

Abstract
Cronobacter (previously known as Enterobacter sakazakii) is a diverse bacterial genus consisting of seven species: C. sakazakii, C. malonaticus, C. turicensis, C. universalis, C. muytjensii, C. dublinensis, and C. condimenti. In this study, we have used a multilocus sequence typing (MLST) approach employing the alleles of 7 genes (atpD, fusA, glnS, gltB, gyrB, infB, and ppsA; total length, 3,036 bp) to investigate the phylogenetic relationship of 325 Cronobacter species isolates. Strains were chosen on the basis of their species, geographic and temporal distribution, source, and clinical outcome. The earliest strain was isolated from milk powder in 1950, and the earliest clinical strain was isolated in 1953. The existence of seven species was supported by MLST. Intraspecific variation ranged from low diversity in C. sakazakii to extensive diversity within some species, such as C. muytjensii and C. dublinensis, including evidence of gene conversion between species. The predominant species from clinical sources was found to be C. sakazakii. C. sakazakii sequence type 4 (ST4) was the predominant sequence type of cerebral spinal fluid isolates from cases of meningitis.
INTRODUCTION
Cronobacter (formerly known as Enterobacter sakazakii) is a genus consisting of Gram-negative, motile, facultatively anaerobic opportunistic bacterial pathogens belonging to the Enterobacteriaceae family (28). The genus was originally identified as yellow-pigmented Enterobacter cloacae. DNA-DNA hybridization results showed the organism to be only 41% and 54% related to Citrobacter freundii and Enterobacter cloacae, respectively, and it was therefore reclassified as an independent species, Enterobacter sakazakii, comprising 15 biogroups (11). Later biotyping and genotyping studies led to its further reclassification into the genus Cronobacter (20). To date, the diverse genus accommodates seven different species: C. sakazakii, C. malonaticus, C. turicensis, C. muytjensii, C. dublinensis, and the two newly defined species C. universalis and C. condimenti (24).
The primary niche of this organism is believed to be plant material (i.e., wheat, rice, herbs, and spices) (18). However, it is brought into contact with humans via food and environmental exposure. These bacteria have been implicated in various life-threatening diseases in humans, such as meningitis, necrotizing enterocolitis, septicemia, and pneumonia, affecting a wide range of age groups (30, 35). Cronobacter species infections are of considerable significance because although human infections are relatively rare, those that do occur are often severe, and because the highest-risk age group is neonates (8, 9, 10), for which some infections have resulted in fatalities (4). There have been Cronobacter species cases in which powdered infant formula (PIF) has been identified as the source of contamination of the organism (46), and hence, it has played an important role in the epidemiology of the organism. Although Cronobacter spp. have been isolated from other mammals and invertebrates, such as rats and flies (14, 34), which may be additional sources of contamination, no disease caused by these routes has been reported.
Research on the organism has considerably progressed in recent years, with improved isolation, identification, and molecular typing methods (22, 38, 40). These methods have detected considerable diversity within the genus, warranting further detailed investigation. This is necessary to ensure reliable and robust methods of detection to control the organism and reduce exposure to vulnerable groups.
Not all Cronobacter species have been linked with infections, and the severity of virulence varies between different strains (4) and in laboratory studies using in vitro tissue culture and rat pup infections (43). Thus, detailed molecular studies will be useful. The virulence traits of Cronobacter spp. are uncertain, and given the ubiquitous occurrence of the bacterium in the environment and foods, it is desirable to distinguish between pathogenic and nonpathogenic variants. The first publication of C. sakazakii genome BAA-894, followed by the C. turicensis z3032 genome (28, 39), has enabled the study of predicted virulence genes, many of which are plasmid borne (13). The presence of a number of proposed virulence traits, such as iron-uptake mechanisms, type six secretion systems, sodA, and pili, across the Cronobacter species has been established (28, 29, 43).
The phylogenetic relationships and diversity of Cronobacter spp. within the Enterobacteriaceae family have been previously analyzed using the 16S rRNA gene and hsp60 sequences (19). Sequence diversity of 16S rRNA genes has been used to define genus (5%) and species (3%) boundaries. However, with respect to very closely related organisms, 16S rRNA gene sequence analysis has limitations due to minimal diversity in the sequences. The sequence diversity between multiple copies of the 16S rRNA gene operon within a bacterium can also introduce discrepancies (1). The partial sequencing of a number of protein-encoding genes of the organism's genome ensures that multiple independent loci are examined and ensures a far larger number of informative characters because these genes typically diverge far faster than the rRNA genes (26, 32). A 7-locus multilocus sequence typing (MLST) scheme was developed for the identification and characterization of C. sakazakii and C. malonaticus (2). This was of particular use since C. malonaticus was initially regarded as a subspecies of C. sakazakii, as the two species could not be distinguished according to 16S rRNA gene sequence analysis (99.7% similarity, full-length sequence); however, DNA-DNA hybridization studies revealed <70% DNA relatedness (20).
The original C. sakazakii and C. malonaticus MLST scheme required the partial sequence analysis of seven housekeeping genes, atpD, fusA, glnS, gltB, gyrB, infB, and ppsA (2). When concatenated, these sequences provided 3,036 nucleotides for analysis. In this study, the MLST scheme was extended from C. sakazakii and C. malonaticus to cover all seven formally recognized species of the Cronobacter genus to better quantitate the intraspecific and interspecific diversity of the genus as well as to potentially characterize the strains according to virulence groupings and source. The Cronobacter MLST database is available online with open access at http://www.pubmlst.org/cronobacter/ and is hosted by the University of Oxford, United Kingdom.
