

Abstract
No antiviral drugs currently exist for the treatment of enterovirus infections, which are often severe and potentially life threatening. Molecular screening of small molecule libraries identified fluoxetine, a selective serotonin reuptake inhibitor, as a potent inhibitor of coxsackievirus replication. Fluoxetine did not interfere with either viral entry or translation of the viral genome. Instead, fluoxetine and its metabolite norfluoxetine markedly reduced the synthesis of viral RNA and protein. In view of its favorable pharmacokinetics and safety profile, fluoxetine warrants additional study as a potential antiviral agent for enterovirus infections.
INTRODUCTION
The human enteroviruses are members of a genus containing more than 100 serologically distinct small nonenveloped RNA viruses responsible for poliomyelitis, encephalitis, myocarditis, fulminate sepsis in newborns, and other life-threatening infections (8). Immunization has all but eliminated the circulation of the polioviruses, the archetype for the genus, but other enteroviruses continue to cause substantial morbidity and mortality in the United States and throughout the world.
Recent outbreaks of enterovirus 71 (EV71) and coxsackievirus B1 (CVB1) highlight the public health dangers posed by enteroviruses. EV71 has been the cause of numerous epidemics of central nervous system infections in Europe and the Asia-Pacific region over the last 15 years (4, 5, 24, 26, 27). Although EV71 infection may be mild or unrecognized, brainstem encephalitis and noncardiogenic pulmonary edema caused many deaths in Asian outbreaks between 1997 and 2010. A recent outbreak of coxsackievirus B1 (CVB1) myocarditis in the United States also highlighted the mutability of enteroviruses and their epidemic potential. CVB1 was initially isolated in 1948 near Coxsackie, NY, but a new variant of CVB1 emerged in 2007 and was detected at nearly 50 sites in the United States. Large clusters of cases occurred in Chicago, IL, and Los Angeles, CA, including cases of sepsis, myocarditis, and deaths among newborns (6, 42, 45). Since then, CVB1 has been the most commonly identified enterovirus in the United States (7).
Enteroviruses exhibit a high degree of genetic variability in their capsid gene sequences, and immunity is serotype specific, precluding a vaccine strategy that would address all of the pathogenic nonpolio enteroviruses. However, enteroviruses exhibit substantial genetic conservation in the internal ribosome entry site (IRES) required for cap-independent translation of the viral genome into a single polyprotein and in the coding domains for the nonstructural viral proteins that are derived from it by autoproteolytic cleavage (30–32). These features and structural conservation of capsid proteins and virion structure of diverse enteroviruses (14) suggest that it may be possible to develop broad-spectrum antienteroviral agents.
No antiviral agents are currently available for these commonly encountered pathogens. None of the dozens of antiviral drugs effective against HIV, hepatitis B or C virus, influenza virus, herpesviruses, or other viruses have any activity against enteroviruses. The investigational antienterovirus agent pleconaril (34) has been dropped from further clinical development and study, apart from an ongoing trial involving 45 newborns with enteroviral sepsis syndrome (Collaborative Antiviral Study Group Trial 106; ClinicalTrials.gov identifier NCT00031512). A clinical trial is under way of a similar capsid-binding drug, BTA-798, for the treatment of asthmatic adults with symptomatic infection with human rhinoviruses, which are now taxonomically incorporated into the Enterovirus genus. Additional compounds have been found and described in the scientific and medical literature that inhibit the growth of enteroviruses, but their utility remains largely unexplored (12, 38). For now, treatment of serious and life-threatening enterovirus infections consists of supportive care, including management of seizures, hemorrhage, and respiratory failure, as needed. Infusions of intravenous immunoglobulin from pooled donors are sometimes given in hopes of limiting virus replication.
In search of additional antiviral agents, we screened various small molecule libraries and identified previously unrecognized inhibitors of enterovirus replication. Interestingly, fluoxetine, a selective serotonin reuptake inhibitor, demonstrated potent antiviral activity against a variety of enterovirus serotypes.
MATERIALS AND METHODS
Cells and virus.
HeLa-RW cells were generously provided by Lindsay Whitton (The Scripps Research Institute, La Jolla, CA). As previously described (29), stocks of CVB-H3 and CVB3 expressing enhanced green fluorescent protein (CVB3-EGFP) were produced by transfecting HeLa-RW cells with a plasmid expressing the T7 polymerase (pAR3126) and plasmid clones of the viral genome (13, 20). CVB3-H3 completes its life cycle very rapidly in these cells, achieving peak viral titers 6 h after infection (20, 36). An isolate of CVB1 recovered during a 2007 outbreak (42, 45) was generously provided by Stan Shulman and Xiaotian Zheng (Northwestern University Feinberg School of Medicine, Chicago, IL). Clinical isolates of CVB2 and CVB3-MCH (21) were provided by the UCLA Clinical Microbiology Laboratory. Virus titers were determined by plaque assays using HeLa-RW cells (29).
Primary screening assay.
