

Abstract
The antimalarial drug artemisinin from Artemisia annua demonstrated remarkably strong activity against Helicobacter pylori, the pathogen responsible for peptic ulcer diseases. In an effort to develop a novel antimicrobial chemotherapeutic agent containing such a sesquiterpene lactone endoperoxide, a series of analogues (2 natural and 15 semisynthetic molecules), including eight newly synthesized compounds, were investigated against clinical and standard strains of H. pylori. The antimicrobial spectrum against 10 H. pylori strains and a few other bacterial and fungal strains indicated specificity against the ulcer causing organism. Of five promising molecules, a newly synthesized ether derivative β-artecyclopropylmether was found to be the most potent compound, which exhibited MIC range, MIC90, and minimum bactericidal concentration range values of 0.25 to 1.0 μg/ml, 1.0 μg/ml, and 1 to 16 μg/ml, respectively, against both resistant and sensitive strains of H. pylori. The molecule demonstrated strong bactericidal kinetics with extensive morphological degeneration, retained functional efficacy at stomach acidic pH unlike clarithromycin, did not elicit drug resistance unlike metronidazole, and imparted sensitivity to resistant strains. It is not cytotoxic and exhibits in vivo potentiality to reduce the H. pylori burden in a chronic infection model. Thus, β-artecyclopropylmether could be a lead candidate for anti-H. pylori therapeutics. Since the recurrence of gastroduodenal ulcers is believed to be mainly due to antibiotic resistance of the commensal organism H. pylori, development of a candidate drug from this finding is warranted.
INTRODUCTION
Helicobacter pylori is the major cause of gastroduodenal infections and has been implicated in the pathogenesis of active and chronic gastritis, peptic ulcer, and gastric carcinoma (29, 30). The organism changes the gastric epithelium directly through bacterial toxicity and indirectly via inflammation-mediated damage (1). Antibiotic therapy has been used successfully with a combination of two or three drugs to eradicate H. pylori infections (12, 32) and also to cure gastroduodenal ulcers (30). However, resistance to amoxicillin (AMX), clarithromycin (CLR), and metronidazole (MNZ) in particular is increasingly becoming widespread due to constant use of these drugs against such infection (7, 33, 34). Development of drugs derived from natural sources is receiving increasing attention particularly because of their potential to keep pathogenic strains sensitive. Sustained efforts worldwide in searching anti-H. pylori leads from natural products have already led to the elaboration of many plant-derived molecules with interesting prospects (10, 23, 36, 48).
Many unanswered questions on how a plant or its product(s) can be successfully used to cure a disease in traditional and/or alternate modes of treatment are now being addressed. Examples include delineation of the stimulating property of tea, the anti-inflammatory and antiseptic properties of turmeric, the analgesic and anti-inflammatory use of willow bark, the antidiabetic potential of Madagascar Periwinkle, the antimalarial property of the plant Artemisia annua, etc. However, recent additional findings regarding the anticancer activity of tea catechins, the antitumor potential of curcumin, the blood-thinning property of the well-known analgesic molecule aspirin, or the therapeutic effect of vinca alkaloids in Hodgkin's lymphoma (see, for example, reference 41) not only brought forth a few unquestioned answers but also opened up innovative spin-offs to revisit the wealth of traditional knowledge through the eyes of modern-day understanding about the pathophysiological targets and therapeutic principles. In a nationwide networked program in India (19), large-scale screening of natural product based molecules is being carried out in appropriate preclinical experimental models, wherein we have been searching for anti-H. pylori principle(s). The rationale has been that many plant materials are being successfully used to cure stomach ailments for centuries; thus, some active principles could act via the anti-H. pylori mechanism as well. Such random screening demonstrated the remarkably high and hitherto-unknown anti-H. pylori activity of artemisinin.
Use of Artemisia annua to treat malaria has been known for at least 1,600 years. Artemisinin, a sesquiterpene lactone endoperoxide, is a secondary metabolite from this plant that is widely dispersed throughout the temperate region (43). The plant has traditionally been grown in China as a medicinal plant and, more recently, in Europe for its aromatic leaves, which are used in flavoring beverages. Besides antimalarial property, artemisinin and its analogues were shown to possess immunomodulatory and antitumor effects (20, 27, 45). Many natural and semisynthetic derivatives of artemisinin and other compounds containing an endoperoxide bridge have been described as biologically active (37, 40). The generation of semisynthetic derivatives such as artesunate, dihydroartemisinin, and arteether, etc., has helped in deciphering the mechanism of action as an antimalarial, antitumor, anti-inflammatory, or immunosuppressive agent (25, 45, 47). The endoperoxide moiety in the chemical structure of artemisinin is thought to be responsible for the bioactivity (25).
