

Abstract
PB1-F2 is a small, 87- to 90-amino-acid-long protein encoded by the +1 alternate open reading frame of the PB1 gene of most influenza A virus strains. It has been shown to contribute to viral pathogenicity in a host- and strain-dependent manner, and we have previously discovered that a serine at position 66 (66S) in the PB1-F2 protein increases virulence of the 1918 and H5N1 pandemic viruses. Recently, we have shown that PB1-F2 inhibits the induction of type I interferon (IFN) at the level of the MAVS adaptor protein. However, the molecular mechanism for the IFN antagonist function of PB1-F2 has remained unclear. In the present study, we demonstrated that the C-terminal portion of the PB1-F2 protein binds to MAVS in a region that contains the transmembrane domain. Strikingly, PB1-F2 66S was observed to bind to MAVS more efficiently than PB1-F2 66N. We also tested the effect of PB1-F2 on the IFN antagonist functions of the polymerase proteins PB1, PB2, and PA and observed enhanced IFN inhibition by the PB1 and PB2 proteins in combination with PB1-F2 but not by the PA protein. Using a flow cytometry-based assay, we demonstrate that the PB1-F2 protein inhibits MAVS-mediated IFN synthesis by decreasing the mitochondrial membrane potential (MMP). Interestingly, PB1-F2 66S affected the MMP more efficiently than wild-type PB1-F2. In summary, the results of our study identify the molecular mechanism by which the influenza virus PB1-F2 N66S protein increases virulence.
INTRODUCTION
Influenza is a global health concern due to its potential to cause pandemics which could affect millions of lives. Studies of the 1918 pandemic influenza virus, which was responsible for 50 million deaths worldwide within a short period of time, revealed that the PB1-F2 protein contributes to its severe pathogenicity (5, 17).
PB1-F2 is a small, approximately 90-amino-acid protein that is expressed from the +1 alternate open reading frame (ORF) of the PB1 gene of most influenza A virus strains. Several mechanisms by which PB1-F2 contributes to virulence have been proposed. Initially, a proapoptotic property was observed for the PB1-F2 protein, which was thought to occur specifically in immune cells in order to evade the host immune response (3). Further studies showed that PB1-F2 localizes to the mitochondria (7, 29), where it decreases the mitochondrial membrane potential (7) and interacts with the VDAC1 and ANT3 proteins (31) or itself to form pores (2) and thus induces cell death. Other reports describe a proinflammatory function for PB1-F2, which is thought to cause severe immunopathology such as seen in patients infected with avian H5N1 influenza virus (16, 17).
We have previously identified a single residue in PB1-F2 which affects the severity of the 1918 pandemic influenza virus and an H5N1 influenza virus (5). Specifically, a serine (S) at position 66 was associated with high pathogenicity, whereas an asparagine (N) at that position resulted in decreased virulence. Microarray analyses on whole mouse lung homogenates showed a decreased induction of interferon (IFN)-regulated genes in animals infected with the PB1-F2 N66S-expressing virus (4). The interferon antagonist function of the PB1-F2 protein was further characterized in vitro, and we demonstrated that PB1-F2 affects the induction of IFN at the level of the MAVS adaptor protein and that the N66S point mutation in PB1-F2 increases the IFN antagonist property (28).
Viral pathogens are sensed by the host cell via three arms of the innate immune response: the Nod-like receptor (NLR) pathway, the Toll-like receptor (TLR) pathway, and the RIG-I-like receptor (RLR) cascade. Influenza virus generates viral RNA species during replication that are sensed by RIG-I in the cytoplasm, which then undergoes a conformational change to expose its N-terminal caspase activation and recruitment domains (CARDs) for subsequent interaction with the CARD of the mitochondrial antiviral signaling protein (MAVS). MAVS (also known as IPS-1, Cardif, or VISA) is located primarily in the outer membrane of mitochondria and is an essential mediator for the production of type I IFNs by activating the interferon regulatory factor (IRF) and nuclear factor κB (NF-κB) transcription factors to drive expression of IFN molecules. Mice deficient in MAVS are highly susceptible to viral infection and fail to mount a robust IFN response (12, 25).
The mechanism by which MAVS propagates the signal for the induction of IFN is still being investigated. It has been demonstrated that MAVS oligomerizes via its transmembrane domain, providing a docking platform for downstream adaptor proteins essential for the induction of IFN (1, 26). Recent reports showed that MAVS forms higher-order complexes which rearrange to create lower-order complexes upon activation of the antiviral response (30) and even induce prion-like aggregates (9). In addition, it was demonstrated that an intact mitochondrial membrane potential is essential for the rearrangement of MAVS complexes (11).
Viral proteins have developed mechanisms to evade the host innate immune response. Since MAVS is a vital protein in the IFN production pathway, it is not surprising that it is targeted by several viruses to inhibit the induction of IFN. The most prominent IFN antagonist to affect MAVS function is the NS3/4A serine protease of hepatitis C virus (HCV). It cleaves MAVS at the transmembrane domain, which leads to the loss of its mitochondrial localization and results in the subsequent inhibition of IFN synthesis (6, 13–15, 18). Proteins of influenza virus have also been shown to inhibit the induction of IFN at the level of the MAVS adaptor protein. The well-characterized IFN antagonist NS1 was shown to form a complex with RIG-I and MAVS and inhibit an IFN-β reporter when stimulated with MAVS (19). In addition, the polymerase proteins PB1, PB2, and PA have been demonstrated to have anti-IFN properties by binding to MAVS (8, 10).
