

Abstract
New drugs to treat malaria must act rapidly and be highly potent against asexual blood stages, well tolerated, and affordable to residents of regions of endemicity. This was the case with chloroquine (CQ), a 4-aminoquinoline drug used for the prevention and treatment of malaria. However, since the 1960s, Plasmodium falciparum resistance to this drug has spread globally, and more recently, emerging resistance to CQ by Plasmodium vivax threatens the health of 70 to 320 million people annually. Despite the emergence of CQ resistance, synthetic quinoline derivatives remain validated leads for new drug discovery, especially if they are effective against CQ-resistant strains of malaria. In this study, we investigated the activities of two novel 4-aminoquinoline derivatives, TDR 58845, N1-(7-chloro-quinolin-4-yl)-2-methyl-propane-1,2-diamine, and TDR 58846, N1-(7-chloro-quinolin-4-yl)-2,N2,N2-trimethylpropane-1,2-diamine and found them to be active against P. falciparum in vitro and Plasmodium berghei in vivo. The P. falciparum clones and isolates tested were susceptible to TDR 58845 and TDR 58846 (50% inhibitory concentrations [IC50s] ranging from 5.52 to 89.8 nM), including the CQ-resistant reference clone W2 and two multidrug-resistant parasites recently isolated from Thailand and Cambodia. Moreover, these 4-aminoquinolines were active against early and late P. falciparum gametocyte stages and cured BALB/c mice infected with P. berghei. TDR 58845 and TDR 58846 at 40 mg/kg were sufficient to cure mice, and total doses of 480 mg/kg of body weight were well tolerated. Our findings suggest these novel 4-aminoquinolines should be considered for development as potent antimalarials that can be used in combination to treat multidrug-resistant P. falciparum and P. vivax.
INTRODUCTION
Approximately 40% of the world population lives in areas of malarial endemicity. Estimates indicate that the disease causes several hundred million cases and about 1.2 million deaths each year (25, 33). The two most prevalent species of Plasmodium that cause malaria in humans are P. falciparum and P. vivax. Severe disease and resistance to antimalarials have been documented for both species (43), and efforts to control malaria have become more challenging in recent years due to widespread drug resistance (12, 16).
Although P. falciparum resistance has been reported for most antimalarials, P. vivax resistance is generally limited to chloroquine (CQ), which was first reported in the late 1980s in Papua New Guinea and Indonesia (2, 9, 46). In addition, P. vivax has shown innate resistance to sulfadoxine but developed additional resistance when in combination with pyrimethamine in many areas (18). Alternative therapies such as artemisinin combinations for treatment of CQ-resistant malaria parasite infections have been developed. However, these therapies do not meet the simplicity of use and low cost of CQ (55).
For many decades, the least expensive and most effective antimalarial was CQ, a 4-aminoquinoline drug. CQ has a rapid onset of action and low toxicity and is well tolerated (54). For these reasons, it was widely used worldwide in countries where malaria is endemic. CQ acts by inhibiting hemozoin aggregation in the digestive vacuole of the malaria parasite (14, 37). The heavy use of CQ resulted in the emergence of CQ resistance starting in Southeast Asia, South America, and Oceania and then reaching almost every corner of areas of malaria endemicity (17). Today more than 80% of isolates of P. falciparum are resistant to CQ due to mutations in the P. falciparum chloroquine-resistant transporter (pfcrt) (13, 15). Parasite resistance to CQ resulted in a switch to the antifolate combination sulfadoxine-pyrimethamine (SP), which encountered resistance within a few years. Later, multidrug resistance (defined as resistance to three or more drugs) appeared in Southeast Asia and South America in response to the switch to either mefloquine (MFQ) or quinine (QUIN) and then in Africa in the late 1970s (6, 8, 53). Recently, the majority of countries have switched to highly efficient artemisinin derivative combinations (ACTs); nevertheless, there is new evidence for clinical resistance to these drugs (11).
Several molecules such as calcium channel blockers (verapamil [VER]) or tricyclic antidepressant (desipramine [DES]) (5, 26), antipsychotic agents (chlorpromazine [CPZ]) (28), and tricyclic histamine (H-1) receptor antagonists (promethazine [PMZ]) (35) have been reported to reverse the CQ resistance in P. falciparum in vitro. Of those, CPZ, prochlorperazine, and DES have been shown to have a similar effect in vivo in monkeys and humans (27, 35). The mechanism for resistance reversal is still a matter of debate. The common molecular characteristics of these compounds are two lipophilic aromatic residues and a basic amino alkyl side chain. It has been proposed that the aryl residues interact with a lipophilic pocket in the substrate binding site of the PfCRT, while the protonated amino group restores the positive charge that repels the CQ dication (3). In addition, some authors have proposed that a decrease in the CQ uptake could account for drug resistance, while others propose that the CQ-resistant parasites rapidly expel the intracellular drug via a pump-mediated mechanism (4, 26, 32).
Even though there is widespread resistance of P. falciparum and P. vivax to CQ, synthetic quinoline derivatives have remained a validated lead class for new drug discovery. Quinoline derivatives such as MFQ and QUIN have strong activity against CQ-resistant strains of P. falciparum, and there have been several studies testing the activity of 4-aminoquinoline derivatives. In particular, among several 4-aminoquinoline derivatives, synthesized AQ-13, a three-carbon chain analog, exhibited activity against parasites resistant to CQ and MFQ (10) and was progressed to clinical trials (34). Recently, new classes of 4-aminoquinolines have been recognized and possess strong activity in vitro with excellent bioavailability (19, 21, 29, 30, 44). Several of these 4-aminoquinolines have been shown to be more active than CQ in CQ-resistant P. falciparum and to have activity in a mouse model in vivo (24, 36).
In this study, two novel 4-aminoquinoline derivatives, TDR 58845, N1-(7-chloro-quinolin-4-yl)-2-methyl-propane-1,2-diamine and TDR 58846, N1-(7-chloro-quinolin-4-yl)-2,N2,N2-trimethylpropane-1,2-diamine, were evaluated for their ability to eliminate malaria parasites in vitro and in vivo. These drugs share the quinoline core of CQ and possess a methylpropane and a trimethylpropane chain (Fig. 1). Both compounds, TDR 58845 and TDR 58846, were among a group of 130 synthesized by Hoffmann la Roche (20) and were found to be active against CQ-sensitive and CQ-resistant P. falciparum. Also, we have evaluated the potency of TDR 58845 and TDR 58846 against the blood and gametocyte stages of P. falciparum. Additionally, we have assessed the curative activities of these two compounds in vivo using a rodent malaria model. We show that these two compounds have excellent activity against different Plasmodium stages in vitro and in vivo, suggesting that 4-aminoquinoline analogs can be considered for development in combination with other drugs as antimalarials.
