

Abstract
Clostridium difficile infection (CDI) causes moderate to severe disease, resulting in diarrhea and pseudomembranous colitis. CDI is difficult to treat due to production of inflammation-inducing toxins, resistance development, and high probability of recurrence. Only two antibiotics are approved for the treatment of CDI, and the pipeline for therapeutic agents contains few new drugs. MBX-500 is a hybrid antibacterial, composed of an anilinouracil DNA polymerase inhibitor linked to a fluoroquinolone DNA gyrase/topoisomerase inhibitor, with potential as a new therapeutic for CDI treatment. Since MBX-500 inhibits three bacterial targets, it has been previously shown to be minimally susceptible to resistance development. In the present study, the in vitro and in vivo efficacies of MBX-500 were explored against the Gram-positive anaerobe, C. difficile. MBX-500 displayed potency across nearly 50 isolates, including those of the fluoroquinolone-resistant, toxin-overproducing NAP1/027 ribotype, performing as well as comparator antibiotics vancomycin and metronidazole. Furthermore, MBX-500 was a narrow-spectrum agent, displaying poor activity against many other gut anaerobes. MBX-500 was active in acute and recurrent infections in a toxigenic hamster model of CDI, exhibiting full protection against acute infections and prevention of recurrence in 70% of the animals. Hamsters treated with MBX-500 displayed significantly greater weight gain than did those treated with vancomycin. Finally, MBX-500 was efficacious in a murine model of CDI, again demonstrating a fully protective effect and permitting near-normal weight gain in the treated animals. These selective anti-CDI features support the further development of MBX 500 for the treatment of CDI.
INTRODUCTION
Clostridium difficile, a Gram-positive, anaerobic, spore-forming, toxin-producing bacterium (2, 19), was originally defined as the cause of antibiotic-associated diarrhea and pseudomembranous colitis in 1978 (5). Since then, research has provided a portfolio of information on the pathophysiology and management of Clostridium difficile infection (CDI) (4). This clinical syndrome has a known cause (toxins A and B produced by C. difficile), established risk factors (antibiotic exposure, hospitalization, and advanced age), validated diagnostic tests (stool toxin detection), and effective treatment (oral vancomycin or metronidazole) (4). C. difficile has some distinctive features: it causes disease almost exclusively in connection with antibiotic therapy, it poses a nosocomial risk, and it produces several toxins in the colon (6). In patients undergoing antibiotic treatment, perturbation of the competing intestinal flora promotes a conversion of spores to vegetative forms that replicate and produce toxins. The clinical signs of disease include watery and bloody diarrhea and cramps, with the characteristic pseudomembranous colitis, a frequently life-threatening condition (6).
During the past several years, there has been renewed interest in CDI, reflecting the recognition of a form of the disease that is more frequent, more severe, and more refractory to standard treatment (19). Based on PCR analysis, the epidemic strain was designated ribotype 27 or NAP1/027 (19), which is distinguished from other strains by increased production of toxins, fluoroquinolone resistance, and production of binary toxin CDT, a third C. difficile toxin that has been associated with increased disease severity (12). Additionally, resistance development to several C. difficile agents has further complicated treatment (9, 21, 23).
Treatment options for CDI traditionally have been to discontinue the implicated antibiotics and switch to vancomycin or metronidazole in patients with moderate or severe CDI. These two antibiotics have been the treatment of choice since the late 1970s, when the etiology of the disease was discovered. In a recent clinical trial, the two agents showed similar response rates in patients with mild infection; however, in patients with severe infection, vancomycin was significantly more effective than metronidazole (30). The high rate of recurrence of CDI is the most significant challenge and occurs after cessation of treatment in 20 to 25% of patients (4), with the frequency increasing in patients who had one recurrence (40%) or more (>60%) (20). Recurrence may result from persistence of spores, reinfection from the environment, or failure to develop a protective immune response (29) but generally not from resistance to antibiotics (16, 22). Patients who have repeated recurrences often take multiple courses of metronidazole or vancomycin, pulsed or tapered courses of vancomycin, or continuous vancomycin (up to 2 years) to control the disease (4, 15), a regimen that can result in selection of vancomycin-resistant enterococci. Alternative experimental strategies using probiotics (26), immunization (27), the administration of nontoxigenic C. difficile (25), or a fecal transplant (1) have been used with mixed results. There are several new treatments for CDI currently being evaluated in clinical trials (17). Additionally, fidaxomicin, a natural product macrocycle antibiotic, was approved by the FDA in 2011 (13), becoming the first drug for CDI to be approved in 20 years (18).
In this report, we demonstrate anti-C. difficile potency of a prototype drug candidate (MBX-500; Fig. 1) which was designed as a hybrid antibacterial molecule linking an anilinouracil (AU) DNA polymerase inhibitor (31) to a fluoroquinolone (FQ) (32) and developed as an agent to treat antibiotic-resistant Gram-positive aerobic pathogens (8). We now show that MBX-500 has activity across a panel of antibiotic-sensitive and -resistant C. difficile isolates, including those of the NAP1/027 ribotype, displays much less activity against a variety of other gut anaerobes, and is active in two experimental animal models of CDI.