We screened for novel inhibitors of enterovirus replication using an assay to monitor cell viability and detect the enterovirus-induced cytopathic effect (CPE) by modifying the assay described by Gong et al. (16). Prior to adding library compounds, 20 μl culture medium per well was dispensed into 384-well microtiter plates (Greiner One) and 0.5 μl of 1 mM test compound solution in dimethyl sulfoxide (DMSO) was added using a 500-nl V&P custom pin tool (San Diego, CA). In negative-control wells, 0.5 μl DMSO alone was added. HeLa-RW cells and CVB3-H3 virus were mixed, and 20 μl was added onto the plates to achieve a ratio of 3,000 cells/well and 20 PFU CVB-H3/well, representing a multiplicity of infection (MOI) of 0.007. Guanidine, a well-known CVB3 inhibitor (12, 33), was used a positive control at a final concentration of 10 mM, which resulted in complete protection from cytopathic effects of CVB3 infection. The plates were incubated in a 37°C cell culture incubator for 48 h. The cytopathic effect (CPE) induced by CBV3-H3 replication was detected and quantified by using ATPLite 1-Step reagent (Perkin Elmer) to assess cell viability. At the end of the incubation period, 25 μl ATPLite 1-Step reagent was added to each well, and the plate was sealed with transparent film. The luminescence signals on the plate were read immediately using a FLUOstar Optima reader (BMG Labtech, Inc., Cary, NC). The Z′ factor of the assay was greater than 0.5, and hits with antiviral activity greater than 50% of antiviral activity of positive controls were selected for follow-up.
Here we present the result from screening the Prestwick Chemical Library. This library contains 1,120 high-purity chemical compounds (including many FDA-approved drugs) selected for structural diversity and a broad spectrum of clinical uses.
Determination of antiviral EC50 and CC50 of fluoxetine and norfluoxetine.
Fluoxetine and norfluoxetine were purchased from Sigma-Aldrich and dissolved in DMSO to achieve an initial concentration of 20 mM, which was further diluted in a series of 2-fold dilutions to the lowest concentration of 38 nM. Using the same ATPLite 1-Step-based assay on a 384-well plate, we also assessed cell viability in the presence of serially diluted chemical compounds to identify a 50% cytotoxic concentration (CC50), defined as 50% reduction in luminescence compared to control wells. The 50% effective concentration (EC50) was defined as the compound concentration that lead to a retention of 50% luminescence values from infected cells. A selectivity index (SI) was calculated for each compound as SI = CC50/EC50.
Assay of IRES-mediated translation.
A bicistronic expression vector was constructed by amplifying the 5′-untranslated region (5′ UTR) of CVB3/28 (39), a myocarditic strain of CVB3, and inserting it into the multiple cloning site of plasmid pDL-N (41), a gift from Asim Dasgupta. In this construct, the cytomegalovirus (CMV) promoter is followed by a Renilla luciferase, which is then followed by a thermostable hairpin, the 5′ UTR sequence, and the firefly luciferase gene. HeLa-RW cells were plated in a 96-well microtiter plate and transfected the next day with this dual-luciferase reporter plasmid, using Bio T transfection reagent (Bioland Scientific) according to the manufacturer's instructions. At 48 h posttransfection, the compounds were added to the transfected cells at a concentration of 6.25 μM, an effective concentration selected based on EC50 and CC50 data. DMSO alone was added to control wells. At 72 h after the transfection, the cells were washed three times with phosphate-buffered saline (PBS), and the measurements of dual-luciferase levels were performed using Promega's dual-luciferase reporter assay system according to the manufacturer's instructions. For stable transfection, a selection medium with 1 mg/ml G418 was used to select cells stably transfected with the plasmid and subsequently maintained in the medium with 1 mg/ml G418.
Determination of antiviral activity by inhibition of EGFP-expressing recombinant CVB3.
HeLa-RW cells were plated at 30,000 cells per well in a 96-well microtiter plate in 100 μl of medium and cultured overnight. The cells were infected on the following day with EGFP-expressing recombinant CVB3 at an MOI of 1. The compounds were added to the cells at 0 h, 1 h, and 2 h postinfection at a concentration of 6.25 μM. DMSO alone was added to control wells. Six hours after inoculation with virus, the cells were fixed with 1% formaldehyde and counterstained with DAPI (4′,6-diamidino-2-phenylindole) for 5 min. The cells were washed 3 times with PBS before being analyzed using an ImageXpress Micro high-content microscope (Molecular Devices).
Test of direct virucidal activity.
Fluoxetine (or DMSO diluent) was added to virus stocks of CVB3-H3 to achieve a final concentration of 10 μM. After incubation at 37°C for 1 h, the preparation was diluted serially, and the virus titer was determined by plaque assay, as described previously (29).
Viral replication assays.
To assess the effect of norfluoxetine and fluoxetine on enteroviral replication, 1 × 105 HeLa-RW cells in 1 ml DMEM with 10% fetal bovine serum (FBS) were placed into each well of 24-well plates and incubated overnight at 37°C. After 24 h, drug compounds were added to wells to achieve the desired concentrations. After 30 min at 37°C, CVB-H3 virus stock was added to achieve an MOI of 1, and the plates were again placed in a 37°C incubator. After 6 h, medium was aspirated from the wells, and cell layers were washed with PBS and then lifted with 100 μl trypsin (2.5%). The trypsin was inactivated with 400 μl Dulbecco's modified Eagle's medium (DMEM)-based medium and transferred into screw-cap tubes. The cells were washed twice and then resuspended in PBS, and aliquots were frozen at −80°C prior to virus titration or reverse transcription-PCR (RT-PCR) and immunoblot analysis.
(i) RT-PCR quantitation of viral RNA.
Viral RNA was extracted from cell pellets using the Qiagen RNEasy commercial kit, and quantified by real-time reverse transcriptase PCR (RT-PCR) using Applied Biosystems SYBR green commercial reagents and primers described by others (10). To produce quantitative standards, viral RNA was synthesized from linearized pH3 (20), quantified by spectrophotometry, and serially diluted.