Here we report that PB1-F2 binds to a region of MAVS that contains the transmembrane domain via its C-terminal region and decreases the mitochondrial membrane potential (MMP), which is essential for MAVS-mediated induction of IFN (11). Furthermore, we show that PB1-F2 enhances the anti-IFN effects of the PB1 and PB2 polymerase proteins and that the N66S mutation in PB1-F2 increases the efficiency of binding to the MAVS adaptor protein and enhances the dissipation of the MMP. Taking our findings together, we provide a molecular mechanism by which the PB1-F2 protein contributes to the pathogenicity of influenza viruses. Furthermore, we describe a novel strategy by which viral proteins can inhibit the induction of IFN by the host, namely, by affecting the mitochondrial membrane potential.
(This paper was submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, Mount Sinai School of Medicine.)
MATERIALS AND METHODS
Cell lines and antibodies.
293T cells were obtained from ATCC (Manassas, VA) and maintained in Dulbecco modified Eagle medium (DMEM) culture medium (Gibco, Invitrogen, San Diego, CA) supplemented with 10% fetal calf serum (HyClone, South Logan, UT) and penicillin/streptomycin (Gibco, Invitrogen). Monoclonal mouse or rabbit antibodies to actin, FLAG, and hemagglutinin (HA) were obtained from Sigma-Aldrich (St. Louis, MO). PB1-F2 protein levels were detected using polyclonal rabbit serum, which has been described previously (31). The polyclonal rabbit MAVS antibody was obtained from Enzo Life Sciences (Plymouth Meeting, PA). The rabbit anti-poly(ADP-ribose) polymerase (anti-PARP) antibody was purchased from Roche Applied Science (Indianapolis, IN). The rabbit antibody against the mitochondrial Tom20 protein was obtained from Santa Cruz Biotechnologies (Santa Cruz, CA). The mouse monoclonal antibody against NP (28D8) was generated in our laboratory.
Rescue of recombinant influenza viruses.
The recombinant A/Puerto Rico/8/1934 (PR8; H1N1) viruses used in this study were rescued as described previously (28). The PR8 PB1-F2 knockout virus was generated by mutating three possible start codons and introducing two stop codons in the PB1-F2 ORF. For this purpose, nucleotide positions 120, 234, and 255 were mutated from T to C in the PB1 gene, and nucleotide positions 153 and 222 were changed from C to G. The PR8 virus expressing an N-terminally truncated form of PB1-F2 (aa 1 to 11) was generated by leaving the first start codon of the PB1-F2 ORF intact (with no mutation at position 120 in the PB1 ORF) in the background of the PB1 PB1-F2 knockout plasmid. All of the mutations performed did not alter the PB1 protein sequence. The PB1-F2 knockout virus shows increased levels of the N40 protein, which may impact the induction of IFN, as discussed previously (28). We thus generated a virus expressing PB1-F2 aa 1 to 11, which has the same N40 protein levels as the wild-type (WT) PB1-F2-expressing virus.
Infection of cells with influenza viruses.
293T cells were seeded onto polylysine-coated tissue culture plates (BD Biosciences, Sparks, MD) and infected with the recombinant PR8 influenza viruses at a multiplicity of infection (MOI) of 2. Cells were washed with phosphate-buffered saline (PBS), virus was diluted in PBS supplemented with 0.3% bovine serum albumin (BSA) and penicillin-streptomycin, and cells were overlaid with the virus preparation for 1 h at 37°C. Cells were subsequently washed, and growth medium was added.
Expression plasmids and cloning.
The pCAGGS vector has been described previously (20). The expression constructs for FLAG-tagged WT and N66S PB1-F2 (PR8), FLAG-tagged RIG-I N, HA-tagged MAVS, FLAG-tagged PB1, FLAG-tagged PB2, FLAG-tagged PA, and FLAG-tagged NP have been described elsewhere (19, 21–23, 28). The PB1-F2 truncation constructs have been described previously (31). The MAVS truncation constructs were generated by PCR amplification and standard recombinant DNA techniques.
Reporter assays.
To determine the induction of type I IFN, 293T cells were transfected with an IFN-β reporter construct that drives the expression of the firefly luciferase and a control Renilla construct using Lipofectamine 2000 (Invitrogen). Cells were lysed at 24 h posttransfection using the lysis buffer of the dual-luciferase assay kit according to the manufacturer's instructions (Promega, Madison, WI). The fold induction was calculated as the ratio of the values for RIG-I N-stimulated and unstimulated samples.
Western blot analysis.
Cells were lysed in urea buffer (6 M urea, 2 M β-mercaptoethanol, 4% SDS), and samples were analyzed for protein expression as described previously (28).
Co-IP studies.
293T cells were transfected with the indicated plasmid DNA using Lipofectamine 2000 (Invitrogen) and lysed the following day with a nondenaturing coimmunoprecipitation (co-IP) lysis buffer containing 50 mM Tris-HCl (pH 7.4), 300 mM NaCl (aqueous), 5 mM EDTA, 0.02% sodium azide, 1% Triton X-100, and complete protease inhibitor cocktail (Roche, Basel, Switzerland). Cell lysates were sonicated 3 times at output level 3.0 for 5 s, centrifuged at 13,000 rpm for 15 min at 4°C, and precleared with protein G-agarose beads (Roche) for 4 h at 4°C. The immunoprecipitation was performed with 1 μg of the indicated antibody at 4°C overnight. The following day, protein G beads were added and left for 2 h to precipitate the protein complexes, and samples were washed 4 times with lysis buffer and 2 times with PBS. Samples were analyzed via SDS-PAGE as described above.