MATERIALS AND METHODS
Bacterial strains.
This study was based on 325 Cronobacter species isolates, which included 226 C. sakazakii strains, 39 C. malonaticus strains, 19 C. turicensis strains, 19 C. dublinensis strains, 17 C. muytjensii strains, 4 C. universalis strains, and the single isolate of the recently defined species C. condimenti. All isolates were either selected from the Nottingham Trent University collection of Cronobacter species strains or recently obtained from collaborators following a review of the literature. Most of the latter strains had been identified only as Enterobacter sakazakii or Cronobacter spp. in previous publications. Strains were chosen on the basis of their species, geographic and temporal distribution, source, and clinical outcome. This included the type strains for each species, infant formula and clinical isolates from across the world, obtained between 1950 and the present, as well as strains from the United States which caused high public concern in December 2011 (5). As part of the diversity study, the 16S rRNA gene sequences submitted as Cronobacter spp. by third parties to GenBank were also analyzed, and any strains of interest were requested from the sequence submitters. The list and details of all the isolates included in this study, along with their MLST profile details, are freely available online at http://www.pubmlst.org/cronobacter.
Citrobacter koseri (GenBank accession number CP000822) and Enterobacter sp. 683 (GenBank accession number CP000653) were used as outgroups, as previously described (2).
DNA isolation and PCR.
Genomic DNA was prepared from 1.5 ml of overnight culture grown in tryptic soy broth using the GenElute bacterial genomic DNA kit (Sigma) according to the manufacturer's instructions. Amplification and nested sequencing primers for the MLST loci were as previously described (2). Reaction conditions for all the primers were as follows: initial denaturation at 94°C for 2 min, 30 cycles of denaturation at 94°C for 1 min, primer annealing at 58°C for 1 min, and extension at 72°C for 2 min, and a final extension step of 72°C for 5 min. Each 25-μl amplification reaction mixture comprised ∼10 ng chromosomal DNA, 20 pmol forward and reverse primer, 1× PCR buffer (Promega, United Kingdom) containing 1.5 mM MgCl2, 0.8 mM deoxynucleotide triphosphates, and 1.25 U Taq (Promega, United Kingdom). The amplified products were then purified using MinElute PCR purification kits (Qiagen) following the manufacturer's protocol. The sequencing primers were used for both the amplification and sequencing of loci for strains if insufficient PCR product was obtained using the amplification primers.
Sequencing.
The sequencing was performed using the purified PCR products (10 ng/μl) on ABI 3730XL sequencing machines by Eurofins MWG Operon (London, United Kingdom) and Source Bioscience (Nottingham, United Kingdom). Using the nested sequencing primers, nucleotide sequences were determined at least once on each DNA strand. The sequence chromatograms were viewed using Chromas Lite (version 2.01; Technelysium Pty Ltd.) for quality control, and the sequences from both strands of a given locus of the same isolate were aligned and trimmed to the desired length using Jalview (version 2+; University of Dundee) (48) in ClustalW.
Analysis of DNA sequences.
Phylogenetic analyses of the concatenated sequences of the 7 loci (3,036 nucleotides) were performed using the maximum likelihood algorithm in MEGA5 (42) and ClonalFrame (version 1.2; School of Public Health, Imperial College London) (7). The allelic profiles identified for the 7 loci were analyzed using the Sequence Type Analysis and Recombinational Tests (START) version 2 software (23). DnaSP (version 5; Universitat de Barcelona) (31) was used to calculate the nucleotide divergence between the type strains of the various Cronobacter species.
Linkage and recombination analysis.
START (version 2; University of Oxford) was also used for calculating the ratio of nonsynonymous substitutions to synonymous substitutions (dN/dS) and the index of association (IA) of the various Cronobacter species for linkage analysis of the population as well as recombination testing (23). A further study of recombination events in the populations was carried out using the DnaSP software (31) and the neighbor-net algorithm in Splitstree (version 4; Universitat Tubingen) (17). The clonality of the population was analyzed using eBURST (version 3; Department of Infectious Disease Epidemiology, Imperial College London) (12).
Evolutionary time scale.
The diversity and branching observed in the Cronobacter MLST sequence data were used to estimate the relative age of the genus Cronobacter and the divergence time of the branches for the individual species. Similar divergence estimation studies using MLST data have previously been carried out for organisms such as Salmonella spp. (33) and Pseudomonas aeruginosa (47), among others. Mean synonymous substitution values (Ds) were calculated for the individual Cronobacter species using the 3,036-bp concatenated sequences in MEGA5 (42) as well as START2 (23). These values were used to calculate the ages of the genus and species by using the formula age = Ds/rate, where “rate” is the synonymous molecular clock rate.
Since this is based on an estimate, a range of dates was calculated by using the substitution rate of 3 × 10−9 for Escherichia coli and Salmonella enterica serovar Typhimurium that was published by Berg and Martelius (3) and a lower rate of 4.5 × 10−9 calculated for E. coli (36).
RESULTS
MLST of the genus Cronobacter.