Fig 1.
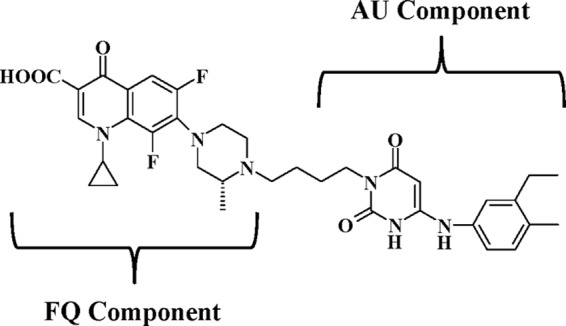
Structure of MBX-500, a hybrid of an anilinuracil (AU) DNA polymerase inhibitor and a fluoroquinolone (FQ) DNA topoisomerase/gyrase inhibitor.
MATERIALS AND METHODS
Materials and bacterial strains.
Clindamycin hydrochloride was purchased from Spectrum Chemicals (New Brunswick, NJ) or Sigma-Aldrich (St. Louis, MO), metronidazole was purchased from Sigma-Aldrich (St. Louis, MO), moxifloxacin was purchased from IHMA (Schaumburg, IL), vancomycin hydrochloride (Vancocin) was purchased from Lilly (Indianapolis, IN), and imipenem was purchased from United States Pharmacopeia. Bacterial strains were purchased from American Type Culture Collection (Manassas, VA) where indicated or represented in-house repositories from Micromyx, LLC, R.M. Alden, Beth Israel Deaconess Medical Center, or Tufts University Veterinary School.
In vitro antibacterial activity.
MICs were determined using the agar dilution reference method as described in CLSI document M11-A7 (11). Isolates were subcultured from frozen samples onto brucella K1 hemin blood agar, incubated at 37°C for 72 h, transferred, and incubated again for 48 h for testing. The comparator antimicrobial agents were reconstituted according to the manufacturers' instructions, serially diluted, and added to molten-supplemented brucella agar. MBX-500 powder was reconstituted with dimethyl sulfoxide (DMSO) to an initial concentration of 3.2 mg/ml, serially diluted in DMSO, and added to molten agar to prepare the plates. Drug-free growth control plates were inoculated before and after each drug set. Colonies were suspended in brucella broth to achieve the turbidity of the 0.5 McFarland standard and pipetted into the wells of the Steers Replicator device. They were applied to the plates with prongs, delivering a final concentration of approximately 105 CFU/spot. After absorption of the inoculum, the plates were passed into an anaerobic incubator and incubated for 44 h. After growth had occurred, the plates were examined and the MIC was determined as the lowest concentration of antimicrobial agent that completely inhibited growth or markedly reduced growth compared to the drug-free growth control.
In vivo efficacy acute hamster model.
Golden Syrian hamsters, housed individually in sterile cages, were treated subcutaneously (s.c.) with 15 mg clindamycin hydrochloride in D5W (5 ml/kg)/kg of body weight. Twenty-four hours later, all were infected by oral gavage with a stock mixture of 3 × 108 CFU C. difficile spores (toxigenic strain VPI 10463, ATCC 43255). Eighteen hours later, groups of 6 animals were treated by oral gavage (10 ml/kg) with 50 mg/kg of vancomycin hydrochloride dissolved in deionized (DI) water or with MBX-500 suspended in DI water or were left untreated. The treatments were repeated twice daily for a total of 3 days. Animals were observed closely for signs and symptoms of disease for 5 days postinfection, when survivors were counted. Necropsies were performed on dead and euthanized animals. The Xpect Clostridium difficile toxin immunochromotographic A/B test (Remel) was used in selected animals. Intestinal fluids were collected and tested for A/B toxins according to kit instructions. Because the kit has not been validated for use in animal CDIs, the results were interpreted qualitatively and not quantitatively.
In vivo efficacy-recurrent hamster model.
Groups (n = 10 to 14) of Golden Syrian hamsters, housed in groups of two, were given a single injection of clindamycin phosphate (10 mg/kg s.c.) on day 0, and 1 day later (day 1) they were infected by oral gavage with 105 spores of C. difficile (toxigenic strain VPI 10463) (3). MBX-500 was suspended in phosphate-buffered saline (PBS) and administered orally at a dose of 200 mg/kg ∼2 h after C. difficile challenge to 14 hamsters and thereafter once daily for 5 days. The control group (n = 10 hamsters) received no oral C. difficile challenge and no treatment, the vehicle-treated control group (n = 10 hamsters) was infected and treated with vehicle, and the vancomycin group (n = 14 hamsters) was infected and received vancomycin 50 mg/kg orally 2 h after challenge and thereafter once daily for 5 days. Animals were weighed daily, monitored for diarrhea, and euthanized when moribund.