Subcellular fractionation.
To isolate mitochondrial and cytosolic fractions from cell preparations, the mitochondrial isolation kit for cultured cells was used (Thermo Scientific, Rockford, IL) according to the manufacturer's instructions. To obtain mitochondrial fractions with fewer lysosomal and peroxisomal contaminants, the supernatant samples were centrifuged at 3,000 × g for 15 min after addition of the mitochondrial isolation reagent. Whole-cell extracts and cytosolic and mitochondrial fractions were analyzed using SDS-PAGE as described above.
TMRE staining for mitochondrial membrane potential.
Cells were stained with a 20 nM preparation of the MitoPT tetramethylrhodamine ethyl ester (TMRE) potentiometric dye according to the manufacturer's instructions (ImmunoChemistry Technologies, Bloomington, MN). Flow cytometry was performed on a BD LSR II flow cytometer using FACSDiva software (BD Biosciences). As a positive control, cells were treated with the uncoupling reagent carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (ImmunoChemistry) at 50 μM for 16 h. The data were analyzed using FlowJo software (Tree Star) and represented as percentage of maximum in the histogram overlays.
Caspase 3 activity assay.
The activation of caspase 3 enzymes in transfected cells was measured using a fluorometric assay according to the manufacturer's instructions (Roche). As a positive control, cells were treated with staurosporine at 1 μM for 12 h. The samples were analyzed in a plate reader (Beckman Coulter DTX 880 instrument) with excitation and emission wavelengths of 400 and 505 nm, respectively.
RESULTS
PB1-F2 binds to MAVS via its C-terminal region, and the N66S mutation confers increased binding efficiency.
In our previous study, we showed that the PB1-F2 protein affects the induction of type I IFN at the level of the MAVS adaptor protein (28). Confocal microscopy studies revealed a striking colocalization of the PB1-F2 and MAVS proteins at the mitochondria (28), prompting us to investigate whether PB1-F2 and MAVS interact with each other. To test this, we transfected 293T cells with MAVS-HA and HA-PB1-F2-green fluorescent protein (GFP) constructs. As can be seen in Fig. 1A, MAVS-HA was pulled down with full-length PB1-F2 and the C-terminal region (aa 38 to 87) of PB1-F2 but not with the N-terminal region (aa 1 to 37). Interestingly, the N66S mutation in both the full-length and C-terminal regions of PB1-F2 lead to an increased binding to MAVS (Fig. 1A). These findings correlate with our previously published results that identify the C-terminal region of PB1-F2 as the “business end” for the IFN antagonist activity and show an increased anti-IFN effect of the PB1-F2 N66S protein (28). To confirm the coimmunoprecipitation (co-IP) data in the context of viral infection, we expressed MAVS-HA in 293T cells and infected the cells 24 h later with recombinant PR8 viruses expressing WT PB1-F2, PB1-F2 N66S, or a truncated nonfunctional form of PB1-F2 (aa 1 to 11). We also infected cells with a PR8 virus that is completely knocked out for PB1-F2 expression. The decreased induction of interferon-stimulated genes by an influenza virus expressing the N66S form of PB1-F2 has been shown in vivo in a previous study (4). To examine the molecular mechanism of this observation in in vitro systems, we chose to use the A/Puerto Rico/8/1934 (H1N1) influenza virus strain due to the availability of reagents (such as antibodies and expression plasmids) for this virus strain. The results shown in Fig. 1B indicate that MAVS is pulled down more efficiently by PB1-F2 N66S in virus-infected cells, even though its protein expression level is lower than that of WT PB1-F2, as observed previously (28). To exclude the possibility that mitochondria are pulled down in the co-IPs, we blotted for Tom20, a mitochondrial membrane protein. We were unable to detect the Tom20 protein in the IP samples, in contrast to the case for whole-cell extract preparations, which show abundant levels of the Tom20 protein (Fig. 1A and B). In summary, these data indicate that PB1-F2 interacts with MAVS and that the N66S point mutation in the PB1-F2 protein leads to stronger binding capability.
Fig 1.

PB1-F2 binds to MAVS via its C-terminal region, and the N66S mutation increases binding efficiency. (A) 293T cells were transfected with a MAVS-HA expression construct and plasmids expressing the indicated proteins. Cell lysates were collected 24 h later for immunoprecipitation using a monoclonal GFP antibody, and the samples were analyzed by Western blotting. The Western blot shown is a representative of at least 5 independent experiments. (B) 293T cells were transfected with a MAVS-HA expression construct and infected with recombinant PR8 viruses expressing the indicated form of the PB1-F2 protein 24 h later. Coimmunoprecipitation and Western blot analyses were performed 12 h postinfection as described for panel A. Whole-cell extract (WCE) from cells infected with the PR8 WT virus was added as a control for the Tom20 antibody. The Western blot shown is a representative of three independent experiments.