From the 325 strains included in this study, 115 sequence types (STs) spanning across the genus Cronobacter have been identified. Of these, 53 STs have been identified for C. sakazakii, which is also the type species of the genus. Seventeen STs have been identified for C. malonaticus, 13 for C. turicensis, 10 for C. muytjensii, and 17 for C. dublinensis. Four STs have been identified for the recently renamed C. universalis (previously known as Cronobacter genomospecies 1), and one has been identified for the recently defined species C. condimenti (24). C. sakazakii ST4 was the most prevalent ST, with 78 isolates. A few strains had been previously misidentified due to biotyping errors or discrepancies in 16S rRNA gene sequences. These misidentifications have mostly been between the closely related species C. sakazakii and C. malonaticus (NTU strains 33 and 101), and a few C. turicensis isolates had been misidentified as C. dublinensis (NTU strains 57 and 564). One of the isolates (NTU strain 96) of the reclassified C. universalis had been previously incorrectly identified as C. turicensis by biotyping.
The average GC content of the seven sequenced genes in all the STs across the Cronobacter genus was of 58%, which is consistent with the overall 57% GC content for the published genome C. sakazakii BAA-894 (28). All the ST and allelic profile numbers were assigned as a continuation of the scheme initially established for C. sakazakii and C. malonaticus (2). All sequences are available for free download at http://www.pubmlst.org/cronobacter. Seven genes were successfully sequenced from all strains. Primers designed to amplify the gltB and ppsA loci failed in less than 5% of cases (mainly C. muytjensii). When these outer primers failed, the sequencing primers were used directly for PCR and for sequencing. Complete sets of data were obtained for all strains (Table 1).
Table 1.
START analysis of the 7 MLST loci of the Cronobacter species isolates
| Gene | Size of fragment analyzed (bp) | No. of alleles identified | No. of polymorphic sites | Proportion of fragment as polymorphic sites (%) | % GC content | dN/dS |
|---|---|---|---|---|---|---|
| atpD | 390 | 52 | 86 | 22.05 | 59.15 | 0.009 |
| fusA | 438 | 51 | 97 | 22.14 | 54.03 | 0.023 |
| glnS | 363 | 56 | 106 | 29.20 | 56.75 | 0.049 |
| gltB | 507 | 68 | 151 | 29.78 | 61.31 | 0.023 |
| gyrB | 402 | 67 | 126 | 31.34 | 56.67 | 0.031 |
| infB | 441 | 67 | 132 | 29.93 | 58.58 | 0.048 |
| ppsA | 495 | 78 | 157 | 31.71 | 62.52 | 0.022 |
Phylogenetic relationships.
A phylogenetic tree based on the concatenated sequences (3,036 nucleotides) and using the maximum likelihood algorithm in MEGA5 showed clear clustering of the various Cronobacter species within the genus (Fig. 1). These clusters corresponded to previously defined species. Analysis of sequences using ClonalFrame (7) indicated that the seven loci supported the taxonomy but did not add support to any particular phylogeny.
Fig 1.
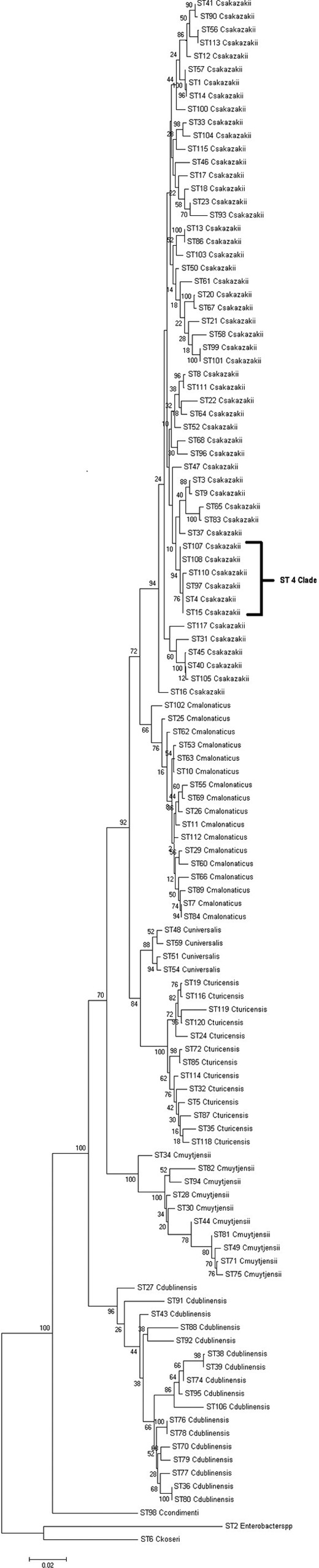
Maximum likelihood tree based on the concatenated sequences (3,036 bp) of the 7 MLST loci for the genus Cronobacter. The STs and the corresponding species are indicated at the tip of each branch. The tree is drawn to scale using MEGA5 with 1,000 bootstrap replicates.
As outgroups, we used the sequences of the 7 MLST loci from the publicly available genomes of Citrobacter koseri (GenBank accession number CP000822) and Enterobacter sp. 683 (GenBank accession number CP000653), two organisms closely related to Cronobacter spp. None of their alleles were found to be shared with those of any of the Cronobacter species isolates, and they were found to be genetically significantly distant from the rest of the Cronobacter genus (Fig. 1).
Clonality and ST groupings.
IA is a measure of the linkage of a population. The IA values for the genus Cronobacter were found to be significantly greater than zero (P < 0.001), indicating the presence of linkage disequilibrium or clonality. eBURST analysis showed the formation of 13 single-locus variant (SLV) clonal complexes among the 115 identified STs for the Cronobacter genus (Table 2). These are the STs which share 6 out of the 7 alleles that comprise a ST. Nine of these clades belong to C. sakazakii, two to C. dublinensis, and one each to C. turicensis and C. malonaticus. C. muytjensii, C. condimenti, and C. universalis did not show the formation of any such clades in this data set. Apart from these SLVs, a number of double-locus variants (DLV) were also observed, and these are the STs which shared 5 out of the 7 definitive allelic profiles. Some of these large clades are especially significant with respect to strain clustering according to their isolation sources.
