

Abstract
During lytic Kaposi's sarcoma-associated herpesvirus (KSHV) infection, host gene expression is severely restricted by a process of global mRNA degradation known as host shutoff, which rededicates translational machinery to the expression of viral proteins. A subset of host mRNAs is spared from shutoff, and a number of these contain cis-acting AU-rich elements (AREs) in their 3′ untranslated regions. AREs are found in labile mRNAs encoding cytokines, growth factors, and proto-oncogenes. Activation of the p38/MK2 signal transduction pathway reverses constitutive decay of ARE-mRNAs, resulting in increased protein production. The viral G-protein-coupled receptor (vGPCR) is thought to play an important role in promoting the secretion of angiogenic molecules from KSHV-infected cells during lytic replication, but to date it has not been clear how vGPCR circumvents host shutoff. Here, we demonstrate that vGPCR activates the p38/MK2 pathway and stabilizes ARE-mRNAs, augmenting the levels of their protein products. Using MK2-deficient cells, we demonstrate that MK2 is essential for maximal vGPCR-mediated ARE-mRNA stabilization. ARE-mRNAs are normally delivered to cytoplasmic ribonucleoprotein granules known as processing bodies (PBs) for translational silencing and decay. We demonstrate that PB formation is prevented during KSHV lytic replication or in response to vGPCR-mediated activation of RhoA subfamily GTPases. Together, these data show for the first time that vGPCR impacts gene expression at the posttranscriptional level, coordinating an attack on the host mRNA degradation machinery. By suppressing ARE-mRNA turnover, vGPCR may facilitate escape of certain target mRNAs from host shutoff and allow secretion of angiogenic factors from lytically infected cells.
INTRODUCTION
Human herpesvirus 8, also known as the Kaposi's sarcoma-associated herpesvirus (KSHV), is the etiologic agent of Kaposi's sarcoma and two rare lymphoproliferative disorders, multicentric Castleman's disease (MCD) and primary effusion lymphoma (PEL) (8, 12, 59). Like all herpesviruses, KSHV is capable of lifelong latent infection of its human host. KS tumors are replete with proliferating, latently infected endothelial cells (ECs) that display an abnormal spindle-shaped morphology and aberrant angiogenic phenotype (recently reviewed in reference 24). During latency, a minimal viral gene expression program ensures maintenance of the viral episome, suppresses the presentation of viral antigens to the immune system, and disrupts cell cycle checkpoints that would otherwise limit the replication of latently infected cells. Therefore, the products of the KSHV latent gene expression program are thought to be of central importance for KS tumorigenesis. However, sporadic lytic replication is commonly observed in KS tumors, and ganciclovir, a drug that selectively disrupts KSHV lytic replication, has been shown to possess some clinical utility against KS (42). KSHV-infected ECs are prone to loss of the viral episome (32), and therefore continued lytic KSHV replication may play an important role in expanding the pool of latently infected spindle cells during KS development.
Accumulating evidence indicates that beyond its role in promoting viral replication and dissemination, the KSHV lytic cycle may further support KS development by directing the expression of oncogenic and angiogenic viral gene products. Many KSHV lytic gene products display angiogenic and/or transforming potential in vitro. Chief among these is the viral G protein-coupled receptor (vGPCR), which shares homology with human CXCR1 and CXCR2 chemokine receptors but differs in that it possesses ligand-independent signaling activity (3, 13, 34). Ectopic expression of vGPCR has been shown to activate PI-3K, p38 mitogen-activated protein kinase (MAPK), and NF-κB pathways and upregulate the expression of many host gene products that promote EC proliferation and angiogenesis, including interleukin 6 (IL-6) and vascular endothelial growth factor (VEGF) (4, 46, 48, 58). vGPCR has demonstrated transforming potential in immortalized fibroblasts and ECs (4), and xenotransplantation of immortal vGPCR-expressing ECs into immunosuppressed rodents causes the progressive systemic development of angiogenic tumors that resemble KS (45). For these reasons, vGPCR has been proposed as a key element in KS pathogenesis.
vGPCR expression is restricted to the KSHV lytic cycle, with delayed-early kinetics (36). Thus, it is expressed only in cells that are fated to die due to the cytotoxicity of lytic replication. The protumorigenic effects of vGPCR are generally thought to be exclusively paracrine, mediated by upregulated host cytokines, chemokines, and growth factors that act on neighboring latently infected ECs (24). This model is complicated by the fact that during lytic replication, production of many host proteins is sharply curtailed by another delayed-early KSHV gene product, the shutoff exonuclease (SOX). SOX directs rapid cytoplasmic degradation of host mRNAs (30) and prevents export of nascent host mRNAs by causing nuclear retention of poly(A) binding protein C (PABPC) (38, 41). A detailed, transcriptome-wide analysis of host shutoff revealed that expression of most transcripts was sharply curtailed during KSHV lytic replication, but approximately 20% of host transcripts decline only slightly, and 2% are upregulated during infection (14). This select group of host mRNAs that evade SOX-mediated host shutoff includes the vGPCR target IL-6, whose production increases steadily throughout the lytic cycle (14, 29). However, other pathogenetically important vGPCR target gene products, like VEGF, are effectively suppressed by host shutoff. Taken together, these studies indicate that host shutoff represents a significant impediment to the oncogenic potential of vGPCR.
The precise details of how certain host mRNAs escape host shutoff remain to be elucidated, but some transcripts have been shown to be refractory to SOX-mediated degradation in vitro, suggesting that cis-acting elements may confer protection (14, 29). Enriched among the host shutoff escapees were transcripts bearing AU-rich elements (AREs) in their 3′ untranslated regions (UTRs). AREs are commonly found in labile mRNAs encoding tightly regulated, potent effector molecules, including many cytokines, chemokines, and proto-oncogenes (5, 16). It is not known precisely how AREs direct constitutive mRNA decay, but transcripts with multiple AREs tend to be subjected to accelerated decay. Host ARE-binding proteins (ARE-BPs) receive inputs from a variety of signal transduction pathways and tightly regulate ARE-mRNA turnover. Perhaps the best characterized ARE-BP is tristetraprolin (TTP), a CCCH zinc finger protein that binds specifically to ARE-mRNAs and directs them to the exosome, an assembly of endo- and exonucleases that digest deadenylated mRNA (15). The rapid degradation of ARE-mRNAs is subject to regulation by the p38 MAPK pathway. Stimulation of p38, long known to play a critical role in inflammation, results in significant increases in the half-lives of ARE-mRNAs and concomitant increases in translation (23). ARE-mRNA stabilization is mediated by the p38 target, mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 (MK2), which phosphorylates several ARE-BPs and disrupts the interactions between ARE-mRNAs and the decay machinery (60, 65).
In response to various stresses, including viral infection, eukaryotic cells arrest translation to prevent the expression of aberrant proteins and promote the expression of stress response proteins (reviewed in references 26 and 64). Four stress-sensing kinases, eukaryotic translation initiation factor 2-α (eIF2α) kinase 1 (HRI), 2 (PKR), 3 (PERK), and 4 (GCN2), respond to distinct stress signals and phosphorylate eIF2α, which prevents translation by disrupting formation of the eIF2-GTP-Met-tRNA ternary complex. Stalled translation initiation complexes are bound by a set of prion-like RNA binding proteins (e.g., TIA-1) that then self-associate and nucleate stress granules (SGs), sites of mRNA triage where stalled mRNPs either (i) await the resolution of stress and the resumption of translation or (ii) are rerouted to processing bodies (PBs) (2). PBs are stress-inducible dynamic aggregates of specific mRNAs and proteins that serve a dual function: first, like SGs, they harbor mRNAs that are translationally silenced, and such mRNAs can exit again from PBs to reengage in translation (7, 10). Second, PBs contain a suite of enzymes that direct rapid mRNA deadenylation, decapping, and exonucleolytic degradation (51). PB-mediated mRNA decay appears to function as a distinct and complementary system to the exosome mRNA decay machinery, which is notably absent from PBs (40, 54). ARE-mRNAs and their associated ARE-BPs have been visualized in both SGs and PBs (22), and fluorescence photobleaching experiments have demonstrated that SGs and PBs transiently interact and exchange cargo (35). The mechanism of PB formation is largely unknown, although their number and size increase when 5′ to 3′ exonucleolytic decay is blocked or when translation initiation is inhibited during stress (35). PBs are highly motile structures that associate with the actin and microtubule cytoskeleton networks (1, 35, 61), and recent work has shown that activation of the actin stress fiber-inducing RhoA subfamily GTPases prevents PB accretion and disrupts ARE-mRNA turnover (62). The accumulating evidence indicates that SGs and PBs are important sites of ARE-mRNA management in stressed cells.