PB1-F2 enhances the anti-IFN effect of the PB1 and PB2 polymerase proteins.
Recent reports describe an IFN antagonism function of the PB1, PB2, and PA polymerase proteins of influenza virus caused by binding to the MAVS protein (8, 10). We thus examined the relationship of the polymerase and PB1-F2 proteins regarding their anti-IFN properties in an IFN-β reporter assay. As shown in Fig. 2A and B, coexpression of PB1 or PB2 and PB1-F2 inhibited RIG-I N-induced reporter activation more efficiently than PB1 or PB2 alone. In contrast, the NP protein did not affect the IFN inhibitory activity of the polymerase proteins (Fig. 2A and B). We used NP as a negative control because we have previously observed that it does not affect the induction of IFN (28). Of note, the PB1-F2 N66S protein was more efficient in enhancing the IFN antagonistic effect of the PB1 and PB2 proteins (Fig. 2A and B). Surprisingly, PB1-F2 did not enhance the IFN inhibitory function of the PA polymerase protein (Fig. 2C). Even when we titrated down the amounts of PA to result in approximately 50% inhibition of the reporter, the PB1-F2 proteins did not exert an additional effect on the IFN antagonist function of the PA protein (data not shown). The expression levels of the epitope-tagged polymerase proteins were comparable to the protein levels seen during influenza virus infection as determined in Western blot analyses using monoclonal antibodies against the PB1, PB2, or PA protein generated in our laboratory (data not shown).
Fig 2.
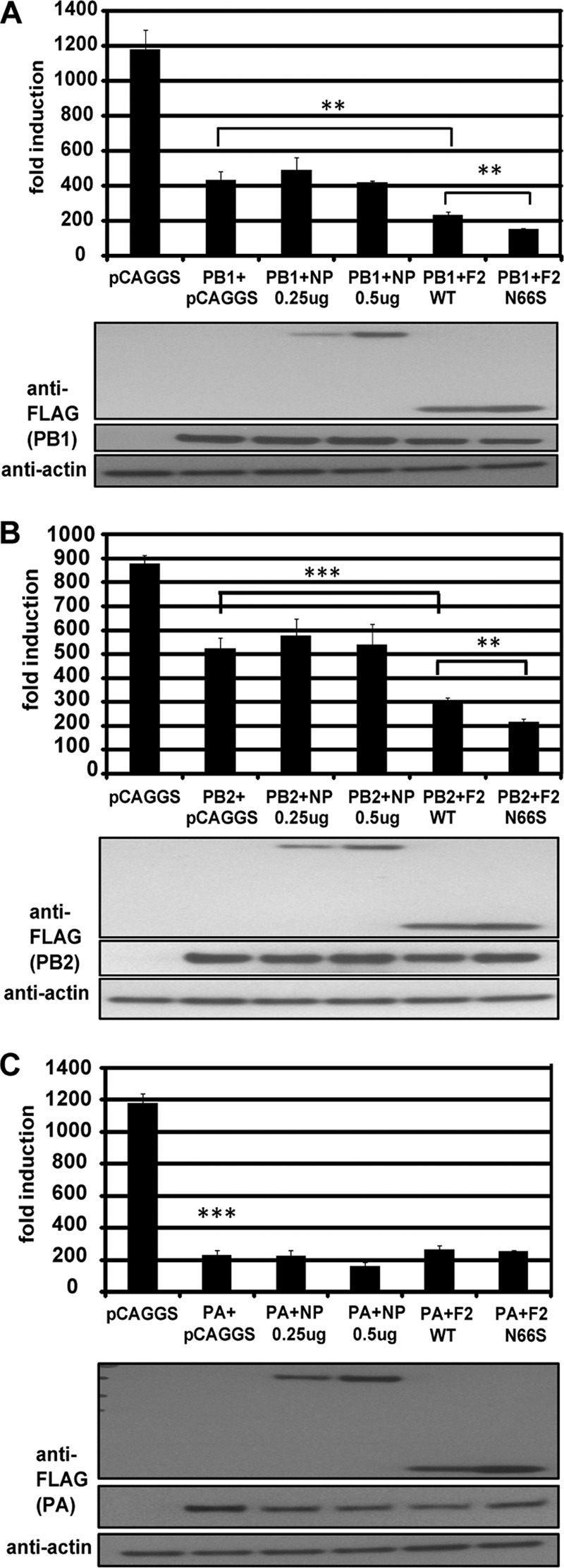
PB1-F2 enhances the anti-IFN effect of the PB1 and PB2 polymerase proteins. 293T cells were transfected with an IFN-β firefly luciferase reporter, a Renilla reporter, and a RIG-I N construct. In addition, the indicated plasmid combinations were transfected. At 24 h posttransfection, lysates were harvested for luminometry and Western blot analyses. The unit on the y axis indicates fold induction of the IFN-β reporter over that with mock transfection (unstimulated, i.e., no RIG-I N transfection). The Western blot panels show protein expression levels in the indicated samples. All data represent means ± standard deviations from one representative experiment (n = 3). Statistical significance was determined using Student's t test. **, P < 0.01; ***, P < 0.001. All data shown are representatives of at least three independent experiments.
PB1-F2 WT and N66S proteins localize to mitochondria with similar efficiencies.
