig. 3A), which resulted in similar coinfection indexes (Fig. 3B). These data suggest that the memory of the first infection persists for at least 24 h in the mechanisms that lead to biased coinfection. The apparently nonrandom susceptibility of cells to coinfection was also studied by mixing DsRed- and GFP-encoding MLV-derived reporter viruses or MLV-DsRed and HIV-GFP viruses. In both instances, coinfection frequencies were similar to that calculated upon separate infection (Fig. 3C) and resulted in coinfection indexes close to 1 (Fig. 3D). Altogether, these results demonstrate that (i) successive incubation of cells with distinct HIV-1 reporters still leads to biased coinfection, (ii) coinfection of cells by MLV is a random process, and (iii) infection by HIV-1 does not render cells more susceptible to concurrent MLV infection.
Fig 2.
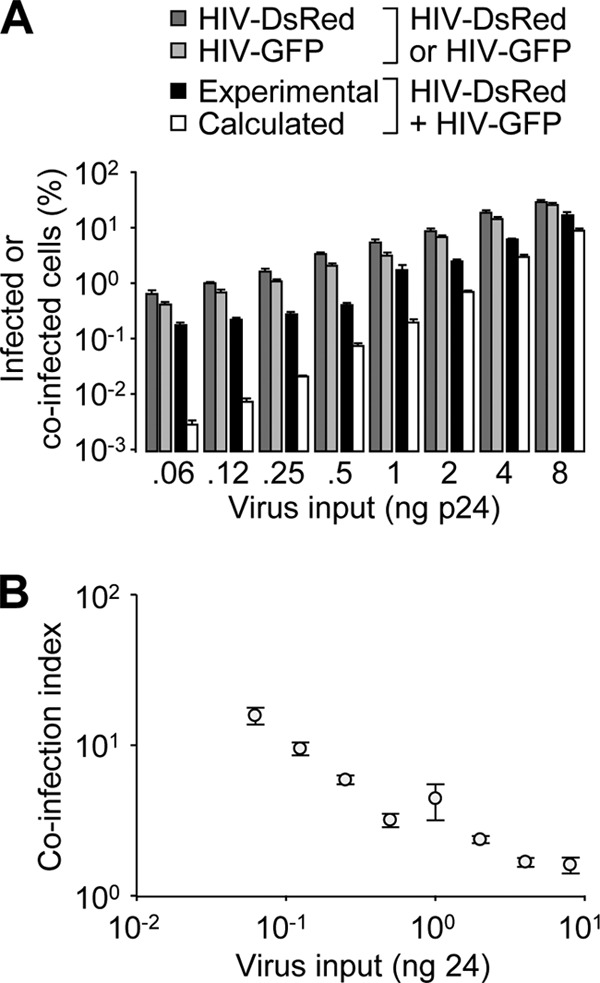
The coinfection index negatively correlates with virus input. (A) HeLa cells were incubated with increasing amounts of DsRed and/or GFP reporter viruses. Shown are the percentages of reporter-positive cells measured and calculated as described in the legend to Fig. 1B. (B) The coinfection index, calculated as the ratio between the experimental and calculated percentages of coinfection events, was plotted as a function of virus input. Representative results of an experiment performed three times in duplicate are shown. The error bars indicate standard deviations.
Fig 3.
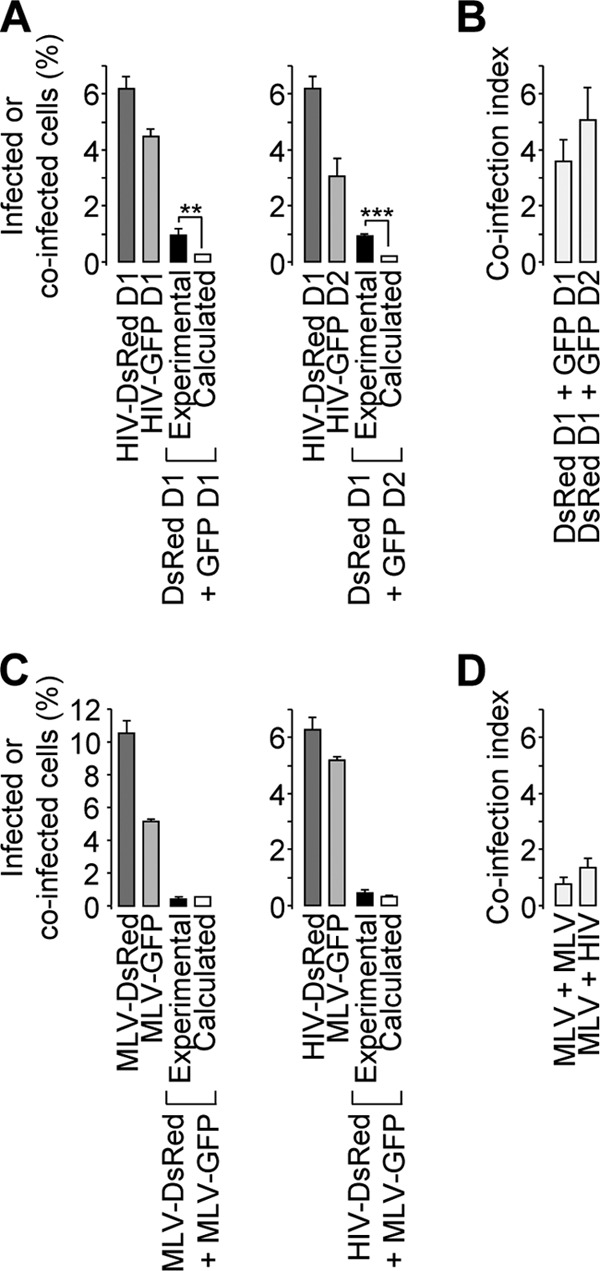
Time course dependence and specificity of the biased productive coinfection events. (A and B) HeLa cells were incubated with the DsRed and/or the GFP reporter HIV-1 on day 1 (A, left, D1) or incubated first with the DsRed reporter virus and/or 24 h later with the GFP reporter virus (A, right, D2). Coinfection indexes calculated from both conditions were plotted (B). (C and D) Same day incubation or coincubation with MLV-DsRed and MLV-GFP or HIV-DsRed and MLV-GFP were conducted and analyzed as for panel A, left. (A and C) Representative results of experiments performed 3 times in duplicate. (B and D) Averages calculated from 3 independent experiments. Two-proportion z test; ***, z > 3.29, P < 0.001; **, z > 2.57, P < 0.01. The error bars indicate standard deviations.