In vivo efficacy mouse model.
C57BL/6 mice, housed in groups of five per cage, were administered a mixture of antibiotics (kanamycin [40 mg/kg/per day], gentamicin [3.5 mg/kg/day], colistin [4.2 mg/kg/day], metronidazole [21.5 mg/kg/day], and vancomycin [4.5 mg/kg/day]) in drinking water for 3 days to overcome the resistance of conventional mice to CDI (10). Mice were left untreated for 2 days and then given a dose of clindamycin (10 mg/kg) intraperitoneally to facilitate C. difficile disease. On the following day, mice were challenged by oral gavage with 1 × 106 CFU of C. difficile (UK1) spores. Groups of mice (n = 10) were treated with 100 mg/kg MBX-500 once daily, 50 mg/kg vancomycin once daily, or vehicle (1% carboxy-methyl cellulose [CMC]) alone, beginning 4 h postchallenge and continuing for 4 days. Clinical signs of CDI, weight loss, and survival were monitored daily for 7 days after treatment ended.
RESULTS
MBX-500 is active against C. difficile but not against most other Gram-positive and Gram-negative anaerobes.
The antibacterial potencies of MBX-500 and several comparator antibiotics were tested against a series of single Gram-positive and Gram-negative anaerobes, species that are normally found populating the gut (Table 1). Among the Gram-positive anaerobes, MBX-500 displayed the greatest relative potency against C. difficile (by a factor of 2- to 32-fold), with the exception of a few species. Interestingly, MBX-500 had no activity (at 32 μg/ml) against one of the strains of the closely related species C. perfringens or against members of the Bifidobacterium and Lactobacillus genera. We also observed little or no potency of MBX-500 against the Gram-negative anaerobes (with the exception of the Fusobacterium species). In contrast, the comparator antibiotics, metronidazole, imipenem, and clindamycin, were very active against both Gram-positive and Gram-negative species, with metronidazole, a clinically relevant C. difficile antibiotic, demonstrating activity against all anaerobes tested. Additionally, most of the Gram-positive species tested were sensitive to vancomycin, another clinically relevant antibiotic.
Table 1.
MICs for MBX-500 and comparator antibiotics against individual Gram-positive and Gram-negative anaerobes
| Anaerobe (ATCC no.) | MIC (μg/ml)a |
||||
|---|---|---|---|---|---|
| MBX-500 | Vanco | Metro | Imipenem | Clinda | |
| Gram positive | |||||
| Bifidobacterium longum (15707) | >32 | 0.5 | 0.06 | 8 | ≤0.03 |
| Bifidobacterium infantis (15702) | >32 | ND | ≤0.03 | 2 | ≤0.03 |
| Bifidobacterium breve (15698) | >32 | 2 | 1 | 2 | ≤0.03 |
| Bifidobacterium bifidum (15696) | >32 | ND | ≤0.03 | 1 | ≤0.03 |
| Clostridium perfringens | >32 | 0.5 | 0.5 | 2 | >16 |
| Clostridium perfringens | 2 | ND | 2 | 0.12 | 2 |
| Clostridium difficile | 4 | 1 | 8 | >16 | 8 |
| Clostridium difficile | 1 | 0.5 | 8 | 0.25 | >16 |
| Eubacterium lentum (43055) | >32 | 2 | 0.5 | 0.25 | 0.12 |
| Lactobacillus acidophilus | >32 | 2 | 0.12 | >16 | 4 |
| Lactobacillus casei | >32 | >16 | 0.25 | >16 | 2 |
| Lactobacillus plantarum (39268) | >32 | ND | 2 | >16 | 0.25 |
| Peptostreptococcus anaerobius | 1 | ND | 0.06 | 0.5 | ≤0.03 |
| Peptostreptococcus micros | 8 | ND | ≤0.03 | >16 | 8 |
| Propionibacterium acnes | >32 | ND | ≤0.03 | >16 | 0.06 |
| Streptococcus constellatus (27823) | 8 | ND | 0.06 | >16 | 0.25 |
| Streptococcus intermedius (27335) | 8 | ND | 0.06 | >16 | >16 |
| Gram negative | |||||
| Bacteroides fragilis | >32 | ND | 0.5 | 1 | 2 |
| Bacteroides ovatus | 2 | ND | 0.25 | 1 | 2 |
| Bacteroides thetaiotaomicron | >32 | ND | 0.5 | 1 | >16 |
| Bacteroides vulgatus | >32 | ND | 0.5 | 2 | 1 |
| Eikenella corrodens (43278) | >32 | ND | 0.25 | >16 | >16 |
| Fusobacterium nucleatum (25586) | 0.06 | ND | ≤0.03 | ≤0.03 | ≤0.03 |
| Fusobacterium necrophorum (25286) | ≤0.03 | ND | ≤0.03 | ≤0.03 | ≤0.03 |
| Porphyromonas asaccharolytica | 2 | ND | ≤0.03 | 1 | 0.12 |
| Prevotella melaninogenica | 32 | ND | ≤0.03 | 0.25 | ≤0.03 |
| Prevotella spp. | 8 | ND | 0.06 | 0.5 | 2 |
| Veillonella parvula (17745) | 4 | ND | ≤0.03 | 1 | 0.06 |
Vanco, vancomycin; Metro, metronidazole; Clinda, clindamycin; ND, not determined.