Fig 1.

Structures of the 4-aminoquinoline derivatives used in this study. (A) TDR 58845, N1-(7-chloro-quinolin-4-yl)-2-methyl-propane-1,2-diamine; (B) TDR 58846, N1-(7-chloro-quinolin-4-yl)-2,N2,N2-trimethylpropane-1,2-diamine; (C) chloroquine, N4-(7-chloro-quinolin-4-yl)-N1,N1-diethyl-pentane-1,4-diamine.
MATERIALS AND METHODS
Parasites.
Strains and isolates of P. falciparum were cultured in human A+ erythrocytes by standard methods under a low-oxygen atmosphere (5% O2, 5% CO2, 90% N2) according to the method of Trager and Jensen, 1976 (51). The culture medium was RPMI 1640 supplemented with 25 mM HEPES buffer, 10 mM glucose, 2 mM glutamine, and AB+ human plasma. Parasites were maintained in fresh human erythrocytes suspended at a 4% hematocrit in complete medium at 37°C. Stock cultures were subpassaged every 3 to 4 days by transfer of infected red cells to a flask containing complete medium and uninfected erythrocytes.
In this study, the following reference clones of P. falciparum were used: NF54 and its clone 3D7 (CQ sensitive, from an airport in the Netherlands), W2 (CQ and SP resistant, from Indochina), D6 (MQ resistant, from Sierra Leone), and TM90C2B (CQ, MQ, and atovaquone resistant, from Thailand). Additionally, we included two recent P. falciparum isolates, Thai6 and Cam6 (multidrug resistant, from Thailand and Cambodia, provided by François Nosten, Shoklo Malaria Research Unit, Thailand, and Arjen M. Dondorp, Mahidol Oxford Tropical Medicine Research Unit).
Chemicals.
The drugs tested in this study were the 4-aminoquinoline derivatives TDR 58845 and TDR 58846 (United Nations' Children's Fund [UNICEF]/UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases, Geneva, Switzerland). Chloroquine diphosphate (MP Biochemicals, Solon, OH) and mefloquine hydrochloride (Sigma, St. Louis, MO) were used as controls in the in vitro studies. Each of the CQ resistance-reversing compounds used were obtained from Sigma (St. Louis, MO): verapamil (VER), desipramine (DES), chlorpromazine (CPZ), promethazine (PMZ), and chlorpheniramine (CPR). Dihydroartemisinin (WRAIR Chemical Inventory, Silver Spring, MD) was used as a positive control for gametocyte studies.
In vitro drug sensitivity assay.
The sensitivity of the parasites in vitro to the different drugs was tested using a SYBR green I fluorescence-based method and confirmed by radioisotopic method as described previously (28, 49). The experiments were set up in 96-well plates with 2-fold dilutions of each drug across the plate in a total volume of 150 μl and at a final red blood cell concentration of 1.5% (vol/vol). Stock solutions of each drug were prepared in dimethyl sulfoxide (DMSO) at 1 mg/ml. All assay procedures were performed by automated pipetting and dilution with the aid of a programmable Biomek 3000 robotic station (Beckman Coulter, Brea, CA). The experiment was started at an initial parasitemia of 0.5% (>80% rings) synchronous parasite-infected red blood cells (PRBC). The plates were incubated for 72 h at 37°C in an atmosphere of 5% CO2, 5% O2, and 90% N2. The SYBR green I dye-lysis mixture (100 μl) was added to 100 μl of the parasites in black plates that were incubated at room temperature for an hour in the dark. The plates were then read using a fluorescence plate reader (Spectramax Gemini-EM; Molecular Diagnostics) at excitation and emission wavelengths of 480 and 535 nm, respectively. The readings, in relative fluorescence units (RFUs), were plotted against the logarithm of the drug concentration and analyzed by nonlinear regression analysis (DataAspects Corporation, CA) to determine the drug concentration that produced 50% of the observed decline from the maximum readings in the drug-free control wells (50% inhibitory concentrations [IC50s]). In addition, a regression coefficient (R2) of >0.9 was considered significant. All the experiments were done in duplicate and repeated twice.
In vitro analysis of 4-aminoquinolines in combination with potentiating compounds.
Dose-response assays were carried out to obtain the IC50 of TDR 58845 and TDR 58846 alone and in combination with CQ resistance-potentiating compounds (VER, DES, CPZ, PMZ, and CPR). A fixed concentration of 0.9 μM potentiating compound was used, while the concentration of TDR 58845, TDR 58846, CQ, or MQ was varied. The IC50s were determined by nonlinear regression analysis of the fluorescence readings against the logarithm of the drug concentration. In order to determine the degree of action of the CQ resistance-potentiating compounds, we used a response modification index (RMI), that is, the ratio of the IC50 of CQ to the IC50 of the potentiating compound in combination with CQ or with one of the 4-aminoquinolines tested (28). An RMI of 1.0 represents no change in the IC50. RMI values of <1.0 represent the degree of potentiation or synergism. All the experiments were done in duplicate and repeated twice.
In vitro P. falciparum gametocyte production and gametocyte drug sensitivity assays.
Asexual and mature gametocytes of NF54 parasites were cultivated in duplicate as previously described (22, 47) in six-well plates. The CQ-sensitive NF54 clone was used because of its ability to form male and female viable gametocytes and to infect mosquitoes. In brief, 0.1% parasitemia cultures were inoculated in 2.5 ml of culture medium and 7.0% hematocrit (day 0) and were grown for 3 days. On day 4 postinoculation (PI), the volume of media was doubled. The plates were kept in 5% O2 and 5% CO2.
On days 7, 8, and 9 PI, when the gametocytes were at stages I, II and III, respectively, the TDR drugs, CQ, or DHA was added to the cultures to reach a final concentration of 0.1 μM or 1 μM. This procedure was used to determine the effect of the drug on early stages of gametocytemia. Similarly, on days 11, 12, and 13 PI, when the gametocytes are usually at stages III, IV, and V, respectively, drug was added in the cultures at the same concentration range to determine the effect of the drug on late stages of gametocytes. Each plate had two untreated controls and four drug-treated wells. Blood smears were made each day starting on day 7 to document the progression of asexual- as well as sexual-stage parasitemia. A minimum of 1,000 cells were counted. Finally, on day 15 PI, the gametocytes were checked for exflagellation as previously described (41). The experiments were repeated twice, and duplicates were used in each case. The t test was used to calculate significance.