Lack of correlation between the cell cycle status and the coinfection index.
It has been suggested that nonrandom productive coinfection with multiple HIV-1 virions might reflect the heterogeneity of cells in respect to their susceptibility to HIV-1 infection (14). Several parameters, such as polarization or activation, might differentiate cells within a population that is considered homogeneous; however, because the coinfection index is >1 in many cell types (this work and references 9, 14, 15, and 22), we hypothesized that a common parameter, such as the cell cycle status, might influence cell susceptibility to coinfection. In order to investigate this hypothesis, HeLa cells were synchronized in G0/G1 by the double thymidine block method and then coincubated with VSV-G-pseudotyped DsRed and GFP reporter HIV-1 at various time points postchase. We reasoned that if HIV-1 targets a subset of cells in a particular phase of the cell cycle, coinfection of cells synchronized in that particular phase should be random and should lead to a coinfection index close to 1. While mock-treated cells were distributed throughout all the phases of the cell cycle, thymidine efficiently synchronized ∼95% of the cells in G0/G1 (Fig. 4A, 0 h, and B). Moreover, the cell cycle resumed once the excess thymidine was washed off, which allowed the recovery of cells enriched in early or late S phase (Fig. 4A, 4 to 8h, and B) or in the G2/M phase (Fig. 4A, 10 h, and B).
Fig 4.

Determination of the coinfection index in synchronized cells. HeLa cells were arrested in G0/G1, and the cell cycle was allowed to resume. (A and B) Nonsynchronized (NS) and synchronized cells were harvested at the indicated postchase time points following a 1-h incubation in the presence of BrdU. Two-channel BrdU/propidium iodide dot plots (A) and cell distribution throughout the cell cycle phases (B) are shown. (C to E) Nonsynchronized and synchronized cells were incubated with HIV-GFP or MLV-GFP at the indicated postchase time points. (C) The percentage of infected cells was determined by flow cytometry and normalized to that of nonsynchronized cells. (D) HIV-GFP and HIV-DsRed coinfection was analyzed as for Fig. 1. The virus input was adjusted to reach ∼5 to 10% productive infection at all time points with either the DsRed or the GFP reporter virus. (E) The coinfection index was calculated under similar conditions. Representative results of experiments performed at least twice in duplicate are shown. The error bars indicate standard deviations.
Fig 8.
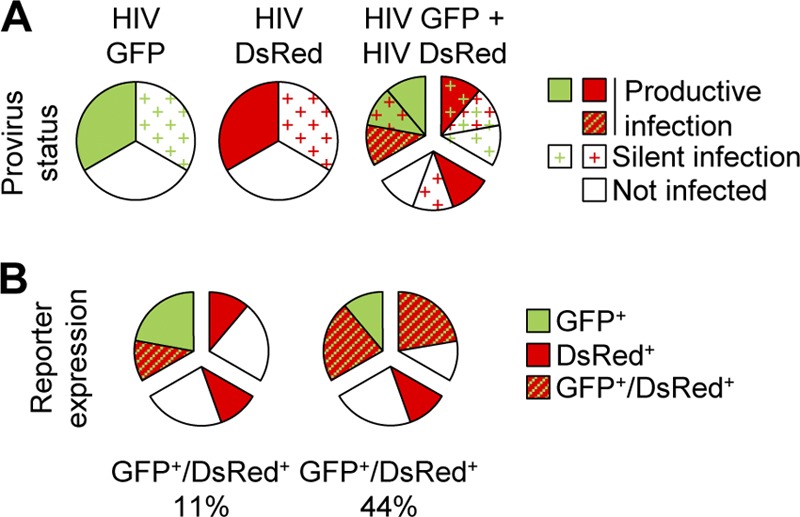
Tat-dependent reactivation of silent proviruses upon coinfection. (A) Target cell incubation with HIV-1 reporters can lead to reporter gene expression or silent infection. When cells are incubated with two distinct viruses, the status of each provirus can be calculated based on stochastic infection. (B) In the absence of cross talk between the GFP and DsRed proviruses (left pie chart), reporter expression takes place only in cells that harbor a provirus with a productive-infection status. In the case of transactivation (right pie chart), proviruses with a productive-infection status can rescue silent proviruses, leading to the expression of both reporter genes. Depending on the strength of transactivation, silent GFP and DsRed proviruses might also transactivate each other and drive the expression of both reporter genes. In this example, single incubation results in 33% reporter-positive cells and 33% infected yet reporter-negative cells. The latter percentage can be estimated with the model described in Materials and Methods from coinfection experiments that result in 44% GFP- and DsRed-positive cells.