Since position 66 lies within the optimal mitochondrial targeting sequence of the PB1-F2 protein, we investigated whether an N66S point mutation would alter the mitochondrial localization. To address this, we performed a subcellular fractionation assay and quantified the protein levels of the PB1-F2 proteins in whole-cell extracts, cytoplasmic fractions, and mitochondrial extracts with or without RIG-I N stimulation. To ensure successful fractionation with minimal contamination, we blotted for MAVS as a marker for the mitochondrial fraction and for tubulin as a marker for the cytosolic fraction. As shown in Fig. 3A and B, the mitochondrial and cytosolic fractions were free of contaminants from the other respective fraction. Furthermore, we observed that levels of the PB1-F2 WT and N66S proteins were equal in all fractions and under both stimulation conditions. Interestingly, the MAVS band pattern was altered upon RIG-I N stimulation and appeared to be different in the whole-cell and mitochondrial extracts, but the significance of this observation is uncertain. The multiple bands may be posttranslationally modified forms of MAVS and MAVS isoforms that differ in their subcellular localization and that may be regulated differentially upon RIG-I stimulation. Overall, these data indicate that the N66S point mutation does not affect the efficiency of localization of the PB1-F2 protein to the mitochondria upon stimulation of the IFN production pathway.
Fig 3.

PB1-F2 WT and N66S proteins localize to mitochondria with similar efficiencies. 293T cells were transfected with PB1-F2 WT or N66S expression plasmids and a RIG-I N expression construct (B) or an empty vector control (A). Twenty-four hours later, cells were collected for subcellular fractionation. In addition, samples were taken for whole-cell extracts (WCE) by lysing the cells with urea buffer. WCE, cytosolic, and mitochondrial fractions were analyzed via Western blot analysis. The Western blots shown are representative of three independent experiments.
PB1-F2 interacts with a portion of MAVS that contains the transmembrane domain.
Three functional domains have been identified in the MAVS adaptor protein: the CARD-like domain, a proline-rich domain, and a C-terminal transmembrane region (24). To understand the functional consequence of the PB1-F2–MAVS interaction, we generated MAVS truncation constructs to identify the functional domain of MAVS with which PB1-F2 interacts. We generated constructs expressing aa 1 to 173 or aa 1 to 153, which contain the CARD and proline-rich domains, aa 103 to 540, which contains the proline-rich and transmembrane domains, and aa 173 to 540, which contains the transmembrane domain (aa 514 to 535) only (Fig. 4A). We made both aa 1 to 153 and aa 1 to 173 MAVS truncations to include or exclude the binding region for TRAF6 (aa 153 to 158) and due to different reports on the mapping of the proline-rich region (aa 103 to 153 and aa 103 to 173). We performed coimmunoprecipitation assays with these MAVS constructs and HA-PB1-F2 N66S-GFP and found that PB1-F2 pulled down full-length MAVS and aa 173 to 540 and aa 103 to 540 of MAVS (Fig. 4B). These data show that PB1-F2 interacts with a region of MAVS that contains the transmembrane domain among the three functional domains known.
Fig 4.
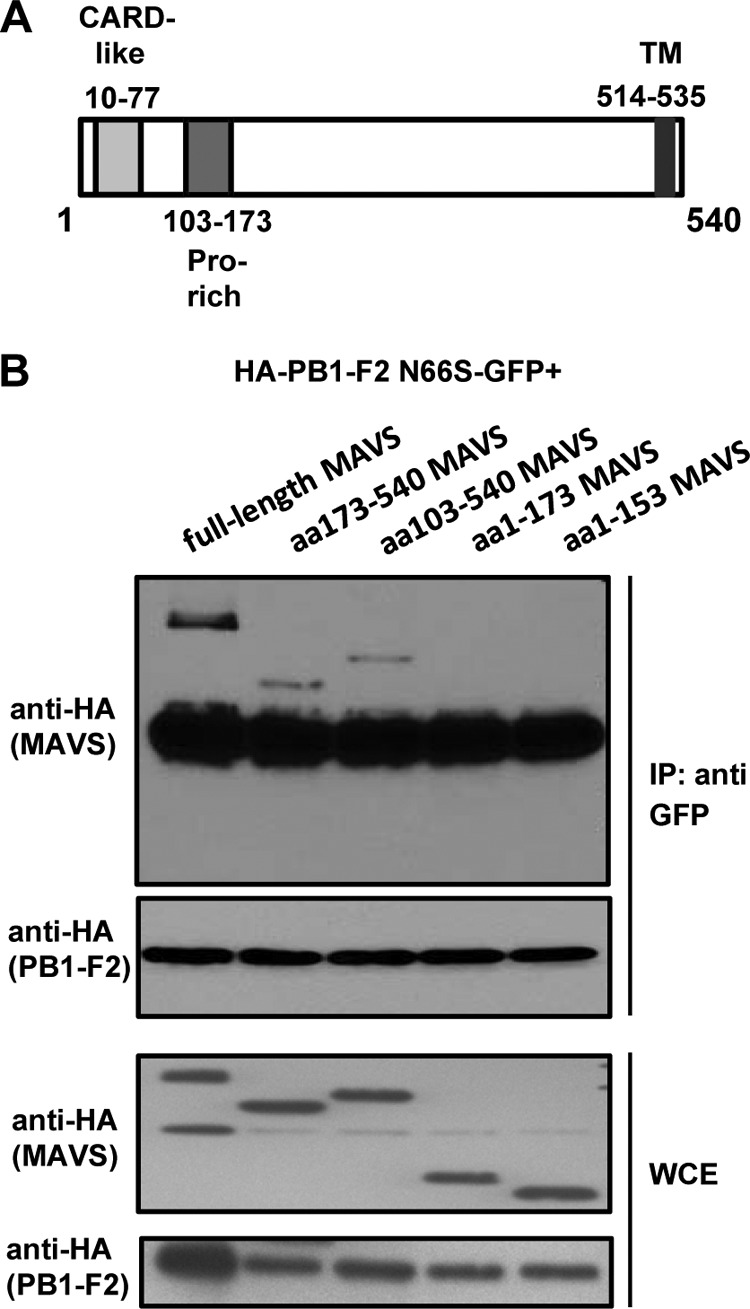
PB1-F2 interacts with a portion of MAVS that contains the transmembrane domain. (A) Schematic of the MAVS adaptor protein and the functional domains: CARD-like domain (aa 10 to 77), proline-rich domain (aa 103 to 173) and transmembrane (TM) domain (aa 514 to 535). (B) 293T cells were transfected with the HA-PB1-F2 N66S-GFP expression plasmid and plasmids expressing the indicated HA-tagged MAVS truncation form or full-length MAVS. Immunoprecipitation was performed using a monoclonal GFP antibody, and samples were analyzed via Western blotting. The two lower blots show whole-cell extract (WCE) preparations. The bands beneath the MAVS proteins in all IP samples are PB1-F2 protein bands upon longer exposure. The Western blots shown are representative of three independent experiments.