The KSHV gene products responsible for modulating ARE-mRNA turnover during lytic replication remain to be identified. Our previous work demonstrated that ARE-mRNAs were stabilized during latent KSHV infection in vitro, a function that mapped to the latent kaposin B protein that directly bound and activated MK2 (44). Kaposin B is also expressed during the lytic cycle (53) and as such represents an attractive candidate protein that may be responsible for the evasion of certain ARE-mRNAs from host shutoff. The viral G-protein-coupled receptor (vGPCR) is thought to play an important role in promoting the secretion of pathogenetically important angiogenic molecules and other cytokines from KSHV-infected cells during lytic replication, but to date it has not been clear how vGPCR achieves this in the context of host shutoff. Here, we demonstrate that vGPCR activates the p38/MK2 pathway and stabilizes ARE-mRNAs, augmenting the levels of their protein products. Using a combination of chemical and genetic approaches, we demonstrate that MK2 activity is essential for maximal stabilization of ARE-mRNAs by vGPCR. We further demonstrate that vGPCR-mediated RhoA activation disrupts the formation of cytoplasmic PBs, sites of ARE-mRNA translational arrest and decay. Together, these data show for the first time that vGPCR impacts host gene expression at the posttranscriptional level. By suppressing ARE-mRNA turnover, vGPCR may facilitate escape of certain target mRNAs from host shutoff and allow secretion of angiogenic factors from lytically infected cells.
MATERIALS AND METHODS
Reagents.
Actinomycin D, doxycycline (Dox), puromycin, and Polybrene were purchased from Sigma Chemicals.
Cells.
HeLa Tet-Off (Clontech), Phoenix (a kind gift from G. Nolan, Stanford), and mouse embryonic fibroblast (MEFs; wild-type and MK2/3−/−; kind gifts from M. Gaestel) or iSLK/iSLK.219 (a kind gift from D. Ganem) cells were maintained at 37°C in a 5% CO2 atmosphere in Dulbecco's modified Eagle's medium containing 100 U of penicillin and streptomycin per ml and 10% heat-inactivated fetal bovine serum. Primary human umbilical vein endothelial (HUVE) cells were purchased from Clonetics. Cultures were expanded in EGM-2 medium (Lonza) on tissue culture plates coated with 0.1% (wt/vol) gelatin (in phosphate-buffered saline [PBS]) and used between passages 5 and 7 for experiments.
Plasmids.
A pcDNA3 expression plasmid for vGPCR containing the 5′ and 3′ untranslated regions (UTRs) was a kind gift from D. Ganem (UCSF). The 5′ and 3′ UTRs of vGPCR were removed by PCR amplification of the open reading frame (ORF) using forward (5′-GAA TTC CAC CAT GGC GGC CGA GGA TTT CCT AAC C) and reverse (5′-CTT TCA TGT CCG GCG CCA CCA CGT AGC TCG AGC) primers that contained flanking EcoRI and XhoI restriction sites, respectively. The resulting amplicon was TA cloned into pCR4-TOPO (Invitrogen), and the vGPCR open reading frame was subcloned into the EcoRI/XhoI sites of pCR3.1 to create pCR3.1-vGPCR. To create pCR3.1-vGPCRR143A, the arginine residue in position 143 was altered to alanine using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. Mutagenic forward (5′-GTG CGT CAG TCT AGT GGC GTA CCT CCT GGT GGC) and reverse (′-GCC ACC AGG AGG TAC GCC ACT AGA CTG ACG CAC) primers are indicated. Both vGPCR and vGPCRR143A open reading frames were subcloned into the EcoRI/XhoI sites of pBMN-IRES-puro (pBMN-IP), a puromycin selectable retroviral expression vector that was generously provided by G. Nolan (Stanford). The expression vector for kaposin B (pCR3.1-kapB) and the ARE-RNA reporter plasmids (pTRE2-Rluc, pTRE2-Fluc-ARE, pTRE2-BBB, pTRE2-BBB-ARE, pTRE2-d1EGFP, and pTRE2-d1EGFP-ARE) have been previously described (18, 44).
Retrovirus preparation and infections.
Retrovirus stocks were produced by calcium phosphate-mediated transfection of the Phoenix amphotropic packing cell line (a kind gift from G. Nolan, Stanford) and collecting virus-containing supernatants after 48 h. These supernatants were spinoculated onto target HUVE cell monolayers for 2 h at 2,000 rpm in the presence of 5 μg/ml Polybrene (Sigma), and after 24 h, 1 μg/ml puromycin was added to select for transductants.
KSHV lytic reactivation assays.
iSLK.219 cells were induced to express RTA by the addition of Dox (1 μg/μl) and/or valproic acid (0.9 mM). iSLK cells, which harbor the Dox-responsive RTA construct but do not contain the KSHV genome, were used as controls.
Luciferase assay.
The luciferase reporter assay for identification of modulators of ARE-RNA decay is described in detail in reference 18. Briefly, 105 HeLa Tet-Off cells were cotransfected with 100 ng of a reporter plasmid master mix (pTRE2-Fluc-ARE and pTRE-2-Rluc, at a ratio of 9:1); 900 ng of an expression vector for either vGPCR, vGPCRR143A, kaposin B, or an empty vector control; and 3 μl of Fugene HD (Roche) according to the instructions of the manufacturer. Twenty-four hours after transfection, Dox was added (1 μg/ml) to stop de novo transcription from the pTRE reporter plasmids. Twenty-four hours after the addition of Dox, transfected cells were lysed in 200 μl of 1× passive lysis buffer, and samples were processed using the dual-luciferase assay kit (Promega) according to the instructions of the manufacturer. Firefly and Renilla luminescence was determined using the GloMax 20/20 luminometer (Promega). Firefly luminescent signal, expressed in relative light units (RLUs), was normalized to that of Renilla luciferase, to eliminate off-target effects of our expression plasmids or the transfection procedure. Results are expressed as normalized luciferase activity.
Northern blotting.
The Northern blotting reporter assay for identification of modulators of ARE-RNA decay is described in detail in reference 18. Briefly, four 35-mm wells containing 2 × 105 HeLa-Tet Off cells were each cotransfected with 125 ng of the pTRE2-BBB, 250 ng of the pTRE2-BBB-ARE reporter plasmids, and 625 ng of an expression plasmid for vGPCR, vGPCRR143A, or an empty vector control using Fugene HD reagent (Promega) according to the instructions of the manufacturer. Eighteen hours after transfection, 1 μg/ml Dox was added and cells were lysed at 0, 0.5, 1, and 2 h after Dox addition using buffer RLT (Qiagen). Total RNA was isolated using the RNeasy miniprep kit (Qiagen) according to the manufacturer's instructions. RNA samples (2 μg) were subjected to 1% agarose-(2.2 M) formaldehyde gel electrophoresis and transferred using Nytran SuPer Charge T TurboBlotter with 10- by 15-cm 0.45-μm-pore-size membranes (Whatman). Blots were hybridized to a biotinylated 0.65-kb probe specific for rabbit β-globin that was generated using the T7 RNA polymerase MAXIscript kit (Ambion) and the linearized pCR4-BBB plasmid as a template and labeled using the BrightStar BioDetect kit (Ambion) according to the manufacturer's instructions. Hybridization was performed at 68°C using ULTRAhyb ultrasensitive hybridization buffer (Ambion), and visualization was performed using the BrightStar BioDetect kit (Ambion). The chemiluminescent signal was detected using the Kodak Image Station 4000 mm PRO with no excitation or emission filter.
Actinomycin D chase and quantitative PCR.