We first measured the ability of VSV-G-pseudotyped HIV-1 to infect cells at each time point indicated in Fig. 4A. Cells synchronized in G0/G1 and untreated cells were equally susceptible to HIV-1 infection; however, cells abruptly became less susceptible upon entry into S phase (Fig. 4C). This was specific to HIV-1, since the susceptibility of cells to MLV infection followed a distinct pattern (Fig. 4C). While this might be at odds with the ability of HIV-1, but not MLV, to infect noncycling cells (56), it should be noted that arrested cells were allowed to reenter the cell cycle prior to infection. Thus, the results presented in Fig. 4C might reflect a unique aspect of HIV and MLV biology.
We then compared the coinfection index within the experimental time points. As shown in Fig. 2, this index varies as a function of virus input; therefore, the virus input was first adjusted so that the percentages of infected cells were similar across all time points. As shown in Fig. 4D, 5 to 10% of cells were productively infected upon incubation with adjusted concentrations of DsRed or GFP reporter viruses. With similar amounts and concentrations of virus, the frequencies of coinfection events did not significantly differ, and the coinfection index remained above 1 (Fig. 4E). These results demonstrate that the phase of the cell cycle impacts the susceptibility of cells to HIV-1 infection yet has no effect on the frequency of coinfection events.
Role of Tat in the nonrandom coinfection phenotype.
Gelderblom et al. demonstrated postentry rescue of replication-deficient viruses by simultaneous infection with wild-type viruses (22). We asked whether viral elements could explain the biased coinfection reported in this work and first focused on possible interactions between GagPol elements of incoming virions. In order to measure coinfection events in the absence of such interactions, the genome of the DsRed reporter virus was delivered to target cells by transfection, after which the cells were infected with a VSV-G-pseudotyped GFP reporter virus (Fig. 5A). As shown in Fig. 5B (left), ∼26% of the cells were efficiently transfected with detectable levels of DsRed expression, and incubation of the cells with a VSV-G-pseudotyped GFP reporter virus resulted in ∼5% infected cells. When the cells were transfected and then infected, the frequency of double-positive cells (7.5%) was significantly higher than that calculated in stochastic transfection/infection (∼1.3%) (Fig. 5B, middle). The ratio of the two values resulted in a coexpression index of ∼5.5 (Fig. 5B, right). A similar experiment conducted with a VSV-G-pseudotyped MLV-based reporter virus resulted in a coexpression index close to 1 and confirmed the specificity of this bias for HIV-1 infection. These data demonstrate that biased coinfection does not require cooperation between Gag/Pol proteins from both viruses and that there is sufficient information in the reporter provirus to trigger such a bias.
Fig 5.

Roles of Gag/Pol and Tat in the biased coinfection events. (A) Schematic representation of the experimental settings. HeLa cells were transfected on day 1 with pHIvec2.DsRed and/or incubated with either a VSV-G-pseudotyped HIV-GFP or MLV-GFP reporter virus on day 2. Psi, HIV-1 packaging signal; cPPT, HIV-1 central polypurine tract. (B) The percentage of transfected or productively infected cells was measured by flow cytometry upon separate transfection/infection (left). The percentage of double-positive cells obtained from the population subjected to both transfection and infection was measured by flow cytometry and compared to the expected results (middle). The coexpression index was obtained by dividing the experimental and expected percentages of double-positive cells (right). Representative results of experiments performed three times in duplicate are shown. Two-proportion z test: ***, z > 3.29, P < 0.001. (C) Schematic representation of the experimental settings. HeLa cells were cotransfected on day 1 with a DsRed-encoding plasmid and either a Tat-encoding plasmid or pUC and infected with either a VSV-G-pseudotyped HIV-GFP or an MLV-GFP reporter virus on day 2. (D) Cells were analyzed by flow cytometry. For each virus, the percentage of GFP-positive cells in the DsRed-positive population was normalized to that in the DsRed-negative population used as an internal control (left). The Tat/pUC ratio was then calculated (right). Representative results and averages of experiments performed 8 times with HIV-GFP and twice with MLV-GFP are shown (left and right, respectively). t test; #, P < 0.005; n = 8). The error bars indicate standard deviations.
The reporter proviruses used in this study encode Vif and Tat. Vif expression is unlikely to account for biased coinfection. Indeed, low levels of APOBEC3G render 293T cells permissive for the production of fully infectious virions even in the absence of Vif (for a review, see reference 11). On the other hand, it has been reported that Tat expression in target cells can rescue replication-deficient viruses (22) or reactivate latent proviruses in J1.1, ACH-2, and Jurkat cell lines (18, 34). We therefore investigated the contribution of Tat to the apparently nonrandom coinfection. Cells were transfected with a Tat-encoding plasmid or a control plasmid prior to infection with a VSV-G-pseudotyped GFP reporter virus. A DsRed-encoding plasmid was also included in the transfection mix in order to track transfected cells (Fig. 5C). In control cells, the proportion of infected cells among the DsRed-positive population was ∼1.5-fold higher than that measured in the DsRed-negative population when cells were incubated with either HIV or MLV (Fig. 5D, left). This likely reflects a nonspecific effect of transfection on cell susceptibility to virus infection. On the other hand, ectopic Tat expression increased the proportion of GFP-positive cells when the cells were incubated with HIV, but not with MLV (Fig. 5D, left). Data normalization showed that Tat expression doubled the proportion of cells infected by HIV but had no effect on MLV infection (Fig. 5D, right). These results clearly indicate that Tat expression might play a role in the apparently nonrandom infection, most likely by activating silent provirus.