(ii) Immunoblot detection of viral protein.
Cell pellets were resuspended in PBS and lysed in Invitrogen NuPAGE lithium dodecyl sulfate (LDS) sample buffer. The lysed cells were then homogenized by passage through small-gauge needles to shear cellular DNA and separated by SDS-PAGE using 12% Bis-Tris gels (Invitrogen NuPAGE). The proteins were transferred to a nylon membrane and blocked in skim milk, and coxsackievirus proteins were detected by immunoblotting with antiserum obtained from ATCC (V030-501-560) using a chemiluminescent substrate (Thermo Scientific SuperSignal; West Pico).
RESULTS
Identification of novel small molecule inhibitors of CVB3 replication.
In an effort to identify small molecule inhibitors of enterovirus infections, we employed a molecular screening assay described previously (16), based on protection of cells from cytotoxic effects of viral infection. HeLa-RW cells were seeded into 384-well plates containing these compounds and infected at a low multiplicity of infection with CVB3-H3, a molecular clone of coxsackievirus B3 (CVB3) (20). CVB3 was chosen as it has consistently appeared among the 15 most commonly reported enteroviruses in the United States, with an estimated case-fatality rate of 5.4% (19). This specific CVB3 strain is known to cause severe myocarditis in mice (20, 36), and we have recently found that it will produce extensive myocarditis and disseminated infection in cynomolgus monkeys (C. E. Cammock et al., submitted for publication).
After 48 h, cell viability was assessed using the luciferase-containing reagent ATPLite 1-Step (Perkin Elmer), which has been optimized for use in high-throughput screening (HTS) assays. The Z′ factor, an assessment of the quality of screening assays (46) was monitored in each plate. Among other small molecule libraries, we screened the Prestwick Chemical Library, consisting of a total of four 384-well microtiter library plates containing 1,120 FDA-approved drugs and other compounds, for antiviral activity. The Z′-factors of screening plates were in the range of 0.5 to 0.7, indicative of robust assay performance. Compounds that strongly protected cells from the lytic effects of CVB3-H3 infection (luminescence values > 50% of positive-control values) were classified as initial hits (Fig. 1).
Fig 1.

Schema of the molecular screening approach. ATP-driven luminescence values are shown for the compounds in the Prestwick Chemical Library. Values are normalized for maximum luminescence observed on positive-control (guanidine HCl) wells in each 384-well plate. Negative-control wells on each plate included DMSO; note the low levels of luminescence. Primary hit compounds are indicated, which produced normalized luminescence levels above a threshold of 50% of the luminescence levels of the positive controls. All compounds were tested at a final concentration of 12.5 μM.
In the primary screen, we identified 8 compounds showing 30% inhibition efficiency and 7 of them showing 50% efficiency, yielding hit rates of 0.7% and 0.5%, respectively. These compounds included mefloquine hydrochloride, dibucaine, fluoxetine hydrochloride, flupentixol dihydrochloride, zuclopenthixol hydrochloride, cycloheximide, and the toxic plant alkaloid lycorine hydrochloride. Fluoxetine was selected for further examination. We also examined the antiviral activity of norfluoxetine, the major metabolite of fluoxetine, as it is present in serum at near equimolar concentrations to fluoxetine during therapy.
Fluoxetine and norfluoxetine inhibit enterovirus replication at established therapeutic concentrations.
Using a modification of our screening assay, we determined the cellular toxicity and antiviral activity of fluoxetine and norfluoxetine over a broad range of concentrations flanking the plasma concentrations achieved during chronic therapy for psychiatric indications (9, 25). A series of 2-fold dilutions were prepared covering a drug concentration range from 380 pM to 200 μM and added to test wells prior to addition of cells and CVB3-H3. Both fluoxetine and norfluoxetine had EC50s of 2.3 μM and a CC50 of 25 μM (Fig. 2A and B), resulting in selectivity index of approximately 10. Peak antiviral activity of both compounds was achieved at a concentration of 6.25 μM.
Fig 2.

Fluoxetine and norfluoxetine antiviral activity. (A) The antiviral EC50s for fluoxetine (Fluox) and norfluoxetine (Norf) were determined by comparison of viabilities 48 h after infection with CVB3 (normalized by comparison to cells infected with CVB3 in the presence of guanidine). A similar assay to the primary HTS assay based on ATPLite 1-Step was applied to determine EC50 and CC50 of the compounds. The final concentration range was 0.38 nM to ∼200 μM (numbers next to symbols represent concentrations in μM), and each concentration was tested in triplicate. Averages and standard deviations of triplicates are shown as symbols with error bars. (B) Similar to the EC50 determination, CC50 determinations for fluoxetine and norfluoxetine were determined by determination of relative luciferase luminescence of uninfected HeLa cells compared to that of untreated control wells (DMSO only). (C) Estimated EC50 for fluoxetine as determined by plaque reduction assay. Cells in replicate wells were infected with CVB3 30 min after addition of fluoxetine at the concentrations indicated. After 6 h, the cells were harvested and the titer of intracellular virus was determined by limiting dilution plaque assay. Symbols represent single determinations, and a representative of two separate experiments is shown. (D) Reduction of viral titer by treatment of infected cells with fluoxetine and norfluoxetine. Cells in replicate wells were infected with CVB3 30 min after addition of the drugs at an effective concentration of 6.25 μM. After 6 h, the cells were harvested and the titer of intracellular virus was determined by limiting dilution plaque assay. Averages and standard deviations of triplicates are shown as shaded bars and error bars, respectively.