In an effort to look into the details of anti-H. pylori profiles of such endoperoxide molecules, a series of artemisinin analogues were investigated that included two isolated molecules, seven known semisynthetic derivatives, and eight newly synthesized compounds. Because artemisinin is thought to be useful against resistant strains of malaria (20, 25), it was of additional interest to see whether antibiotic-resistant strains of H. pylori can also be killed by such natural-product-based molecules. Based on primary screening through a disc diffusion sensitivity assay and MIC/minimum bactericidal concentration (MBC) studies using both standard strains and clinical isolates and also based on investigations of the general antimicrobial features, including both antibacterial and antifungal activities, we selected five molecules as potential anti-H. pylori candidates. Such molecules were further evaluated for their acid stability (because these are supposed to act under stomach acidic pH), bactericidal kinetics, and sensitivity/resistance profile. The most potent compound was further examined for its capacity to induce morphological deformity, show synergism with anti-H. pylori antibiotics, and reduce the H. pylori burden in vivo. Typically, the objective and the approach plan have been to synthesize newer derivatives upon observing anti-H. pylori activity profile of some molecules, so as to be able to bring out compounds endowed with desired attributes. It is concluded that β-artecyclopropylmether could be a lead candidate for anti-H. pylori therapeutics.
MATERIALS AND METHODS
Test compound preparation.
Isolation of secondary metabolites artemisinin and artemisinic acid from the plant Artemisia annua, grown at the Central Institute of Medicinal and Aromatic Plants (CIMAP) experimental farms, Lucknow, India, was carried out essentially according to the patented procedure (18). A schematic outline for the extraction protocol is provided in Fig. 1A. Figure 1B summarizes the outline for the preparation of a series of new and known derivatives of artemisinin. Briefly, artemisinin (GRB-17) was reduced with sodium borohydride at 0 to −5°C to prepare dihydroartemisinin (GRB-8) in 85% (wt/wt) yield in ∼2 h. After crystallization and drying, dihydroartemisinin was alkylated with different substituted alcohols in 1,4-dioxan in the presence of the catalyst chlorotrimethylsilane (Sigma-Aldrich) at room temperature to produce a series of ether derivatives (GRB-1 to GRB-6, GRB-9 to GRB-11, and GRB-16). Methanol and ethanol were used as the alkylating agent, as well as the reaction solvent, in preparing artemethers (GRB-3 and GRB-4) and arteether (GRB-2), respectively, whereas other alcohols were reacted using 1,4-dioxan as the reaction solvent to prepare other known and new ethers (3, 4). For the purpose of achieving diversity, dihydroartemisinin was also acylated to obtain different esters (GRB-7, GRB-14, and GRB-15). Further, dehydration of dihydroartemisinin afforded anhydrodihydroartemisinin (GRB-13). The chemical structures, compound names, and the code numbers of the 17 molecules, two natural isolates (artemisinin [GRB-17] and artemisinic acid [GRB-12]), and 15 semisynthetic derivatives are provided in Table 1. The syntheses of GRB-2, GRB-3, GRB-4, GRB-8, GRB-11, GRB-13, and GRB-14 were carried out as described earlier by us and others (3, 4, 15, 18, 37, 40), while the synthetic outlines and spectral data for the newer ether derivatives (GRB-1, GRB-5, GRB-6, GRB-9, GRB-10, and GRB-16) and the ester derivatives (GRB-7 and GRB-15) are presented below.
Fig 1.

Extraction of artemisinin and synthesis of different artemisinin derivatives there from. (A) Outline of extraction protocol (see reference 18 for details). (B) Summary of the synthetic steps involved for the preparation of different analogues from artemisinin.
Table 1.