In order to verify this hypothesis, coinfection experiments were conducted with the HIV-1 DsRed reporter virus and a virus lacking the Tat ORF and encoding GFP under the control of the PGK promoter. Using starting quantities of GFP and DsRed reporter viruses that ensured similar percentages of productive infection, each virus was serially diluted and incubated with cells, either separately or in combination. For each dilution, the proportion of coinfected cells among the total infected cells was plotted as shown in Fig. 6. In agreement with the apparently biased coinfection reported in this study, experimental values of HPB-ALL and HeLa cell coinfection with Tat-dependent DsRed and GFP reporter viruses did not match the coinfection calculated assuming stochastic infection (Fig. 6, top and middle). On the contrary, when coinfection was investigated with HIV and HIV-PGK, experimental and calculated values were almost identical (Fig. 6, bottom), confirming that infection with HIV-1 is stochastic. Of note, although the amounts of reporter viruses were adjusted to ensure similar percentages of productive infection, small differences explain the gap between calculated frequencies and the Poisson model of coinfection that was calculated based on identical titers of GFP and DsRed viruses.
The ability of Tat to reveal infected yet reporter-negative cells is reminiscent of the negative effect of Tat on the establishment of latency in Jurkat cells recently described by Donahue et al. (18). In order to confirm the link between latency and apparently nonrandom coinfection, we addressed the effect of TNF-α, which reactivates latent proviruses (18), on the coinfection index. As shown in Fig. 7, coincubation of VSV-G-pseudotyped reporter viruses on HeLa cells induced a higher percentage of coinfection events than that calculated from single-infection percentages (Fig. 7A, left). When TNF-α was added 24 h postinfection, the percentage of productive single infection doubled while the percentage of double-positive events did not change (Fig. 7A, right), which decreased the coinfection index from ∼4.5 to ∼2.2 (Fig. 7B). These results confirm that suboptimal provirus transcription explains apparently nonrandom cell coinfection.
Fig 7.
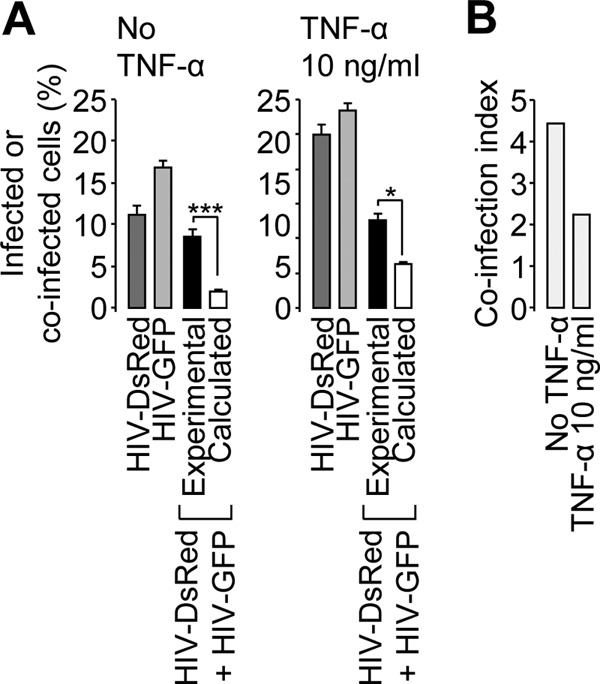
Effect of TNF-α treatment on apparently nonrandom coinfection. HeLa cells were incubated with a GFP or DsRed reporter or a VSV-G-pseudotyped virus or coincubated with both viruses. The cells were treated with 10 ng/ml of TNF-α or vehicle 24 h postinfection and analyzed by flow cytometry. (A) The percentages of reporter-positive cells were analyzed as for Fig. 1. (B) The coinfection index was calculated as for Fig. 2. Two-proportion z test; ***, z > 3.29, P < 0.001; *, z > 1.65, P < 0.05. The error bars indicate standard deviations.
Silent-infected-cell proportion estimated from productive coinfection events.
Together, our findings indicate that when cells are incubated with a reporter virus, a fraction of reporter-negative cells harbor a latent provirus (Fig. 8A). In the case of coinfection, and in the absence of cross talk between two reporters, latent proviruses remain silent, and the phenotypes of cells coincubated with distinct viruses match that estimated on the basis of random infection (Fig. 8B, left). On the other hand, when cross talk is possible, a transcriptionally active provirus might activate latent proviruses in coinfected cells, explaining the discrepancy between the experimental and calculated results, suggestive of nonrandom coinfection (Fig. 8B, right). We took advantage of the rescue of latent proviruses that operates in coinfected cells to better document virus infectivity and to determine the total infection potency of a virus stock: productive infection (reporter positive) and silent infection (reporter negative). Based on the assumption that all silent proviruses can be rescued by either a transcriptionally active provirus or another silent provirus, the infection potency was calculated from double-positive events as described in Materials and Methods and shown in Table 1 and Fig. 9. When separate incubation of HeLa cells led to 5 to 10% reporter-positive cells, the calculated infection potency of the viral stocks was ∼10 to 20%, suggesting ∼5 to 10% infected yet reporter-negative cells. In HPB-ALL cells, the proportion of silent infected cells was twice that of reporter-positive cells. Of note, the phenotype of activated primary CD3+ T cells was closer to that of HeLa cells than that of HPB-ALL cells. In addition, the extents of silent infection were similar whether entry was analyzed with VSV-G- or HXBc2-pseudotyped viruses (Fig. 9, compare open and closed symbols).