We used a virus titer reduction assay to confirm these results. Conventional plaque assays were employed to quantify the amount of CVB3-H3 produced by HeLa-RW cells following pretreatment with fluoxetine and norfluoxetine prior to infection at an MOI of 1. Guanidine, a known potent inhibitor of enterovirus replication (2), was used as a positive control. Fluoxetine suppressed CVB3 replication with an estimated EC50 of approximately 2 μM (Fig. 2C), and both fluoxetine and norfluoxetine reduced viral titers by more than 3 orders of magnitude. Fluoxetine and norfluoxetine also protected Vero cells from cytolysis following CVB3-H3 infection (data not shown).
We added fluoxetine directly to virus stocks and incubated them for 1 h at 37°C to determine if the compound had a direct virucidal effect. The infectious titer, as determined by plaque assays, was not altered by this exposure (identical results in two separate experiments; data not shown).
Inhibition of viral RNA and viral protein accumulation by fluoxetine and norfluoxetine.
To better understand the inhibition of enterovirus replication by fluoxetine and norfluoxetine, we assessed their global impact on the synthesis of viral protein and RNA. We infected cells at a high multiplicity of infection with CVB3-H3 and harvested total cellular RNA and protein after 6 h. Immunoblotting revealed a total abrogation of detectable viral protein (Fig. 3A). RT-PCR analysis revealed that both compounds markedly reduced the amount of CVB3 RNA at this time point by more than 1,000-fold compared to untreated cells (Fig. 3B) These effects were comparable in magnitude to that of guanidine HCl, which interferes with the activity of viral protein 2C, thereby suppressing the formation of negative viral RNA (2, 33).
Fig 3.
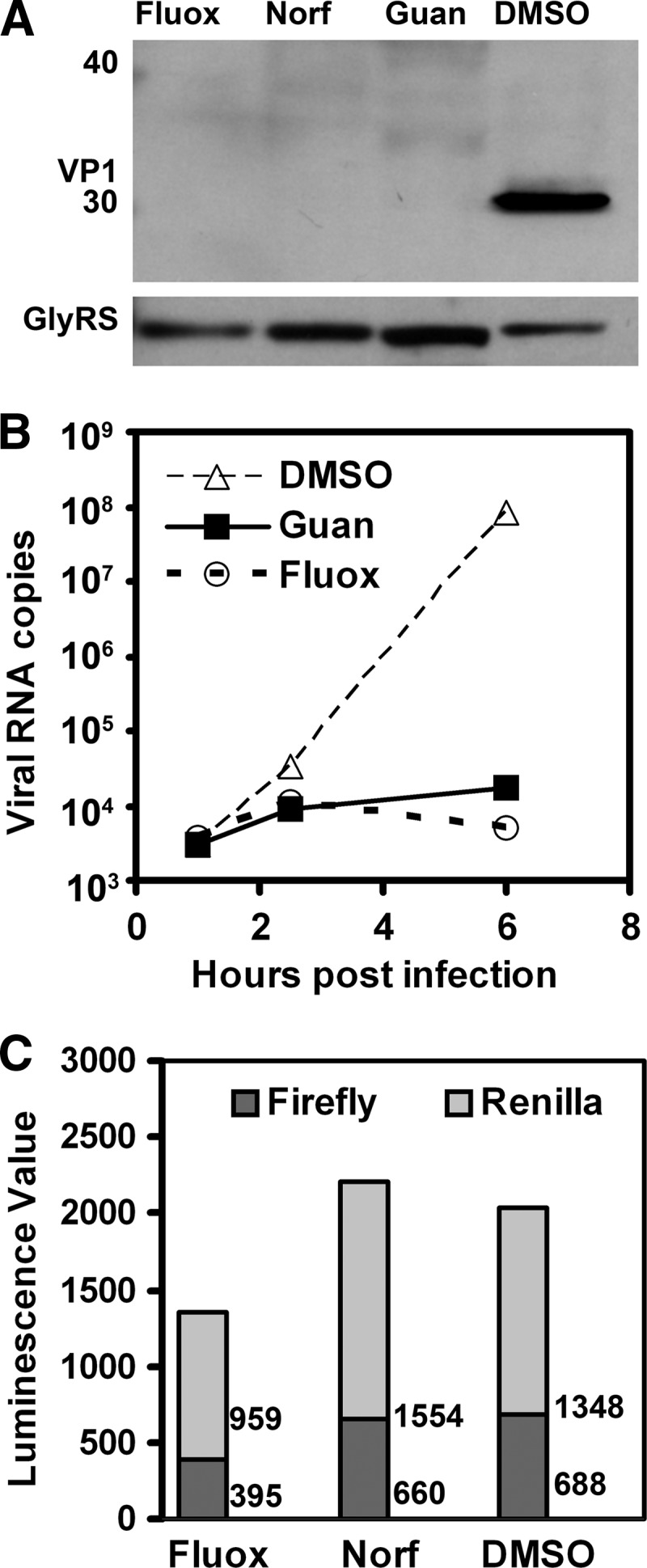
Assays of antiviral activity. (A) Immunoblot detection of viral capsid protein 6 h after infection with CVB3-H3; no viral protein is detected in cells treated with fluoxetine (Fluox) and norfluoxetine (Norf). Immunoblot detection of glycyl-tRNA synthease protein (GlyRS) (a housekeeping protein) is shown as a loading control. (B) RT-PCR detection of CVB3 RNA in cells treated with guanidine (Guan) or fluoxetine beginning 30 min before inoculation of cell cultures. Symbols represent single determinations, and a representative of two separate experiments is shown. (C) Dual-luciferase assay of Renilla and firefly luciferase in cells transfected with plasmid encoding a bicistronic RNA with firefly luciferase expressed under the control of the CVB3 IRES. The values next to the bars are average luminescence readings of duplicate samples for Renilla luciferase and firefly luciferase, respectively. The values of mock-transfected HeLa cells are 0.3 and 0.1 for Renilla luciferase and firefly luciferase, respectively. A representative of two separate experiments with similar results is shown.