Transduced and selected HUVE or MEF cells were treated with actinomycin D to stop de novo transcription. Cells were lysed at 0, 1, 2, and 3 h after actinomycin D addition. Total RNA was isolated using the RNeasy mini-prep kit (Qiagen) according to the manufacturer's instructions, and the cDNA synthesis was performed using the Qiagen QuantiTecht cDNA synthesis kit on 0.5 μg of each RNA sample. Quantitative PCR (qPCR) analysis was performed using Stratagene MX6000P unit and Qiagen QuantiFast SYBR green qPCR mastermix. Primers for cDNA amplifications were as follows: human GAPDH forward (5′-GAG TCA ACG GAT TTG GTC GT) and reverse (5′-TTG ATT TTG GAG GGA TCT CG), human IL-6 forward (5′-GAA GCT CTA TCT CGC CTC CA) and reverse (5′-TTT TCT GCC AGT GCC TCT TT), human PTGS2 (Cox-2) forward (5′-CTG TTG CGG AGA AAG GAG TC) and reverse (5′-TCA TGG AAG ATG CAT TGG AA), mouse GAPDH forward (5′-AAC TTT GGC ATT GTG GAA GG) and reverse (5′-ACA CAT TGG GGG TAG GAA CA), and mouse Cox-2 forward (5′-TGG TGC CTG GTC TGA TGA TG) and reverse (5′-GTG GTA ACC GCT CAG GTG TTG). Relative initial template quantities were determined using the standard curve method and normalized to GAPDH. For each biological replicate, qPCRs were done in duplicate and mean cycle threshold (CT) values were used.
ELISA.
Human IL-6 and IL-8 were analyzed via an in-house enzyme-linked immunosorbent assay (ELISA). ELISA involved capture antibodies (IL-6 monoclonal antibody clone 5IL6 [Endogen, Woburn, MA] and IL-8 monoclonal antibody 208 [R&D Systems, Minneapolis, MN]). Nonspecific binding was blocked by using 1% bovine serum albumin (BSA) in phosphate-buffered saline for 1 h at 37°C. Matched biotinylated detection antibodies were for 7IL6 (Endogen) for IL-6 and BAF 208 (R&D Systems) for IL-8. A commercial ELISA amplification system (Life Technologies) was used for detection.
Protein preparation and Western blotting.
Six-well plates of cells were washed once with phosphate-buffered saline and lysed directly in 1× sodium dodecyl sulfate (SDS) sample buffer. Equivalent amounts of protein (10 or 25 μg) were subjected to SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amersham). Membranes were blocked in Tris-buffered saline–Tween-20 (TBST) containing 2.5% bovine serum albumin and probed overnight at 4°C using anti-vGPCR (1:2,000, a kind gift from Gary Hayward, Johns Hopkins School of Medicine), anti-cox2 (1:500, Santa Cruz), anti-phospho(ser82)Hsp27 (1:1,000), or anti-β-actin (1:5,000) antibody. Horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse immunoglobulin secondary antibodies were used at a 1:2,500 dilution. All antibodies were purchased from Cell Signaling Technologies unless otherwise indicated. Secondary antibody was detected using ECL Plus detection reagents (Amersham Biosciences) according to the manufacturer's instructions. The chemiluminescent signal was detected using the Kodak Image Station 4000 mm PRO with no excitation or emission filter.
Chemical inhibitors and activators.
To activate RhoA, HUVE cells were treated with 5 μg/ml of Rho activator II (Cytoskeleton Inc.) for 4 h in serum-free (SF) medium, 1 μg/ml of lysophosphatidic acid (LPA) for 30 min, or 125 ng/ml of nocodazole for 30 min. To activate the formation of stress granules and processing bodies (PBs), cells were subjected to stress by treatment with 0.25 mM sodium arsenite for 30 min. To irreversibly inhibit the GTPase RhoA, HUVE cells were treated with 1 μg/ml C3 transferase (Rho inhibitor I; Cytoskeleton Inc.) for 6 h in SF medium. Serum-free conditions are important to minimize baseline RhoA activity in the context of C3 exposure. However, since RhoA inhibition by C3 is irreversible after treatment, C3 and serum-free control cells were incubated for 1 h in normal medium to restore baseline PB levels. To inhibit the Rho-associated kinase (ROCK), downstream of RhoA subfamily GTPase activation, HUVE cells were treated with 10 μM Y27632 (EMD) for 1 h. To inhibit the kinases p38 and MK2, HeLa Tet-Off cells were treated with the inhibitors SB203580 and compound III (1 and 10 μM; both from EMD) for 1 h before Dox addition and for the remainder of the incubation with Dox.
Immunofluorescent staining and microscopy.
After transduction and puromycin selection, 60,000 HUVE cells were seeded on coverslips for microscopy. Twenty-four hours later, cells were either not treated or treated with the appropriate inhibitors or activators as described above and in the figure legends. After treatment, cells were fixed at room temperature in 4% paraformaldehyde for 10 min and then permeabilized with 0.1% Triton X-100 for 10 min. Cells were subsequently washed 3 times with PBS and then blocked in 1% human AB serum in PBS for 1 h at room temperature. To stain PB resident proteins, fixed cells were incubated with anti-Hedls antibody (1:1,000; Santa Cruz), anti-DDX6 antibody (C terminus, 1:1,000; Bethyl Laboratories), or both in 1% human AB serum overnight at 4°C. When appropriate, cells were subsequently incubated with anti-vGPCR antibody (1:1,000 in 1% human AB serum) for 30 min at room temperature before being washed (three washes of PBS for 5 min each). Goat anti-rabbit Alexa 555, chicken anti-mouse Alexa 488, or goat anti-rabbit Alexa 647 secondary antibodies were added for 1 h in the dark. After being washed as described above, cells were incubated with phalloidin (conjugated to either Alexa 555 or 647; Molecular Probes). Finally, cells were washed 3 more times and mounted on microscope slides with ProLong Gold antifade mounting medium (Invitrogen) and visualized using a Zeiss LSM 510 META laser-scanning confocal microscope and the 40× objective.
Quantification of PBs.
HUVE cells transduced with vector were examined at ×400 magnification, and the number of cells per field of view that contained normal-sized (approximately 1 μm in diameter; see Fig. 5A for a visual) PBs was counted. After counting between 100 and 200 cells (usually 4 fields of view), the percentage of cells containing normal PBs was determined. For treatments with RhoA activators, results are displayed as the average fold reduction in cells with PBs compared to untreated vector control (±standard error [SE]; n = 2 experiments). The effect of vGPCR expression on PB accretion was determined as described above except that only cells that were determined positive by immune fluorescence for vGPCR were counted. Results are displayed as the average fold reduction in cells with PBs compared to that of the untreated vector control (±SE; n = 5 experiments for wild-type vGPCR, n = 2 experiments for vGPCRR143A).
Fig 5.


vGPCR disrupts the formation of cytoplasmic PBs in a Rho-dependent fashion. (A) HUVE cells were treated with arsenite for 30 min to induce stress and the formation of PBs. After fixation and permeabilization, PBs were immunostained with two antibodies directed against resident proteins: anti-hedls antibody (green), which also binds to nuclear S6K, and anti-DDX6 antibody (red). (B and C) HUVE cells, transduced with empty vector and selected as described in Fig. 2, were treated with the RhoA activators lysophosphatidic acid (LPA) (B, C), nocodazole (C), or Rho activator II (C) as described in Materials and Methods. After fixation, cells were stained with anti-hedls antibody (green) to visualize PBs, and Alexa 555-conjugated phalloidin (red) to visualize actin. (C and D) To quantify the effect of Rho activation on PB accretion, the number of cells with normal (approximately 1 μm in diameter)-sized PBs per each field of view was counted. After transduction with vGPCR, the number of vGPCR-positive cells with normal PBs was counted. For each treatment, 100 to 200 cells were counted from three independent experiments. The fold reduction in the number of cells with PBs was calculated as described in Materials and Methods. (D to F) After transduction with vGPCR (wild type or signaling-inactive mutant R143A) or control retroviruses, selected cells were either mock treated in serum-free medium or treated with 1 μg/ml of C3 transferase, an irreversible inhibitor of RhoA subfamily GTPases, for 6 h, followed by 1 h in normal medium. Additional wells were treated with an inhibitor of the RhoA-associated kinase, ROCK, for 1 h, or not treated. These cells were fixed and triple stained with antibodies to the PB resident hedls protein (green) and vGPCR (falsely colored blue) and Alexa 647-conjugated phalloidin (red) to visualize actin stress fibers. Images are representative of two (R143A mutant) or five (vector and wild-type vGPCR) independent experiments (scale bar = 10 μm). Insets clearly indicate the presence or absence of PBs in vGPCR-expressing cells under different conditions.