Table 2.
Clades resulting among the 115 STs of the Cronobacter MLST database using eBURST
| Clade | Species | ST | No. of isolates | Source(s) | Geographic distribution | Period of isolation |
|---|---|---|---|---|---|---|
| 1 | C. sakazakii | 1 | 36 | Clinical, PIF, formula, food, environment | UK, Australia, USA, Germany, China, Brazil, Czech Republic, Switzerland, Turkey, Russia, Netherlands | 1979-2010 |
| 14 | 3 | PIF | France | 1994 | ||
| 2 | C. malonaticus | 7 | 15 | Food, clinical, PIF, weaning food | USA, New Zealand, France, Czech Republic, Brazil | 1973-2007 |
| 84 | 2 | Clinical | Czech Republic | Unknown | ||
| 89 | 1 | Clinical | Czech Republic | Unknown | ||
| 3 | C. sakazakii | 18 | 2 | Clinical, mouse | UK, Netherlands | 1953 |
| 23 | 2 | Food | Czech Republic, China | 2010 | ||
| 93 | 1 | Clinical | Czech Republic | 1979 | ||
| 4 | C. sakazakii | 4 | 78 | Clinical, PIF, milk powder, weaning food, chocolate, washing brush, environment, prepared formula, foot wound | UK, USA, France, China, Canada, Netherlands, Germany, Russia, Czech Republic, Switzerland, Slovakia, New Zealand, Saudi Arabia, Bangladesh | 1950-2011 |
| 15 | 1 | Clinical | Canada | 2003 | ||
| 97 | 1 | Milk powder factory | Australia | 2007 | ||
| 107 | 1 | Clinical | USA | 2011 | ||
| 108 | 1 | PIF | USA | 2011 | ||
| 5 | C. dublinensis | 38 | 1 | Follow-up formula | Korea | 2008 |
| 39 | 1 | Follow-up formula | Korea | 2008 | ||
| 74 | 2 | Follow-up formula, food | Korea | 2008-2011 | ||
| 6 | C. sakazakii | 40 | 6 | Food, spices | UK | 2007-2010 |
| 45 | 1 | PIF | Russia | 1988 | ||
| 105 | 1 | Milk powder factory | Australia | 2007 | ||
| 7 | C. sakazakii | 20 | 3 | Food, spices | China, UK | 2007-2010 |
| 67 | 1 | Herbs | UK | 2010 | ||
| 8 | C. sakazakii | 13 | 12 | Clinical, PIF, food, herbs, spices | France, Germany, China, Turkey | 1988-2010 |
| 86 | 1 | Clinical | France | 1994 | ||
| 9 | C. sakazakii | 65 | 1 | PIF | USA | 1988 |
| 83 | 2 | Milk powder | Australia | 2007 | ||
| 10 | C. sakazakii | 99 | 5 | Environmental | Germany | 2006 |
| 101 | 1 | Environmental | Germany | 2006 | ||
| 11 | C. sakazakii | 56 | 1 | PIF | Brazil | 2007 |
| 113 | 1 | PIF | Brazil | 2007 | ||
| 12 | C. turicensis | 119 | 1 | Milk powder factory | Australia | 2007 |
| 120 | 1 | Milk powder | Australia | 2007 | ||
| 13 | C. dublinensis | 36 | 1 | Unknown | Unknown | Unknown |
| 80 | 1 | Unknown | Czech Republic | Unknown |
Clade 1 comprises C. sakazakii sequence types 1 (ST1) and 14 (ST14). ST1 is a dominant ST consisting of 36 strains isolated from across the world over a period of more than 25 years. These have been isolated mainly from PIF and clinical cases and also more recently from milk powder-processing factories in Germany and Australia (6, 21) but also include a few food isolates. Three ST14 strains were isolated in 1994 from infant formula used during a neonatal intensive care unit Cronobacter species outbreak in France. According to pulsed-field gel electrophoresis, those strains did not match isolates from the infected neonates (4). Also linked to this clonal complex is ST57, which is a PIF isolate obtained from Denmark in 1988. This is a DLV to ST1.
Clade 2 comprises C. malonaticus STs 7, 84, and 89. Among C. malonaticus isolates, ST7 has the highest frequency (15/41) and has a mixture of clinical and PIF isolates obtained over 30 years. STs 84 and 89 comprise clinical isolates from the Czech Republic. These clinical isolates were recovered mainly from fecal, sputum, and blood samples. Apart from these, there were also food and weaning food isolates in this clade.
Clade 3 comprises C. sakazakii STs 18, 23, and 93 and had been isolated between 1953 and 2010. These strains included clinical and food isolates and one from a mouse.