Antibacterial susceptibility of artemisinin and its derivatives against H. pyloria
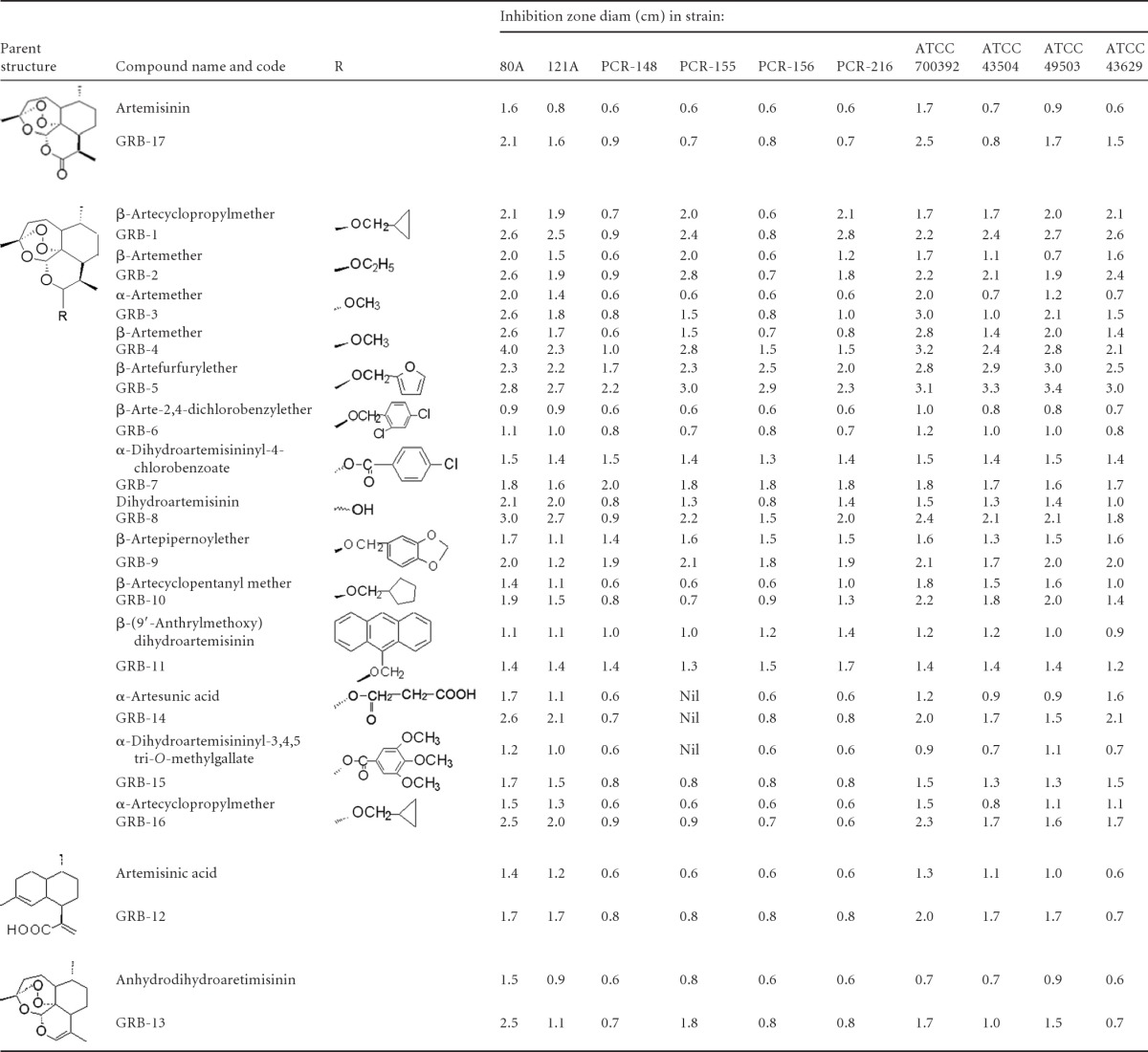
Discs were impregnated with 0.5 μg (upper row) and 1.0 μg (lower row) of the compounds, except for GRB-12, wherein the respective doses were 40 and 80 μg/disc.
Dihydroartemisinin (1.0 g) was dissolved in 1,4-dioxan (20 ml), stirred for 2 to 4 h in the presence of appropriate alkylating/acylating reagents, viz., cyclopropyl methanol (for GRB-1 and GRB-16), cyclopentanyl methanol (for GRB-10), pipernoyl alcohol (for GRB-9), furfuryl alcohol (for GRB-5), 2,4-dichlorobenzyl alcohol (for GRB-6), and 4-chlorobenzoyl chloride (for GRB-7) and the catalyst chlorotrimethylsilane. After completion of the reaction (as monitored by thin-layer chromatography [TLC]), the reaction mixture was quenched with cooled 1% aqueous NaHCO3 (50 ml) and extracted with ethyl acetate (three times with 50 ml each time). The combined extract was washed with water till neutral, dried (Na2SO4), and concentrated to get the crude mixture of α/β products (1.2 to 1.8 g). The mixture was subjected to silica gel column chromatography to obtain the pure β-isomer, followed by the α-isomer.