Fluoxetine and norfluoxetine target an early step of CVB3 infection after viral entry.
RT-PCR analysis was used to determine if the two compounds block the entry of CVB3 into cells or translation of the viral genome, as these are key initial steps in the enterovirus life cycle (44). We found that similar amounts of viral RNA were initially detected in cells pretreated with fluoxetine 30 min before infection, compared to untreated cells (Fig. 3B). Subsequently, viral RNA markedly increased in untreated cells, while viral RNA levels remained fairly constant within cells treated with guanidine or fluoxetine. Both fluoxetine and norfluoxetine inhibited CVB3 replication, even if added 2 h postinfection. Similar results were found when we used CVB3-H3 virus in a plaque reduction assay (data not shown).
We used a dual-luciferase reporter system to examine the possibility that fluoxetine and norfluoxetine interfere with translation of the viral RNA genome under the control of the viral internal ribosome entry site (IRES). We added the compounds to HeLa cells 24 h after transfection with a bicistronic construct in which the Renilla and firefly luciferases are expressed from cap-dependent and IRES-mediated ribosome mechanisms, respectively. At 48 h after transfection, neither fluoxetine nor norfluoxetine reduced the expression of firefly luciferase (Fig. 3C). Similarly, the relative expression of firefly luciferase and Renilla luciferase in cells stably transfected with this plasmid construct was not affected by addition of norfluoxetine or fluoxetine to the culture medium (data not shown).
These data indicate that CVB3 entry and translation of the viral RNA are unaffected by norfluoxetine or fluoxetine and that virus replication is being impeded at a subsequent intracellular step in virus replication. As an additional confirmatory assay, we infected HeLa-RW cells with CVB3-EGFP and added either norfluoxetine or fluoxetine coincident with or after infection. Both compounds ablated expression of EGFP from this recombinant virus, even when added 2 h after inoculation (Table 1).
Table 1.
EGFP fluorescence of HeLa cells infected with CVB3-EGFP
| Compound | % of GFP+ cells at addition time postinfectiona: |
||
|---|---|---|---|
| 0 h | 1 h | 2 h | |
| Fluoxetine | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Norfluoxetine | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| DMSO only | 33.8 ± 0.0 | 31.3 ± 2.3 | 29.9 ± 4.4 |
Percentages of GFP-positive cells after 8 h of infection with CVB3-EGFP viruses are shown as means ± standard deviations from duplicate wells. Similar results were observed after 14 h of infection. Shown are results from a representative of two separate experiments with similar results.
Fluoxetine and norfluoxetine have antiviral activity against other group B coxsackieviruses.
To confirm that the antiviral activity of fluoxetine is not limited to the CVB3 strain used in our screening assay, we performed virus titer reduction assays using additional coxsackievirus B isolates (B1, B2, and B3), including two strains known to be myocarditic in humans. Fluoxetine consistently reduced the amount of virus produced following cell infection by 3 to 4 orders of magnitude (Fig. 4).
Fig 4.
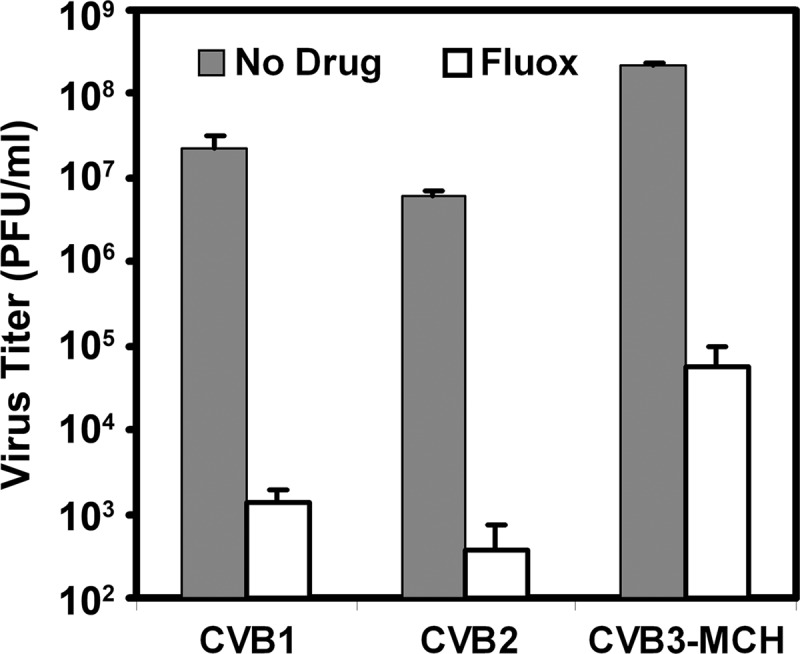
Fluoxetine inhibits replication of clinical isolates of coxsackieviruses B1 to B3. HeLa-RW cells were infected with clinical isolates of three serotypes of coxsackieviruses at a multiplicity of infection of approximately 1 with or without pretreatment 30 min prior with 6 μM fluoxetine (Fluox). After 6 h, cells were harvested and the titer of intracellular virus was determined by limiting dilution plaque assay. Averages of triplicate determinations are shown in the bar graph, with standard deviations shown by error bars.