In vivo efficacy study tests using the murine model P. berghei NK65 (modified Thompson test).
To test the TDR compounds for their in vivo activity, P. berghei NK65-infected BALB/c mice (Jackson Laboratories, Bar Harbor, ME) were used in a modified Thompson test (50). Endpoints for efficacy included survivability of mice to 30 days and suppression of parasitemia on day 6 postinfection following administration of the drug. In brief, 5 mice were randomly assigned to each dosage group, and 1 × 106 P. berghei-infected erythrocytes (NK65 strain) were inoculated into the intraperitoneal cavity of female mice weighing 15 to 20 g. On day 3 postinfection, parasitemia was around 1%. Each drug was suspended in 0.5% hydroxyethylcellulose (HEC)–0.1% Tween 80 and was administered orally (p.o.) at 40, 80, and 160 mg/kg of body weight once daily for 3 days on days 3 to 5 postinfection. Blood films were made from tail vein puncture on days 3, 6, 9, 13, 20, 27, and 30 postinfection, and a minimum of a 1,000 cells were counted to monitor and determine parasitemia progression. Mice without a parasitemia on day 30 postinfection were considered cured. To evaluate the toxicity of the drugs, we looked for signs of morbidity or death compared to the untreated controls. The study was approved by the Institutional Animal Care and Use Committee for the University of South Florida.
RESULTS
4-Aminoquinoline derivatives are potent against asexual erythrocytic stages of P. falciparum.
The efficacy of TDR 58845 and TDR 58846 was assessed against a series of lab lines and field isolates of P. falciparum that possess a range of susceptibilities and resistance to antimalarial drugs (Table 1). Both 4-aminoquinolines tested had low-nM IC50s against the CQ-sensitive lines 3D7 and D6. In these parasites, the activities of TDR 58845 and TDR 58846 were equivalent to the activities of CQ for a CQ-sensitive parasite (IC50s < 12 nM). TDR 58845 was more active than CQ against the Southeast Asian CQ-resistant laboratory lines W2 and C2B (89.8 and 35.5 nM, respectively). The IC50s of TDR 58845 for Thailand-Cambodia multidrug-resistant isolates were considerably lower than CQ IC50s but higher than for CQ-sensitive lines (<25 nM). Conversely, TDR 58846 was equipotent against all laboratory lines and field isolates tested (<25 nM) and was significantly more active than CQ. A trend for cross-resistance with CQ was evident for TDR 58845 but was not observed with TDR 58846.
Table 1.
IC50 and IC90 of P. falciparum laboratory lines and field isolates exposed to 4-aminoquinoline derivatives, chloroquine, and mefloquinea
| Strain | TDR58845 |
TDR58846 |
Chloroquine |
Mefloquine |
||||
|---|---|---|---|---|---|---|---|---|
| IC50 | IC90 | IC50 | IC90 | IC50 | IC90 | IC50 | IC90 | |
| 3D7b | 7.75 (4.81) | 11.2 (3.73) | 10.3 (11.4) | 15.3 (9.50) | 8.36 (6.16) | 10.2 (5.88) | 19.3 (2.15) | 44.1 (22.2) |
| W2c | 89.8 (45.6) | 124 (60.5) | 23.1 (10.6) | 32.9 (13.2) | 271 (81.2) | 332 (82.8) | 10.0 (3.01) | 20.1 (7.46) |
| D6b | 9.22 (3.63) | 11.3 (3.12) | 11.8 (9.56) | 14.9 (8.97) | 9.78 (4.77) | 11.1 (4.93) | 26.3 (22.3) | 41.1 (25.3) |
| C2Bc | 35.5 (4.51) | 57.9 (9.69) | 16.3 (12.1) | 21.0 (9.84) | 112 (50.1) | 277 (78.9) | 17.4 (13.7) | 51.9 (3.62) |
| Cam6c | 22.4 (11.8) | 126 (149) | 10.3 (8.08) | 17.1 (7.33) | 160 (95.9) | 287 (268) | 39.5 ( 22.5) | 229 (39.8) |
| Thai6c | 7.65 (0.50) | 13.4 (2.39) | 5.52 (2.71) | 10.4 (3.41) | 26.0 (3.56) | 49.6 (9.92) | 58.1 (34.7) | 135 (94.4) |
Values are in nM (±standard deviation).
CQ sensitive.
CQ resistant.
TDR 58845 is synergistic with verapamil, desipramine, and chlorpromazine.
Previous studies have established the potential of VER and other compounds to reverse CQ resistance in vitro. Response modification indexes (RMI) were used to quantify the synergism of this resistance reversal. RMIs decrease in CQ-resistant parasites when treated with reversal compounds (28, 31, 35). Our data show that in combination with 0.9 μM CQ resistance-reversing compounds, there was an increased sensitivity of TDR 58845 in CQ-resistant isolates, indicating synergism of TDR 58845 with VER, DES, and CPZ (Tables 2 to 4). In addition, the RMIs decreased in combinations of TDR 58845 with VER, DES, or CPZ. The IC50 and RMI values of CQ and TDR 58845 were reduced 2- to 3-fold in W2, C2B, and the multidrug-resistant isolates Thai6 and Cam6 (Tables 2 to 4). In addition, TDR 58845 was synergistic with the CQ resistance reversal drugs chlorpheniramine (CPR) and PMZ (data not shown). Conversely, these compounds were not synergistic with mefloquine. Interestingly, the activity of TDR 58846 is not enhanced by VER, DES, CPZ, CPR, or PMZ; rather, it is reduced slightly by the resistance reversal compounds (Tables 2 to 4).
Table 2.