MBX-500 exhibits potency comparable to that of vancomycin and metronidazole against multiple C. difficile isolates.
When tested against 16 C. difficile clinical isolates, MBX-500 displayed a MIC90 value of 2 μg/ml and a narrow potency range of 0.5 to 2 μg/ml (Table 2). MBX-500 was equipotent with vancomycin and metronidazole and more potent than clindamycin and imipenem. The isolates used in this experiment were not typed, so there was no indication of whether any were toxigenic and/or fluoroquinolone resistant. To assess more clinically relevant isolates, MBX-500 was then tested against 30 defined moxifloxacin-resistant isolates, including 14 NAP1/027 types and 16 isolates that were resistant to one or more other antibiotic in addition to moxifloxacin (13 were erythromycin resistant, 13 were clindamycin resistant, and 8 were linezolid resistant) (Table 3). The results in Table 3 demonstrate that MBX-500 displayed excellent potency against the panel of 30 isolates, including NAP1/027 isolates as well as isolates carrying various antibiotic-resistant phenotypes, with only a 2-fold increase in the MIC90 value compared to that of noncharacterized isolates (Table 2). More specifically, MBX-500 maintained the same MIC90 for the NAP1/027 strains (Table 3) (14 of 30 isolates), all of which are moxifloxacin resistant, compared with that of the nonresistant strains. These results demonstrate that MBX-500, although part fluoroquinolone in structure, maintains good potency against fluoroquinolone-resistant C. difficile isolates. These results also suggest that hybrid antibacterials will not play a role in providing a selective advantage for the epidemic NAP1/027 strains, a role that fluoroquinolones have played in the emergence of this strain (19).
Table 2.
MICs for 16 C. difficile isolates
| Micromyx strain | MIC (μg/ml) |
||||
|---|---|---|---|---|---|
| MBX-500 | Vancomycin | Metronidazole | Clindamycin | Imipenem | |
| QC strain ATCC 4381 | 1 | 1 | 0.5 | 4 | 2 |
| 3579 | 2 | 1 | 0.25 | 16 | 4 |
| 3580 | 2 | 1 | 0.5 | 8 | 4 |
| 3581 | 1 | 1 | 1 | >16 | 4 |
| 3582 | 2 | 0.25 | 0.25 | 0.5 | 0.5 |
| 3584 | 1 | 0.5 | 0.5 | 8 | 4 |
| 3585 | 2 | 0.5 | 0.5 | 8 | 4 |
| 3586 | 1 | 0.5 | 0.5 | 8 | 4 |
| 3587 | 2 | 1 | 1 | 8 | 4 |
| 3588 | 0.5 | 2 | 0.5 | 8 | 1 |
| 3589 | 2 | 0.5 | 1 | >16 | 4 |
| 3590 | 1 | 1 | 0.5 | 8 | 4 |
| 3591 | 1 | 2 | 0.5 | 8 | 4 |
| 3593 | 2 | 0.5 | 0.5 | 8 | >8 |
| 3594 | 1 | 1 | 1 | >16 | 4 |
| 3595 | 1 | 1 | 1 | >16 | 8 |
| 1209 | 2 | 1 | 0.5 | 4 | NDa |
| MIC90 | 2 | 2 | 1 | >16 | 8 |
| MIC50 | 1 | 1 | 0.5 | 8 | 4 |
| MIC range | 0.5–2 | 0.25–2 | 0.25–1 | 0.5–>16 | 0.5–>8 |
ND, not determined.
Table 3.
