

Abstract
Hepatitis C virus (HCV) infection is a major global health burden and is associated with an increased risk of liver cirrhosis and hepatocellular carcinoma. There remains an unmet medical need for efficacious and safe direct antivirals with complementary modes of action for combination in treatment regimens to deliver a high cure rate with a short duration of treatment for HCV patients. Here we report the in vitro inhibitory activity, mode of action, binding kinetics, and resistance profile of TMC647055, a novel and potent nonnucleoside inhibitor of the HCV NS5B RNA-dependent RNA polymerase. In vitro combination studies with an HCV NS3/4A protease inhibitor demonstrated potent suppression of HCV RNA replication, confirming the potential for combination of these two classes in the treatment of chronic HCV infection. TMC647055 is a potent nonnucleoside NS5B polymerase inhibitor of HCV replication with a promising in vitro biochemical, kinetic, and virological profile that is currently undergoing clinical evaluation.
INTRODUCTION
Hepatitis C is caused by a chronic viral infection with the hepatitis C virus (HCV) and may ultimately result in severe liver diseases, including fibrosis, cirrhosis, and hepatocellular carcinoma (12). HCV is transmitted via the blood or contaminated blood products, and although blood screening has considerably reduced the rate of new infections (40), an estimated 170 million people are currently infected worldwide (13). There are six HCV genotypes (1 to 6) and multiple subtypes thereof, and genotype 1 is the predominant genotype in Europe, North America, Japan, and China (38).
Until recently, patients were treated with weekly injections of pegylated alpha interferon (IFN-α) in combination with twice-daily oral ribavirin for 48 weeks. This treatment regimen results in a sustained virological response (SVR) in approximately 80% of patients infected with genotype 2 or 3 but only 40 to 50% of those infected with genotype 1 (39). Moreover, this regimen is associated with a range of side effects, including flu-like symptoms, anemia, and depression (31). Hence, considerable efforts are being focused on the discovery and development of direct-acting antivirals (DAAs) for hepatitis C. It is believed that inhibitors of HCV NS3/4A protease, NS5A, and NS5B polymerase will be effective antivirals with fewer side effects than the combination of pegylated IFN-α and ribavirin.
In 2011, the HCV NS3/4A protease inhibitors telaprevir (17, 44) and boceprevir (2, 35) were approved by the U.S. Food and Drug Administration and the European Medicines Agency for the treatment of genotype 1 chronic hepatitis C in combination therapies with pegylated IFN-α and ribavirin. Treatment with telaprevir and boceprevir in combination with pegylated IFN-α and ribavirin significantly increased the SVR rate in treatment-naïve and experienced patients infected with genotype 1 virus and resulted in shortened treatment durations for a substantial number of treatment-naïve patients (17, 35, 44). Nevertheless, multiple DAAs with complementary modes of action are needed to minimize viral resistance, reduce the treatment duration time; increase the cure rates of hard-to-treat patient populations, such as prior nonresponders to an IFN-based regimen; and establish all-oral IFN-free treatment regimens with fewer side effects, thereby providing therapeutic options for IFN-intolerant patients.
HCV NS5B is an RNA-dependent RNA polymerase (RdRp) that is responsible for the synthesis of both positive-strand genomic RNA and negative-strand RNA (5, 29). The three-dimensional structure of HCV NS5B is similar to those of other RdRps, comprising a right-hand motif with thumb and finger domains and a palm domain containing the active site (26). Several classes of NS5B inhibitors have been reported (3, 19). The nucleoside NS5B polymerase inhibitors (NI) compete with the natural ribonucleoside triphosphate substrates of NS5B at the active site of the enzyme, where chain elongation is catalyzed. Nonnucleoside inhibitors (NNI) belong to diverse chemical families and usually bind to one of four distinct binding sites named NNI-1 to NNI-4, in locations distal from the catalytic center of the NS5B polymerase, thereby interfering with the initiation and/or elongation step of RNA synthesis. The NNI-1 binding site, also known as the thumb pocket I site, is located at the interface of the finger and thumb domains of the NS5B polymerase and is occupied by the flexible fingertip λ1 loop when the NS5B polymerase is in a closed active conformation (11). Binding of inhibitors to NNI-1 is purported to displace the flexible λ1 loop from its thumb domain binding site, perturbing the interaction between the NS5B polymerase finger and thumb domains and thereby locking the enzyme in an open inactive conformation. Small molecules with benzimidazole (4, 16, 41) and indole chemical scaffolds (15) were identified as NNI-1-directed NS5B inhibitors.
A proof-of-concept study of HCV genotype 1-infected subjects showed that monotherapy with the NNI-1 inhibitor BI 207127 has antiviral activity against HCV genotype 1 (23). Zeuzem et al. reported on an orally administered triple combination treatment for 4 weeks with BI 207127, the HCV NS3/4A protease inhibitor BI 201335, and ribavirin without pegylated IFN-α in treatment-naïve patients infected with HCV genotype 1 (45). This triple combination demonstrated potent early antiviral activity against HCV genotype 1 with good safety and tolerability.
We now report the in vitro inhibitory activity, mode of action, resistance profile, and genotypic coverage of TMC647055, a novel indole class NNI-1-directed NS5B inhibitor. Furthermore, we investigated the affinity of TMC647055 for a panel of HCV NS5B polymerases representing diverse genotypes. TMC647055-NS5B complex stability accounts for the potent inhibitory activity of TMC647055 against the majority of HCV genotypes.
MATERIALS AND METHODS
Inhibitor synthesis.
TMC647055 and TMC435 were synthesized in-house as described elsewhere (36, 43).
Cells and plasmids.
Huh7-Luc cells containing the genotype 1b Con-1 bicistronic subgenomic HCV replicon (clone ET) with cell culture adaptive mutations E1202G, T1280I, and K1846T in NS3 and NS4B and encoding a firefly luciferase reporter (28, 30) were kindly provided by R. Bartenschlager, University of Heidelberg, Heidelberg, Germany. Huh7-SG-Con1b cells (i.e., genotype 1b Con1-based subgenomic replicon clone with the S2204I cell culture adaptive mutation in NS5A) and Huh7-SG-1a cells (i.e., a genotype 1a H77-based subgenomic replicon clone with the P1496L and S2204I cell culture adaptive mutations in NS3 and NS5A) were obtained from Apath LLC (St. Louis, MO) (6, 7). Replicon-containing cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Sigma D-5546 medium supplemented with 10% fetal calf serum [FCS], 1% l-glutamine, 0.04% [50 mg/ml] gentamicin) containing 500 to 750 μg/ml G418. Human hepatoma Huh7-lunet (obtained from R. Bartenschlager), VeroE6, MRC-5, HEK293T, and HepG2 cells were maintained in DMEM.
Cloning, expression, and purification of NS5B.