Table 1.
Silent-infected-cell estimation from coincubation experiments
| Target cell | Repeat (donor) | Separate incubationa |
% Coincubation double positiveb | Infection potency (%)c |
|||
|---|---|---|---|---|---|---|---|
| % Reporter 1 positive | % Reporter 2 positive | Reporter 2/1 ratio | Reporter 1 | Reporter 2 | |||
| T CD3+ | 1 | 7.3 ± 0.1 | 5.8 ± 1.5 | 0.8 ± 0.2 | 1.7 ± 0.6 | 14.6 | 11.7 |
| 2 | 5.3 ± 0.0 | 3.0 ± 0.1 | 0.6 ± 0.0 | 0.9 ± 0.0 | 12.7 | 7.1 | |
| HPB | 1 | 7.8 ± 0.1 | 3.8 ± 0.3 | 0.5 ± 0.0 | 2.9 ± 0.0 | 24.1 | 11.9 |
| 2 | 5.3 ± 0.4 | 2.2 ± 0.0 | 0.4 ± 0.0 | 1.4 ± 0.0 | 18.0 | 7.6 | |
| 3 | 5.0 ± 0.4 | 4.0 ± 0.4 | 0.8 ± 0.1 | 4.9 ± 0.4 | 24.9 | 19.8 | |
| HeLa | 1 | 6.5 ± 0.4 | 4.5 ± 0.3 | 0.7 ± 0.1 | 0.8 ± 0.3 | 11.0 | 7.6 |
| 2 | 6.2 ± 0.1 | 4.7 ± 0.0 | 0.8 ± 0.0 | 1.0 ± 0.1 | 11.5 | 8.7 | |
| 3 | 5.8 ± 0.2 | 4.2 ± 0.2 | 0.7 ± 0.0 | 1.1 ± 0.2 | 12.1 | 8.8 | |
| HeLad | 1 | 6.5 ± 0.4 | 2.8 ± 0.1 | 0.4 ± 0.0 | 0.9 ± 0.0 | 14.3 | 6.2 |
| 2 | 6.2 ± 0.1 | 3.8 ± 0.3 | 0.6 ± 0.1 | 0.9 ± 0.1 | 12.4 | 7.6 | |
| 3 | 5.8 ± 0.2 | 2.6 ± 0.1 | 0.4 ± 0.0 | 0.9 ± 0.1 | 14.5 | 6.5 | |
Cells were incubated separately with viruses encoding either reporter 1 or reporter 2; the percentages of reporter-positive cells measured by flow cytometry and the reporter 2/reporter 1 ratio are indicated ± standard deviation (SD).
Cells were incubated with both viruses, and the percentages of double-positive cells are indicated ± SD.
Theoretical percentages of reporter-positive cells upon separate incubation were calculated from double-positive cells as described in Materials and Methods.
Cells were incubated with reporter 1 and/or 24 h later with reporter 2.
Fig 9.
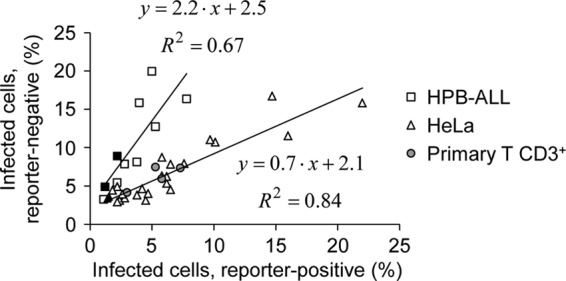
The proportion of reporter-negative infected cells is cell type dependent. HeLa, HPB-ALL, or primary CD3+ T cells were incubated separately or coincubated with a GFP and a DsRed reporter virus, and reporter-positive cells were scored by flow cytometry as for Fig. 1. The infection potency was calculated for each virus input from the frequency of coinfection events as described in Materials and Methods, and the percentages of infected yet reporter-negative cells were plotted as a function of reporter-positive cells. The closed and open symbols represent experiments conducted with HIV-1HXBc2 or VSV envelope glycoprotein-pseudotyped viruses. Primary T cells were infected with VSV-G-pseudotyped viruses. The results shown are from 7, 3, and 2 independent experiments performed on HeLa, HPB, and primary CD3+ T cells, respectively. Trend lines were calculated for HeLa and HPB-ALL cells and are represented together with the corresponding equations. The trend line for primary cells is as follows: y = 0.7 · x + 2.3; R = 0.70.
DISCUSSION
In the present study, we demonstrate that coincubation of cells with two distinct HIV-1-derived reporter viruses leads to higher frequencies of coinfection events than would be expected from stochastic infection, regardless of the target cell type and the route of virus entry. This bias, also reported by others (9, 14, 15, 22), was not noticed in MLV coinfection assays or when HIV-1 and MLV were coincubated on target cells. One hypothesis to account for such a bias is the preferred coinfection of a subpopulation of target cells with HIV-1. Our investigations confirmed that target cells are not equally susceptible to HIV-1 infection, due in part to their distribution throughout the cell cycle phases. However, the bias reported here was independent of the cell susceptibility to HIV-1 infection, since the coinfection index was >1 regardless of the phase of the cell cycle at which cells were infected. In fact, we report that experimental values obtained by coincubating cells with a Tat-dependent and a Tat-independent reporter HIV-1 match expected results calculated on the basis of random infection.