PB1-F2 decreases the mitochondrial membrane potential upon RIG-I N and MAVS stimulation.
It has been recently reported that the mitochondrial membrane potential (MMP) is essential for MAVS-mediated IFN induction (11). PB1-F2 has been shown to localize to mitochondria, which prompted us to investigate whether PB1-F2 affects the MMP in the presence of RIG-I N or MAVS. To test this, we transfected 293T cells with HA-PB1-F2-GFP constructs used in the coimmunoprecipitation assays (Fig. 1A) and quantified the MMP using the potentiometric dye tetramethylrhodamine ethyl ester (TMRE), which accumulates in the membrane of healthy mitochondria but diffuses into the cytoplasm upon loss of the MMP. We compared the TMRE signals in cells expressing the GFP-tagged PB1-F2 constructs (GFP hi) and cells expressing small amounts of or no GFP-PB1-F2 proteins (GFP lo). Cells expressing the N-terminal portion of PB1-F2 did not show a loss in the MMP, even when the N-terminally truncated PB1-F2 peptide was fused to a mitochondrial targeting sequence (MTS) (Fig. 5A). The C-terminally truncated and full-length PB1-F2 proteins, in contrast, decreased the TMRE signal, which indicates a loss of the MMP (Fig. 5B and C). Of note, the N66S mutation enhanced the decrease in the MMP by approximately 2-fold (Fig. 5B to D), which correlates with the 2-fold enhancement of the anti-IFN activity of PB1-F2 by the N66S mutation (28). The ability of the PB1-F2 proteins to decrease the MMP was also observed with MAVS stimulation (Fig. 5D). To demonstrate that the decrease in MMP is not due to apoptosis in cells expressing PB1-F2, we tested whether the PB1-F2 proteins induce apoptosis upon stimulation with RIG-I N. We found that there was no caspase 3 activation or poly(ADP-ribose) polymerase (PARP) cleavage detectable in RIG-I N- and PB1-F2-transfected cells, in contrast to the case for cells treated with the apoptosis inducer staurosporine (Fig. 5E). To test whether PB1-F2 N66S decreases the MMP in the context of viral infection, we infected cells with PR8 viruses expressing either WT PB1-F2 or PB1-F2 N66S and stained the cells with TMRE at 8 h postinfection (hpi). As shown in Fig. 5F, the PB1-F2 N66S-expressing virus decreased the TMRE signal compared to the WT PB1-F2-expressing virus. These observations indicate that PB1-F2 decreases the induction of IFN by dissipating the MMP (Fig. 6), similar to what has been reported for the ionophore CCCP (11).
Fig 5.

PB1-F2 decreases the mitochondrial membrane potential upon RIG-I N and MAVS stimulation. (A to C) 293T cells were transfected with RIG-I N and the indicated PB1-F2 expression plasmid. At 24 h posttransfection, the cells were stained with TMRE and analyzed for the MMP by flow cytometry. The cells were separated into GFP hi (red) and GFP lo (blue) cells, and the TMRE intensities are shown in the histogram overlays. To better display the data in the histograms, the cells in the two subpopulations are represented as percentage of maximum (FlowJo software). (D) The TMRE signal was calculated by generating the ratio of the TMRE signal of the GFP hi to the TMRE signal of the GFP lo cell populations. For example, the absolute mean TMRE signal of the GFP hi population is 1,786 and that of the GFP lo population is 5,537 for the PB1-F2 N66S sample (panel B, right panel, PB1-F2 N66S). Hence, the ratio is 1,786/5,537 ≈ 0.32. This was done for the experiments performed using RIG-I N (A to C) and MAVS (not shown) stimuli. All samples were duplicates. (E) 293T cells were transfected with RIG-I N and WT or N66S PB1-F2 or treated with staurosporine. The following day, cell lysates were analyzed by Western blotting or a fluorometric caspase 3 activity assay. The lysate from the staurosporine sample was diluted 1:5 with assay buffer. The data represent means ± standard deviations from one representative experiment (n = 3). Statistical significance was determined using Student's t test. **, P < 0.01. All data shown are representatives of two independent experiments. (F) 293T cells were infected with PR8 viruses expressing WT or N66S PB1-F2. At 8 hpi, cells were analyzed with the TMRE dye via fluorescence-activated cell sorting (FACS) as described for panels A to C. The data represent means ± standard deviations from one representative experiment (n = 3). Statistical significance was determined using Student's t test. *, P < 0.05. All data shown are representatives of two independent experiments.