Clade 4 comprises C. sakazakii STs 4, 15, 97, 107, and 108. This is a key clade with respect to Cronobacter species epidemiology. ST4 is the most dominant ST in this MLST study, with 78 isolates, and is also the most frequent clinical ST. In an earlier study of clinical C. sakazakii strains, ST4 was identified as a genetic signature for C. sakazakii meningitis, with 75% of the isolates being linked to meningitis cases over a period of 50 years in six different countries (25). Out of 81 clinical C. sakazakii isolates in the current study, 50% were ST4. This included the strain from the fatal Lebanon, IN, infection case which received considerable publicity in December 2011 (5). In contrast to the correlation of ST4 with meningitis, correlation with ST4 or other STs was not found for other clinical presentations, such as necrotizing enterocolitis (25). Twelve strains of C. sakazakii that were isolated from PIF collected in 12 countries were shown to be ST4. Fifteen strains which had been isolated from milk-processing factories, including roller dryers, tanker bays, and spray dryers, in Australia and Germany (6, 21) were revealed to be ST4. Other nonclinical sources included isolates from weaning food, chocolate (44), and a washing brush (46). ST15 has a lone isolate from a Canadian clinical case (37). ST97 has one of the strains isolated from a tanker bay at a milk powder-processing factory in Australia (6). ST107 and ST108 are isolates from Cronobacter species cases in the United States in 2011, with one from cerebrospinal fluid and the other from an opened pediatric infant formula tin, respectively. ST110 is related to ST4, as they share 4 out of the 7 alleles; the ST110 strain was a cerebrospinal fluid isolate obtained in the United States in 2011. Due to the strong association of ST4 and locus variants (15, 97, 107, 108, and 110) with severe cases of meningitis, which is of high clinical significance, this cluster of STs will be referred to as the “ST4 clade” as an extension of the previous ST4 association (25).
In addition to the four important clades of C. sakazakii and C. malonaticus, described above, some of the other SLVs and DLVs, such as clade 8 from food and feed, also showed clustering patterns depending on the source.
Interspecies recombination events.
In addition to the close similarities among many isolates of some species, especially C. sakazakii, there was also evidence of homologous recombination (gene conversion) events having occurred in the evolution of the genus. Splitstree4 was used to visualize recombination events and evolutionary relationships in the data set. Figure 2 represents a neighbor net generated for the Cronobacter genus using the 7-locus MLST data. This shows the tight clustering of the C. sakazakii and C. malonaticus species. It also reveals a higher diversity in the C. muytjensii and C. dublinensis species, with the formation of parallelograms denoting the possibility of recombination events.
Fig 2.

Neighbor net of the concatenated 7-locus sequence alignment generated for the Cronobacter MLST data set, indicating diversity and recombination events. The figure has been drawn to scale using Splitstree4. The numbers at the tips of the branches indicate STs. The formation of parallelograms indicates possible recombination events.
There were some instances of allelic profiles being shared between two species. Among the seven genes, gltB had the most (seven) allelic profiles shared between species. This sharing was observed mostly between clades, such as C. sakazakii with C. muytjensii and C. sakazakii with C. dublinensis species. In order to eliminate the possibility of these recombination events having influenced the overall genus phylogeny, a phylogenetic tree was constructed with the concatenated sequences of 6 loci, excluding gltB. This tree confirmed the robustness of the taxonomic divisions within the genus.
Among the seven loci, fusA was observed to be the most stable with the least number of shared alleles among species, and none of the profiles were shared between two or more species (Fig. 3).
Fig 3.

Maximum likelihood tree of the fusA alleles (438 bp) of the Cronobacter MLST data set. The numbers at the end of each branch indicate the allelic profiles. The tree is drawn to scale using MEGA5 with 1,000 bootstrap replicates.
Intraspecies genetic diversity.
The Cronobacter genus phylogenetic tree (Fig. 1) showed that some of the species clusters (mainly C. muytjensii and C. dublinensis) contained some individual branches that were more genetically distant from the main cluster. This is a phenomenon that has been described as “fuzzy” species in the MLST of Neisseria spp. (16). Strain 1330 (ST98) was one such candidate, seen as a lineage branching out from the C. dublinensis cluster which, following further phenotypic and DNA-DNA hybridization studies, was confirmed to be a previously unrecognized independent species, C. condimenti (24). Divergent strains are also evident in the C. muytjensii and C. dublinensis clusters. The diversity observed in the branching of the C. muytjensii and C. dublinensis clusters (Fig. 2) was further investigated using nucleotide divergence among populations in the MLST sequence data. Table 3 contains the DNA divergence values for the concatenated 7 loci that were calculated for the individual type strains of the Cronobacter species. The lowest divergence value was 2.8% (3,036 bp), observed between the type strains of C. sakazakii (NCTC 11467 [ST8]) and C. malonaticus (CDC 1058-77 [ST7]), and this value was used as a minimum cutoff value for identifying potential candidates for new species. As described earlier, these strains are only 0.3% different by full-length 16S rRNA gene sequences. However, when the DNA divergence values between the individual isolates in the C. muytjensii and C. dublinensis clusters were calculated, many of them were found to be above this cutoff value (data not shown), a further indication of the wide diversity within these species.
Table 3.
Nucleotide divergence values of type strains of the Cronobacter species
| Organism | Nucleotide divergence value (%) between strainsa |
|||||
|---|---|---|---|---|---|---|
| C. malonaticus CDC 1058-77T (ST7) | C. turicensis LMG 23827T (ST19) | C. dublinensis LMG 23823T (ST106) | C. universalis NCTC 9529T (ST54) | C. condimenti LMG 26250T (ST98) | C. muytjensii ATCC 51329T (ST81) | |
| C. sakazakii NCTC 11467T (ST8) | 2.8 | 4.2 | 7.5 | 3.8 | 8.7 | 7.4 |
| C. malonaticus CDC 1058-77T (ST7) | 4.5 | 7.6 | 3.5 | 8.8 | 7.7 | |
| C. turicensis LMG 23827T (ST19) | 7.4 | 3.1 | 8.7 | 7.6 | ||
| C. dublinensis LMG 23823T (ST106) | 7.3 | 7.9 | 5.0 | |||
| C. universalis NCTC 9529T (ST54) | 8.9 | 7.8 | ||||
| C. condimenti LMG 26250T (ST98) | 8.5 | |||||
Based on 3,036 bp of the concatenated sequences of the seven MLST loci.