Susceptibility of P. falciparum to chloroquine, TDR 58845, TDR 58846, and mefloquine in the absence and presence of verapamil
| Parasite isolate | VERa (0.9 μM) | Chloroquine |
TDR 58845 |
TDR 58846 |
Mefloquine |
||||
|---|---|---|---|---|---|---|---|---|---|
| IC50b (SD) | RMIc | IC50 (SD) | RMI | IC50 (SD) | RMI | IC50 (SD) | RMI | ||
| 3D7 | − | 8.36 (6.16) | 7.75 (4.81) | 10.3 (11.4) | 19.3 (2.15) | ||||
| + | 9.44 (8.12) | 1.13 | 10.2 (4.30) | 1.32 | 16.1 (9.41) | 1.55 | 13.5 (0.28) | 0.70 | |
| W2 | − | 271 (81.2) | 89.8 (45.6) | 23.1 (10.6) | 10.0 (3.01) | ||||
| + | 47.7 (20.8) | 0.18 | 22.9 (2.72) | 0.25 | 22.2 (3.65) | 0.96 | 4.70 (0.43) | 0.47 | |
| D6 | − | 9.78 (4.77) | 9.22 (3.63) | 11.8 (9.56) | 26.3 (22.3) | ||||
| + | 11.8 (5.09) | 1.20 | 11.4 (2.33) | 1.24 | 15.0 (11.5) | 1.27 | 34.5 (29.8) | 1.31 | |
| C2B | − | 112 (50.1) | 35.5 (4.51) | 16.3 (12.1) | 17.4 (13.7) | ||||
| + | 35.4 (7.64) | 0.31 | 15.1 (3.63) | 0.42 | 12.3 (8.91) | 0.75 | 8.65 (10.8) | 0.59 | |
| Cam6 | − | 160 (95.8) | 22.4 (11.8) | 10.3 (8.08) | 39.5 (22.5) | ||||
| + | 59.1 (24.7) | 0.37 | 18.6 (0.24) | 0.83 | 13.5 (6.40) | 1.32 | 30.5 (12.8) | 0.77 | |
| Thai6 | − | 26.0 (3.56) | 7.65 (0.50) | 5.52 (2.71) | 58.1 (34.7) | ||||
| + | 13.1 (10.1) | 0.51 | 4.57 (0.33) | 0.60 | 6.71 (0.70) | 1.22 | 28.8 (0.19) | 0.50 | |
+, with VER; −, without VER.
IC50 values are in nM.
RMI is the ratio of IC50 with VER to IC50 without VER.
Table 4.
Susceptibility of P. falciparum to chloroquine, TDR 58845, TDR 58846, and mefloquine in the absence and presence of chlorpromazine
| Parasite isolate | CPZa (0.9 μM) | Chloroquine |
TDR 58845 |
TDR 58846 |
Mefloquine |
||||
|---|---|---|---|---|---|---|---|---|---|
| IC50b (SD) | RMIc | IC50 (SD) | RMI | IC50 (SD) | RMI | IC50 (SD) | RMI | ||
| 3D7 | − | 8.36 (6.16) | 7.75 (4.81) | 10.3 (11.4) | 19.3 (2.15) | ||||
| + | 12.2 (4.02) | 1.46 | 13.5 (0.22) | 1.73 | 13.7 (13.3) | 1.33 | 19.1 (2.32) | 0.99 | |
| W2 | − | 271 (81.2) | 89.8 (45.6) | 23.1 (10.6) | 10.0 (3.01) | ||||
| + | 31.8 (9.86) | 0.12 | 18.8 (7.87) | 0.21 | 23.8 (8.37) | 1.03 | 3.44 (0.34) | 0.34 | |
| D6 | − | 9.78 (4.77) | 9.22 (3.63) | 11.8 (9.56) | 26.3 (22.3) | ||||
| + | 11.7 (4.89) | 1.19 | 11.7 (2.02) | 1.27 | 16.1 (10.2) | 1.37 | 35.3 (26.3) | 1.34 | |
| C2B | − | 112 (50.1) | 35.5 (4.51) | 16.3 (12.1) | 17.4 (13.7) | ||||
| + | 24.7 (16.4) | 0.22 | 11.6 (6.37) | 0.33 | 14.2 (14.6) | 0.87 | 9.91 (11.43) | 0.67 | |
| Cam6 | − | 160 (95.9) | 22.4 (11.8) | 10.3 (8.08) | 39.5 (22.5) | ||||
| + | 35.3 (1.16) | 0.22 | 14.2 (0.28) | 0.63 | 13.0 (7.27) | 1.27 | 28.6 (23.0) | 0.72 | |
| Thai6 | − | 26.0 (3.56) | 7.65 (0.5) | 5.52 (2.71) | 58.1 (34.7) | ||||
| + | 7.76 (0.80) | 0.30 | 6.58 (3.71) | 0.86 | 9.17 (4.84) | 1.66 | 38.9 (17.0) | 0.67 | |
+, with CPZ; −, without CPZ.
IC50 values are in nM.
RMI is the ratio of IC50 with CPZ to IC50 without CPZ.
Table 3.
Susceptibility of P. falciparum to chloroquine, TDR 58845, TDR 58846, and mefloquine in the absence and presence of desipramine
| Parasite isolate | DESa (0.9 μM) | Chloroquine |
TDR 58845 |
TDR 58846 |
Mefloquine |
||||
|---|---|---|---|---|---|---|---|---|---|
| IC50b (SD) | RMIc | IC50 (SD) | RMI | IC50 (SD) | RMI | IC50 (SD) | RMI | ||
| 3D7 | − | 8.36 (6.16) | 7.75 (4.81) | 10.3 (11.4) | 19.3 (2.15) | ||||
| + | 10.5 (6.69) | 1.26 | 11.9 (2.05) | 1.53 | 14.5 (12.3) | 1.40 | 18.6 (1.45) | 0.96 | |
| W2 | − | 271 (81.2) | 89.8 (45.6) | 23.1 (10.6) | 10.0 (3.01) | ||||
| + | 18.9 (1.28) | 0.07 | 10.4 (5.71) | 0.12 | 15.3 (6.41) | 0.66 | 2.44 (1.04) | 0.24 | |
| D6 | − | 9.78 (4.77) | 9.22 (3.63) | 11.8 (9.56) | 26.3 (22.3) | ||||
| + | 11.2 (5.65) | 1.14 | 11.3 (2.60) | 1.23 | 11.0 (5.14) | 0.94 | 34.8 (25.4) | 1.32 | |
| C2B | − | 112 (50.1) | 35.5 (4.51) | 16.3 (12.1) | 17.4 (13.7) | ||||
| + | 21.5 (17.3) | 0.19 | 9.84 (6.21) | 0.28 | 11.7 (8.82) | 0.72 | 10.6 (4.38) | 0.72 | |
| Cam6 | − | 160 (95.9) | 22.4 (11.8) | 10.3 (8.08) | 39.5 (22.5) | ||||
| + | 31.6 (3.93) | 0.20 | 13.0 (0.88) | 0.58 | 12.2 (4.53) | 1.19 | 26.9 (8.73) | 0.68 | |
| Thai6 | − | 26.0 (3.56) | 7.65 (0.5) | 5.52 (2.71) | 58.1 (34.7) | ||||
| + | 6.51 (1.58) | 0.25 | 5.03 (2.50) | 0.66 | 5.36 (0.06) | 0.97 | 26.8 (3.61) | 0.46 | |
+, with DES; −, without DES.
IC50 values are in nM.
RMI is the ratio of IC50 with DES to IC50 without DES.