RESULTS
KSHV vGPCR prevents ARE-mRNA decay.
The reasons for the escape of certain host ARE-mRNAs from shutoff during lytic KSHV infection remain obscure, but accumulating evidence implicates control at the posttranscriptional level. vGPCR stimulates the transcription of a wide variety of genes that contain AREs, but its effects on the stability of these ARE-mRNAs have not been investigated to date. To determine whether vGPCR can stabilize ARE-mRNAs, we performed a battery of tests that have been developed in our lab to measure ARE-mRNA turnover (18). First, we cotransfected HeLa Tet-Off cells with (i) an empty plasmid vector or (ii) a vGPCR expression vector, along with a doxycycline (Dox)-responsive reporter plasmid encoding firefly luciferase linked to a canonical ARE derived from the labile CSF2 transcript. After 24 h, transcription was arrested by the addition of Dox, and 24 h later, lysates were harvested for luciferase assays. A cotransfected Dox-responsive Renilla luciferase reporter lacking an ARE served as a normalization control. In lysates derived from the empty-vector-transfected cells, relative luciferase activity was low, reflecting the inherent instability of the ARE-mRNA encoding firefly luciferase (Fig. 1A). In contrast, vGPCR expression caused a striking increase in relative luciferase activity, comparable to the levels achieved by kaposin B expression. Disruption of vGPCR signaling activity with a point mutation (R143A) known to eliminate constitutive signaling from the molecule (34) and verified to be expressed at levels similar to that of the wild-type protein (Fig. 1B) returned relative luciferase activity levels to baseline. Thus, vGPCR can increase the production of protein from ARE-mRNAs, perhaps via ARE-mRNA stabilization.
Fig 1.
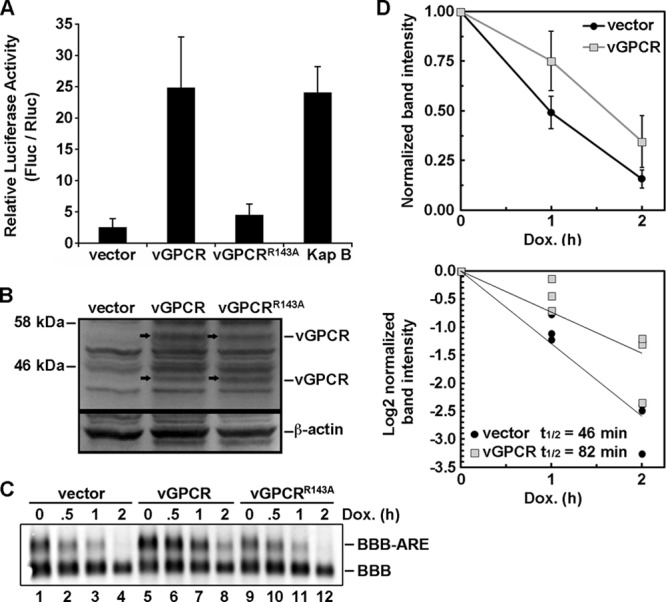
vGPCR stabilizes reporter ARE-containing mRNA. (A) HeLa Tet-Off cells were cotransfected with pTRE2-Rluc (no ARE), pTRE2-Fluc-ARE, and an expression plasmid for kaposin B, vGPCR, a signaling inactive mutant of vGPCR (vGPCRR143A), or an empty vector control. At 24 h posttransfection, Dox was added to halt reporter gene transcription. Twenty-four hours after Dox addition, cell lysates were harvested and analyzed for firefly, Renilla, or normalized (firefly/Renilla) luciferase activity as described in Materials and Methods. Results are displayed in relative light units (RLUs) and are the averages from three independent experiments ± the standard error. (B) Lysates from transfected HeLa Tet-Off cells were harvested and immunoblotted with anti-vGPCR (1:1,000 dilution) or anti-β-actin (1:5,000 dilution) antibody. (C) HeLa Tet-Off cells were cotransfected with pTRE2-BBB and pTRE2-BBB-ARE, with or without an expression plasmid for vGPCR. At 18 h posttransfection, Dox was added to halt reporter gene transcription, and total RNA was harvested after 0, 0.5, 1, and 2 h. The amounts of reporter RNAs were detected simultaneously with a probe to the β-globin ORF. (D) Log2 values of the band intensities from panel B were plotted versus time and fitted with the linear regression curves. From the linear regression analyses, the slope value m was obtained. The inverted negative values of each m value are the calculated half-life values shown on the plot (t1/2 = −1/m).
To directly test whether vGPCR could affect the stability of ARE-mRNAs, we examined the stability of a reporter ARE-mRNA over a time course in control and vGPCR-expressing cells. HeLa Tet-Off cells were cotransfected with empty vector or vGPCR expression vector, along with two Dox-responsive reporter plasmids encoding β-globin or β-globin linked to an ARE. After adding doxycycline to stop transcription, RNA was harvested at defined intervals over the course of 2 h and Northern blotted using a β-globin-specific probe. vGPCR expression significantly stabilized the longer β-globin-ARE mRNA compared to vector- and R143A-transfected controls, without significantly affecting the turnover of the corresponding β-globin mRNA lacking an ARE (Fig. 1C). Densitometric analysis revealed that vGPCR expression increased the half-life of the ARE-mRNA reporter approximately 2-fold compared to that of the vector control (Fig. 1D). Taken together, these findings provide compelling evidence that vGPCR signaling can prevent ARE-mRNA turnover and increase the production of ARE-mRNA-encoded proteins.
KSHV vGPCR stabilizes host ARE-mRNAs in primary endothelial cells.
The driving force of KS oncogenesis is the KSHV-infected spindle cell. Abundant evidence indicates that spindle cells are endothelial in origin, and several different types of ECs have been usefully employed for in vitro studies of KSHV infection, including human umbilical vein endothelial (HUVE) cells. HUVE cells support KSHV latency and adopt the hallmark spindle-shaped morphology, and following treatment with phorbol esters and/or histone deacetylase inhibitors can support productive lytic replication (21, 25). For these reasons, we decided to further investigate the properties of vGPCR in HUVE cells. HUVE cells were transduced with retroviral vectors encoding vGPCR or the R143A mutant (34) and selected with puromycin to ensure expression of vGPCR proteins in 100% of the cells. vGPCR expressing HUVE cells displayed a characteristic cell morphology change (Fig. 2A) (4) and upregulated the expression of the ARE-containing vGPCR target gene products cyclooxygenase-2 (Cox-2), IL-6, and IL-8 (Fig. 2C to G), consistent with previous reports (56). The signaling mutant vGPCRR143A failed to exhibit any of these properties. Importantly, although lysates from retrovirally transduced HUVE cells contained reduced steady-state levels of the R143A mutant compared to that of wild-type vGPCR (Fig. 2C, right), significant amounts of the mutant protein could be visualized at the plasma membrane by immunofluorescence staining (Fig. 2B). These observations validate the R143A mutant as a useful negative control in these experiments.
Fig 2.