These divergence patterns were reflected in the phylogenetic tree (Fig. 1) of the genus as well, thus splitting the C. muytjensii species into two main clusters and a lone diverse branch (ST34). The C. dublinensis species showed higher diversity, with two major clusters and a few independent branches distant from each other. Previously, this species had been divided into subspecies based on phenotypic tests (20).
Current taxonomic standards for recognizing new species by the International Journal for Systematic and Evolutionary Microbiology require a polyphasic analysis, including DNA-DNA hybridization and phenotypic testing for biochemical traits. Phenotypic testing and DNA-DNA hybridization analysis of these clusters of isolates that were conducted by us in the laboratory have not yet produced biochemical traits that can distinguish between the independent branches within C. muytjensii and C. dublinensis (S. Joseph and S. J. Forsythe, unpublished data). The boundaries for defining a bacterial species have always been a topic of debate (27) and are ultimately a matter of taxonomic convenience. Hence, a whole-genome level of analysis should improve our understanding of this intraspecies diversity.
Evolutionary time scale.
An evolutionary analysis of the MLST data set predicts that the Cronobacter genus split from its closest ancestor in the Enterobacteriaceae family approximately 45 to 68 million years ago, which was the Paleogene period of the Cenozoic era (Fig. 4). This is when early flowering plants evolved and coincides with the suspected natural plant habitat of the organism. Compared with the ages of other members of the Enterobacteriaceae family as estimated by similar MLST studies, the evolution of distinguishable Cronobacter species may have occurred over the same period as the divergence of the Salmonella species and subspecies after its split from E. coli (33). The earliest branches of the genus led to C. dublinensis and C. muytjensii. The estimates of the divergence dates of the individual species were also similarly calculated and have been denoted on the branches of the tree in Fig. 4. C. condimenti had to be excluded from this data set because only one isolate has been identified for this species to date, and hence, it was not possible to calculate the Ds values for the species.
Fig 4.
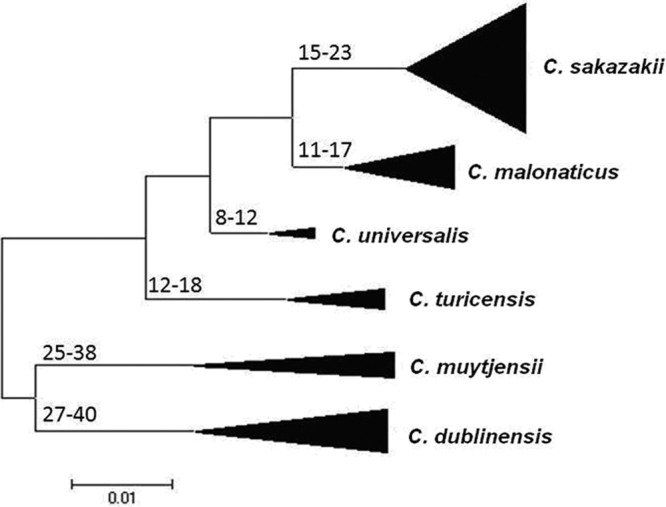
Maximum likelihood tree of the Cronobacter MLST data set, indicating ranges of the hypothetical divergence dates of each species node, measured in millions of years before the present. The tree has been drawn to scale using MEGA5. The bases of the triangles indicate the numbers of isolates used for the analysis, while the heights indicate the diversity of each branch. The single C. condimenti isolate has been excluded from this analysis.
DISCUSSION
The Cronobacter genus has come to prominence recently due to its association with life-threatening infections of neonates, necrotizing enterocolitis, septicemia, and meningitis (4, 5). Our current knowledge of the virulence and epidemiology of this organism is limited, and therefore, an improved understanding of the diversity of the genus is warranted.
The use of a set of seven housekeeping genes in MLST has greater sequence diversity located at seven different loci around the genome than 16S rRNA gene sequencing and has been applied to many bacteria (45). We have used a similar approach to study the diversity of this genus.
Overall, the phylogeny of the genus showed the members of the species C. sakazakii and C. malonaticus sampled to date to be tightly clustered and genetically closely related to each other yet clearly distinguishable, a resolution not previously possible using 16S rRNA gene sequence analysis (2). The dominance of the meningitis-related strains in a single ST (ST4) of C. sakazakii, as well as the clustering of isolates from milk powder-processing environments in specific STs, also opens up the possibility of using MLST as a typing tool for quality control checks in the industry. C. malonaticus and C. sakazakii also contained other STs (such as ST8) that are associated with clinical isolates but not so strongly associated with neonates or PIF. This may reflect alternative routes of infection, such as other foods, commensal colonization in the throat, and the environment (15).
None of the fusA profiles were shared between two or more species (Fig. 3). Consequently, this locus can be used with two PCR primer sets to define species of Cronobacter without the ambiguity of 16S rRNA gene sequence analysis.
Four strains of recently recognized C. universalis were characterized here and were found to cluster with C. turicensis. C. universalis was previously known as Cronobacter genomospecies 1, which had been defined based on only two isolates (20, 24). The recently defined C. condimenti branches as an independent lineage most closely related to C. dublinensis.