The coding sequences of the different NS5B isolates lacking 21 C-terminal residues (NS5BΔC21) were amplified and subcloned into the pET21b plasmid as described previously (32). The NS5BΔC21 expression constructs were then transformed into Escherichia coli Rosetta 2 (DE3) (Novagen, Madison, WI) for NS5B production, and protein was purified as reported before (32).
RdRp assay.
A primer-dependent transcription assay enabled the determination of 50% inhibitory concentrations (IC50s) essentially as described previously (32). Briefly, following a 15-min preincubation with the inhibitor, 20 nM purified Con1b NS5BΔC21 polymerase was incubated for 10 min with a mixture of 150 nM 5′-biotinylated oligonucleotide (rG13) primer, 15 nM poly(rC) template, 19 mM Tris-HCl, 5 mM MgCl2, 17 mM NaCl, 21 mM KCl, and 2.5 mM dithiothreitol (DTT). Then, 600 nM GTP and 0.13 μCi of [3H]GTP were added to the 40-μl mixture to initiate the reaction; the mixture was then incubated at room temperature for 2 h before the reaction was stopped by the addition of 40 μl streptavidin-coated SPA beads.
Anti-replicon activity and cytotoxicity of TMC647055.
The luciferase reporter replicon assay and the replicon assays with a quantitative real-time PCR (qRT-PCR) readout were performed as described previously (27). Briefly, Huh7-Luc replicon cells and counterscreen cell lines (Huh7-CMV-Luc and MT4-LTR-Luc) were incubated with serial dilutions of TMC647055 for 3 days, after which the luciferase activity was determined or HCV replicon RNA levels were measured and normalized to a cellular reference mRNA (the ribosomal subunit RPL13 gene). The 50% effective concentrations (EC50s) and n-fold changes in the EC50s were calculated for the inhibitor assayed in triplicate unless stated otherwise. The cytotoxicity of TMC647055 for different cell types (MRC-5, HEK-293T, HepG2, and VeroE6 cells) was analyzed after 3 days of inhibitor exposure by incubation with resazurin (Sigma-Aldrich, St. Louis, MO), followed by resorufin fluorescence detection in a fluorescence reader. Dose-dependent inhibition curves were used to calculate 50% cytotoxic concentrations (CC50s). In addition, CC50s were determined in an Huh7 hepatoma cell line expressing a luciferase reporter under the control of a human cytomegalovirus (CMV) major immediate-early promoter (Huh7-CMV-Luc) and in an MT4 T lymphocyte cell line (MT4-LTR-Luc) encoding a luciferase reporter under the control of the human immunodeficiency virus type 1 (HIV-1) long terminal repeat by measuring luciferase luminescence after 3 days of incubation in the presence of increasing concentrations of the compound.
Transient replicon assay: mutant replicons.
The drug susceptibility of mutant replicons was assessed as described previously (25, 32). Mutant replicons were generated by the introduction of site-directed mutations into the pFKi341Luc_NS3-3′_ET backbone, obtained from R. Bartenschlager (22). Based on luciferase luminescence measurements of inhibitor-treated wells and nontreated-cell controls, dose-response curves and EC50s were calculated.
Transient replicon assay: NS5B chimeric replicons.
NS5B chimeric replicons were generated as described previously (32). The sensitivity of the chimeric replicons to TMC647055 was tested in a transient replicon assay as described previously (30).
Selectivity of TMC647055 for HCV.
Antiviral activities against a broad range of DNA (CMV, adenovirus, hepatitis B virus, vaccinia virus) and RNA (coxsackie virus, dengue virus, influenza virus, yellow fever virus) viruses were determined as described previously (1, 14, 18, 21, 27).
Determination of affinity and dissociation constants of TMC647055 binding to NS5B.
Interactions between NS5BΔC21 polymerase isolates and inhibitors were measured via surface plasmon resonance (SPR) using the T100 Biacore system as described previously (32). Briefly, purified, HIS6-tagged polymerases were immobilized using noncovalent, reversible capture on nitrilotriacetic acid sensor chips (GE Healthcare) in immobilization buffer (20 mM morpholinepropanesulfonic acid [MOPS; pH 7.4], 500 mM NaCl, 0.005% Tween 20, 1 mM DTT, 50 μM EDTA). Immobilization levels of 5,000 to 20,000 resonance units were routinely reached. Interaction studies were all performed at 25°C with a flow rate of 10 μl/min. Inhibitors were serially diluted in running buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 50 μM EDTA, 1 mM DTT, 0.005% Tween 20) containing 5% dimethyl sulfoxide (DMSO). Single-cycle kinetics were used in which five increasing concentrations of inhibitor were injected for a period of 300 s each in one single cycle, and dissociation was monitored for a period of 1,200 s. The sensor surface was regenerated by sequentially injecting regeneration buffer 1 {350 mM EDTA, 10% DMSO, 500 mM NaCl, 0.2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 0.1% soluble carboxymethyl-dextran} and regeneration buffer 2 (0.05% SDS in phosphate-buffered saline). Data were analyzed using simultaneous nonlinear regression analysis (global fitting) adapted for single-cycle kinetics with Biacore T100 BiaEval evaluation software 2.0 (GE Healthcare) as described previously (32).
Colony formation assay with Huh7-Luc replicon cells.
A colony formation assay was performed as described previously (25). Briefly, Huh7-Luc replicon cells were seeded at a concentration of 2 × 104/10-cm dish and treated with different concentrations of a single inhibitor or a combination of two different inhibitors, respectively, in the presence of 1,000 μg/ml G418. Medium was refreshed twice weekly, and fresh inhibitor was added. After 2 to 3 weeks, when significant cell death was visible, the remaining cell colonies were stained with neutral red and counted.
In vitro selection of resistant replicons.
Huh7-Luc replicon cells were seeded at a concentration of 3 × 105/10-cm dish and treated with different concentrations of compound in the presence of 250 μg/ml G418. TMC647055 concentrations of 375 nM, 750 nM, and 1.875 μM (approximately 5, 10, and 25 times the EC50, respectively) were used for genotype 1b resistance selection experiments. For the genotype 1a resistance selection experiments, 750 nM and 1,500 nM TMC647055 (approximately 5 and 10 times the EC50, respectively) were used. Medium was refreshed twice weekly, and fresh compound was added. Cells were subcultured when necessary, and significant cell death typically occurred after 2 to 3 weeks. Surviving cell colonies were individually picked or pooled and expanded. As soon as the quantity of cells obtained was sufficient, RNA was extracted and amplified and the NS5B region was sequenced by standard Sanger sequencing at a population level.
HCV replicon clearance and rebound assay.
For the HCV replicon clearance and rebound assay, Huh7-Luc replicon cells were seeded at a concentration of 3 × 105/10-cm dish and treated with different concentrations of a single inhibitor or a combination of two inhibitors for 2 weeks in the absence of G418 (clearance phase). Cells were subcultured twice per week during the clearance phase upon reaching 70 to 80% confluence, and cell pellets were collected for RNA extraction, followed by cDNA synthesis and RT-PCR to monitor HCV replicon RNA levels. After 2 weeks, cells were incubated in the absence of inhibitor for a further period of 3 weeks in the presence of 250 μg/ml G418 to enable a rebound to occur where residual HCV replicon RNA was present after drug treatment. The G418-containing cell culture medium was refreshed every 3 to 4 days, and cells were subcultured when 70 to 80% confluence was reached. Cell pellets of each passage during the rebound phase were collected and subjected to RNA extraction, cDNA generation, and RT-PCR to monitor HCV replicon RNA levels.