vGPCR increases the half-life of endogenous ARE-containing mRNAs. (A to E) HUVE cells were transduced with recombinant retroviruses expressing vGPCR, a signaling inactive mutant of vGPCR (vGPCRR143A), or the empty vector control virus. After 2 days of selection with puromycin and 1 day of recovery, cell monolayers were examined by light microscopy (scale bar in panel A = 200 μm). Transduced cell monolayers were also analyzed as follows: cells were seeded on coverslips overnight and then fixed and immunostained using anti-vGPCR antibody (scale bar = 10 μm, thick arrows indicate high-expressing cells while thin arrows indicate low-expressing cells) (B); cells were lysed in protein sample buffer, and 25 μg of protein was analyzed by SDS-PAGE and immunoblotting with anti-Cox-2 antibody at 1:500, anti-vGPCR antibody at 1:2,000, or anti-β-actin antibody at 1:5,000 (C); ELISA analysis of cell-free supernatants was done for the presence of human IL-8 and IL-6, which are encoded by ARE-containing RNAs (D); cells were harvested for total RNA isolation, and the steady-state levels of the Cox-2 and IL-6 transcripts were determined by RT-qPCR (E). Error bars represent standard deviations (n = 3 experiments). (F) HUVE cells transduced as described in the legends to panels A to E were either treated with actinomycin D, which stops de novo transcription, for 1, 2, or 3 h, or not treated and harvested for total RNA. Each sample was analyzed by RT-qPCR to determine the decay rates of the Cox-2 transcript. Values are presented as fractions of the transcript levels at the time of actinomycin D addition, and the error bars represent standard deviations (n = 3 experiments). (G) Log2 values of the data from panel E were plotted versus time independently for all three replicates and fitted with the linear regression curves. From the linear regression analyses, the slope value m was obtained. The inverted negative values of each m value are the calculated half-life values shown on the plot (t1/2 = −1/m).
To determine whether vGPCR can stabilize endogenous ARE-mRNAs, HUVE cells transduced with either control retroviral vector, vGPCR, or vGPCRR143A were treated with actinomycin D, a well-described inhibitor of RNA polymerase II-driven transcription, and RNA was harvested over the course of 2 h. Reverse transcription (RT)-qPCR analysis revealed that the Cox-2 ARE-mRNA possesses a half-life of 44 min in vector-transduced cells and a similar decay rate in vGPCRR143A transduced cells (Fig. 2F, G). In contrast, vGPCR expression significantly extended the half-life of Cox-2 ARE-mRNA to 102 min. These experiments clearly demonstrate that vGPCR can stabilize ARE-mRNAs and suggest that posttranscriptional mechanisms play a role in the reprogramming of host gene expression by vGPCR.
MK2 activity is essential for vGPCR-induced ARE-mRNA stabilization.
The rapid degradation of ARE-mRNAs can be reversed by activation of the p38/MK2 pathway (23, 65). Previous reports have shown that vGPCR activates p38 in simian COS-7 cells (58) and human HEK293T epithelial cells (4). Here, we show that vGPCR-expressing HUVE cells display sharp increases in the levels of phosphorylation of the MK2 target protein Hsp27 (Fig. 3A), clearly demonstrating the activity of the MK2 pathway. To determine whether this pathway plays a significant role in vGPCR-mediated stabilization of ARE-mRNAs, we employed selective chemical inhibitors of p38 (SB203580; denoted p38i) and MK2 (MK2 inhibitor III; denoted MK2i). Treatment of HeLa Tet-Off cells with the MK2 inhibitor caused significant decreases in the vGPCR-mediated stabilization of the luciferase-ARE reporter in a concentration-dependent fashion, whereas vehicle control had no effect (Fig. 3B) Treatment with the p38 inhibitor had only minimal effect (Fig. 3B). The decreased ability of vGPCR to stabilize firefly luciferase after MK2 inhibition mirrors the decrease in the amount of phosphorylated Hsp27 detected after the same treatment (Fig. 3A).
Fig 3.
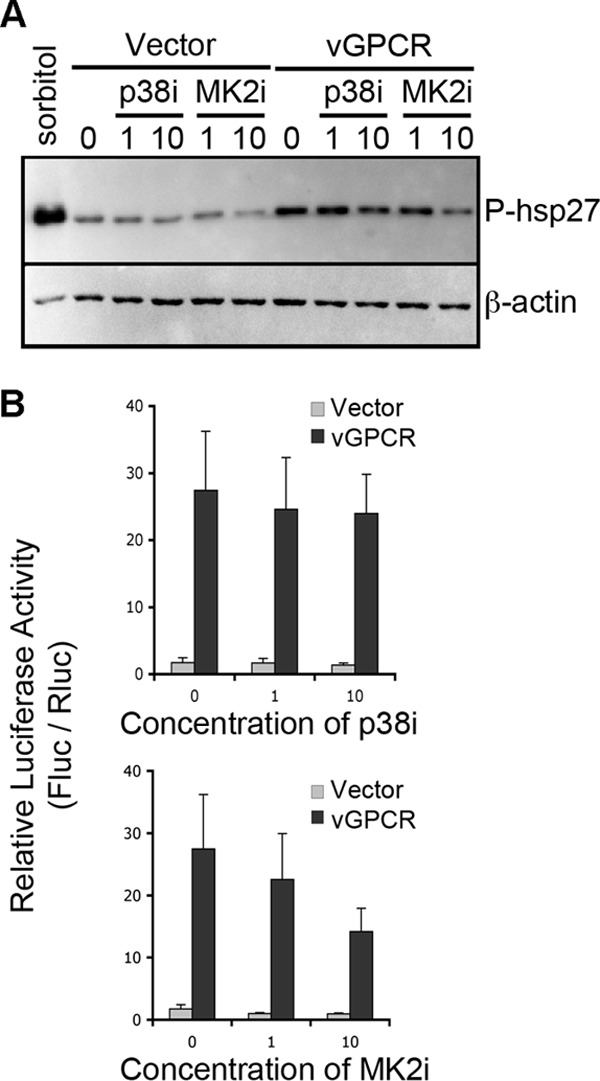
MK2 inhibition disrupts vGPCR-mediated ARE-mRNA stabilization. (A) HUVE cells were transduced with recombinant retroviruses expressing vGPCR or the empty vector control virus and selected as described for Fig. 2. Transduced cells were treated with chemical inhibitors of the kinases p38 (SB203580; 1 or 10 μM), MK2 (compound III; 1 or 10 μM), or dimethyl sulfoxide (DMSO) as a vehicle control for 1 h before lysis in 1× protein sample buffer. Additional wells of cells were treated with sorbitol (0.4 M), to activate the stress-inducible p38 MAPK pathway. A total of 10 μg of total protein was subjected to 12% SDS-PAGE and immunoblotted with antibodies against the phosphorylated form of heat shock protein 27 (Hsp27) (ser82; 1:1,000) and β-actin (1:5,000). One representative experiment of three is shown. (B) HeLa Tet-Off cells were cotransfected with pTRE2-Rluc (no ARE), pTRE2-Fluc-ARE, and an expression plasmid for vGPCR or an empty vector control. At 23 h posttransfection, cells were treated with chemical inhibitors of the kinases p38 (SB203580; 10 μM), MK2 (compound III; 10 μM), or DMSO for 1 h. At 24 h posttransfection, Dox was added to halt reporter gene transcription. Twenty-four hours after Dox addition, cells were lysed and analyzed for firefly, Renilla, and normalized (firefly/Renilla) activity as described in Materials and Methods. Results are displayed in relative light units (RLUs) and are the averages from three independent experiments ± the standard errors.
Next, ARE-mRNA turnover was measured in mouse embryo fibroblasts (MEFs) derived from mice with genetic lesions in MK2 and MK3 (52). Parental and MK2/3-knockout MEFs were transduced with vGPCR and treated with actinomycin D to stop transcription as described above. RNA harvested over a time course was examined via RT-qPCR using oligonucleotide primers specific for murine Cox-2. In parental MEFs, vGPCR expression caused 4- and 3-fold increases in the steady-state level and half-life of the Cox-2 ARE-mRNA, respectively, compared to those of controls (Fig. 4A, B, D). In contrast, vGPCR transduction had a modest effect on the turnover of the Cox-2 ARE-mRNA in the MK2/3-null MEFs, increasing the half-life approximately 1.5-fold compared to controls (Fig. 4C and E). Together, these findings highlight the importance of the p38/MK2 pathway in vGPCR function and indicate that activation of MK2 is important for vGPCR to influence ARE-mRNA turnover.
Fig 4.

MK2/3 knockout cells do not support vGPCR-mediated ARE-RNA stability. Wild-type (wt) or MK2/3 knockout (MK2/3−/−) mouse embryonic fibroblasts (MEFs) were transduced with recombinant retroviruses expressing vGPCR or the empty vector control virus and selected as described in Fig. 2. Postselection, cells were treated with actinomycin D to stop de novo transcription and harvested for total RNA isolation after 0, 1, 2, and 3 h. Each sample was analyzed by RT-qPCR to determine the decay rates of the Cox-2 transcript during the chase. Values are presented as fractions of the transcript levels at the time of actinomycin D addition.
vGPCR disrupts cytoplasmic PBs in an RhoA subfamily GTPase-dependent fashion.