4-Aminoquinoline derivatives are active against gametocyte stages of P. falciparum.
To compare the effect of 4-aminoquinoline derivatives against the sexual stages of P. falciparum, we treated stage I to III or stage III to V gametocytes for 3 days with 0.1 μM or 1 μM TDR 58845, TDR 58846, CQ, or DHA. DHA eliminates early-stage gametocytes; therefore, it was used as a control in this experiment. Our findings show that CQ (1 μM) kills early-stage gametocytes, as demonstrated in the reduction of stage V gametocytes at day 14 compared to the control (Fig. 2). Both 4-aminoquinoline derivatives TDR 58845 and TDR 58846 are effective at eliminating the early gametocyte stages at 1 μM but were not effective at significantly decreasing the number of stage V gametocytes at 0.1 μM (P = 0.13 and P = 0.16) (Fig. 2). As expected, DHA (0.1 μM) completely inhibited the development of early-stage gametocytes to stage V. The 4-aminoquinoline derivatives tested in this study did not completely eliminate P. falciparum gametocytes but significantly decreased the stage V gametocytemia when gametocyte cultures in stages III to V were treated with 1 μM concentrations (P = 0.02 and P = 0.01). Only 1 μM DHA completely blocked development to stage V gametocytes when treated in stages III to V (Fig. 3).
Fig 2.
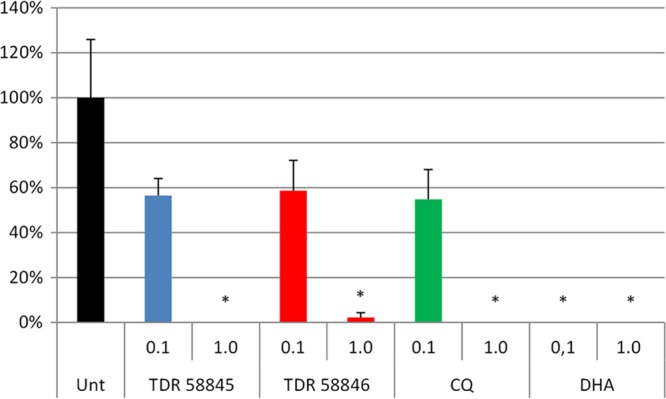
P. falciparum stage V gametocytemia (as a percentage of the control) that develops after treatment of stage I to III gametocytes with TDR 58845, TDR 58846, CQ, or DHA. P. falciparum gametocytes were treated on days 7, 8, and 9 postinoculation with 0.1 μM or 1 μM concentration of the compound, and the percentage of stage V gametocytes was calculated on day 14. CQ, TDR 58845, and TDR 58846 treatments showed significantly fewer stage V gametocytes than the control (Unt) at 1 μM (P = 0.0015). DHA (0.1 μM) was more potent and completely prevented formation of stage V gametocytes. *, statistically significant compared to the untreated control (t test).
Fig 3.

P. falciparum stage V gametocytemia (as a percentage of the control) that developed after stage III to V gametocytes were treated with TDR 58845, TDR 58846, CQ, or DHA. P. falciparum (NF54) gametocytes were treated on days 11, 12, and 13 postinoculation with a 0.1 μM or 1 μM concentration of the compound, and the percentage of stage V gametocytes was calculated on day 14 postinoculation. No decrease in gametocytemia was observed in CQ-treated parasites. TDR 58845- and TDR 58846-treated gametocytes showed a decrease in stage V gametocytemia that was significant at 1.0 μM (P = 0.02 and P = 0.01). No stage V gametocytes were found in 1 μM DHA-treated cultures. *, statistically significant compared to the untreated control (Unt) by the t test.
4-Aminoquinoline drugs (TDR 58845 and TDR 58846) cure P. berghei infection in mice.
The activity of the 4-aminoquinoline derivatives was tested in vivo using a modified Thompson test (3 consecutive days of dosing) with P. berghei-infected mice. A summary of the in vivo results is presented in Table 5. Both 4-aminoquinolines (TDR 58845 and TDR 58846) showed potent activity in vivo against P. berghei. In fact, 100% suppression of parasitemia was observed on day 6 postinfection, and both compounds produced cures at all doses tested (40, 80, and 160 mg/kg). In particular, 40 mg/kg of TDR 58845 cured 4 of 5 mice over the course of 30 days, and only one mouse had delayed parasitemia on day 30 postinfection (Fig. 4A). An 80-mg/kg dose of TDR 58846 was completely curative in all the experimental mice, and recrudescence was observed in only one mouse at 40 mg/kg dose of TDR 58846. No toxicity was observed at 160 mg/kg of TDR 58846 (total dose, 480 mg/kg; Fig. 4B). In summary, our results demonstrate remarkable efficacy and tolerability of 4-aminoquinoline derivatives in the P. berghei model in vivo.
Table 5.
Summary of activities of TDR 58845 and TDR 58846 against blood stage infections with P. berghei in vivo
| Drug | Dose (mg/kg/day for 3 days) | Vehicle | Route | Outcome or no. of days to death | Activity |
|---|---|---|---|---|---|
| None | None | None | None | 7–8 | NAa |
| TDR 58845 | 40 | 0.5% HEC–0.1% Tween 80 | p.o. | 30 | Curative |
| 80 | All mice survived | Curative | |||
| 160 | 4/5 mice survived | Curative | |||
| TDR 58846 | 40 | 0.5% HEC–0.1% Tween 80 | p.o. | 27 (4/5 survived) | Curative |
| 80 | All mice survived | Curative | |||
| 160 | All mice survived | Curative |
NA, not applicable.
Fig 4.
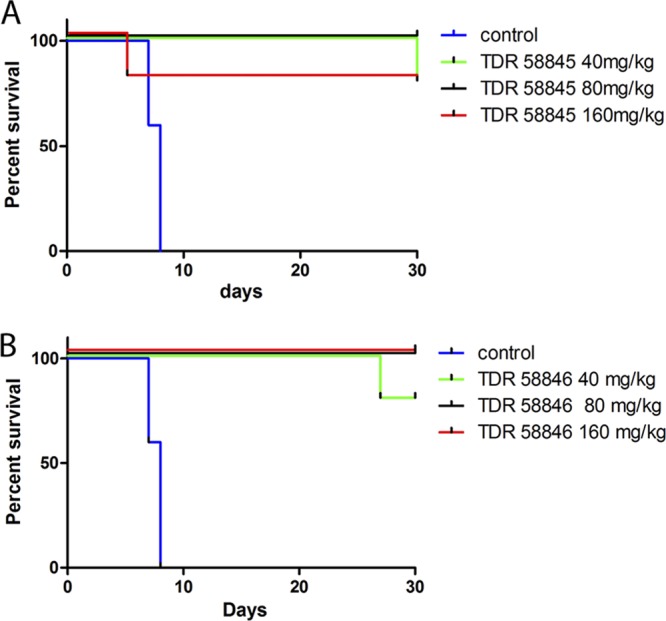
Survival curves of mice untreated or treated with the 4-aminoquinoline analogs TDR 58845 and TDR 58846. (A) TDR 58845 treatment. All mice at doses of 40 and 80 mg/kg survived until the end of the experiment (day 30), although one mouse had parasitemia at day 30 and one mouse died for unknown reasons in the 160-mg/kg group on day 5. (B) TDR 58846 treatment. Recrudescence was observed only in the lower-dose group. All the mice in groups receiving 80 and 160 mg/kg survived and showed no sign of parasitemia.