Our results support the hypothesis that when cells are incubated with a reporter virus, some cells score negative for reporter expression because they harbor a silent provirus. These cells are revealed in the presence of Tat (either expressed from a plasmid upon transfection or from a second transcriptionally active provirus), which transactivates the LTR of the silent provirus, thus driving reporter gene expression. As a consequence, the fact that not all infected cells are reporter positive following incubation with a GFP or a DsRed reporter virus causes underestimation of the expected coinfection frequencies, calculated as percent GFP positive × percent DsRed positive/100, and this artificially increases the coinfection index. The significance of these results is that cell coinfection with HIV-1 is a random phenomenon that reveals infection events falsely scored as negative upon single infection.
Unintegrated viral DNA from one virus was shown to affect cell infection with another virus (22; for a review, see reference 51). One question raised by our observation is whether the rescue of silent proviruses requires viral-DNA integration. The coinfection index was >1 even when cells were incubated at 24-h intervals with DsRed and GFP reporter viruses. Given that unintegrated viral DNA is a labile intermediate, postintegration interference likely accounts for the reactivation of silent proviruses. Several factors might explain this postintegration latency: the silencing of viral-gene expression; the unfavorable structure of the chromatin in the vicinity of the provirus; the low level of transcription initiation due to low levels of endogenous transcription factors, such as NFAT and NF-κB; and premature transcription termination due to suboptimal levels of Tat (for a review, see references 24 and 31). In our system, poor transcription initiation and premature transcription termination both contribute to the presence of infected yet reporter-negative cells. Indeed, there was a 2-fold increase in the percentages of HIV-1-infected cells when target cells overexpressed Tat or when target cells were incubated with TNF-α. Of note, both silencing mechanisms contributed to apparently biased coinfection, since experimental coinfection frequencies were close to that calculated on the grounds of stochastic infection when Tat-LTR cross talk was prevented and when target cells were incubated in the presence of TNF-α.
The proportion of cells harboring a silent provirus upon incubation with a GFP or DsRed reporter virus was estimated from coinfection experiments. We found a positive linear correlation between the percentage of reporter-positive cells and that of cells harboring a silent provirus when productive infection was in a 0 to 25% range. This proportion did not vary significantly whether entry was mediated by HIV-1HXBc2 or VSV envelope glycoprotein, confirming that, unlike the nuclear import pathway, the entry pathway does not significantly influence the integration of viral DNA into more or less active regions of the host genome (48). On the contrary, target cell types appeared differentially susceptible to provirus silencing/reactivation. In HeLa cells or IL-2/CD28-activated CD3+ T cells, when 5 to 10% of reporter-positive cells were detected upon separate infection with either GFP or DsRed reporter viruses, reactivation of silent proviruses by coinfection revealed an additional 5 to 10% of the cells harboring a silent provirus, which is consistent with the ability of Tat overexpression and TNF-α treatment to double the percentage of reporter-positive cells. In HPB-ALL cells, the percentage of silent cells calculated from coinfection was 2- to 4-fold higher than that of productively infected cells detected upon separate infection, suggesting that the concentrations of Tat required for full provirus transactivation might be higher in HPB-ALL cells than in HeLa or activated T cells. Of note, our model is based on the optimistic assumption that coinfection reactivates all silent proviruses, and therefore, the proportion of silent infected cells is probably higher than that documented in this study.
Del Portillo et al. also reported that coinfection frequencies are higher than expected when target cells are incubated with donor cells expressing both GFP and DsRed reporter HIV (15). This reflects the ability of infected cells to transfer more than one virus through the immunological synapse and is different from biased coinfection. Intriguingly, the study reported biased coinfection neither when target cells were coincubated with cell-free GFP and DsRed reporter viruses nor when GFP or DsRed reporter virus-expressing donor cells were pooled and incubated on target cells. One possible explanation is that the MT4 cells used in the earlier study are highly susceptible to HIV-1 infection and less prone to provirus silencing than the HeLa, HPB-ALL, and primary CD3+ T cells used in the present study or the Hut/CCR5, Jurkat, and primary CD4+ T cells used by others (9, 14, 22).
Regardless of the mechanism that leads to cell coinfection in vivo, our work suggests that multiple infection increases the frequency of each provirus expression compared to singly infected cells. Thus, while HIV has evolved strategies that render productively infected cells less susceptible to reinfection, it also seems to take advantage of coinfection by rescuing silent proviruses and promoting the biogenesis of heterozygous virions and subsequent recombination during RT. The presence of distinct proviruses per cell can be explained by simultaneous infection of a cell with distinct viruses or reinfection in the short time after the first virus has entered and prior to efficient cell surface CD4 downregulation by Nef, Env, and Vpu. One could also speculate that such cells might be targeted by viruses with Env variants capable of using suboptimal levels of cell surface CD4. Alternatively, successive infection might be possible in cases where HIV-1-infected T cells revert to a memory state in which viral DNA transcription is almost switched off and CD4 levels are similar to those of uninfected cells (25, 32). This suggests that HIV-1 reservoirs not only are an obstacle to HIV eradication, they probably also nurture the emergence of drug-resistant recombinant viruses by hosting several proviruses, leading to the biogenesis of heterozygous virions.