Accumulating evidence indicates that the ARE-mRNA decay machinery is spatially organized in the cell. One discrete location of ARE-mRNA turnover is the PB, a stress-induced dynamic assembly of nontranslating mRNPs and degradative enzymes. Little is known about the signals that govern PB assembly, but PBs are associated with the cytoskeleton, and recent work has implicated the cytoskeleton remodeling RhoA subfamily of small GTPases in negatively regulating PB accretion and function (62). Because vGPCR has been previously shown to activate RhoA (43, 57), we hypothesized that vGPCR may disrupt the formation of PBs. To visualize PBs, we subjected HUVE cells to oxidative stress by treating with sodium arsenite, a known inducer of translational arrest and PB formation (2), and performed immunofluorescent staining for two PB resident proteins, hedls and DDX6 (Fig. 5A). After arsenite treatment, all HUVE cells displayed multiple cytoplasmic granules that stained with both antibodies, indicating robust PB formation. We then examined the ability of RhoA to deregulate PB formation using three chemical activators of RhoA (lysophosphatidic acid [LPA], nocodazole, and Rho activator II). After activating treatments, vector-transduced HUVE cells displayed a 2- to 4-fold reduction in the steady-state number of cells that contained normal PBs (Fig. 5B and C). Treated cells displayed strong parallel actin stress fibers, confirming RhoA activation (Fig. 3B and data not shown).
To test the effect of vGPCR expression on PB accretion, vGPCR, vGPCRR143A, or vector-transduced HUVE cells were fixed and PBs were detected by staining for the PB-resident protein hedls (20). Approximately two-thirds of control cells contained normal PBs at steady state, as did vGPCRR143A-expressing cells, while only one-third of untreated vGPCR-expressing cells stained positive for PBs, indicating that vGPCR expression caused a 2-fold reduction in the number of cells capable of PB formation. vGPCR expression also resulted in lower numbers of PB-containing cells after stress induction (data not shown) and after treatment with serum-free medium (Fig. 5D and F). vGPCR expression also caused significant stress fibers and massive morphological changes (Fig. 2A and 5E and F), two cellular processes regulated by RhoA activity. To determine if RhoA activation by vGPCR was responsible for its effect on PB equilibrium, vGPCR-expressing cells were treated with Botulinum C3 exotoxin, an irreversible and specific inhibitor of RhoA subfamily GTPases, and Y-27632, a chemical inhibitor of the downstream kinase, Rho-associated kinase (ROCK). C3 treatment of vGPCR-expressing cells decreased actin stress fiber formation and partially restored the steady-state level of PBs to that observed in control cells (Fig. 5D and F). However, even though inhibition of ROCK eliminated the appearance of actin stress fibers in vGPCR-expressing cells, it did not have any effect on PB balance (Fig. 5D and F). These data suggest that RhoA activation is responsible for altering the dynamics of both actin stress fibers and PBs but that ROCK plays no role in controlling PB equilibrium. Consistent with these findings, a previous study reported that overexpression of ROCK did not alter PB dynamics or ARE-RNA stabilization, while RhoA altered both (62). Taken together, these experiments provide solid evidence that vGPCR inhibits ARE-mRNA turnover, in part by altering PB accretion, and that activation of p38/MK2 and RhoA signaling pathways play important roles in this process.
Induction of KSHV lytic gene expression in endothelial cells eliminates cytoplasmic PBs.
vGPCR expression is restricted to the KSHV lytic cycle, with delayed-early kinetics (36). For this reason, we hypothesized that PB formation should be disrupted as vGPCR levels rise following reactivation from latency. To examine PB dynamics during KHSV infection, we employed a Dox-inducible KSHV producer cell line known as iSLK.219 (49). In this system, SLK endothelial cells were engineered to express RTA from a Dox-responsive promoter and subsequently infected with a recombinant KSHV known as rKSHV.219 that constitutively expresses a puromycin resistance gene and green fluorescent protein (GFP). Unlike many other KSHV-infected cell lines, iSLK.219 cells display tight control of latency, with no appreciable spontaneous lytic reactivation. The addition of Dox enforces expression of RTA and promotes reactivation from latency and expression of red fluorescent protein (RFP) from an RTA-responsive promoter. Thus, latently infected iSLK.219 cells are GFP positive, whereas RTA expression following the addition of Dox or a mixture of Dox and the HDAC inhibitor valproic acid (VPA) leads to lytic reactivation and robust expression of the RFP transgene.
Using this inducible system, we examined PB dynamics in latently and lytically infected cells. Robust RFP expression was observed 24 h after induction with Dox and VPA, indicating engagement of lytic replication (Fig. 6B). This correlated with PB disruption in the RFP-positive cells but not in the neighboring GFP-positive cells that fail to reactivate from latency (Fig. 6B). In latently infected iSLK.219 cells (no induction, Dox alone, or VPA alone), PB size and distribution were similar to control levels observed in other ECs (60 to 80% of cells contain normal PBs; see Fig. 5). Furthermore, PB size and distribution were unperturbed in the parental iSLK cell line following treatment with Dox and VPA, confirming that these drugs have no significant impact on PB dynamics (Fig. 6A). Taken together, these data indicate that robust initiation of the KSHV lytic cycle is required to impact PB dynamics and suggest that the accumulation of vGPCR during the lytic cycle may contribute to PB disruption.
Fig 6.

Induction of KSHV lytic gene expression in endothelial cells eliminates cytoplasmic PBs. Control iSLK cells (A) or iSLK.219 cells latently infected with rKSHV.219 (B) were either not treated or treated with Dox and/or valproic acid to induce RTA expression and reactivation from latency. Twenty-four hours after induction, cells were fixed and immunostained for PBs using anti-DDX6 antibody. Scale bars = 10 μm. GFP expression is driven by a constitutive CMV promoter and marks all infected cells, whereas RFP is driven by an RTA-responsive promoter and identifies cells supporting lytic KSHV infection (indicated by white arrows).
DISCUSSION
All viruses are obligate intracellular parasites that depend upon host translational machinery for the expression of viral gene products. For KSHV and a number of related herpesviruses, host shutoff serves as a mechanism to gain control over the translational machinery and rededicate it to the expression of viral gene products. The effects of host shutoff are dramatic, and yet it has become increasingly clear in recent years that it often fails to completely block host gene expression. KSHV host shutoff is mediated by the SOX protein (30), which promotes mRNA degradation (28) as well as the ability to relocalize PABPC1 to the nucleus, causing hyperadenylation and nuclear retention of nascent transcripts (38, 41). A detailed, transcriptome-wide analysis of host shutoff revealed that expression of most transcripts was sharply curtailed during KSHV lytic replication, but approximately 20% of host transcripts decline only slightly, and 2% are upregulated during infection (14). It remains unclear precisely how certain transcripts evade host shutoff, but transcripts that contain AREs are overrepresented among escapees during lytic KSHV replication. How might ARE-mRNAs escape host shutoff? Ectopic expression of SOX in 293T cells reveals that most ARE-mRNAs are not intrinsically resistant to SOX (17), suggesting that additional KSHV lytic gene products may play important roles in escape. Kaposin B has been shown to directly bind and activate MK2, resulting in the stabilization of ARE-mRNAs (44). Kaposin B expression is regulated by multiple promoter elements: a latent promoter directs low-level expression during latency, while an RTA-responsive lytic promoter directs potent upregulation of kaposin B expression during the lytic cycle. The impact of kaposin B on ARE-mRNA stabilization during the lytic cycle remains to be tested, but nevertheless, it has been hypothesized that kaposin B may facilitate the escape of certain ARE-mRNAs from host shutoff. Our data clearly demonstrates that vGPCR also possesses MK2-dependent ARE-mRNA-stabilizing activity. In light of these findings, we speculate that kaposin B and vGPCR, as well as other KSHV lytic gene products that stimulate p38/MK2 signaling (e.g., ORF49 [31]), may play a significant role in the posttranscriptional control of host gene expression during lytic KSHV replication.