Fig 6.
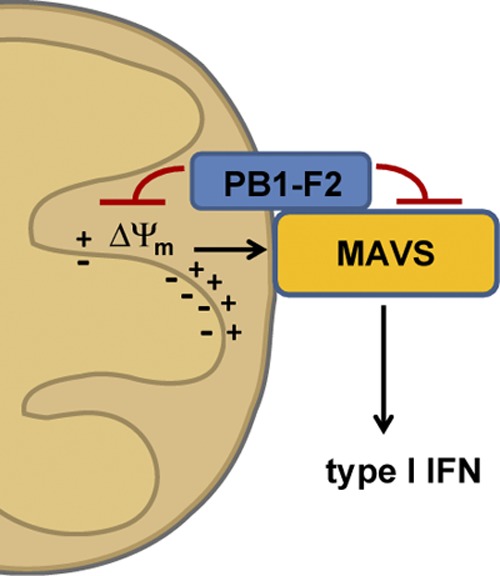
Proposed model for the IFN antagonist function of the influenza virus PB1-F2 protein. The PB1-F2 protein interacts with the MAVS adaptor protein via its C-terminal region and dissipates the mitochondrial membrane potential (Δψm), which is essential for MAVS-mediated interferon production. It is not clear whether these two effects caused by PB1-F2 are linked or executed separately from each other. The schematic is not drawn to scale.
DISCUSSION
In our previous report we showed that the influenza virus PB1-F2 protein has an anti-IFN effect and that the N66S form of PB1-F2, which is associated with increased virulence, exerts a stronger IFN antagonist function (28). Here, we identify the mechanism by which the PB1-F2 protein inhibits MAVS-mediated IFN synthesis.
We demonstrate that PB1-F2 binds to the MAVS adaptor protein via its C-terminal region (Fig. 1A) and that the N66S point mutation enhances the efficiency of binding of PB1-F2 with MAVS both in an overexpression system (Fig. 1A) and in the context of influenza virus infection (Fig. 1B), with similar mitochondrial localization patterns (Fig. 3). Of note, the C-terminal region of PB1-F2 has previously been shown to mediate the anti-IFN function (28). The coimmunoprecipitation data shown in Fig. 1A indicate that the region from aa 38 to 87 of PB1-F2 may not be optimal for MAVS binding, as the binding efficiency of the C-terminal truncation is reduced compared to that of the full-length PB1-F2 protein. It is possible that the C-terminally truncated PB1-F2 construct (aa 38 to 87) does not fold optimally or that additional residues are needed for binding to the MAVS protein. The N-terminal region of PB1-F2 (aa 1 to 37) only minimally pulls down MAVS, which was also observed with the N-terminal region fused to the mitochondrial targeting sequence of human cytochrome c oxidase (data not shown). Additionally, we observed an enhanced interaction of PB1-F2 N66S with MAVS during viral infection (Fig. 1B) even though the PB1-F2 N66S protein is less stable or expressed less efficiently than the WT PB1-F2 protein, as observed previously (28). The increased binding efficiency exerted by the N66S point mutation in the PB1-F2 protein may be due to a higher binding affinity to the MAVS protein compared to that of WT PB1-F2, but we have no molecular understanding of this phenomenon.
The mechanism by which MAVS mediates downstream signaling for the induction of type I IFNs is still being investigated. PB1-F2 interacts with a region in MAVS that mediates self-association and oligomerization as well as interaction with adaptor proteins such as tumor necrosis factor receptor-associated factor 3 (TRAF3) (26), TRAF6 (9), and other signaling molecules. It is possible that PB1-F2 disrupts the complex formation or rearrangement of MAVS with other proteins and/or the downstream communication of MAVS with adaptor proteins. It will be interesting to investigate whether other proteins can be found in this interaction complex between PB1-F2 and MAVS and whether the interaction is direct. The MMP has been demonstrated to be important for the orchestration of an active MAVS complex with numerous other proteins and the rearrangement of this complex upon stimulation (11). We found that PB1-F2 dissipates the MMP upon stimulation with RIG-I N or MAVS when exogenously expressed and that the N66S mutation in PB1-F2 enhances this effect (Fig. 5A to D). We also observed a significant decrease of the MMP caused by the PR8 PB1-F2 N66S virus compared to the isogenic WT PR8 PB1-F2 virus (Fig. 5F). Based on the findings that PB1-F2 binds to MAVS (Fig. 1) and affects the MMP (Fig. 5), we propose that PB1-F2 may interfere with the formation of an active MAVS-containing protein complex or alter the composition and/or rearrangement of this complex (Fig. 6). Future studies will be aimed at investigating this hypothesis. It is not clear whether the binding of PB1-F2 to MAVS initiates the decrease of the MMP or whether these two phenomena are independent from each other. Of note, the PB1-F2 proteins did not induce the activation of the cell death pathway upon stimulation with RIG-I N as determined by caspase 3 activity and PARP cleavage assays (Fig. 5E). These observations indicate that the anti-IFN and proapoptotic effects of PB1-F2 are independent from each other in the scenario of an activated IFN synthesis pathway. Future studies will focus on dissecting the proapoptotic and anti-IFN properties of the PB1-F2 protein (27).