MICs for 30 C. difficile isolates with antibiotic resistancesa
| R.M. Alden strain | Phenotype | MIC (μg/ml) |
|||
|---|---|---|---|---|---|
| MBX-500 | Moxi | Vanco | Metro | ||
| QC strain | Sensitive | 2 | 2 | 1 | 0.5 |
| 8826 | Err Clr Ter | 4 | 32 | 1 | 0.25 |
| 8827 | Err Clr Syr | 4 | 32 | 1 | 0.25 |
| 8830 | Err Clr Ter | 4 | 32 | 1 | 0.25 |
| 8831 | Err | 4 | 2 | 1 | 0.25 |
| 8832 | Err Clr Syr Lzr | 2 | 2 | 1 | 0.25 |
| 8834 | Err Clr Syr Imr Ter Lzr | 4 | 32 | 1 | 0.5 |
| 9400 | Err Clr Syr Ter | 2 | 32 | 1 | 0.25 |
| 10540 | Err Clr Ter | 1 | 2 | 1 | 0.5 |
| 11788 | Err Imr | 2 | 32 | 4 | 1 |
| 12019 | Ter | 2 | 2 | 0.5 | 0.25 |
| 13879 | Err Clr Syr Imr Ter | 4 | 32 | 1 | 0.5 |
| 16277 | Err Clr Syr | 4 | 32 | 4 | 0.25 |
| 16278 | Err Clr Syr Lzr | 2 | 32 | 4 | 0.25 |
| 16282 | Clr Imr Lzr | 4 | 32 | 1 | 0.25 |
| 16286 | Err Clr Syr Lzr | 2 | 32 | 1 | 0.25 |
| 16458 | Clr Lzr | 2 | 32 | 1 | 0.5 |
| 19556 | NAP1/027 | 2 | 32 | 1 | 2 |
| 19560 | NAP1/027 | 2 | 64 | 1 | 2 |
| 19562 | NAP1/027 | 2 | 64 | 1 | 2 |
| 19565 | NAP1/027 | 2 | 32 | 1 | 1 |
| 19566 | NAP1/027 | 2 | 32 | 1 | 1 |
| 19567 | NAP1/027 | 2 | 16 | 1 | 1 |
| 19569 | NAP1/027 | 2 | 32 | 1 | 1 |
| 19571 | NAP1/027 | 2 | 32 | 2 | 1 |
| 19574 | NAP1/027 | 2 | 64 | 1 | 2 |
| 19575 | NAP1/027 | 2 | 64 | 1 | 2 |
| 19643 | NAP1/027 | 2 | 32 | 1 | 1 |
| 19645 | NAP1/027 | 4 | 16 | 4 | 1 |
| 19646 | NAP1/027 | 2 | 32 | 1 | 4 |
| 19647 | NAP1/027 | 2 | 32 | 1 | 1 |
| MIC90 | 4 | 64 | 4 | 2 | |
| MIC50 | 2 | 32 | 1 | 1 | |
| MIC range | 1–4 | 2–64 | 0.5–4 | 0.25–4 | |
r, resistant; Er, erythromycin; Cl, clindamycin; Te, tetracycline; Sy, synercid; Lz, linezolid; Im, imipenem; NAP1/027, North American pulse-field type 1, ribotype 27; Moxi, moxifloxacin; Vanco, vancomycin; Metro, metronidazole.
MBX-500 displays efficacy in both acute and recurrent CDI in hamsters.
In vivo efficacy of MBX-500 in acute infections was assessed in Golden Syrian hamsters. Clindamycin-pretreated hamsters were infected by oral gavage with C. difficile, and 18 h later antibiotic treatment was initiated. C. difficile-infected and untreated animals displayed the symptoms of colitis (e.g., bloated abdomen, wet tail, diarrhea, and low-activity level); all animals died by 136 h postinfection, and stool samples collected from dead hamsters were positive for toxins A and B (data not shown). When dosed with vancomycin or MBX-500, all animals survived for 6 days postinfection and 3 days beyond the end of treatment (Fig. 2). All drug-treated hamsters were healthy and completely free of clinical symptoms of the disease throughout the experiment. Additionally, stool samples collected from MBX-500 or vancomycin-treated hamsters at the end of the study were completely negative for toxins A and B (data not shown).
Fig 2.

Percentage of survival over 6 days of acute C. difficile infection in hamsters. Diamonds, vehicle-treated control; squares, vancomycin, 50 mg/kg; triangles, MBX-500, 50 mg/kg.
Efficacy of MBX-500 was subsequently tested in a 30-day model of recurrent CDI, in which disease is initially cured, and once treatment has been stopped, infection may recur. Similar to the previous experiment, in the acute phase of the infection (up to day 6), MBX-500, dosed orally at 200 mg/kg daily, provided 100% protection from death, as did the control antibiotic, vancomycin (Fig. 3A). In the second phase of infection, recurrence was measured with death as an outcome. Four of 14 animals treated with MBX-500 succumbed to recurrent CDI on day 9 (Fig. 3A), and the remainder (71%) survived until the end of the study on day 29. Similarly, the vancomycin-treated animals exhibited a 71% survival rate after recurrence occurred on days 15 to 20. The secondary measurement during the 29-day study was weight gain. Daily weights, plotted with error bars (Fig. 3B), in the early days of the study (i.e., up to day 10) showed that weight gain in uninfected control animals was significantly greater than in the MBX-500- and vancomycin-treated animals (P < 0.001), while the MBX-500 and vancomycin-treated groups were not significantly different from each other (P = 0.14). Interestingly, by the last day of the experiment, both the MBX-500 and vancomycin-treated groups were gaining weight. However, the weight gain for MBX-500 was significantly higher than that for vancomycin (P < 0.001), although still significantly different from the uninfected/untreated control group (P < 0.05). In the past, an observation of improved weight gain has correlated with less severe disease as quantified by histopathological injury (3). These results suggest that MBX-500 may be as efficacious as vancomycin at the selected doses.
Fig 3.