RESULTS
TMC647055 is a cell-permeating, selective HCV NS5B inhibitor.
TMC647055 is a macrocyclic indole inhibitor (Fig. 1) that showed itself to be a potent inhibitor of the HCV NS5B polymerase (genotype 1b, Con1b), eliciting a mean IC50 of 34 nM, as assessed in the RdRp primer-dependent transcription assay. In the stable replicon system, TMC647055 inhibited the replication of a genotype 1b replicon (clone ET) in the Huh7-Luc cell line with median EC50s of 77 nM (luciferase readout) and 139 nM (quantitative reverse transcription-PCR readout) (Table 1). The median EC50s in Huh7-SG-Con1b, a stable genotype 1b cell line, and in Huh7-SG-1a, a stable genotype 1a cell line, were, respectively, 74 nM and 166 nM (RT-PCR readout). To investigate the effect on replicon activity of binding of the compound to human serum proteins, we added 40% human serum to the Huh7-Luc cell line. This resulted in an ∼10-fold increase in the median EC50 to 740 nM (luciferase readout). The mean CC50s for Huh7 and MT4 cells in a luciferase reporter assay were 42.1 μM (n = 5) and 28.9 μM (n = 16), respectively. Cytotoxicity measurements in a panel of additional cell lines (MRC-5, HEK-293T, HepG2, and VeroE6) using a resazurin assay all resulted in CC50s in excess of 50 μM (see Table S1 in the supplemental material).
Fig 1.
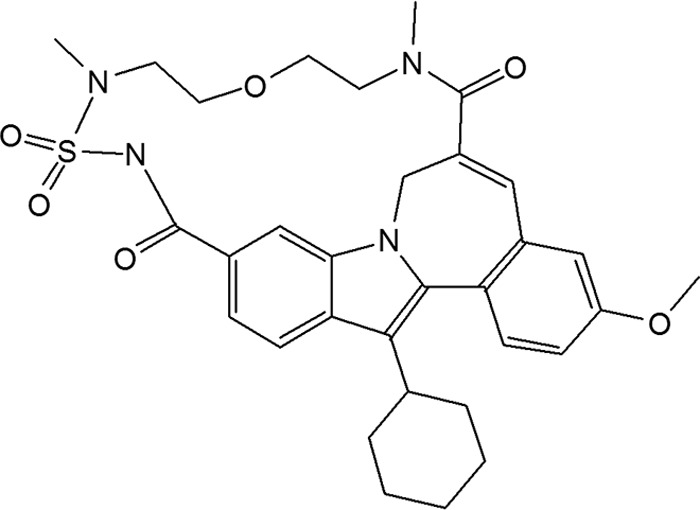
Structure of TMC647055.
Table 1.
Antiviral activity of TMC647055 in stable replicon-containing cell lines
| Replicon cell line (readout) | Subtype | EC50 (μM)a | EC50 IQRb (Q1-Q3) | nc | EC90 (μM)a | EC90 IQRb (Q1-Q3) | nc |
|---|---|---|---|---|---|---|---|
| Huh7-Luc (luciferase) | 1b | 0.077 | 0.052–0.158 | 109 | 0.339 | 0.266–0.454 | 109 |
| Huh7-Luc (qRT-PCR) | 1b | 0.139 | 0.103–0.140 | 6 | 0.395 | 0.317–0.454 | 6 |
| Huh7-SG-Con1b (qRT-PCR) | 1a | 0.074 | 0.052–0.165 | 5 | 0.407 | 0.308–0.483 | 6 |
| Huh7-SG-1a (qRT-PCR) | 1a | 0.166 | 0.041–0.180 | 8 | 0.465 | 0.110–0.495 | 8 |
| Huh7-Luc + 40% human serum (luciferase) | 1b | 0.740 | 0.503–1.035 | 59 | 3.70 | 1.88–5.02 | 58 |
EC50s and EC90s are expressed as median values.
IQR, interquartile range.
n, number of independent measurements.
TMC647055 showed high selectivity for HCV when evaluated with a broad panel of human DNA and RNA viruses. At concentrations of up to 100 μM, no antiviral activity was observed for CMV, adenovirus, vaccinia virus, coxsackie virus, influenza virus, and yellow fever virus and the compound showed no effect on dengue virus up to the highest tested concentration of 25 μM (data not shown). The EC50 in the hepatitis B virus antiviral assay was 86 μM (data not shown).
In order to confirm that the inhibitor inhibited the NS5B polymerase via interaction with the NNI-1 pocket, the antiviral activity of TMC647055 was tested on a mutant panel in the transient replicon assay. TMC647055 showed median EC50 changes in the replicon assay of 9-, 3-, and 371-fold for L392I, V494A, and P495L, respectively, three mutations that are associated with reduced sensitivity to NNI-1 inhibitors (37) (Table 2). Consistent with its mode of action, TMC647055 activity was not affected by mutations associated with reduced sensitivity to NNI-2, NNI-3, NNI-4, HCV NS5B nucleoside inhibitors, HCV NS3/4A protease inhibitors, and HCV NS5A inhibitors (20).
Table 2.
Effects of NS3/4A, NS5A, and NS5B mutations in the reference genotype 1b (clone ET) replicon on TMC647055 susceptibility
| Inhibitor classa and mutation | EC50 (μM)b | EC50 range (μM) | Fold changec | Fold change range | nd |
|---|---|---|---|---|---|
| None (wild-type clone ET) | 0.051 | 0.008–0.295 | 140 | ||
| NS5B nonnucleoside inhibitor | |||||
| L392I (NNI-1) | 1.039 | 0.814–1.65 | 8.8 | 8.2–13.3 | 5 |
| V494A (NNI-1) | 0.208 | 0.073–0.311 | 3.3 | 2.9–3.9 | 7 |
| P495L (NNI-1) | 17.8 | 14.7–>31.5 | 371.2 | 175.4–772.1 | 8 |
| M423T (NNI-2) | 0.067 | 0.014–0.084 | 0.8 | 0.6–1.1 | 8 |
| M414T (NNI-3) | 0.072 | 0.050–0.085 | 0.9 | 0.9–0.9 | 3 |
| C316Y (NNI-4) | 0.024 | 0.008–0.050 | 0.5 | 0.3–0.6 | 6 |
| NS5B nucleoside inhibitor | |||||
| S96T | 0.099 | 0.078–0.191 | 1.3 | 0.9–2.2 | 9 |
| S282T | 0.073 | 0.037–0.109 | 0.9 | 0.7–1.2 | 12 |
| NS3/4A protease inhibitor | |||||
| R155K | 0.085 | 0.031–0.137 | 1.2 | 1.0–1.5 | 5 |
| A156V | 0.081 | 0.076–0.087 | 2.8 | 2.7–3.0 | 2 |
| D168V | 0.055 | 0.019–0.072 | 0.8 | 0.7–1.1 | 5 |
| NS5A inhibitor | |||||
| L31V | 0.040 | 0.015–0.075 | 1.0 | 0.6–1.6 | 4 |
| Y93H | 0.052 | 0.015–0.083 | 0.9 | 0.6–1.5 | 5 |
For each inhibitor class, major mutations associated with reduced susceptibility to this class of inhibitors were engineered into the genotype 1b wild-type (clone ET) replicon.