How might vGPCR disrupt ARE-mRNA decay? In our working model (Fig. 7), vGPCR employs a “belt and suspenders” approach to negatively impact ARE-mRNA turnover via coordinate activation of p38/MK2 and RhoA. We show that vGPCR potently activates p38, consistent with previous reports, and further demonstrate activation of the p38 target MK2. Recent work has shown that vGPCR interacts with transforming growth factor beta (TGF-β)-activated kinase 1 (TAK1), recruiting it to the plasma membrane and causing lysine 63-linked polyubiquitination and kinase activation (9). TAK1 is known to phosphorylate and activate the MAPKKs, MKK3, and MKK6 (47), transducing a signal to p38, which results in MK2 activation and ARE-mRNA stabilization. At the same time, vGPCR associates with the Gα12/13 family of heterotrimeric G proteins (43, 57), which activate RhoA through the guanine nucleotide exchange factor p115RhoGEF (33, 37). Furthermore, there is intriguing evidence that MK2 may indirectly regulate RhoA via the phosphorylation of Hsp27 and the formation of active p-Hsp27/p115RhoGEF complexes (27), suggesting that vGPCR may activate RhoA via multiple pathways. Recent work implicated RhoA in regulating PB assembly and function (62), which inspired us to investigate the consequences of vGPCR expression on these processes. In our experimental system, vGPCR lowers basal levels of PBs. These effects can be partially reversed by inhibition of RhoA activity with C3 exotoxin. Together, these observations strongly implicate PB disruption as part of the mechanism of vGPCR-mediated ARE-mRNA stabilization and are consistent with the defective PB dynamics we observe during lytic KSHV replication. Intriguingly, RhoA has been shown to be activated at early times postinfection as a consequence of KSHV binding and entry into ECs (50, 55, 63), suggesting that PB dynamics may impact host gene expression at different stages of infection, even the earliest moments.
Fig 7.

Model of the mechanism of vGPCR-mediated ARE-mRNA stabilization. In this working model, vGPCR stimulates two signal transduction pathways that negatively impact ARE-mRNA decay. vGPCR has been reported to promote the phosphorylation and polyubiquitination of TAK1 (9), which initiates a signal transduction pathway involving activation of MKK3 and/or MKK6 (47), p38 MAPK, and the p38 substrate MK2. MK2 phosphorylates destabilizing ARE-binding proteins like tristetraprolin (TTP), generating binding sites for cytoplasmic 14-3-3 scaffolding proteins that disrupt recruitment of a complex of endo- and exonucleases known as the exosome (15, 39). In addition, MK2 phosphorylates the heat shock protein 27 (Hsp27), which has been shown to form a complex with p115GEF to activate RhoA (27). In response to a variety of stresses, including viral infection, eukaryotic translation initiation factor 2α (eIF2α) is phosphorylated, which prevents initiation by limiting the availability of eIF2-GTP-tRNAmet. Specific RNA binding proteins bind to these stalled translation preinitiation complexes and nucleate the formation of large cytoplasmic mRNP aggregates known as stress granules (SGs), in which translationally inactive transcripts are triaged and routed to sites of reinitiation or degradation (2). TTP has been reported to transport bound ARE-mRNAs to processing bodies (PBs), sites of translational repression, and mRNA degradation (35). vGPCR activates RhoA (43, 57), and recent work has shown that RhoA activation disrupts the stress-induced rearrangement of PBs and stabilizes ARE-mRNAs (62). We provide evidence that vGPCR impacts both MK2 activity and PB dynamics, thereby potently stabilizing ARE-mRNAs and increasing the production of proteins encoded by ARE-mRNAs (e.g., Cox-2, IL-6). These studies provide the first evidence for vGPCR-mediated reprogramming of host gene expression at the posttranscriptional level, which may facilitate secretion of angiogenic growth factors from lytically infected cells.
ARE-mRNA cargo is thought to be delivered to PBs via SGs, but the status of SGs and their dynamic interactions with PBs have not yet been investigated during KSHV lytic replication. Two lytic cycle proteins, vIRF2 and vIRF3, have been reported to inhibit PKR (11, 19) and may therefore negatively impact PKR-mediated SG formation during lytic infection. vGPCR has not been investigated for PKR inhibition, but the vGPCR homolog from Epstein-Barr virus (EBV), BILF1, inhibits PKR activation in COS-7 cells (6). In any case, even if lytic gene products suppress PKR, other eIF2α kinases may be competent to initiate SG formation during infection. Because KSHV relies on the cap-dependent translation machinery for the expression of viral proteins and SOX-mediated host shutoff would be predicted to severely limit the number of cytoplasmic host mRNAs that could be translationally stalled and subsequently nucleate SGs, any SGs that form during the lytic cycle would be expected to harbor significant numbers of stalled viral mRNAs. It will be interesting to test whether SGs form at any time during lytic KSHV replication and whether the stalled cargo is host, viral, or both. Furthermore, we speculate that during the stress of KSHV lytic replication, SGs may transiently form and sequester ARE-mRNAs. In this scenario, vGPCR-mediated disruption of PBs would prevent transfer of ARE-mRNA cargo from SGs, protecting these ARE-mRNAs and perhaps allowing them to be reinitiated later during infection. The precise fate of ARE-mRNAs in lytically infected cells remains to be elucidated, but clearly, a detailed investigation of SG and PB dynamics over the course of the KSHV lytic cycle is warranted.
Herein, we provide evidence for the selective stabilization of ARE-mRNAs by vGPCR and link this observation to known vGPCR signaling activities. This study focuses attention, for the first time, on potential contribution of vGPCR to posttranscriptional control of gene expression during lytic KSHV replication. The emerging picture is that the escape of certain pathogenetically important host gene products from host shutoff is likely the result of the cumulative efforts of several viral gene products. Doubtless, the mechanism will be complicated, and the control of ARE-mRNA turnover will be only part of the story. Indeed, even though ARE-mRNAs are overrepresented in escapees, they do not all escape shutoff. Much work remains to be done in identifying additional cis-acting elements and trans-acting factors that regulate the escape of ARE-mRNAs from host shutoff.
ACKNOWLEDGMENTS
We thank members of the McCormick lab for helpful discussions and critical manuscript review. We thank Don Ganem (Novartis Institutes of Biomedical Research), Matthias Gaestel (Hannover Medical School), and Garry Nolan (Stanford University) for reagents and Steve Whitefield for his assistance with fluorescence microscopy.
This study was supported by an operating grant from the Canadian Institutes of Health Research (CIHR-MOP-84554). Three trainees were supported by awards administered by the Beatrice Hunter Cancer Research Institute; an award to J.A.C. was funded by The Terry Fox Foundation Strategic Health Research Training Program in Cancer Research at CIHR; and awards to D.P.C. and B.P.J. were funded by The Canadian Cancer Society, Nova Scotia Division.