Percentage of survival (A) and mean relative weight (B) measurements over 29 days of acute and recurrent C. difficile infection in hamsters. Diamonds, uninfected, untreated control; squares, vehicle-treated control; triangles, vancomycin, 50 mg/kg; circles, MBX-500, 200 mg/kg. Error bars indicate standard errors of the data in panel B, and statistical analysis was performed using the Student t test set for the “two-sample assuming unequal variances” assumption.
MBX-500 treatment at 100 mg/kg reduces disease severity and prevents mortality in mice.
Mice develop typical CDI after multiple-antibiotic treatment and C. difficile inoculation (10, 28). Using this murine CDI model, we evaluated the therapeutic impact of MBX-500 on CDI. MBX-500 was administered to 1 group of mice at 100 mg/kg dosed once per day, and MBX-500-treated mice were compared to vehicle-treated or vancomycin-treated (50 mg/kg once per day) mice. All drugs were given 4 h after C. difficile oral challenge and continued over 4 days. All vehicle-treated mice developed severe diarrhea within 24 h after challenge, and 40% of them succumbed within 48 h. MBX-500 treatment (100 mg/kg) delayed the onset of the diarrhea as well as the severity of the disease, with 100% survival (Fig. 4A). Vancomycin prevented mice from developing CDI during the entire course of treatment, but the mice started to display severe CDI and weight loss, and 20% of mice succumbed within 4 to 6 days after stopping the treatment (Fig. 4A). Consistent with the disease manifestation, vancomycin prevented weight loss during the treatment, but all these mice experienced sharp weight loss after withdrawing the antibiotic (Fig. 4B). In contrast, the MBX-500 treatment group lost weight initially but then began to regain weight 4 days postchallenge and continued to gain weight after the last dose had been administered.
Fig 4.

Outcome over 12 days of acute and recurrent C. difficile infection in mice. (A) Percent survival; (B) mean weight. Diamonds, untreated control; squares, MBX-500, 100 mg/kg once daily; circles, vancomycin, 50 mg/kg once daily.
DISCUSSION
Current antibiotics for C. difficile treatment, including vancomycin and metronidazole, are suboptimal, with significant rates of treatment failures and recurrence. Add to that worsening clinical outcomes, including increasing mortality, the emergence of resistance to metronidazole, and rising incidence of vancomycin resistance in enterococci, and the need for new anti-C. difficile agents is clear (17). Indeed, the recent approval of fidaxomicin is evidence of the therapeutic need. Desirable characteristics of an antimicrobial agent for treating CDI include selectivity for C. difficile, a high local concentration in the colon, and low rates of resistance. Based on the results presented here and other unpublished findings, MBX-500 appears to meet those criteria. For example, MBX-500 is not a broad-spectrum antibacterial agent and therefore will be less likely than vancomycin and metronidazole to cause recurrence since it should not decimate the native intestinal microflora. In addition, MBX-500 demonstrates very low oral bioavailability (T. Bowlin, unpublished observation), which is expected to provide higher concentrations in the gut at the location of C. difficile infection. In this regard, MBX-500 offers a clear advantage over drugs which are absorbed systemically, such as metronidazole. Finally, we have shown that treatment with the hybrid antibacterial MBX-500 is less likely to select resistance mutations since the hybrid mimics the administration of two drugs targeting three separate bacterial enzymes: the replicative DNA polymerase, topoisomerase, and gyrase (8). The results of this study provide details on the in vitro and in vivo potencies of MBX-500 for CDI.
The in vitro MIC values for MBX-500 against individual isolates of a wide range of anaerobic gut pathogens suggest that the compound is rather selective for C. difficile. In addition, MBX-500 has broad activity across C. difficile isolates (Tables 2 and 3), including those with resistance to multiple antibiotics. When potencies for antibiotic-sensitive (Table 2) and antibiotic-resistant (Table 3) isolates were compared, the fact that the MBX-500 MIC90 values increased only slightly, from 2 to 4 μg/ml, within experimental error for this type of assay, was noteworthy due to the fact that at least 26 of the isolates were fluoroquinolone (FQ) (moxifloxacin) resistant and the activity of MBX-500 is, at least in part, due to its FQ component. We have previously shown in other Gram-positive species that cross resistance among anilinouracil (AU)- and FQ-resistant mutants and double mutants indicates that the hybrid acts in the intact cell by targeting both the polymerase and topoisomerase/gyrase activities (8). This appears to hold true for C. difficile, although it would be prudent to generate an AU-resistant isolate to fully evaluate cross-resistance. Furthermore, MBX-500 displayed the same MIC90 for the NAP1/027 strains (Table 3, 14 of 30 isolates) as it did for the nontoxigenic strains. These results suggest that this hybrid antibacterial may not play a role in providing a selective advantage for the epidemic NAP1/027 strains, a role that FQs have played in the emergence of this strain (19).