EC50s are expressed as median values.
Fold changes in EC50s compared to the wild-type (clone ET) replicon were calculated for every single experiment, and the median values are shown.
n, number of independent measurements. For n = 1 or 2, the observed value or the mean of the two observed values is, respectively, reported.
Inhibitory activity of TMC647055 on HCV genotypes.
The genotypic coverage of TMC647055 was investigated in a transient replicon assay using chimeric replicons based on the genotype 1b replicon (clone ET) in which the C-terminal part of NS5A (amino acids 440 to 447) and full-length NS5B (amino acids 1 to 591) were replaced with the corresponding sequence derived from samples from patients infected with HCV genotype 1a (n = 14), 1b (n = 10), 2b (n = 2), 3a (n = 2), 4a (n = 1), or 6a (n = 2); each chimeric replicon represented the NS5B sequence from a different patient. The genotype 5a chimera did not replicate, preventing the determination of an EC50. For genotype 2a, the corresponding sequence of JFH-1 was used.
The median EC50 of TMC647055 on NS5B sequences derived from clinical isolates of genotypes 1a, 1b, 3a, 4a, and 6a ranged between 27 nM and 113 nM (Table 3). However, the genotype 2a and 2b chimeric replicons exhibited a more-than-200-fold reduction in susceptibility to TMC647055 in comparison with the reference genotype 1b (clone ET) replicon.
Table 3.
Antiviral activity of TMC647055 against chimeric replicons containing the NS5B region derived from patient isolates assessed in a transient replicon assaya
| NS5B genotype in transient replicon | No. of isolates | Median EC50 (μM) | EC50 range (μM) |
|---|---|---|---|
| Wild-type clone ET | 0.051 | ||
| 1a | 14 | 0.048 | 0.016–0.185 |
| 1b | 10 | 0.027 | 0.014–0.051 |
| 2a | 1 | 12.530 | |
| 2b | 2 | NDb | 28.7–>98.4 |
| 3a | 2 | 0.088 | 0.050–0.127 |
| 4a | 1 | 0.097 | |
| 6a | 2 | 0.113 | 0.082–0.143 |
The median EC50 across the different isolates within one genotype, the number of different isolates per genotype, and the range of the EC50s of the different isolates within one genotype are shown. For n = 1 or 2, the observed value or the mean of the two observed values is, respectively, reported.
ND, value not determined because the median EC50 for one genotype 2b isolate was 28.7 μM and that for the other genotype 2b isolate was >98.4 μM.
The affinity, binding kinetics, and dissociation constants of TMC647055 and HCV NS5B.
Previous studies have demonstrated a good correlation between the cellular inhibitory activity of an inhibitor (EC50) and the affinity of the inhibitor for its target protein as measured by SPR (10, 32, 33, 42). This technique enables accurate determination of inhibitor binding kinetics, i.e., the association rate constant (kon), dissociation rate constant (koff), affinity (KD = koff/kon), and complex stability (t1/2 = ln2/koff).
TMC647055 displayed high-affinity interaction (median KD, <35 nM) with NS5B across all genotypes, except for genotype 2b, for which the median KD of 880 nM was more than 20 times higher than those for the other genotypes (Table 4). This reduced affinity for genotype 2b NS5B was due mainly to a >9-fold increased dissociation rate and concomitant >9-fold decreased inhibitor-NS5B complex stability. Genotype 1b samples displayed the highest complex stability (median t1/2, 121 min). The median n-fold change in the KD of the NS5B enzymes of different genotypes compared to that of the Con1b wild-type enzyme, as measured in SPR, was shown to be concordant with the median n-fold change in the EC50s of NS5B sequences of different genotypes, compared to that of the wild-type genotype 1b (clone ET) replicon, as assessed in a transient replicon assay (Fig. 2).
Table 4.
Kinetic parameters of TMC647055 interactions with NS5B clinical isolates of different genotypesa
| NS5B genotype of purified protein | No. of isolates |
kon (1/Ms) |
koff (1/s) |
KD (M) |
t1/2 (min) |
||||
|---|---|---|---|---|---|---|---|---|---|
| Median | Range | Median | Range | Median | Range | Median | Range | ||
| Wild-type Con1b | 2.2E+04 | 8.9E−05 | 4.1E−09 | 130.5 | |||||
| 1a | 5 | 7.5E+04 | (3.9–10.9)E+04 | 4.7E−04 | (3.1–8.2)E−04 | 7.3E−09 | (2.9–10.5)E−09 | 24.5 | 14.1–37.2 |
| 1b | 5 | 3.1E+04 | (2.3–3.7)E+04 | 9.5E−05 | (4.9–14.1)E−05 | 3.9E−09 | (2.3–5.1)E−09 | 121.4 | 82.1–234.0 |
| 2b | 3 | 1.5E+04 | (4.4–42.9)E+03 | 1.5E−02 | (1.2–1.7)E−02 | 8.8E−07 | (2.8–38.5)E−07 | 0.8 | 0.7–1.0 |
| 3a | 3 | 6.0E+04 | (2.5–9.5)E+04 | 3.6E−04 | (3.6–4.5)E−04 | 6.1E−09 | (4.0–19.1)E−09 | 31.7 | 25.5–32.1 |
| 4a | 3 | 2.1E+05 | (1.8–2.9)E+05 | 4.4E−04 | (3.9–6.0)E−04 | 2.0E−09 | (1.4–3.8)E−09 | 26.2 | 19.1–29.6 |
| 5a | 1 | 4.0E+04 | 9.7E−04 | 2.1E−08 | 11.9 | ||||
| 6a | 1 | 5.1E+04 | 1.6E−03 | 3.0E−08 | 7.3 | ||||
The interaction was analyzed using the SPR reversible capture assay for HCV NS5B.
Fig 2.