DISCUSSION
In this study, we assessed the activity of two novel 4-aminoquinoline derivatives (TDR 58845 and TDR 58846) against the erythrocytic and gametocyte stages of P. falciparum in vitro and in vivo. Our results indicate that the 4-aminoquinoline derivatives show excellent potential as a treatment for erythrocytic stages of CQ-sensitive and -resistant P. falciparum, as well as gametocyte stages of a CQ-sensitive line. In addition, both 4-aminoquinolines cured P. berghei-infected mice at concentrations as low as 40 mg/kg per day and were well tolerated at a total dose of 480 mg/kg over 3 days.
One of the most important findings of this study is that both novel 4-aminoquinolines are more potent than CQ against CQ-resistant P. falciparum. In particular, TDR 58846 produced consistently low IC50s when tested against CQ-resistant parasites. The highest IC50 for TDR 58846 was 23 nM for the CQ-resistant W2, while none of the IC50s in the multidrug-resistant isolates exceeded 11 nM. These values are much lower than reported in previous studies (45) of four different 4-aminoquinoline analogs (related to the compounds in this study) against the K1 CQ-resistant line (IC50s ranged between 46 and 61 nM). Interestingly, overcoming CQ cross-resistance was not complete for TDR 58845. The IC50s of TDR 58845 against CQ-resistant laboratory lines and field isolates have a positive correlation with the IC50s of CQ in these parasites, suggesting that TDR 58845 shares cross-resistance with CQ. Our TDR 58845 data are in accordance with the results of previous studies by Ridley et al. (45) that show cross-resistance of the 4-aminoquinolines Ro 47-0543, Ro 41-3118, Ro 47-9396, and Ro 48-0346 with CQ. In particular, in our study the highest IC50 for TDR 58845 was 90 nM in the CQ-resistant W2, yet IC50s were lower than 23 nM for the recent multidrug-resistant field isolates. These results indicate a potential role of the novel 4-aminoquinolines in this study to treat CQ-resistant malaria and demonstrate the potential of these drugs to treat multidrug-resistant malaria.
Here, we report potency of two novel 4-aminoquinolines that is similar to activities reported for AQ-13 in the CQ-resistant lines Dd2, TM91-C235, and TM92-C815 (10). In comparison to CQ, both TDR compounds and AQ-13 have shorter alkyl side chains that are likely to affect cross-resistance with CQ (10). Interestingly, the two compounds containing a tertiary amine at the end of their side chains (AQ-13 and TDR 58846) are the most potent and possess no cross-resistance with CQ in vitro. In contrast, TDR 58845, with its side chain terminated by a primary amine, is potent, yet cross-resistance with CQ was evident. Similarly, the activities of AQ-13 and TDR58846 are not potentiated by resistance reversal drugs VER, CPR, and DES (reference 10 and this study). The position of the N2 in the alkyl chain should affect the pK of the compounds, with TDR 58846 expected to be more basic by approximately 1.5 pK units.
Since the initial observation on the CQ-reversing potential of VER and DES (5, 26), more than 40 compounds that enhance the activity of CQ have been identified (42, 52). We examined the classical series of CQ reversal drugs in combination with TDR 58845 and TDR 58846. We found that the combination of TDR 58845 with VER, DES, CPZ, PMZ, and CPR increases the sensitivity in vitro to this drug with the CQ-resistant lines (W2 and C2B) and the field isolates Cam6. The activity of TDR 58845 in Thai6 was slightly improved by the CQ reversal drugs. Ridley found the activity of four 4-aminoquinolines with shortened side chains was not potentiated by DES (45). The fact that TDR 58845 activity is potentiated by CQ reversal compounds is in agreement with the observation of TDR 58845 showing cross-resistance with CQ and suggests a similar mechanism of action for these two drugs. More importantly, we found that activity of TDR 58846 was not improved by any of the CQ resistance-reversing compounds, nor was there evidence of cross-resistance with CQ. This observation is in accordance to what has been reported for AQ-13 and other aminoquinoline derivatives when combined with verapamil (10). In this work, we included CQ-resistant parasites with multiple unique pfcrt alleles, and TDR 58846 was equally effective against them all. These data suggest TDR 58846 may not interact with mutant pfcrt. This may be due to structural differences caused by the variation in the diamine groups and in the number of methyl groups between the two tested compounds. We hypothesize that TDR 58846 is unable to interact with the CQ transporter receptor, which would imply that this compound has great promise as a CQ replacement drug.
CQ has been reported to kill early-stage gametocytes in vitro at concentrations greater than 1 nM (48). To determine if TDR 58845 and TDR 58846 have gametocytocidal activity, we tested two different concentrations of the drugs in an in vitro gametocyte assay. We found that in comparison to DHA, which eliminates gametocytes at 0.1 μM, both TDR 58845 and TDR 58846 completely eliminate early-stage gametocytes of P. falciparum NF54 at 1 μM. Other studies have observed an increase in the number of gametocytes when subcurative doses of CQ were used. Interestingly, these studies did not show a significant difference between CQ-sensitive and CQ-resistant laboratory strains in gametocyte production (7). In our studies, we did not observe an increase in the number of gametocytes when treated in early stages with 0.1 μM CQ, TDR 58845, or TDR 58846. The observation that CQ, TDR 58845, and TDR 58846 eliminate gametocytes at 1 μM suggests a mode of action similar to that of CQ against early-stage gametocytes. In addition, we found that 1 μM TDR 58845 or TDR 58846 significantly decreases the number of mature gametocytes when added to late-stage gametocytes (stages III to V). The fact that these 4-aminoquinoline derivatives affect both early and late gametocyte stages as well as erythrocytic stages makes them attractive candidates for further development.