We have previously observed an enhanced IFN antagonist function of the influenza virus NS1 protein in the presence of PB1-F2 (28), and we found a similar effect with the polymerase proteins PB1 and PB2 (Fig. 2). PB1-F2 expression did not appear to enhance the anti-IFN property of the PA polymerase protein (Fig. 2). This may be due to the possibility that the PA protein uses a mechanism to inhibit the induction of IFN that differs from that of the PB1 and PB2 proteins and that is not enhanced by PB1-F2. Confocal microscopy studies revealed that PB1-F2 does not seem to alter the subcellular localization of these proteins when exogenously expressed in HeLa cells (data not shown). It is possible that PB1-F2 enhances the inhibitory effect on IFN induction of the polymerase proteins by binding to a different region in MAVS than the polymerase proteins. In the future, it will be of interest to unravel the interplay of the multiple interferon antagonists of influenza viruses and the possible cell-type- and strain-specific differences. Taking our findings together, we have identified the molecular mechanism by which an N66S mutation in the PB1-F2 protein evades the IFN response and thus increases the pathogenicity of influenza viruses.
ACKNOWLEDGMENTS
We thank M. T. Sanchez-Aparicio for assistance with the bimolecular YFP complementation assay and helpful discussions. We are grateful to Christopher Seibert for providing monoclonal PB1, PB2, and PA antibodies. We also acknowledge Ryan Langlois for assistance with the flow cytometry.
This study was partly funded by the Center of Research in Influenza Pathogenesis (CRIP) (NIAID contract HHSN266200700010C) and NIAID (http://www.niaid.nih.gov/) grant P01AI058113. B.M. is supported by NIH/NIAID K99 Pathway to Independence award (1K99AI095320-01).
Footnotes
Published ahead of print 6 June 2012
REFERENCES
- 1. Baril M, Racine ME, Penin F, Lamarre D. 2009. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J. Virol. 83:1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bruns K, et al. 2007. Structural characterization and oligomerization of PB1-F2, a proapoptotic influenza A virus protein. J. Biol. Chem. 282:353–363 [DOI] [PubMed] [Google Scholar]
- 3. Chen W, et al. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 7:1306–1312 [DOI] [PubMed] [Google Scholar]
- 4. Conenello GM, et al. 2011. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J. Virol. 85:652–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. 2007. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 3:1414–1421 doi:10.1371/journal.ppat.0030141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foy E, et al. 2005. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. U. S. A. 102:2986–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW. 2003. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 77:7214–7224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graef KM, et al. 2010. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 84:8433–8445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hou F, et al. 2011. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146:448–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iwai A, et al. 2010. Influenza A virus polymerase inhibits type I interferon induction by binding to interferon beta promoter stimulator 1. J. Biol. Chem. 285:32064–32074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. 2011. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal. 4:ra7 doi:10.1126/scisignal.2001147 [DOI] [PubMed] [Google Scholar]
- 12. Kumar H, et al. 2006. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 203:1795–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li K, et al. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U. S. A. 102:2992–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin R, et al. 2006. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 80:6072–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loo YM, et al. 2006. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 103:6001–6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McAuley JL, et al. 2010. PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 6:e1001014 doi:10.1371/journal.ppat.1001014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McAuley JL, et al. 2007. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meylan E, et al. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172 [DOI] [PubMed] [Google Scholar]
- 19. Mibayashi M, et al. 2007. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 81:514–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199 [DOI] [PubMed] [Google Scholar]
- 21. Park MS, et al. 2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol. 77:1501–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perez JT, et al. 2010. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc. Natl. Acad. Sci. U. S. A. 107:11525–11530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prins KC, Cardenas WB, Basler CF. 2009. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 83:3069–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682 [DOI] [PubMed] [Google Scholar]
- 25. Sun Q, et al. 2006. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24:633–642 [DOI] [PubMed] [Google Scholar]
- 26. Tang ED, Wang CY. 2009. MAVS self-association mediates antiviral innate immune signaling. J. Virol. 83:3420–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Varga ZT, Palese P. 2011. The influenza A virus protein PB1-F2: killing two birds with one stone? Virulence 2:542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Varga ZT, et al. 2011. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 7:e1002067 doi:10.1371/journal.ppat.1002067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamada H, Chounan R, Higashi Y, Kurihara N, Kido H. 2004. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 578:331–336 [DOI] [PubMed] [Google Scholar]
- 30. Yasukawa K, et al. 2009. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2:ra47 doi:10.1126/scisignal.2000287 [DOI] [PubMed] [Google Scholar]
- 31. Zamarin D, GarcíA-Sastre A, Xiao X, Wang R, Palese P. 2005. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1:e4 doi:10.1371/journal.ppat.0010004 [DOI] [PMC free article] [PubMed] [Google Scholar]