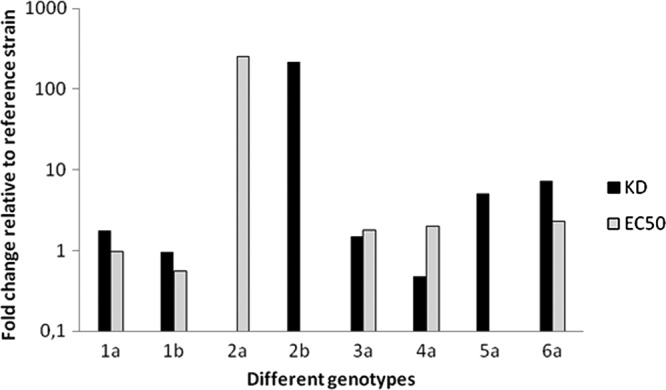
Comparison of n-fold changes in the KD and EC50 of TMC647055 between different genotypes. The n-fold changes in median KD values (Table 4) were calculated by using the Con1b reference strain, while for n-fold changes in median EC50s (Table 3), the 1b clone ET reference strain was used.
In agreement with the data from the transient replicon studies, the affinity of TMC647055 for NS5B was affected by mutations in the NNI-1 binding pocket (Table 5) and not by mutations in other NNI and NI binding pockets (data not shown). The L392I and V494A mutations resulted in, respectively, 13- and 4-fold reductions in affinity, mainly due to an increased dissociation rate constant (koff) and commensurately reduced complex stability. The P495L mutant elicited a >1,000-fold reduction in affinity, through a combination of a 6-fold reduced kon and a >200-fold increased koff (Table 5 and Fig. 3).
Table 5.
Kinetic parameters of TMC647055 interactions with different NS5B mutantsa
| Mutation | kon (1/Ms) | koff (1/s) | KD (M) | t1/2 (min) | n |
|---|---|---|---|---|---|
| Con1b wild type | 2.2E+04 | 8.9E−05 | 4.1E−09 | 130.5 | 11 |
| L392I (NNI-1) | 1.8E+04 | 9.7E−04 | 5.5E−08 | 11.9 | 2 |
| V494A (NNI-1) | 5.5E+04 | 8.4E−04 | 1.6E−08 | 13.8 | 2 |
| P495L (NNI-1) | 3.4E+03 | 2.2E−02 | 6.5E−06 | 0.5 | 2 |
The interaction was analyzed using the SPR reversible capture assay for HCV NS5B. For each inhibitor class, major mutations associated with reduced susceptibility to this class of inhibitors were engineered into the genotype 1b backbone. n, number of independent measurements. Values are averages of at least two independent experiments.
Fig 3.

Sensorgrams for the interaction of TMC647055 with two selected NS5B isolates (Con1b wild type in the left panel and Con1b P495L mutant in the right panel), as analyzed using the SPR reversible capture assay for HCV NNS5B. The series of increasing concentrations of TMC647055 that were injected onto the immobilized NS5B proteins are indicated. Gray traces represent the double-reference subtracted data, while the red traces represent the fitted values.
Frequency of resistant colony formation.
The frequency of resistant colony formation was determined in a colony formation assay using Huh7-Luc replicon cells incubated in the presence of different concentrations of TMC647055 and TMC435, a potent, once-daily HCV NS3/4A protease inhibitor (27), alone or in combination (see Fig. S1 in the supplemental material). After 2 to 3 weeks, the control dish containing untreated Huh7-Luc replicon cells showed widespread cell growth, whereas a dose-dependent reduction of cell colony formation was observed under TMC435 (>100 colonies at 80 nM and 19 colonies at a 200 nM concentration) or TMC647055 (30 colonies at a 750 nM concentration) pressure (see Fig. S1 in the supplemental material). Interestingly, no resistant replicon cell colonies were observed with 1.5 μM TMC647055 alone, indicating complete clearance of the replicon from the cells. Combination of TMC435 with TMC647055 suppressed the formation of resistant colonies, even at the lowest concentrations used (see Fig. S1 in the supplemental material). No long-term cytotoxic effects in the absence of the G418 selection agent were observed at combinations of the highest concentrations tested (data not shown).
In vitro resistance selection with TMC647055.
Six independent resistance selection experiments were performed with TMC647055 and genotype 1b replicon (Huh7-Luc) cells, and two independent experiments were performed with genotype 1a replicon cells. In all of the experiments, resistant colonies were observed and the HCV replicon RNA was sequenced. NS5B sequences were obtained from 70 TMC647055-treated replicon colonies or pools (21 of genotype 1a and 49 of genotype 1b). Eight sequences were obtained from untreated controls. Only amino acid substitutions that were observed in at least three NS5B sequences from experiments in the presence of TMC647055, as well as any observed TMC647055 binding pocket variations, were further profiled in the transient replicon assay. Notably, none of the sequences analyzed from the control experiments incubated in the absence of TMC647055 contained amino acid changes in the NS5B region.
In genotype 1b resistance selection experiments, mutations were most frequently observed at NS5B residues L392 (L392I) and P495 (P495S/T/L), while in genotype 1a experiments, mutations at NS5B residues P495 (P495S/L), Q309 (Q309R/K), F574 (F574V/S/A), and C575 (C575F/S) were most frequently selected (Fig. 4 and Table 6). When the corresponding mutations were introduced into the genotype 1b ET transient replicon backbone, NNI-1 binding pocket mutations L392I, P495S, P495T, and P495L showed EC50 changes of 9-, 51-, 45-, and 371-fold, respectively. Another mutation that was identified, although at a low frequency (observed in two NS5B sequences from genotype 1b selection experiments), and was previously shown to be associated with reduced sensitivity to NNI-1 inhibitors was V494A. This mutation caused a 3-fold change in sensitivity to TMC647055 (37). None of the mutations outside the NNI-1 binding pocket that were identified in the resistance selection studies and tested in the transient replicon assay (i.e., K79R, N110S, Q309R, T418A, W574V [F574V in genotype 1a], and C575F) displayed an EC50 change of >2-fold compared to reference genotype 1b (Table 6). However, it needs to be taken into account that the F574V and C575F mutations were selected only in the genotype 1a replicon but phenotyped in a genotype 1b transient replicon. The lack of a change in EC50 in the genotype 1b backbone supports the finding that those mutations are not selected in the genotype 1b background.
Fig 4.

Summary of results of the in vitro resistance selection experiment with TMC647055. Shown are the frequencies of changes observed at different amino acid positions in NS5B sequences from TMC647055-treated replicon colonies or pools (21 of genotype 1a, 49 of genotype 1b). Positions at which mutations were present in at least three sequences are shown. Gray bars, genotype 1a; black bars, genotype 1b.
Table 6.