Our experiments in vitro indicate that the 4-aminoquinoline derivatives are efficacious against several multidrug-resistant P. falciparum strains. In addition, both TDR 58845 and 58846 cured 100% of P. berghei-infected mice at doses of 80 mg/kg/day and cured 80% of mice dosed at 40 mg/kg/day. As a result, both drugs are more efficacious than CQ in this model, where daily doses of 160 mg/kg/day clear parasitemia by day 6 but do not cure infections (data not shown) (23, 38).
Our findings illustrate the enormous potential of these 4-aminoquinoline derivatives as antimalarials. In fact, TDR 58845 can overcome parasite resistance through the use of CQ resistance-reversing compounds in vitro. More importantly, TDR 58846, to our knowledge, is one of the first 4-aminoquinoline derivatives that are potent in vitro and in vivo and do not have cross-resistance with CQ. Given the fact that CQ has not been used for many years in areas of the world, these novel 4-aminoquinolines could be part of a combination therapy to efficiently treat malaria. Our results suggest that these novel compounds, particularly TDR 58846, should be considered as partner drugs in antimalarial combinations to treat multiple-drug-resistant P. falciparum and P. vivax malaria.
ACKNOWLEDGMENTS
We thank Hoffmann la Roche for providing the 4-aminoquinoline derivatives TDR 58845 and TDR 58846. We thank François Nosten and Arjen Dondorp for supplying P. falciparum isolates from Thailand and Cambodia. We thank Naresh Singh and Sandra Kennedy for supplying the mosquitoes and thank Alexis LaCrue, Anupam Pradhan, and Anuradha Srivastava for critical reviews of the manuscript.
This research was funded by the University of South Florida, College of Public Health.
Footnotes
Published ahead of print 18 June 2012
REFERENCES
- 1. Reference deleted.
- 2. Baird JK, et al. 1991. Resistance to chloroquine by Plasmodium vivax in Irian Jaya, Indonesia. Am. J. Trop. Med. Hyg. 44:547–552 [DOI] [PubMed] [Google Scholar]
- 3. Batra S, Srivastava P, Roy K, Pandey VC, Bhaduri AP. 2000. A new class of potential chloroquine-resistance reversal agents for Plasmodia: syntheses and biological evaluation of 1-(3′-diethylaminopropyl)-3-(substituted phenylmethylene)pyrrolidines. J. Med. Chem. 43:3428–3433 [DOI] [PubMed] [Google Scholar]
- 4. Bayoumi RA, et al. 1993. Drug response and genetic characterization of Plasmodium falciparum clones recently isolated from a Sudanese village. Trans. R. Soc. Trop. Med. Hyg. 87:454–458 [DOI] [PubMed] [Google Scholar]
- 5. Bitonti AJ, et al. 1988. Reversal of chloroquine resistance in malaria parasite Plasmodium falciparum by desipramine. Science 242:1301–1303 [DOI] [PubMed] [Google Scholar]
- 6. Black F, et al. 1981. Fansidar resistant falciparum malaria acquired in South East Asia. Trans. R. Soc. Trop. Med. Hyg. 75:715–716 [DOI] [PubMed] [Google Scholar]
- 7. Buckling A, Ranford-Cartwright LC, Miles A, Read AF. 1999. Chloroquine increases Plasmodium falciparum gametocytogenesis in vitro. Parasitology 118(Pt 4):339–346 [DOI] [PubMed] [Google Scholar]
- 8. Campbell CC, Chin W, Collins WE, Teutsch SM, Moss DM. 1979. Chloroquine-resistant Plasmodium falciparum from East Africa: cultivation and drug sensitivity of the Tanzanian I/CDC strain from an American tourist. Lancet ii:1151–1154 [DOI] [PubMed] [Google Scholar]
- 9. Collignon P. 1991. Chloroquine resistance in Plasmodium vivax. J. Infect. Dis. 164:222–223 [DOI] [PubMed] [Google Scholar]
- 10. De D, Krogstad FM, Cogswell FB, Krogstad DJ. 1996. Aminoquinolines that circumvent resistance in Plasmodium falciparum in vitro. Am. J. Trop. Med. Hyg. 55:579–583 [DOI] [PubMed] [Google Scholar]
- 11. Dondorp AM, et al. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361:455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fidock DA. 2010. Drug discovery: priming the antimalarial pipeline. Nature 465:297–298 [DOI] [PubMed] [Google Scholar]
- 13. Fidock DA, et al. 2000. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6:861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fitch CD. 2004. Ferriprotoporphyrin IX, phospholipids, and the antimalarial actions of quinoline drugs. Life Sci. 74:1957–1972 [DOI] [PubMed] [Google Scholar]
- 15. Ginsburg H, Stein WD. 2005. How many functional transport pathways does Plasmodium falciparum induce in the membrane of its host erythrocyte? Trends Parasitol. 21:118–121 [DOI] [PubMed] [Google Scholar]
- 16. Greenwood BM, et al. 2008. Malaria: progress, perils, and prospects for eradication. J. Clin. Invest. 118:1266–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hastings IM. 2004. The origins of antimalarial drug resistance. Trends Parasitol. 20:512–518 [DOI] [PubMed] [Google Scholar]
- 18. Hastings MD, et al. 2004. Dihydrofolate reductase mutations in Plasmodium vivax from Indonesia and therapeutic response to sulfadoxine plus pyrimethamine. J. Infect. Dis. 189:744–750 [DOI] [PubMed] [Google Scholar]
- 19. Hocart SJ, et al. 2011. 4-Aminoquinolines active against chloroquine-resistant Plasmodium falciparum: basis of antiparasite activity and quantitative structure-activity relationship analyses. Antimicrob. Agents Chemother. 55:2233–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hofheinz W, Jaquet C, Jolidon S. 1995. Aminochinoline derivatives useful in the treatment of malaria. European patent EP0656353
- 21. Hwang JY, et al. 2011. Synthesis and evaluation of 7-substituted 4-aminoquinoline analogues for antimalarial activity. J. Med. Chem. 54:7084–7093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ifediba T, Vanderberg JP. 1981. Complete in vitro maturation of Plasmodium falciparum gametocytes. Nature 294:364–366 [DOI] [PubMed] [Google Scholar]
- 23. Ishih A, Suzuki T, Muregi FW, Matsui K, Terada M. 2006. Chloroquine efficacy in Plasmodium berghei NK65-infected ICR mice, with reference to the influence of initial parasite load and starting day of drug administration on the outcome of treatment. Southeast Asian J. Trop. Med. Public Health 37:13–17 [PubMed] [Google Scholar]