For the purpose of identification, the reaction mixtures and the crystallized products were monitored by co-TLC with the starting material artemisinin or dihydroartemisinin in the solvent system n-hexane–ethyl acetate (70:30). Purity of the compounds was assumed if a single spot developed on TLC. Infrared (IR) spectra were recorded using a Perkin-Elmer Bx infrared spectrophotometer with a KBr window. 1H and 13C nuclear magnetic resonance (NMR) spectra were determined in CDCl3 on Bruker Avance spectrometer operating at 300 and 75 MHz, respectively. The chemical shifts are reported in ppm. Spectra were interpreted from DEPT-90 and DEPT-35 (distortionless enhancement by polarization transfer) and COSY (correlation spectroscopy) results. Electrospray ionization (ESI) mass spectra were recorded on a JEOL-AccuTOF JMS-T100LC mass spectrometer and using the Shimadzu LC-MS (liquid chromatography-mass spectrometry) system, respectively.
For β-artecyclopropylmether (GRB-1), the column was eluted with n-hexane–ethyl acetate (98:2) to yield GRB-1, 76% (wt/wt), Rf = 0.54 (7:3 n-hexane–ethyl acetate), colorless plates (n-hexane), melting point (mp) 59 to 60°C; IR νmax = 3,089, 3,010 (cyclopropane), 2,870, 1,597, 1,453, 1,377, 1,102, and 1,025 cm−1; 1H NMR = δ 0.18 and 0.46 (2H each, m, H2-3′/H2-4′), 0.89 (3H, d, J = 7.2 Hz, H3-13), 0.93 (3H, d, J = 6.3 Hz, H3-14), 1.02 (1H, m, H-2′), 1.40 (3H, s, H3-15), 3.32 (1H, dd, J = 10.5, 6.9 Hz, Ha-1′), 3.55 (1H, dd, J = 10.5, 3.9 Hz, Hb-1′), 4.80 (1H, d, J = 3.3 Hz, αH-12), and 5.38 (1H, m, H-5); 13C NMR = δ 2.7 and 3.2 (C-3′ and C-4′), 10.9 (C-2′), 13.2 (C-13), 20.6 (C-14), 24.8 (C-2), 25.1 (C-8), 26.5 (C-15), 31.3 (C-11), 35.2 (C-9), 36.9 (C-3), 37.9 (C-10), 45.0 (C-7), 53.1 (C-1), 72.7 (C-1′), 81.4 (C-6), 88.2 (C-5), 101.9 (C-12), and 104.3 (C-4). ESI-MS (m/z) = 339 [M+H]+, 361[M+Na]+ (C19H30O5).
For α-artecyclopropylmether (GRB-16), column elution with n-hexane–ethyl acetate (98:2), followed by preparative TLC in n-hexane–ethyl acetate (95:5), provided the viscous compound GRB-16, 14% (wt/wt), Rf = 0.49 (7:3 n-hexane–ethyl acetate); IR νmax = 3083 (cyclopropane), 2,873, 1,596, 1,454, 1,378, 1,102, and 1,027 cm−1; 1H NMR = δ 0.22 and 0.48 (2H each, m, H2-3′/H2-4′), 0.90 (3H, d, J = 7.5 Hz, H3-13), 0.95 (3H, d, J = 5.7 Hz, H3-14), 1.03 (1H, m, H-2′), 1.43 (3H, s, H3-15), 3.33 (1H, dd, J = 10.5, 6.9 Hz, Ha-1′), 3.74 (1H, dd, J = 10.5, 3.6 Hz, Hb-1′), 4.80 (1H, d, J = 9.3 Hz, βH-12), and 5.32 (1H, m, H-5); 13C NMR = δ 2.6 and 3.2 (C-3′ and C-4′), 10.4 (C-2′), 12.6 (C-13), 20.2 (C-14), 22.1 (C-2), 24.7 (C-8), 26.0 (C-15), 32.4 (C-11), 34.2 (C-9), 36.3 (C-3), 37.3 (C-10), 45.3 (C-7), 51.6 (C-1), 73.1 (C-1′), 80.3 (C-6), 91.2 (C-5), 99.4 (C-12), and 104.2 (C-4). ESI-MS (m/z) = 339 [M+H]+ (C19H30O5).