MBX-500 was as efficacious as vancomycin (50 mg/kg once daily) in acute (<5 days) infections, with doses of 100 mg/kg once daily (hamster) and 50 mg/kg once daily (mouse) providing complete protection (Fig. 2 and 4A). The required efficacious dose for MBX-500 is expected to be lower once an optimal formulation (solution instead of suspension) has been identified.
In the recurrent hamster model, the recurrence rates for MBX-500 and vancomycin were identical (29%), although the timing was delayed for vancomycin (Fig. 3A). Interestingly, although hamsters treated with MBX-500 and vancomycin demonstrated lower weight gains than the uninfected control animals during the acute phase of CDI (Fig. 3B), the animals treated with MBX-500 gained weight at the same rate as uninfected controls during later stages of the study. In the recurrent murine infection, MBX-500 (100 mg/kg, once daily) outperformed vancomycin, providing complete protection from recurrence for 12 days postinfection (Fig. 4A). In contrast, vancomycin dosed at a similar level (50 mg/kg once daily) provided only 80% protection. Weight observations in the mice were similar to those in the hamster model. After withdrawal of treatment, the vancomycin-treated animals continued to lose weight, whereas the MBX-500-treated mice gradually regained weight. In previous studies, observations of improved weight gain have correlated with less severe disease as quantified by histopathological injury (3). These results suggest that MBX-500 may be useful as a first-line agent or in salvage therapy for recurrent infections.
The fact that MBX-500 demonstrates a decrease in recurrence rates over vancomycin merits further studies. The newly approved CDI antibiotic fidaxomicin exhibits a statistically significant reduction in recurrence of CDI for non-NAP1/027 strains of C. difficile compared to vancomycin (7.8% versus 25.5%); however, there was no difference in recurrence rates for those patients infected with the NAP1/027 ribotype (24.4% versus 23.6%), designating fidaxomicin as noninferior to vancomycin rather than superior (18). MBX-500 has the potential to increase treatment options for recurrent CDI caused by toxigenic strains, especially those with the NAP1/027 ribotype, based on the promising in vitro results with the NAP1/027 isolates and its in vivo potency against other toxigenic isolates.
In summary, we have identified a novel hybrid antibacterial, MBX-500, representing dual mechanism activities of an AU DNA polymerase inhibitor and an FQ topoisomerase/gyrase inhibitor. Although hybrid molecules containing an FQ moiety have been evaluated in the past (7, 14, 24), no group has linked a novel, unexploited DNA polymerase inhibitor to the FQ moiety. Further refinement of this new class of antibiotics selectively targeting C. difficile may provide a new weapon against the highly virulent, toxigenic NAP1/027 strain that is causing epidemics in U.S. hospitals and worldwide. MBX-500 is ideally suited for this application since it is administered orally, is locally active with no systemic absorption, and has a narrow spectrum of activity, avoiding the decimation of normal gut microflora, a key risk factor for recurrence of CDI. Finally, use of a triple-target agent such as MBX-500 will likely reduce the rate of resistance compared to that of a single-target agent.
ACKNOWLEDGMENTS
This study was sponsored in part by NIH grant 9 R44 AI068349-04-06 (G.E.W., principal investigator).
We thank Donald Moir for excellent editorial assistance.
All animal studies have complied with federal guidelines and institutional policies.
Footnotes
Published ahead of print 25 June 2012
REFERENCES
- 1. Aas J, Gessert CE, Bakken JS. 2003. Recurrent Clostridium difficile colitis: case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin. Infect. Dis. 36:580–585 [DOI] [PubMed] [Google Scholar]
- 2. Allen SD, Emery CL, Siders JA. 1999. Clostridium, p 654–671 In Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH. (ed), Manual of clinical microbiology, 7th ed ASM Press, Washington, DC [Google Scholar]
- 3. Anton PM, et al. 2004. Rifalazil treats and prevents relapse of Clostridium difficile-associated diarrhea in hamsters. Antimicrob. Agents Chemother. 48:3975–3979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartlett JG. 2006. New drugs for Clostridium difficile infection. Clin. Infect. Dis. 43:428–431 [DOI] [PubMed] [Google Scholar]
- 5. Bartlett JG, Chang TW, Gurwith M, Gorbach SL, Onderdonk AB. 1978. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N. Engl. J. Med. 298:531–534 [DOI] [PubMed] [Google Scholar]
- 6. Bartlett JG, Perl TM. 2005. The new Clostridium difficile-–what does it mean? N. Engl. J. Med. 353:2503–2505 [DOI] [PubMed] [Google Scholar]
- 7. Bryskier A. 1997. Dual beta-lactam-fluoroquinolone compounds: a novel approach to antibacterial treatment. Expert Opin. Invest. Drugs 6:1479–1499 [DOI] [PubMed] [Google Scholar]