Footnotes
Published ahead of print 13 June 2012
REFERENCES
- 1. Aizer A, et al. 2008. The dynamics of mammalian P body transport, assembly, and disassembly in vivo. Mol. Biol. Cell 19:4154–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson P, Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33:141–150 [DOI] [PubMed] [Google Scholar]
- 3. Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. 1997. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 385:347–350 [DOI] [PubMed] [Google Scholar]
- 4. Bais C, et al. 1998. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391:86–89 [DOI] [PubMed] [Google Scholar]
- 5. Bakheet T, Williams BR, Khabar KS. 2006. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 34:D111–D114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beisser PS, et al. 2005. The Epstein-Barr virus BILF1 gene encodes a G protein-coupled receptor that inhibits phosphorylation of RNA-dependent protein kinase. J. Virol. 79:441–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. 2006. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125:1111–1124 [DOI] [PubMed] [Google Scholar]
- 8. Boshoff C, et al. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1:1274–1278 [DOI] [PubMed] [Google Scholar]
- 9. Bottero V, et al. 2011. Phosphorylation and polyubiquitination of transforming growth factor beta-activated kinase 1 are necessary for activation of NF-kappaB by the Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. J. Virol. 85:1980–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brengues M, Teixeira D, Parker R. 2005. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 310:486–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burysek L, Pitha PM. 2001. Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J. Virol. 75:2345–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 13. Cesarman E, et al. 1996. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi's sarcoma and malignant lymphoma. J. Virol. 70:8218–8223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chandriani S, Ganem D. 2007. Host transcript accumulation during lytic KSHV infection reveals several classes of host responses. PLoS One 2:e811 doi:10.1371/journal.pone.0000811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen CY, et al. 2001. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 107:451–464 [DOI] [PubMed] [Google Scholar]
- 16. Chen CY, Shyu AB. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20:465–470 [DOI] [PubMed] [Google Scholar]
- 17. Clyde K, Glaunsinger BA. 2011. Deep sequencing reveals direct targets of gammaherpesvirus-induced mRNA decay and suggests that multiple mechanisms govern cellular transcript escape. PLoS One 6:e19655 doi:10.1371/journal.pone.0019655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corcoran JA, Khaperskyy DA, McCormick C. 2011. Assays for monitoring viral manipulation of host ARE-mRNA turnover. Methods 55:172–181 [DOI] [PubMed] [Google Scholar]
- 19. Esteban M, et al. 2003. The latency protein LANA2 from Kaposi's sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J. Gen. Virol. 84:1463–1470 [DOI] [PubMed] [Google Scholar]
- 20. Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. 2005. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol. Cell 20:905–915 [DOI] [PubMed] [Google Scholar]
- 21. Flore O, et al. 1998. Transformation of primary human endothelial cells by Kaposi's sarcoma-associated herpesvirus. Nature 394:588–592 [DOI] [PubMed] [Google Scholar]
- 22. Franks TM, Lykke-Andersen J. 2007. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev. 21:719–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frevel MA, et al. 2003. p38 mitogen-activated protein kinase-dependent and -independent signaling of mRNA stability of AU-rich element-containing transcripts. Mol. Cell. Biol. 23:425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120:939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gao SJ, Deng JH, Zhou FC. 2003. Productive lytic replication of a recombinant Kaposi's sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 77:9738–9749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garcia MA, Meurs EF, Esteban M. 2007. The dsRNA protein kinase PKR: virus and cell control. Biochimie 89:799–811 [DOI] [PubMed] [Google Scholar]
- 27. Garcia MC, et al. 2009. Arachidonic acid stimulates cell adhesion through a novel p38 MAPK-RhoA signaling pathway that involves heat shock protein 27. J. Biol. Chem. 284:20936–20945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glaunsinger B, Chavez L, Ganem D. 2005. The exonuclease and host shutoff functions of the SOX protein of Kaposi's sarcoma-associated herpesvirus are genetically separable. J. Virol. 79:7396–7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glaunsinger B, Ganem D. 2004. Highly selective escape from KSHV-mediated host mRNA shutoff and its implications for viral pathogenesis. J. Exp. Med. 200:391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glaunsinger B, Ganem D. 2004. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 13:713–723 [DOI] [PubMed] [Google Scholar]
- 31. Gonzalez CM, et al. 2006. Identification and characterization of the Orf49 protein of Kaposi's sarcoma-associated herpesvirus. J. Virol. 80:3062–3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Invest. 113:124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hart MJ, et al. 1998. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science 280:2112–2114 [DOI] [PubMed] [Google Scholar]
- 34. Ho HH, Ganeshalingam N, Rosenhouse-Dantsker A, Osman R, Gershengorn MC. 2001. Charged residues at the intracellular boundary of transmembrane helices 2 and 3 independently affect constitutive activity of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. J. Biol. Chem. 276:1376–1382 [DOI] [PubMed] [Google Scholar]
- 35. Kedersha N, et al. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169:871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kirshner JR, Staskus K, Haase A, Lagunoff M, Ganem D. 1999. Expression of the open reading frame 74 (G-protein-coupled receptor) gene of Kaposi's sarcoma (KS)-associated herpesvirus: implications for KS pathogenesis. J. Virol. 73:6006–6014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kozasa T, et al. 1998. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science 280:2109–2111 [DOI] [PubMed] [Google Scholar]
- 38. Kumar GR, Glaunsinger BA. 2010. Nuclear import of cytoplasmic poly(A) binding protein restricts gene expression via hyperadenylation and nuclear retention of mRNA. Mol. Cell. Biol. 30:4996–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lai WS, et al. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 19:4311–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lebreton A, Tomecki R, Dziembowski A, Seraphin B. 2008. Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature 456:993–996 [DOI] [PubMed] [Google Scholar]
- 41. Lee YJ, Glaunsinger BA. 2009. Aberrant herpesvirus-induced polyadenylation correlates with cellular messenger RNA destruction. PLoS Biol. 7:e1000107 doi:10.1371/journal.pbio.1000107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martin DF, et al. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N. Engl. J. Med. 340:1063–1070 [DOI] [PubMed] [Google Scholar]
- 43. Martin MJ, et al. 2007. The Galpha12/13 family of heterotrimeric G proteins and the small GTPase RhoA link the Kaposi sarcoma-associated herpes virus G protein-coupled receptor to heme oxygenase-1 expression and tumorigenesis. J. Biol. Chem. 282:34510–34524 [DOI] [PubMed] [Google Scholar]
- 44. McCormick C, Ganem D. 2005. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307:739–741 [DOI] [PubMed] [Google Scholar]
- 45. Montaner S, et al. 2003. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell 3:23–36 [DOI] [PubMed] [Google Scholar]
- 46. Montaner S, Sodhi A, Pece S, Mesri EA, Gutkind JS. 2001. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 61:2641–2648 [PubMed] [Google Scholar]
- 47. Moriguchi T, et al. 1996. A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J. Biol. Chem. 271:13675–13679 [DOI] [PubMed] [Google Scholar]
- 48. Munshi N, Ganju RK, Avraham S, Mesri EA, Groopman JE. 1999. Kaposi's sarcoma-associated herpesvirus-encoded G protein-coupled receptor activation of c-Jun amino-terminal kinase/stress-activated protein kinase and lyn kinase is mediated by related adhesion focal tyrosine kinase/proline-rich tyrosine kinase 2. J. Biol. Chem. 274:31863–31867 [DOI] [PubMed] [Google Scholar]
- 49. Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Naranatt PP, Krishnan HH, Smith MS, Chandran B. 2005. Kaposi's sarcoma-associated herpesvirus modulates microtubule dynamics via RhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to deliver its DNA to the nucleus. J. Virol. 79:1191–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parker R, Sheth U. 2007. P bodies and the control of mRNA translation and degradation. Mol. Cell 25:635–646 [DOI] [PubMed] [Google Scholar]
- 52. Ronkina N, et al. 2007. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol. Cell. Biol. 27:170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schmid M, Jensen TH. 2008. The exosome: a multipurpose RNA-decay machine. Trends Biochem. Sci. 33:501–510 [DOI] [PubMed] [Google Scholar]
- 55. Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. 2004. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 78:4207–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shelby BD, et al. 2007. Kaposi's sarcoma associated herpesvirus G-protein coupled receptor activation of cyclooxygenase-2 in vascular endothelial cells. Virol. J. 4:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shepard LW, et al. 2001. Constitutive activation of NF-kappa B and secretion of interleukin-8 induced by the G protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus involve G alpha(13) and RhoA. J. Biol. Chem. 276:45979–45987 [DOI] [PubMed] [Google Scholar]
- 58. Sodhi A, et al. 2000. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 60:4873–4880 [PubMed] [Google Scholar]
- 59. Soulier J, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 60. Stoecklin G, et al. 2004. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 23:1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sweet TJ, Boyer B, Hu W, Baker KE, Coller J. 2007. Microtubule disruption stimulates P-body formation. RNA 13:493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Takahashi S, et al. 2011. RhoA activation participates in rearrangement of processing bodies and release of nucleated AU-rich mRNAs. Nucleic Acids Res. 39:3446–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Veettil MV, et al. 2006. RhoA-GTPase facilitates entry of Kaposi's sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J. Virol. 80:11432–11446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Williams BR. 1999. PKR: a sentinel kinase for cellular stress. Oncogene 18:6112–6120 [DOI] [PubMed] [Google Scholar]
- 65. Winzen R, et al. 1999. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18:4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]