For β-artecyclopentanylmether (GRB-10), the column was eluted with n-hexane–ethyl acetate (98:2) to yield GRB-10, 84% (wt/wt), viscous, Rf = 0.57 (7:3 n-hexane–ethyl acetate); IR νmax = 2,950, 2,869, 2,873, 1,595, 1,457, 1,380, 1,103, and 1,027 cm−1; 1H NMR = δ 0.90 (3H, d, J = 7.5 Hz, H3-13), 0.95 (3H, d, J = 6.0 Hz, H3-14), 1.24 (4H, m, H2-4′, H2-5′), 1.44 (3H, s, H3-15), 1.72 (4H, m, H2-3′, H2-6′), 2.15 (1H, t, J = 7.5 Hz, H-2′), 3.25 (1H, dd, J = 9.3, 9.3 Hz, Ha-1′), 3.72 (1H, dd, J = 9.3, 7.5 Hz, Hb-1′), 4.77 (1H, d, J = 3.0 Hz, αH-12), and 5.39 (1H, s, H-5); 13C NMR = 13.0 (C-13), 20.4 (C-14), 24.4/24.7 (C-2/C-8), 26.2 (C-15), 26.6 (C-4′ and C-5′), 29.4/29.8 (C-3′/C-6′), 31.0 (C-11), 34.7 (C-9), 36.4 (C-3), 37.5 (C-10), 39.5 (C-2′), 44.5 (C-7), 52.6 (C-1), 72.9 (C-1′), 81.2 (C-6), 87.9 (C-5), 102.0 (C-12), and 104.0 (C-4); COSY (1H-1H correlations): correlations of Ha-1′, Hb-1′ (3.25, 3.72) with H-2′ (2.15), H-2′with H2-3′, H2-6′ (1.72) and H2-3′, H2-6′ with H2-4′, H2-5′ (1.24). ESI-MS (m/z) = 389 [M+Na]+, 267 [M−C6H11O]+ (C21H34O5).
For β-artepipernoylether (GRB-9), the column was eluted with n-hexane–ethyl acetate (95:5) to yield derivative GRB-9, 68% (wt/wt), Rf = 0.47 (7:3 n-hexane–ethyl acetate), colorless needles (n-hexane), mp = 118 to 120°C; IR νmax = 1,595 and 927 (pipernoyl group) cm−1;1H NMR = δ 0.90 (3H, d, J = 7.2 Hz, H3-13), 0.95 (3H, d, J = 5.7 Hz, H3-14), 1.46 (3H, s, H3-15), 4.42 (1H, d, J = 12 Hz, Ha-1′), 4.78 (1H, d, J = 12 Hz, Hb-1′), 4.89 (1H, d, J = 3.3 Hz, αH-12), 5.46 (1H, s, H-5), 5.96 (2H, s, H2-8′), 6.77 (1H, s, H-3′), and 6.81 (2H, d, J = 8.1 Hz, H-6′ and H-7′); 13C NMR = 13.4 (C-13), 20.6 (C-14), 24.9 (C-8), 25.1 (C-2), 26.5 (C-15), 31.3 (C-11), 35.0 (C-9), 36.8 (C-3), 37.8 (C-10), 44.8 (C-7), 53.0 (C-1), 70.01 (C-1′), 81.5 (C-6), 88.3 (C-5), 101.2, 101.5 (C-8′, C-12), 104.4 (C-4), 108.3 (C-3′), 108.5 (C-6′), 121.2 (C-7′), 132.6 (C-2′), 147.3 (C-5′), and 148.0 (C-4′). ESI-MS (m/z) = 441 [M+Na]+ (C23H30O7), 267 [M−C8H7O3]+.