Summary of results of in vitro resistance selection experiments with TMC647055
| NS5B mutationa | No. (%)a of observations in: |
EC50 (μM)b | Fold change in EC50b,c | |
|---|---|---|---|---|
| GT1a replicon | GT1b replicon | |||
| Wild-type clone ET | 0.051 | |||
| L10P | 4 (8.2) | NDd | ND | |
| P19S | 1 (2.0) | ND | ND | |
| Q19H | 2 (9.5) | ND | ND | |
| K79R | 5 (10.2) | 0.144 | 1.0 | |
| N110S | 7 (14.3) | 0.133 | 1.2 | |
| Q309R | 7 (33.3) | 1 (2.0) | 0.047 | 0.8 |
| Q309K | 1 (4.8) | ND | ND | |
| L392I | 4 (19.1) | 17 (34.7) | 1.039 | 8.8 |
| M414V | 3 (14.3) | ND | ND | |
| T418A | 2 (9.5) | 7 (14.3) | 0.178 | 1.7 |
| T418S | 2 (9.5) | ND | ND | |
| E440G | 3 (6.1) | ND | ND | |
| V485M | 3 (14.3) | 1 (2.0) | ND | ND |
| P495S/L | 1 (4.8) | ND | ND | |
| P495S | 5 (23.8) | 3 (6.1) | 6.553 | 51.0 |
| P495L | 3 (14.3) | 5 (10.2) | 17.785 | 371.2 |
| P495T | 2 (4.1) | 3.634 | 44.7 | |
| W571R | 3 (14.3) | 2 (4.1) | ND | ND |
| F574S | 3 (14.3) | ND | ND | |
| F574V | 4 (19.1) | 0.047e | 0.7e | |
| F574A/S/V | 1 (4.8) | ND | ND | |
| C575F | 6 (28.6) | 0.036 | 0.6 | |
| C575S | 2 (9.5) | ND | ND | |
| Y586C | 3 (6.1) | ND | ND | |
| Y586H | 1 (2.0) | ND | ND | |
| Total no. of sequenced colonies/pools | 21 | 49 | ||
NS5B sequences were obtained from 70 TMC647055-treated replicon colonies or pools (21 of genotype 1a and 49 of genotype 1b). Mutations are shown for NS5B residues at which changes were observed in at least three sequences. The frequency (in parentheses) was calculated for each subtype.
EC50s and fold changes in EC50s were measured in a mutant genotype 1b replicon (clone ET) panel in at least three independent experiments. Fold changes in EC50s compared to the wild-type (clone ET) replicon were calculated for every single experiment, and the median values are shown.
Compared to the wild-type replicon (clone ET).
ND, not determined/tested.
In the genotype 1b replicon used for determination of the EC50 and the fold change in the EC50, this corresponds to a W574V change.
In vitro clearance of HCV replicon RNA in genotype 1b replicon.
The in vitro clearance of HCV genotype 1b replicon RNA was determined in an HCV replicon clearance and rebound assay using Huh7-Luc replicon cells incubated in the presence of different concentrations of TMC647055 alone or in combination with the HCV NS3/4A protease inhibitor TMC435 (Fig. 5). HCV replicon RNA was monitored by RT-PCR. The median EC50 of TMC435 was 13 nM (27), compared with 139 nM for TMC647055 in a 3-day assay in Huh7-Luc replicon cells with HCV RNA readout. In the untreated Huh7-Luc control cells, the HCV replicon RNA content was relatively constant over the whole time of the experiment, with a maximum fluctuation of ∼0.3 log10.
Fig 5.

Rebound of HCV replicon RNA after a 14-day treatment with the NNI TMC647055 alone or in combination with the HCV NS3/4A protease inhibitor TMC435 in the absence of G418. After 14 days of treatment, the inhibitors were withdrawn and 250 μg/ml was added to enrich the remaining HCV replicon RNA-containing cells that are capable of growing in the presence of G418 (rebound). The cultures were monitored for another 21 days in the presence of G418, and cell samples were collected whenever the cells were split. The level of HCV replicon RNA in the cells was determined by a qRT-PCR assay.
A fast dose-dependent reduction of HCV replicon RNA was detected upon incubation with 1.5 μM and 3.75 μM TMC647055 during the clearance phase in week 1, after the first passage of the cells. A maximal reduction of HCV replicon RNA was observed after the second passage with a decline of 3.1 log10 for the 1.5-μM-treated cells and 3.7 log10 for the 3.75 μM-treated cells.
Treatment of the replicon-containing cells with a combination of 1.5 or 3.75 μM TMC647055 with 100 nM TMC435 resulted in a rapid decline of HCV replicon RNA within the first two passages of the cells with a maximal reduction of 4.5 log10. HCV replicon RNA remained below the limit of quantification during the remainder of the clearance phase, suggesting efficient removal of the replicon and suppression of resistance. These data were in line with the complete suppression of the formation of resistant colonies by the combination of TMC647055 and TMC435.
DISCUSSION
Before the introduction of NS3/4A protease inhibitors in HCV treatment regimens, cure rates resulting from pegylated IFN-α and ribavirin were moderate, with only 40 to 50% of patients infected with HCV genotype 1 achieving an SVR. The inclusion of telaprevir or boceprevir in regimens with pegylated IFN-α and ribavirin strongly improves the SVR. However, the side effects from the pegylated IFN-α treatment and the limited response in treatment-experienced nonresponders highlight the need for the development of other DAAs to be used in DAA combination treatment regimens.
Combination therapies with DAAs with different modes of action, e.g., NS3/4A protease and NS5B polymerase inhibitors, will likely become the future standard of care for the treatment of chronic hepatitis C with improved treatment outcomes and the potential to develop an IFN-free treatment regimen with fewer side effects. TMC647055 was identified as a novel NS5B inhibitor (43) that binds to the NNI-1 allosteric site of the NS5B polymerase. The inhibitor demonstrated potent inhibition in a biochemical RdRp assay with the genotype 1b NS5B enzyme, as well as in genotype 1a and 1b replicon systems. A 10-fold shift in replicon activity was observed in the presence of 40% human serum, which could be expected given the fact that this compound shows a high degree of binding to human serum proteins (data not shown). Additionally, no cytotoxicity (CC50) was observed in a panel of cell lines with concentrations of TMC647055 of up to 50 μM, while it was a very specific HCV replication inhibitor.
The activity of TMC647055 against NS5B sequences of different HCV genotypes was determined using a transient assay with chimeric replicons based on the genotype 1b clone ET replicon backbone. The inhibitor displayed high potency against genotypes 3a, 4a, and 6a comparable to its activity against genotype 1, suggesting that the overall shape of the NNI-1 pocket is well conserved among these different genotypes. Variability in susceptibility to TMC647055 was observed within the genotype 1a chimeric replicons with a EC50 range of 0.016 to 0.185 μM. The observed variability could not be associated with potential differences in the TMC647055 binding pocket residues, as evaluated by population sequencing of the constructs (data not shown). Activity in the genotype 2a and 2b chimeric replicons was reduced compared to that in the reference genotype 1b clone ET replicon. Likewise, loss of susceptibility of a genotype 2b NS5B polymerase to the Merck indole-N-acetamide class NNI-1 inhibitors has been reported (37), suggesting that this may be a common property of NNI-1 inhibitors. The genotype 5a chimera did not replicate, and hence, TMC647055 could not be assessed against this genotype in the chimeric replicon assay. Key mutations at amino acid positions 392, 494, and 495 known to be selected in the presence of NNI-1 inhibitors were identified during in vitro resistance selection experiments with TMC647055 (37). Linkage studies of the observed mutations in resistance selection studies with TMC647055 were not performed at this stage.