DISCUSSION
We used a molecular screening approach to search for small molecule enterovirus inhibitors. In the primary screen, we used a low MOI of 0.007 for the infection and a 48-h incubation period, favoring compounds with low toxicity and moderate to low antiviral activities. Of the 1,120 compounds in the Prestwick Chemical Library, seven [lycorine hydrochloride, dibucaine, mefloquine hydrochloride, flupentixol dihydrochloride cis-(Z), zuclopenthixol hydrochloride, cycloheximide, and fluoxetine hydrochloride] showed at least 50% inhibition of cytopathic effect in our screening assay.
Lycorine hydrochloride, a plant alkaloid, was previously shown to inhibit poliovirus replication in vitro (17, 43) but is known to be highly toxic to mammalian cells. Cycloheximide is also known to inhibit poliovirus replication but is a potent inhibitor of cellular translation (40). Dibucaine and mefloquine are quinolone derivatives. Dibucaine (also known as cinchocaine) is a potent inhibitor of plasma cholinesterase and is currently employed as a topically applied anesthetic cream for relief of hemorrhoid-associated pain, while mefloquine is a widely used antimalarial drug. Flupentixol (also known as flupenthixol) dihydrochloride and zuclopenthixol hydrochloride are closely related thioxanthene antipsychotic medications which act as antagonists of dopamine, serotonin, adrenaline, and histamine receptors. While effective as antipsychotics, chronic use of these neuroleptic agents carries a risk of blood dyscrasias and antipyramidal side effects. Fluoxetine is classified as a selective serotonin uptake inhibitor, although it also engages membrane receptors of other neurotransmitters. Fluoxetine is approved by the U.S. Food and Drug Administration for major depressive disorder, obsessive compulsive disorder, bulimia nervosa, and panic disorder, and its safety profile in long-term use is well known.
We applied four separate secondary screen assays to confirm the antiviral activities of fluoxetine and norfluoxetine. Specifically, our data demonstrated that both fluoxetine and its metabolite norfluoxetine markedly reduced replication of the viral genome and completely blocked viral protein synthesis. Viral replication appeared to be totally abrogated in an assay based on detection of green fluorescent protein expression from a recombinant CVB3 variant (CVB3-EGFP). Most importantly, the production of infectious CVB3 virions, as detected by plaque assays, was reduced by approximately 4 orders of magnitude. This antiviral activity was also seen with three other coxsackievirus B strains, including a CVB1 strain isolated during the 2007 nationwide outbreak of cases involving newborn infants (42) and a myocarditic CVB3 variant isolated from a child with fatal myocarditis (21). We note that fluoxetine and its in vivo metabolite norfluoxetine achieved EC50s comparable to the serum concentrations achieved during steady-state dosing of fluoxetine. Specifically, Chouinard measured mean steady-state serum concentrations of 1.6 μM and 1.0 μM during chronic therapy for depression (9). In view of the very long half-lives of fluoxetine (1 to 3 days) and norfluoxetine (7 to 15 days) at steady state (25), inhibitory concentrations of both compounds would likely be present throughout the dosing interval.
The mechanism of the antiviral activity of fluoxetine is unclear. The compound (RS)-N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]propan-1-amine) does not share structural homology with other known inhibitors of enteroviruses or genera within the picornavirus family (12, 37). One might speculate that fluoxetine and norfluoxetine could block viral entry, based on their engagement of the cell surface G protein-coupled receptors for dopamine, serotonin, and other neurotransmitters (3). However, RT-PCR detection of viral RNA and time-of-addition experiments indicated that virus entry was not impeded by the compound, indicating an intracellular site of action. At this time, the basis for the antiviral effects of fluoxetine remains unclear, although it has been shown to affect the transcription of a wide variety of cellular genes and alter microRNA levels (23, 28).
The earliest distinct step after virus entry is IRES-mediated translation of a single polyprotein from the viral genome. Fluoxetine did not interfere with translation from a bicistronic mRNA, suggesting that subsequent steps in the picornavirus life cycle might be affected, including the rearrangement of cellular membranes by the viral 2BC or 3A proteins or synthesis of viral RNA by inhibition of the NTPase/helicase activity of protein 2C (2), among other possibilities.
Our study has several limitations, including our study of only a limited subset of enteroviruses. While coxsackieviruses B1, B2, and B3 represent 11% of all enteroviruses detected at sites of the National Enterovirus Surveillance System in the years between 1970 and 2005 (19) and 25% of all serotypes reported between 2006 and 2008 (7), they are members of the Human enterovirus B (HEV-B) genus; additional studies will be required to determine the true breadth of antiviral activity of fluoxetine.
Moreover, attempts to “repurpose” fluoxetine as an antiviral may be limited by nonneurological effects of its inhibition of serotonin uptake and pharmacological off-target effects. Serotonin selective uptake inhibitors have also been associated with an enhanced risk of bleeding when given with inhibitors of platelet function, likely because of the involvement of serotonin in platelet activation (1, 11, 22). This could prove to be a liability, since enterovirus infections have been associated with hemorrhage, particularly in newborns. Fluoxetine is also associated with biochemical activities other than inhibition of serotonin uptake, including anti-inflammatory effects and inhibition of cytokine release in response to Toll-like receptor stimulation (35). Fortunately, there do not appear to be reports of exacerbation of viral infections by concomitant use of fluoxetine, which is frequently used in individuals with HIV infection (15, 18).