The species C. muytjensii and C. dublinensis showed diverse branching within each species, with the possibility of high numbers of recombination events having occurred. Also, according to the estimated divergence times (Fig. 4), it appears that the earliest branches of the genus would have been C. dublinensis and C. muytjensii. Hence, it can be proposed that high levels of recombination led to the splits in the Cronobacter genus, which later stabilized and became clonal in the more-recently evolved species like C. sakazakii and C. malonaticus. These latter two species are also the ones most associated with human clinical cases, suggesting a role of host adaptation in driving forward the evolution of the genus Cronobacter (41). Of course, one must keep in mind a possible bias in this analysis, since C. sakazakii is the most dominant species of the data set and therefore influences the numbers. Also, most of the C. muytjensii and C. dublinensis STs represent single strains and may therefore influence the weak clustering.
MLST has proven to be an effective and robust typing scheme for the Cronobacter genus and has exhibited a high level of discrimination between the isolates. The data obtained have revealed considerable diversity within the genus as well as in interspecies relationships. The scheme has also revealed a genetic signature for neonatal meningitis, highlighting its significance for epidemiological studies. Taking into account the stability and portable nature of the MLST scheme, this database can play an important role in future Cronobacter species research.
ACKNOWLEDGMENTS
We thank Nottingham Trent University for their financial support. M.M. and P.D. were supported in part by NIH grants AI052237, AI075093, and AI083646.
We thank the many contributors to the Cronobacter PubMLST database (http://www.pubMLST.org/cronobacter), especially Judith Noble-Wang, Anna Bowen, Matthew Arduino, Heather Craven, Philipp Hammer, and Rosana Santos.
Footnotes
Published ahead of print 11 July 2012
REFERENCES
- 1. Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF. 2004. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186:2629–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baldwin A, et al. 2009. Multilocus sequence typing of Cronobacter sakazakii and Cronobacter malonaticus reveals stable clonal structures with clinical significance which do not correlate with biotypes. BMC Microbiol. 9:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berg OG, Martelius M. 1995. Synonymous substitution-rate constants in Escherichia coli and Salmonella typhimurium and their relationship to gene expression and selection pressure. J. Mol. Evol. 41:449–456 [DOI] [PubMed] [Google Scholar]
- 4. Caubilla-Barron J, et al. 2007. Genotypic and phenotypic analysis of Enterobacter sakazakii strains from an outbreak resulting in fatalities in a neonatal intensive care unit in France. J. Clin. Microbiol. 45:3979–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. CDC 2011. CDC update: investigation of Cronobacter infections among infants in the United States. http://www.cdc.gov/foodsafety/diseases/cronobacter/investigation.html/
- 6. Craven HM, McAuley CM, Duffy LL, Fegan N. 2010. Distribution, prevalence and persistence of Cronobacter (Enterobacter sakazakii) in the nonprocessing and processing environments of five milk powder factories. J. Appl. Microbiol. 109:1044–1052 [DOI] [PubMed] [Google Scholar]
- 7. Didelot X, Falush D. 2007. Inference of bacterial microevolution using multilocus sequence data. Genetics 175(3):1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. FAO, WHO 2004. Workshop on Enterobacter sakazakii and other microorganisms in powdered infant formula, Geneva, 2-5 February 2004. http://www.who.int/foodsafety/micro/jemra/meetings/feb2004/en/index.html
- 9. FAO, WHO 2006. Expert meeting on Enterobacter sakazakii and Salmonella in powdered infant formula, Rome, 16-20 January 2006. http://www.who.int/foodsafety/micro/jemra/meetings/jan2006/en/index.html
- 10. FAO, WHO 2008. Enterobacter sakazakii (Cronobacter spp.) in powdered follow-up formulae. Microbiological Risk Assessment Series no. 15. http://www.who.int/foodsafety/publications/micro/mra_followup/en/
- 11. Farmer JJ, III, et al. 1980. Enterobacter sakazakii: a new species of “Enterobacteriaceae” isolated from clinical specimens. Int. J. Syst. Bacteriol. 30:569–584 [Google Scholar]
- 12. Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186:1518–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Franco AA, et al. 2011. Characterization of putative virulence genes on the related RepFIB plasmids harbored by Cronobacter spp. Appl. Environ. Microbiol. 77:3255–3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gakuya FM, Kyule MN, Gathura PB, Kariuki S. 2001. Antimicrobial resistance of bacterial organisms isolated from rats. East Afr. Med. J. 78(12):646–649 [DOI] [PubMed] [Google Scholar]
- 15. Hagg U, Kaveewatcharanont P, Samaranayake YH, Samaranayake LP. 2004. The effect of fixed orthodontic appliances on the oral carriage of Candida species and Enterobacteriaceae. Eur. J. Orthod. 26:623–629 [DOI] [PubMed] [Google Scholar]
- 16. Hanage WP, Fraser C, Spratt BG. 2005. Fuzzy species among recombinogenic bacteria. BMC Biol. 3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267 [DOI] [PubMed] [Google Scholar]
- 18. Iversen C, Forsythe S. 2003. Risk profile of Enterobacter sakazakii, an emergent pathogen associated with infant milk formula. Trends Food Sci. Technol. 14:443–454 [Google Scholar]