Mutations in the NNI-2, NNI-3, and NNI-4 binding pockets and the catalytic site did not affect the affinity of TMC647055, nor did mutations that confer resistance to other classes of HCV inhibitors, i.e., NS3/4A protease and NS5A inhibitors (20), suggesting that combination therapy with other classes of NS5B inhibitors and HCV NS3/4A protease and NS5A inhibitors is possible.
TMC647055 at a concentration of 1.5 μM completely suppressed the formation of resistant replicon colonies in a colony formation assay using genotype 1b Huh7-Luc replicon cells. At 750 nM, a combination with the HCV NS3/4A protease inhibitor TMC435 at 80 nM was needed to suppress the formation of resistant colonies, suggesting that combination of the two inhibitors is beneficial in suppressing the emergence of resistant mutants in vitro. This finding is in line with earlier findings in the HIV field, where combinations of antivirals with different mechanisms of action are used to reach sustained viral suppression and to prevent the development of resistance (34).
In an HCV replicon clearance and rebound assay using Huh7-Luc replicon cells, a fast dose-dependent reduction but incomplete clearance of HCV replicon RNA was detected upon incubation with 3.75 μM TMC647055 during the clearance phase of the assay, and after cessation of TMC647055 treatment, a rebound of HCV replicon RNA was observed. Hence, TMC647055 alone was not sufficient to clear the replicon completely from the cells in this experimental setting. Interestingly, combination of 1.5 μM TMC647055 with 100 nM TMC435 resulted in a fast decline of HCV replicon RNA to below the level of quantification during the clearance phase and prevention of the rebound of HCV replicon RNA during the rebound phase. These results further substantiate the thesis that a combination of two DAAs with different mechanisms of action is favored over monotherapy in terms of HCV resistance development and virus suppression. However, for each combination, further studies are needed to exclude a potential antagonistic effect of the combination. Currently, many clinical studies are ongoing that evaluate the use of DAA combinations in the treatment of HCV-infected patients. In particular, the combination of a protease inhibitor and an NNI with ribavirin has shown clinical efficacy in HCV-infected patients with high rates of rapid virologic response and SVR, especially in genotype 1b-infected patients (46).
SPR enabled delineation of the kinetics of TMC647055 binding to multiple NS5BΔC21 polymerase clinical isolates and isolates with site-directed mutations. In general, high-affinity interactions between TMC647055 and NS5B were observed for all genotypes, except for genotype 2b, with genotype 1b complexes displaying the greatest complex stability. The reduced affinity for the genotype 2b enzyme derived from an increased dissociation rate and the resultant decreased complex stability; the association rate for the genotype 2b enzyme was similar to that observed for other genotypes.
Similar trends in affinity (KD) and inhibitory activity (EC50) were observed for TMC647055 across the tested genotypes and mutants. This makes it possible to predict the likely drug susceptibility of clinical isolates even when these isolates are not compatible with replication in the replicon system, as was the case for a genotype 5a chimera.
SPR enables the determination of protein-inhibitor complex half-lives, which may be an important factor in clinical efficacy (9). It was recently reported that BI207127 and MK-3281, two NNI-1 inhibitors currently in clinical development, exhibit lower clinical efficacy in genotype 1a-infected patients than in genotype 1b-infected patients (8, 23). While it remains to be determined whether these observations are related to reduced complex half-lives for the genotype 1a NS5B polymerase complexes, our results suggest that it may be useful to evaluate complex half-life as an additional selection criterion during NNI-1 discovery and optimization. After demonstrating the safety and tolerability of single and repeated doses of TMC647055 in healthy volunteers, its antiviral activity was studied by treating HCV genotype 1a- and 1b-infected patients with doses of 500 or 1,000 mg twice daily for 5 days with a morning dose on day 6. A potent antiviral response was observed in HCV genotype 1b-infected patients (median maximum decrease in HCV RNA from baselines of, respectively, 3.3 and 3.4 log10 IU/ml) (24). In HCV genotype 1a-infected patients, the antiviral activity was dose dependent with a median maximum decrease in HCV RNA from a baseline of 1.4 log10 IU/ml at 500 mg twice daily and of 2.4 log10 IU/ml at 1,000 mg twice daily (24). These first clinical efficacy data support the further development of TMC647055 for the treatment of hepatitis C disease.
In summary, we report that TMC647055 is a potent and selective NS5B NNI-1 inhibitor with activity against most HCV genotypes. The contribution of TMC647055 to in vitro virus eradication and prevention of the emergence of resistant virus mutants when combined with the HCV NS3/4A protease inhibitor TMC435 supports its further clinical evaluation for the treatment of HCV infections.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the excellent technical assistance of Geneviève Vandercruyssen, Katrien Vermeiren, Frédéric Delouvroy, and Inky De Baere, and we appreciate the help of Luc Geeraert and Roberto Martinez-Neira in preparing the manuscript.
Jan Martin Berke, Leen Vijgen, Pascale Dehertogh, Erna Cleiren, Liesbeth van der Helm, Abdellah Tahri, Katie Amssoms, Oliver Lenz, Maxwell D. Cummings, Sandrine Vendeville, Pierre Raboisson, Kenneth A. Simmen, Gregory C. Fanning, and Tse-I Lin are Johnson & Johnson employees. Benoit Devogelaere, Els Fransen, Origène Nyanguile, and Reginald F. Clayton are former Johnson & Johnson employees.