- 24. Iwaniuk DP, et al. 2009. Synthesis and antimalarial activity of new chloroquine analogues carrying a multifunctional linear side chain. Bioorg. Med. Chem. 17:6560–6566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kappe SH, Vaughan AM, Boddey JA, Cowman AF. 2010. That was then but this is now: malaria research in the time of an eradication agenda. Science 328:862–866 [DOI] [PubMed] [Google Scholar]
- 26. Krogstad DJ, et al. 1987. Efflux of chloroquine from Plasmodium falciparum: mechanism of chloroquine resistance. Science 238:1283–1285 [DOI] [PubMed] [Google Scholar]
- 27. Kyle DE, Milhous WK, Rossan RN. 1993. Reversal of Plasmodium falciparum resistance to chloroquine in Panamanian Aotus monkeys. Am. J. Trop. Med. Hyg. 48:126–133 [DOI] [PubMed] [Google Scholar]
- 28. Kyle DE, Oduola AM, Martin SK, Milhous WK. 1990. Plasmodium falciparum: modulation by calcium antagonists of resistance to chloroquine, desethylchloroquine, quinine, and quinidine in vitro. Trans. R. Soc. Trop. Med. Hyg. 84:474–478 [DOI] [PubMed] [Google Scholar]
- 29. Madrid PB, Liou AP, DeRisi JL, Guy RK. 2006. Incorporation of an intramolecular hydrogen-bonding motif in the side chain of 4-aminoquinolines enhances activity against drug-resistant P. falciparum. J. Med. Chem. 49:4535–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Madrid PB, et al. 2005. Synthesis of ring-substituted 4-aminoquinolines and evaluation of their antimalarial activities. Bioorg. Med. Chem. Lett. 15:1015–1018 [DOI] [PubMed] [Google Scholar]
- 31. Martin SK, Oduola AM, Milhous WK. 1987. Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science 235:899–901 [DOI] [PubMed] [Google Scholar]
- 32. Millet J, et al. 2004. Polymorphism in Plasmodium falciparum drug transporter proteins and reversal of in vitro chloroquine resistance by a 9,10-dihydroethanoanthracene derivative. Antimicrob. Agents Chemother. 48:4869–4872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murray CJ, et al. 2012. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379:413–431 [DOI] [PubMed] [Google Scholar]
- 34. Mzayek F, et al. 2007. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin. Trials 2:e6 doi:10.1371/journal.pctr.0020006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oduola AM, et al. 1998. In vitro and in vivo reversal of chloroquine resistance in Plasmodium falciparum with promethazine. Am. J. Trop. Med. Hyg. 58:625–629 [DOI] [PubMed] [Google Scholar]
- 36. Omodeo-Sale F, et al. 2009. Novel antimalarial aminoquinolines: heme binding and effects on normal or Plasmodium falciparum-parasitized human erythrocytes. Antimicrob. Agents Chemother. 53:4339–4344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pagola S, Stephens PW, Bohle DS, Kosar AD, Madsen SK. 2000. The structure of malaria pigment beta-haematin. Nature 404:307–310 [DOI] [PubMed] [Google Scholar]
- 38. Peters W. 1975. The chemotherapy of rodent malaria, XXII. The value of drug-resistant strains of P. berghei in screening for blood schizontocidal activity. Ann. Trop. Med. Parasitol. 69:155–171 [PubMed] [Google Scholar]
- 39. Reference deleted.
- 40. Reference deleted.
- 41. Ponnudurai T, Meuwissen JH, Leeuwenberg AD, Verhave JP, Lensen AH. 1982. The production of mature gametocytes of Plasmodium falciparum in continuous cultures of different isolates infective to mosquitoes. Trans. R. Soc. Trop. Med. Hyg. 76:242–250 [DOI] [PubMed] [Google Scholar]
- 42. Pradines B, Pages JM, Barbe J. 2005. Chemosensitizers in drug transport mechanisms involved in protozoan resistance. Curr. Drug Targets Infect. Disord. 5:411–431 [DOI] [PubMed] [Google Scholar]
- 43. Price RN, et al. 2007. Vivax malaria: neglected and not benign. Am. J. Trop. Med. Hyg. 77:79–87 [PMC free article] [PubMed] [Google Scholar]
- 44. Ray S, et al. 2010. Development of a new generation of 4-aminoquinoline antimalarial compounds using predictive pharmacokinetic and toxicology models. J. Med. Chem. 53:3685–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ridley RG, et al. 1996. 4-Aminoquinoline analogs of chloroquine with shortened side chains retain activity against chloroquine-resistant Plasmodium falciparum. Antimicrob. Agents Chemother. 40:1846–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rieckmann KH, Davis DR, Hutton DC. 1989. Plasmodium vivax resistance to chloroquine? Lancet ii:1183–1184 [DOI] [PubMed] [Google Scholar]
- 47. Saenz FE, Balu B, Smith J, Mendonca SR, Adams JH. 2008. The transmembrane isoform of Plasmodium falciparum MAEBL is essential for the invasion of Anopheles salivary glands. PLoS One 3:e2287 doi:10.1371/journal.pone.0002287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smalley ME. 1977. Plasmodium falciparum gametocytes: the effect of chloroquine on their development. Trans. R. Soc. Trop. Med. Hyg. 71:526–529 [DOI] [PubMed] [Google Scholar]
- 49. Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 48:1803–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thompson PE, Bayles A, Olszewski B, Waitz JA. 1965. Quinine-resistant Plasmodium berghei in mice. Science 148:1240–1241 [DOI] [PubMed] [Google Scholar]
- 51. Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675 [DOI] [PubMed] [Google Scholar]
- 52. van Schalkwyk DA, Egan TJ. 2006. Quinoline-resistance reversing agents for the malaria parasite Plasmodium falciparum. Drug Resist. Updat. 9:211–226 [DOI] [PubMed] [Google Scholar]
- 53. Warhurst DC, Hall AP, Tjokrosonto S. 1985. RI quinine-Fansidar resistant falciparum malaria from Malawi. Lancet ii:330. [DOI] [PubMed] [Google Scholar]
- 54. Wellems TE, Plowe CV. 2001. Chloroquine-resistant malaria. J. Infect. Dis. 184:770–776 [DOI] [PubMed] [Google Scholar]
- 55. Whitty CJ, Chandler C, Ansah E, Leslie T, Staedke SG. 2008. Deployment of ACT antimalarials for treatment of malaria: challenges and opportunities. Malar. J. 7(Suppl 1):S7 doi:10.1186/1475-2875-7-S1-S7 [DOI] [PMC free article] [PubMed] [Google Scholar]