In conclusion, these studies reveal unexpected antiviral activity by the widely used drug fluoxetine and its major metabolite, norfluoxetine. Understanding their mechanisms of action against coxsackieviruses will add to our understanding of enterovirus replication and lead to assessment of their potential clinical utility for the treatment of serious enterovirus infections.
ACKNOWLEDGMENTS
This work was supported by grants from the Today's and Tomorrow's Children's Fund and the UCLA Department of Pediatrics Nanopediatrics Program.
We acknowledge the valuable technical assistance of Winnie Wong and Jongsang Lee and thank Laura Sheehan for assistance in manuscript preparation.
Footnotes
Published ahead of print 2 July 2012
REFERENCES
- 1. Alderman CP, Moritz CK, Ben-Tovim DI. 1992. Abnormal platelet aggregation associated with fluoxetine therapy. Ann. Pharmacother. 26:1517–1519 [DOI] [PubMed] [Google Scholar]
- 2. Barton DJ, Flanegan JB. 1997. Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J. Virol. 71:8482–8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beasley CM, Masica DN, Potvin JH. 1992. Fluoxetine: a review of receptor and functional effects and their clinical implications. Psychopharmacology (Berl.) 107:1–10 [DOI] [PubMed] [Google Scholar]
- 4. Brown BA, Oberste MS, Alexander JP, Jr, Kennett ML, Pallansch MA. 1999. Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J. Virol. 73:9969–9975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cardosa MJ, et al. 2003. Molecular epidemiology of human enterovirus 71 strains and recent outbreaks in the Asia-Pacific region: comparative analysis of the VP1 and VP4 genes. Emerg. Infect. Dis. 9:461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Centers for Disease Control and Prevention 2008. Increased detections and severe neonatal disease associated with coxsackievirus B1 infection—United States, 2007. MMWR Morb. Mortal. Wkly. Rep. 57:553–556 [PubMed] [Google Scholar]
- 7. Centers for Disease Control and Prevention 2010. Nonpolio enterovirus and human parechovirus surveillance—United States, 2006–2008. MMWR Morb. Mortal. Wkly. Rep. 59:1577–1580 [PubMed] [Google Scholar]
- 8. Cherry JD, Krogstad PK. 2009. Enteroviruses and parechoviruses, p 1984–2041 In Feigin RD, Cherry JD, Demmler GJ, Kaplan S. (ed), Textbook of pediatric infectious diseases, 6th ed WB Saunders Co., Philadelphia, PA [Google Scholar]
- 9. Chouinard G. 1985. A double-blind controlled clinical trial of fluoxetine and amitriptyline in the treatment of outpatients with major depressive disorder. J. Clin. Psychiatry 46:32–37 [PubMed] [Google Scholar]
- 10. Corless CE, et al. 2002. Development and evaluation of a ‘real-time’ RT-PCR for the detection of enterovirus and parechovirus RNA in CSF and throat swab samples. J. Med. Virol. 67:555–562 [DOI] [PubMed] [Google Scholar]
- 11. Dalton SO, et al. 2003. Use of selective serotonin reuptake inhibitors and risk of upper gastrointestinal tract bleeding: a population-based cohort study. Arch. Intern. Med. 163:59–64 [DOI] [PubMed] [Google Scholar]
- 12. De Palma AM, Vliegen I, De Clercq E, Neyts J. 2008. Selective inhibitors of picornavirus replication. Med. Res. Rev. 28:823–884 [DOI] [PubMed] [Google Scholar]
- 13. Feuer R, Mena I, Pagarigan R, Slifka MK, Whitton JL. 2002. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J. Virol. 76:4430–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fry EE, Stuart DI. 2010. Virion structure, p 59–72 In Ehrenfeld DE, Roos RP. (ed), The picornaviruses. ASM Press, Washington, DC [Google Scholar]
- 15. Furlanut M, Soardo G, Donnini D, Sechi L, Franceschi L. 2010. Fluoxetine disposition in patients with chronic hepatitis C treated with interferon-alpha. Clin. Pharmacokinet. 49:767–772 [DOI] [PubMed] [Google Scholar]
- 16. Gong E, Ivens T, Van den Eynde C, Hallenberger S, Hertogs K. 2008. Development of robust antiviral assays for profiling compounds against a panel of positive-strand RNA viruses using ATP/luminescence readout. J. Virol. Methods 151:121–125 [DOI] [PubMed] [Google Scholar]
- 17. Hwang YC, Chu JJ, Yang PL, Chen W, Yates MV. 2008. Rapid identification of inhibitors that interfere with poliovirus replication using a cell-based assay. Antiviral Res. 77:232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jong E, et al. 2010. Predictors and treatment strategies of HIV-related fatigue in the combined antiretroviral therapy era. AIDS 24:1387–1405 [DOI] [PubMed] [Google Scholar]
- 19. Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. 2006. Enterovirus surveillance—United States, 1970–2005. MMWR Surveill. Summ. 55:1–20 [PubMed] [Google Scholar]
- 20. Knowlton KU, Jeon ES, Berkley N, Wessely R, Huber S. 1996. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J. Virol. 70:7811–7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krogstad P, Hammon R, Halnon N, Whitton JL. 2008. Fatal neonatal myocarditis caused by a recombinant human enterovirus-B variant. Pediatr. Infect. Dis. J. 27:668–669 [DOI] [PubMed] [Google Scholar]