Footnotes
Published ahead of print 18 June 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Andries K, et al. 2003. Substituted benzimidazoles with nanomolar activity against respiratory syncytial virus. Antiviral Res. 60:209–219 [DOI] [PubMed] [Google Scholar]
- 2. Bacon BR, et al. 2011. Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1207–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beaulieu PL. 2009. Recent advances in the development of NS5B polymerase inhibitors for the treatment of hepatitis C virus infection. Expert Opin. Ther. Pat. 19:145–164 [DOI] [PubMed] [Google Scholar]
- 4. Beaulieu PL, et al. 2004. Non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase: discovery and preliminary SAR of benzimidazole derivatives. Bioorg. Med. Chem. Lett. 14:119–124 [DOI] [PubMed] [Google Scholar]
- 5. Behrens S-E, Tomei L, De Francesco R. 1996. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 15:12–22 [PMC free article] [PubMed] [Google Scholar]
- 6. Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brainard DM, et al. 2009. Safety, tolerability, and pharmacokinetics after single and multiple doses of MK-3281 in healthy subjects. 44th Annu. Meet. EASL European Association for the Study of the Liver, Geneva, Switzerland: http://www.natap.org/2009/EASL/EASL_28.htm [Google Scholar]
- 9. Copeland RA, Pompliano DL, Meek TD. 2006. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 5:730–739 [DOI] [PubMed] [Google Scholar]
- 10. Dierynck I, et al. 2007. Binding kinetics of darunavir to human immunodeficiency virus type 1 protease explain the potent antiviral activity and high genetic barrier. J. Virol. 81:13845–13851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Marco S, et al. 2005. Interdomain communication in hepatitis C virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J. Biol. Chem. 280:29765–29770 [DOI] [PubMed] [Google Scholar]
- 12. Ghobrial RM, et al. 2001. A 10-year experience of liver transplantation for hepatitis C: analysis of factors determining outcome in over 500 patients. Ann. Surg. 234:384–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Global Burden of Hepatitis C Working Group 2004. Global burden of disease (GBD) for hepatitis C. J. Clin. Pharmacol. 44:20–29 [DOI] [PubMed] [Google Scholar]
- 14. Gong E, Ivens T, Van den Eynde C, Hallenberger S, Hertogs K. 2008. Development of antiviral assays for profiling compounds against a panel of positive-strand RNA viruses using ATP/luminescence readout. J. Virol. Methods 151:121–125 [DOI] [PubMed] [Google Scholar]
- 15. Harper S, et al. 2005. Development and preliminary optimization of indole-N-acetamide inhibitors of hepatitis C virus NS5B polymerase. J. Med. Chem. 48:1314–1317 [DOI] [PubMed] [Google Scholar]
- 16. Ishida T, et al. 2006. Benzimidazole inhibitors of hepatitis C virus NS5B polymerase: identification of 2-[(4-diarylmethoxy)phenyl]-benzimidazole. Bioorg. Med. Chem. Lett. 16:1859–1863 [DOI] [PubMed] [Google Scholar]
- 17. Jacobson IM, et al. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 18. Jochmans D, et al. 2006. Indolopyridones inhibit human immunodeficiency virus reverse transcriptase with a novel mechanism of action. J. Virol. 80:12283–12292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koch U, Narjes F. 2007. Recent progress in the development of inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. Curr. Top. Med. Chem. 7:1302–1329 [DOI] [PubMed] [Google Scholar]
- 20. Koev G, Kati W. 2008. The emerging field of HCV drug resistance. Expert Opin. Investig. Drugs 17:303–319 [DOI] [PubMed] [Google Scholar]
- 21. Korba BE, Gerin JL. 1992. Use of a standardized cell culture assay to assess activities of nucleoside analogs against hepatitis B virus replication. Antiviral Res. 19:55–70 [DOI] [PubMed] [Google Scholar]
- 22. Krieger N, Lohmann V, Bartenschlager R. 2001. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75:4614–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larrey D, et al. 2009. BI 207127 is a potent HCV RNA polymerase inhibitor during 5 days monotherapy in patients with chronic hepatitis C. 60th Annu. Meet. AASLD American Association for the Study of Liver Diseases, Alexandria, VA: http://www.natap.org/2009/AASLD/AASLD_10.htm [Google Scholar]
- 24. Leempoels J, et al. 2011. Human safety, pharmacokinetics and antiviral activity of TMC647055, a novel HCV non-nucleoside polymerase inhibitor. 62nd Annu. Meet. AASLD American Association for the Study of Liver Diseases, Alexandria, VA: http://www.natap.org/2011/AASLD/AASLD_04.htm [Google Scholar]
- 25. Lenz O, et al. 2010. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob. Agents Chemother. 54:1878–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lesburg CA, et al. 1999. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6:937–943 [DOI] [PubMed] [Google Scholar]
- 27. Lin T-I, et al. 2009. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob. Agents Chemother. 53:1377–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lohmann V, Korner F, Herian U, Bartenschlager R. 1997. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 71:8416–8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lohmann V, et al. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 31. Manns MP, et al. 2007. The way forward in HCV treatment—finding the right path. Nat. Rev. Drug Discov. 6:991–1000 [DOI] [PubMed] [Google Scholar]
- 32. Nyanguile O, et al. 2010. 1a/1b subtype profiling of nonnucleoside polymerase inhibitors of hepatitis C virus. J. Virol. 84:2923–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nyanguile O, et al. 2008. 1,5-Benzodiazepines, a novel class of hepatitis C virus polymerase non-nucleoside inhibitors. Antimicrob. Agents Chemother. 52:4420–4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pomerantz RJ, Horn DL. 2003. Twenty years of therapy for HIV-1 infection. Nat. Med. 9:867–873 [DOI] [PubMed] [Google Scholar]
- 35. Poordad F, et al. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Raboisson P, et al. 2008. Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350. Bioorg. Med. Chem. Lett. 18:4853–4858 [DOI] [PubMed] [Google Scholar]
- 37. Rydberg EH, et al. 2009. Structural basis for resistance of the genotype 2b hepatitis C virus NS5B polymerase to site A non-nucleoside inhibitors. J. Mol. Biol. 390:1048–1059 [DOI] [PubMed] [Google Scholar]
- 38. Simmonds P. 2004. Genetic diversity and evolution of hepatitis C virus–15 years on. J. Gen. Virol. 85:3173–3188 [DOI] [PubMed] [Google Scholar]
- 39. Strader DB, Wright T, Thomas DL, Seeff LB, American Association for the Study of Liver Diseases 2004. Diagnosis, management, and treatment of hepatitis C. Hepatology 39:1147–1171 [DOI] [PubMed] [Google Scholar]
- 40. Takano S, et al. 1996. Prospective assessment of donor blood screening for antibody to hepatitis C virus by first- and second-generation assays as a means of preventing posttransfusion hepatitis. Hepatology 23:708–712 [DOI] [PubMed] [Google Scholar]
- 41. Tomei L, et al. 2003. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 77:13225–13231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vandyck K, et al. 2009. Structure-based design of a benzodiazepine scaffold yields a potent allosteric inhibitor of hepatitis C NS5B RNA polymerase. J. Med. Chem. 52:4099–4102 [DOI] [PubMed] [Google Scholar]
- 43. Vendeville S, et al. 2012. Finger loop inhibitors of the HCV NS5b polymerase. Part II. Optimization of tetracyclic indole-based macrocycle leading to the discovery of TMC647055. Bioorg. Med. Chem. Lett. 22:4437–4443 [DOI] [PubMed] [Google Scholar]
- 44. Zeuzem S, et al. 2011. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 364:2417–2428 [DOI] [PubMed] [Google Scholar]
- 45. Zeuzem S, et al. 2011. Efficacy of the protease inhibitor BI 201335, polymerase inhibitor BI 207127, and ribavirin in patients with chronic HCV infection. Gastroenterology 141:2047–2055 [DOI] [PubMed] [Google Scholar]
- 46. Zeuzem S, et al. 2012. SVR4 and SVR12 with an interferon-free regimen of BI 201335 and BI 207127, +/− ribavirin, in treatment-naive patients with chronic genotype-1 HCV infection: interim results of SOUND-C2. 47th Annu. Meet. EASL European Association for the Study of the Liver, Geneva, Switzerland: http://www.natap.org/2012/EASL/EASL_38.htm [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.