- 8. Butler MM, et al. 2007. Antibacterial activity and mechanism of action of a novel anilinouracil-fluoroquinolone hybrid compound. Antimicrob. Agents Chemother. 51:119–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. CDC 2010. Fluoroquinolone resistance and Clostridium difficile, Germany. CDC, Atlanta, GA: http://wwwnc.cdc.gov/eid/article/16/4/09-0859_article.htm [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen X, et al. 2008. A mouse model of Clostridium difficile-associated disease. Gastroenterology 135:1984–1992 [DOI] [PubMed] [Google Scholar]
- 11. CLSI 2007. Methods for antimicrobial susceptibility testing of anaerobic bacteria; approved standard M11-A7. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 12. Geric B, et al. 2006. Binary toxin-producing, large clostridial toxin-negative Clostridium difficile strains are enterotoxic but do not cause disease in hamsters. J. Infect. Dis. 193:1143–1150 [DOI] [PubMed] [Google Scholar]
- 13. Hardesty JS, Juang P. 2011. Fidaxomicin: a macrocyclic antibiotic for the treatment of Clostridium difficile infection. Pharmacotherapy 31:877–886 [DOI] [PubMed] [Google Scholar]
- 14. Hubschwerlen C, et al. 2003. Structure-activity relationship in the oxazolidinone-quinolone hybrid series: influence of the central spacer on the antibacterial activity and the mode of action. Bioorg. Med. Chem. Lett. 13:4229–4233 [DOI] [PubMed] [Google Scholar]
- 15. Kelly CP, LaMont JT. 2008. Clostridium difficile—more difficult than ever. N. Engl. J. Med. 359:1932–1940 [DOI] [PubMed] [Google Scholar]
- 16. Kokkotou E, et al. 2008. Comparative efficacies of rifaximin and vancomycin for treatment of Clostridium difficile-associated diarrhea and prevention of disease recurrence in hamsters. Antimicrob. Agents Chemother. 52:1121–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koo HL, Garey KW, Dupont HL. 2010. Future novel therapeutic agents for Clostridium difficile infection. Expert Opin. Invest. Drugs 19:825–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lancaster JW, Matthews SJ. 2012. Fidaxomicin: the newest addition to the armamentarium against Clostridium difficile infections. Clin. Ther. 34:1–13 [DOI] [PubMed] [Google Scholar]
- 19. McDonald LC, et al. 2005. An epidemic, toxin gene-variant strain of Clostridium difficile. N. Engl. J. Med. 353:2433–2441 [DOI] [PubMed] [Google Scholar]
- 20. McFarland LV, Elmer GW, Surawicz CM. 2002. Breaking the cycle: treatment strategies for 163 cases of recurrent Clostridium difficile disease. Am. J. Gastroenterol. 97:1769–1775 [DOI] [PubMed] [Google Scholar]
- 21. Noren T, et al. 2006. Frequent emergence of resistance in Clostridium difficile during treatment of C. difficile-associated diarrhea with fusidic acid. Antimicrob. Agents Chemother. 50:3028–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pelaez T, et al. 2002. Reassessment of Clostridium difficile susceptibility to metronidazole and vancomycin. Antimicrob. Agents Chemother. 46:1647–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pelaez T, et al. 2008. Metronidazole resistance in Clostridium difficile is heterogeneous. J. Clin. Microbiol. 46:3028–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robertson GT, et al. 2008. In vitro evaluation of CBR-2092, a novel rifamycin-quinolone hybrid antibiotic: studies of the mode of action in Staphylococcus aureus. Antimicrob. Agents Chemother. 52:2313–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seal D, et al. 1987. Treatment of relapsing Clostridium difficile diarrhoea by administration of a nontoxigenic strain. Eur. J. Clin. Microbiol. 6:51–53 [DOI] [PubMed] [Google Scholar]
- 26. Segarra-Newnham M. 2007. Probiotics for Clostridium difficile-associated diarrhea: focus on Lactobacillus rhamnosus GG and Saccharomyces boulardii. Ann. Pharmacother. 41:1212–1221 [DOI] [PubMed] [Google Scholar]
- 27. Sougioultzis S, et al. 2005. Clostridium difficile toxoid vaccine in recurrent C. difficile-associated diarrhea. Gastroenterology 128:764–770 [DOI] [PubMed] [Google Scholar]
- 28. Sun X, et al. 2011. Mouse relapse model of Clostridium difficile infection. Infect. Immun. 79:2856–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilcox MH, Fawley WN, Settle CD, Davidson A. 1998. Recurrence of symptoms in Clostridium difficile infection—relapse or reinfection? J. Hosp. Infect. 38:93–100 [DOI] [PubMed] [Google Scholar]
- 30. Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. 2007. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin. Infect. Dis. 45:302–307 [DOI] [PubMed] [Google Scholar]
- 31. Zhi C, et al. 2005. Synthesis and antibacterial activity of 3-substituted-6-(3-ethyl-4-methylanilino)uracils. J. Med. Chem. 48:7063–7074 [DOI] [PubMed] [Google Scholar]
- 32. Zhi C, et al. 2006. Hybrid antibacterials. DNA polymerase-topoisomerase inhibitors. J. Med. Chem. 49:1455–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]