- 22. Labos C, Dasgupta K, Nedjar H, Turecki G, Rahme E. 2011. Risk of bleeding associated with combined use of selective serotonin reuptake inhibitors and antiplatelet therapy following acute myocardial infarction. CMAJ 183:1835–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lauterbach EC. 2012. Psychotropic drug effects on gene transcriptomics relevant to Parkinson's disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 38:107–115 [DOI] [PubMed] [Google Scholar]
- 24. Lee MS, et al. 2010. An investigation of epidemic enterovirus 71 infection in Taiwan, 2008: clinical, virologic, and serologic features. Pediatr. Infect. Dis. J. 29:1030–1034 [DOI] [PubMed] [Google Scholar]
- 25. Lemberger L, et al. 1985. Fluoxetine: clinical pharmacology and physiologic disposition. J. Clin. Psychiatry 46:14–19 [PubMed] [Google Scholar]
- 26. Liu CC, Tseng HW, Wang SM, Wang JR, Su IJ. 2000. An outbreak of enterovirus 71 infection in Taiwan, 1998: epidemiologic and clinical manifestations. J. Clin. Virol. 17:23–30 [DOI] [PubMed] [Google Scholar]
- 27. Ma E, Chan KC, Cheng P, Wong C, Chuang SK. 2010. The enterovirus 71 epidemic in 2008—public health implications for Hong Kong. Int. J. Infect. Dis. 14:e775–e780 [DOI] [PubMed] [Google Scholar]
- 28. Millan MJ. 2011. MicroRNA in the regulation and expression of serotonergic transmission in the brain and other tissues. Curr. Opin. Pharmacol. 11:11–22 [DOI] [PubMed] [Google Scholar]
- 29. Miller JP, Geng Y, Ng HL, Yang OO, Krogstad P. 2009. Packaging limits and stability of HIV-1 sequences in a coxsackievirus B vector. Vaccine 27:3992–4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oberste M. 2008. Comparative genomics of the coxsackie B viruses and related enteroviruses. Curr. Top. Microbiol. Immunol. 323:33–47 [DOI] [PubMed] [Google Scholar]
- 31. Oberste MS, Maher K, Pallansch MA. 2004. Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes. J. Virol. 78:855–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oberste MS, Penaranda S, Pallansch MA. 2004. RNA recombination plays a major role in genomic change during circulation of coxsackie B viruses. J. Virol. 78:2948–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pfister T, Wimmer E. 1999. Characterization of the nucleoside triphosphatase activity of poliovirus protein 2C reveals a mechanism by which guanidine inhibits poliovirus replication. J. Biol. Chem. 274:6992–7001 [DOI] [PubMed] [Google Scholar]
- 34. Rotbart HA, Webster AD. 2001. Treatment of potentially life-threatening enterovirus infections with pleconaril. Clin. Infect. Dis. 32:228–235 [DOI] [PubMed] [Google Scholar]
- 35. Sacre S, Medghalchi M, Gregory B, Brennan F, Williams R. 2010. Fluoxetine and citalopram exhibit potent antiinflammatory activity in human and murine models of rheumatoid arthritis and inhibit toll-like receptors. Arthritis Rheum. 62:683–693 [DOI] [PubMed] [Google Scholar]
- 36. Slifka MK, Pagarigan R, Mena I, Feuer R, Whitton JL. 2001. Using recombinant coxsackievirus B3 to evaluate the induction and protective efficacy of CD8+ T cells during picornavirus infection. J. Virol. 75:2377–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thibaut HJ, De Palma AM, Neyts J. 2012. Combating enterovirus replication: state-of-the-art on antiviral research. Biochem. Pharmacol. 83:185–192 [DOI] [PubMed] [Google Scholar]
- 38. Thibaut HJ, et al. 2011. Towards the design of combination therapy for the treatment of enterovirus infections. Antiviral Res. 90:213–217 [DOI] [PubMed] [Google Scholar]
- 39. Tracy S, et al. 2002. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J. Virol. 76:12097–12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Urzainqui A, Carrasco L. 1989. Degradation of cellular proteins during poliovirus infection: studies by two-dimensional gel electrophoresis. J. Virol. 63:4729–4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Venkatesan A, Sharma R, Dasgupta A. 2003. Cell cycle regulation of hepatitis C and encephalomyocarditis virus internal ribosome entry site-mediated translation in human embryonic kidney 293 cells. Virus Res. 94:85–95 [DOI] [PubMed] [Google Scholar]
- 42. Verma NA, et al. 2009. Outbreak of life-threatening coxsackievirus B1 myocarditis in neonates. Clin. Infect. Dis. 49:759–763 [DOI] [PubMed] [Google Scholar]
- 43. Vrijsen R, Vanden Berghe DA, Vlietinck AJ, Boeye A. 1986. Lycorine: a eukaryotic termination inhibitor? J. Biol. Chem. 261:505–507 [PubMed] [Google Scholar]
- 44. Whitton JL, Cornell CT, Feuer R. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3:765–776 [DOI] [PubMed] [Google Scholar]
- 45. Wikswo ME, et al. 2009. Increased activity of Coxsackievirus B1 strains associated with severe disease among young infants in the United States, 2007–2008. Clin. Infect. Dis. 49:e44–e51 [DOI] [PubMed] [Google Scholar]
- 46. Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73 [DOI] [PubMed] [Google Scholar]