ACKNOWLEDGMENTS
We thank Séverine Pechberty, Marija Zivojnovic, and Corinne Cordier for technical assistance in cell cycle analyses and Chenxia He-Kong for primary cell isolation and activation. We are indebted to Malo Jaffré for expert assistance in statistical analysis.
This work was supported by Inserm, Université Paris Descartes, and grants from the Agence Nationale de la Recherche sur le SIDA et les Hepatites Virales (ANRS) and Université Paris Descartes to S.B. C.B. is supported by a Ph.D. fellowship from the French Ministry of Research. S.B. received support from Inserm and ANRS successively.
Footnotes
Published ahead of print 13 June 2012
REFERENCES
- 1. Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. 2010. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 84:9864–9878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bailes E, et al. 2003. Hybrid origin of SIV in chimpanzees. Science 300:1713. [DOI] [PubMed] [Google Scholar]
- 3. Balagam R, Singh V, Sagi AR, Dixit NM. 2011. Taking multiple infections of cells and recombination into account leads to small within-host effective-population-size estimates of HIV-1. PLoS One 6:e14531 doi:10.1371/journal.pone.0014531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Batorsky R, et al. 2011. Estimate of effective recombination rate and average selection coefficient for HIV in chronic infection. Proc. Natl. Acad. Sci. U. S. A. 108:5661–5666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bocharov G, et al. 2005. A genetic-algorithm approach to simulating human immunodeficiency virus evolution reveals the strong impact of multiply infected cells and recombination. J. Gen. Virol. 86:3109–3118 [DOI] [PubMed] [Google Scholar]
- 6. Bretscher MT, Althaus CL, Muller V, Bonhoeffer S. 2004. Recombination in HIV and the evolution of drug resistance: for better or for worse? Bioessays 26:180–188 [DOI] [PubMed] [Google Scholar]
- 7. Buonaguro L, Tornesello ML, Buonaguro FM. 2007. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and therapeutic implications. J. Virol. 81:10209–10219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Charpentier C, Nora T, Tenaillon O, Clavel F, Hance AJ. 2006. Extensive recombination among human immunodeficiency virus type 1 quasispecies makes an important contribution to viral diversity in individual patients. J. Virol. 80:2472–2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen J, et al. 2005. Mechanisms of nonrandom human immunodeficiency virus type 1 infection and double infection: preference in virus entry is important but is not the sole factor. J. Virol. 79:4140–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen J, et al. 2009. High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. Proc. Natl. Acad. Sci. U. S. A. 106:13535–13540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiu YL, Greene WC. 2008. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 26:317–353 [DOI] [PubMed] [Google Scholar]
- 12. Clewley JP. 2004. Enigmas and paradoxes: the genetic diversity and prevalence of the primate lentiviruses. Curr. HIV Res. 2:113–125 [DOI] [PubMed] [Google Scholar]
- 13. Coffin JM. 1979. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 42:1–26 [DOI] [PubMed] [Google Scholar]
- 14. Dang Q, et al. 2004. Nonrandom HIV-1 infection and double infection via direct and cell-mediated pathways. Proc. Natl. Acad. Sci. U. S. A. 101:632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Del Portillo A, et al. 2011. Multiploid inheritance of HIV-1 during cell-to-cell infection. J. Virol. 85:7169–7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Delviks-Frankenberry K, et al. 2011. Mechanisms and factors that influence high frequency retroviral recombination. Viruses 3:1650–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dixit NM, Perelson AS. 2004. Multiplicity of human immunodeficiency virus infections in lymphoid tissue. J. Virol. 78:8942–8945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Donahue DA, Kuhl BD, Sloan RD, Wainberg MA. 2012. The viral protein Tat can inhibit the establishment of HIV-1 latency. J. Virol. 86:3253–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dull T, et al. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fraser C. 2005. HIV recombination: what is the impact on antiretroviral therapy? J. R. Soc. Interface 2:489–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Galetto R, Giacomoni V, Veron M, Negroni M. 2006. Dissection of a circumscribed recombination hot spot in HIV-1 after a single infectious cycle. J. Biol. Chem. 281:2711–2720 [DOI] [PubMed] [Google Scholar]
- 22. Gelderblom HC, et al. 2008. Viral complementation allows HIV-1 replication without integration. Retrovirology 5:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gratton S, Cheynier R, Dumaurier MJ, Oksenhendler E, Wain-Hobson S. 2000. Highly restricted spread of HIV-1 and multiply infected cells within splenic germinal centers. Proc. Natl. Acad. Sci. U. S. A. 97:14566–14571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Han Y, Wind-Rotolo M, Yang HC, Siliciano JD, Siliciano RF. 2007. Experimental approaches to the study of HIV-1 latency. Nat. Rev. Microbiol. 5:95–106 [DOI] [PubMed] [Google Scholar]
- 25. Hermankova M, et al. 2003. Analysis of human immunodeficiency virus type 1 gene expression in latently infected resting CD4+ T lymphocytes in vivo. J. Virol. 77:7383–7392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hofmann W, et al. 1999. Species-specific, postentry barriers to primate immunodeficiency virus infection. J. Virol. 73:10020–10028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jetzt AE, et al. 2000. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 74:1234–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jung A, et al. 2002. Recombination: multiply infected spleen cells in HIV patients. Nature 418:144. [DOI] [PubMed] [Google Scholar]