- 19. Iversen C, Waddington M, On SLW, Forsythe S. 2004. Identification and phylogeny of Enterobacter sakazakii relative to Enterobacter and Citrobacter. J. Clin. Microbiol. 42:5368–5370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iversen C, et al. 2007. The taxonomy of Enterobacter sakazakii: proposal of a new genus Cronobacter gen. nov. and descriptions of Cronobacter sakazakii comb. nov., Cronobacter sakazakii subsp. sakazakii comb. nov., Cronobacter sakazakii subsp. malonaticus subsp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov. and Cronobacter genomospecies 1. BMC Evol. Biol. 7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jacobs C, Braun P, Hammer P. 2011. Reservoir and routes of transmission of Enterobacter sakazakii (Cronobacter spp.) in a milk powder-producing plant. J. Dairy Sci. 94:3801–3810 [DOI] [PubMed] [Google Scholar]
- 22. Jarvis KG, et al. 2011. Molecular characterization of Cronobacter lipopolysaccharide O-antigen gene clusters and development of serotype-specific PCR assays. Appl. Environ. Microbiol. 77:4017–4026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jolley KA, Feil EJ, Chan MS, Maiden MC. 2001. Sequence type analysis and recombinational tests (START). Bioinformatics 17:1230–1231 [DOI] [PubMed] [Google Scholar]
- 24. Joseph S, et al. 2011. Cronobacter condimenti sp. nov., isolated from spiced meat and Cronobacter universalis sp. nov., a novel species designation for Cronobacter sp. genomospecies 1, recovered from a leg infection, water, and food ingredients. Intl. J. Syst. Evol. Microbiol. 62:1277–1283 [DOI] [PubMed] [Google Scholar]
- 25. Joseph S, Forsythe SJ. 2011. Predominance of Cronobacter sakazakii ST4 with neonatal infections. Emerg. Infect. Dis. 17:1713–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Konstantinidis KT, Ramette A, Tiedje JM. 2006. Toward a more robust assessment of intraspecies diversity, using fewer genetic markers. Appl. Environ. Microbiol. 72:7286–7293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Konstantinidis KT, Ramette A, Tiedje JM. 2006. The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361:1929–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kucerova E, et al. 2010. Genome sequence of Cronobacter sakazakii BAA-894 and comparative genomic hybridization analysis with other Cronobacter species. PLoS One 5:e9556 doi:10.1371/journal.pone.0009556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kucerova E, Joseph S, Forsythe S. 2011. The Cronobacter genus: ubiquity and diversity. Qual. Assur. Saf. Crop. 3:104–122 [Google Scholar]
- 30. Lai KK. 2001. Enterobacter sakazakii infections among neonates, infants, children, and adults. Case reports and a review of the literature. Medicine 80:113–122 [DOI] [PubMed] [Google Scholar]
- 31. Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452 [DOI] [PubMed] [Google Scholar]
- 32. Maiden MC, et al. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U. S. A. 95:3140–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McQuiston JR, et al. 2008. Molecular phylogeny of the salmonellae: relationships among Salmonella species and subspecies determined from four housekeeping genes and evidence of lateral gene transfer events. J. Bacteriol. 190:7060–7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mramba F, Broce A, Zurek L. 2006. Isolation of Enterobacter sakazakii from stable flies, Stomoxys calcitrans L. (Diptera: Muscidae). J. Food. Prot. 69:671–673 [DOI] [PubMed] [Google Scholar]
- 35. Muytjens HL, et al. 1983. Analysis of eight cases of neonatal meningitis and sepsis due to Enterobacter sakazakii. J. Clin. Microbiol. 18:115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ochman H, Elwyn S, Moran NA. 1999. Calibrating bacterial evolution. Proc. Natl. Acad. Sci. U. S. A. 96:12638–12643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pagotto FJ, Nazarowec-White M, Bidawid S, Farber JM. 2003. Enterobacter sakazakii: infectivity and enterotoxin production in vitro and in vivo. J. Food. Prot. 66:370–375 [DOI] [PubMed] [Google Scholar]
- 38. Seo KH, Brackett RE. 2005. Rapid, specific detection of Enterobacter sakazakii in infant formula using a real-time PCR assay. J. Food. Prot. 68:59–63 [DOI] [PubMed] [Google Scholar]
- 39. Stephan R, Lehner A, Tischler P, Rattei T. 2011. Complete genome sequence of Cronobacter turicensis LMG 23827, a food-borne pathogen causing deaths in neonates. J. Bacteriol. 193:309–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun Y, et al. 2011. Development of an O-antigen serotyping scheme for Cronobacter sakazakii. Appl. Environ. Microbiol. 77:2209–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Talarico S, Whitefield SE, Fero J, Haas R, Salama NR. 2012. Regulation of Helicobacter pylori adherence by gene conversion. Mol. Microbiol. 84:1050–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Bio. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Townsend SM, Hurrell E, Forsythe S. 2008. Virulence studies of Enterobacter sakazakii isolates associated with a neonatal intensive care unit outbreak. BMC Microbiol. 8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Turcovský I, Kuniková K, Drahovská H, Kaclíková E. 2011. Biochemical and molecular characterization of Cronobacter spp. (formerly Enterobacter sakazakii) isolated from foods. Antonie Van Leeuwenhoek 99:257–269 [DOI] [PubMed] [Google Scholar]
- 45. Urwin R, Maiden MC. 2003. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 11:479–487 [DOI] [PubMed] [Google Scholar]
- 46. van Acker J, et al. 2001. Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in powdered milk formula. J. Clin. Microbiol. 39:293–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Waine DJ, Honeybourne D, Smith EG, Whitehouse JL, Dowson CG. 2009. Cross-sectional and longitudinal multilocus sequence typing of Pseudomonas aeruginosa in cystic fibrosis sputum samples. J. Clin. Microbiol. 47:3444–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]