- 29. Kharytonchyk SA, Kireyeva AI, Osipovich AB, Fomin IK. 2005. Evidence for preferential copackaging of Moloney murine leukemia virus genomic RNAs transcribed in the same chromosomal site. Retrovirology 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laguette N, Benichou S, Basmaciogullari S. 2009. HIV-1 Nef incorporation into virions does not increase infectivity. J. Virol. 83:1093–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lassen K, Han Y, Zhou Y, Siliciano J, Siliciano RF. 2004. The multifactorial nature of HIV-1 latency. Trends Mol. Med. 10:525–531 [DOI] [PubMed] [Google Scholar]
- 32. Lassen KG, Bailey JR, Siliciano RF. 2004. Analysis of human immunodeficiency virus type 1 transcriptional elongation in resting CD4+ T cells in vivo. J. Virol. 78:9105–9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U. S. A. 101:4204–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Macias D, Oya R, Saniger L, Martin F, Luque F. 2009. A lentiviral vector that activates latent human immunodeficiency virus-1 proviruses by the overexpression of tat and that kills the infected cells. Hum. Gene Ther. 20:1259–1268 [DOI] [PubMed] [Google Scholar]
- 35. Malim MH, Emerman M. 2001. HIV-1 sequence variation: drift, shift, and attenuation. Cell 104:469–472 [DOI] [PubMed] [Google Scholar]
- 36. Mansky LM, Temin HM. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Michel N, Allespach I, Venzke S, Fackler OT, Keppler OT. 2005. The Nef protein of human immunodeficiency virus establishes superinfection immunity by a dual strategy to downregulate cell-surface CCR5 and CD4. Curr. Biol. 15:714–723 [DOI] [PubMed] [Google Scholar]
- 38. Moore MD, et al. 2009. Probing the HIV-1 genomic RNA trafficking pathway and dimerization by genetic recombination and single virion analyses. PLoS. Pathog. 5:e1000627 doi:10.1371/journal.ppat.1000627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neher RA, Leitner T. 2010. Recombination rate and selection strength in HIV intra-patient evolution. PLoS. Comput. Biol. 6:e1000660 doi:10.1371/journal.pcbi.1000660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nishimura Y, et al. 2011. Recombination-mediated changes in coreceptor usage confer an augmented pathogenic phenotype in a nonhuman primate model of HIV-1-induced AIDS. J. Virol. 85:10617–10626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nora T, et al. 2007. Contribution of recombination to the evolution of human immunodeficiency viruses expressing resistance to antiretroviral treatment. J. Virol. 81:7620–7628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Onafuwa-Nuga A, Telesnitsky A. 2009. The remarkable frequency of human immunodeficiency virus type 1 genetic recombination. Microbiol. Mol. Biol. Rev. 73:451–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parolin C, Dorfman T, Palu G, Gottlinger H, Sodroski J. 1994. Analysis in human immunodeficiency virus type 1 vectors of cis-acting sequences that affect gene transfer into human lymphocytes. J. Virol. 68:3888–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Preston BD, Poiesz BJ, Loeb LA. 1988. Fidelity of HIV-1 reverse transcriptase. Science 242:1168–1171 [DOI] [PubMed] [Google Scholar]
- 45. Ramirez BC, Simon-Loriere E, Galetto R, Negroni M. 2008. Implications of recombination for HIV diversity. Virus Res. 134:64–73 [DOI] [PubMed] [Google Scholar]
- 46. Rasmussen SV, Pedersen FS. 2006. Co-localization of gammaretroviral RNAs at their transcription site favours co-packaging. J. Gen. Virol. 87:2279–2289 [DOI] [PubMed] [Google Scholar]
- 47. Rhodes T, Wargo H, Hu WS. 2003. High rates of human immunodeficiency virus type 1 recombination: near-random segregation of markers one kilobase apart in one round of viral replication. J. Virol. 77:11193–11200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schaller T, et al. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7:e1002439 doi:10.1371/journal.ppat.1002439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schlub TE, Smyth RP, Grimm AJ, Mak J, Davenport MP. 2010. Accurately measuring recombination between closely related HIV-1 genomes. PLoS Comput. Biol. 6:e1000766 doi:10.1371/journal.pcbi.1000766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shriner D, Rodrigo AG, Nickle DC, Mullins JI. 2004. Pervasive genomic recombination of HIV-1 in vivo. Genetics 167:1573–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sloan RD, Wainberg MA. 2011. The role of unintegrated DNA in HIV infection. Retrovirology 8:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sodroski J, Goh WC, Rosen C, Campbell K, Haseltine WA. 1986. Role of the HTLV-III/LAV envelope in syncytium formation and cytopathicity. Nature 322:470–474 [DOI] [PubMed] [Google Scholar]
- 53. Tebit DM, Nankya I, Arts EJ, Gao Y. 2007. HIV diversity, recombination and disease progression: how does fitness “fit” into the puzzle? AIDS Rev. 9:75–87 [PubMed] [Google Scholar]
- 54. Terwilliger E, et al. 1988. The art gene product of human immunodeficiency virus is required for replication. J. Virol. 62:655–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wildum S, Schindler M, Munch J, Kirchhoff F. 2006. Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J. Virol. 80:8047–8059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yamashita M, Emerman M. 2004. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J. Virol. 78:5670–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhuang J, et al. 2002. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J. Virol. 76:11273–11282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zufferey R, Donello JE, Trono D, Hope TJ. 1999. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 73:2886–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zufferey R, et al. 1998. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 72:9873–9880 [DOI] [PMC free article] [PubMed] [Google Scholar]