β-Artefurfurylether (GRB-5) was obtained from the column with n-hexane–ethyl acetate (95:5), yield = 61% (wt/wt), Rf = 0.51 (7:3 n-hexane–ethyl acetate), colorless amorphous solid (n-hexane–ethyl acetate), mp = 145 to 147°C; IR νmax = 1,595, 874, and 747 (furan skeleton) cm−1; 1H NMR = δ 0.84 (3H, d, J = 7.5 Hz, H3-13), 0.94 (3H, d, J = 6.0 Hz, H3-14), 1.45 (3H, s, H3-15), 4.52 (1H, d, J = 12.9 Hz, Ha-1′), 4.75 (1H, d, J = 12.9 Hz, Hb-1′), 4.89 (1H, d, J = 3.3 Hz, αH-12), 5.46 (1H, s, H-5), 6.31 (1H, dd, J = 6.3, 1.2 Hz, H-3′), 6.33 (1H, d, J = 6.3Hz, H-4′), and 7.39 (1H, brs, H-5′); 13C NMR = 13.0 (C-13), 20.5 (C-14), 24.7 (C-8), 24.9 (C-2), 26.4 (C-15), 31.0 (C-11), 34.8 (C-9), 36.6 (C-3), 37.6 (C-10), 44.6 (C-7), 52.8 (C-1), 61.8 (C-1′), 81.4 (C-6), 88.2 (C-5), 100.9 (C-12), 104.3 (C-4), 109.1 (C-4′), 110.3 (C-3′), 142.8 (C-5′), and 151.9 (C-2′). FABMS (positive) = 365 [M+H]+ (C20H28O6).
For β-arte-2,4-dichlorobenzylether (GRB-6), elution of the column with n-hexane–ethyl acetate (95:5) yielded the oily compound GRB-6, 93% (wt/wt), Rf = 0.55 (7:3 n-hexane–ethyl acetate); IR νmax = 1,592, 1,102, and 753 (Cl groups) cm−1; 1H NMR = δ 0.92 (3H, d, J = 7.2 Hz, H3-13), 0.95 (3H, d, J = 6.0 Hz, H3-14), 1.45 (3H, s, H3-15), 4.48 (1H, d, J = 13.2 Hz, Ha-1′), 4.96 (1H, d, J = 13.2 Hz, Hb-1′), 4.94 (1H, d, J = 3.3 Hz, αH-12), 5.47 (1H, s, H-5), 7.24 (1H, dd, J = 8.4, 1.8 Hz, H-6′), 7.35 (1H, d, J = 8.4 Hz, H-7′), and 7.37 (1H, brs, H-4′); 13C NMR = δ 13.3 (C-13), 20.5 (C-14), 24.7, 24.9 (C-2, C-8), 26.2 (C-15), 31.1 (C-11), 34.8 (C-9), 36.5 (C-3), 37.6 (C-10), 44.5 (C-7), 52.8 (C-1), 67.4 (C-1′), 81.2 (C-6), 88.3 (C-5), 100.2 (C-12), 104.4 (C-12), 127.2 (C-7′), 129.3 (C-4′), 129.8 (C-6′), 130.2 (C-3′), 133.8 (C-5′), and 135.0 (C-2′). FABMS (positive) = 443 [M+H]+, 465 [M+Na]+ (C22H28O5Cl2).
α-Dihydroartemisininyl-4-chlorobenzoate (GRB-7) was obtained from the column with n-hexane–ethyl acetate (95:5), yield = 91% (wt/wt), Rf = 0.52 (7:3 n-hexane–ethyl acetate), colorless plates (n-hexane), mp = 98 to 100°C; IR νmax = 1733 (ester CO), 1,592, 1,090, and 758 (Cl group) cm−1; 1H NMR = δ 0.95 (3H, d, J = 7.2 Hz, H3-13), 0.98 (3H, d, J = 5.7 Hz, H3-14), 1.42 (3H, s, H3-15), 5.52 (1H, s, H-5), 5.98 (1H, d, J = 9.9 Hz, βH-12), 7.41 (2H, d, J = 8.4 Hz, H-4′, H-6′), 8.01 (1H, d, J = 8.4 Hz, H-7′), and 8.08 (1H, d, J = 8.4 Hz, H-3′); 13C NMR = δ 12.9 (C-13), 20.2 (C-14), 22.1 (C-8), 24.5 (C-2), 25.9 (C-15), 31.9 (C-11), 34.1 (C-9), 36.2 (C-3), 37.3 (C-10), 45.3 (C-7), 51.6 (C-1), 80.1 (C-6), 91.6 (C-5), 92.7 (C-12), 104.4 (C-4), 128.7 (C-4′, C-6′), 131.5 (C-3′, C-7′), 131.9 (C-2′), 139.8 (C-5′), and 164.4 (C-1′). FABMS (positive) = 423 [M+H]+, 445 [M+Na]+ (C22H27O6Cl).
For α-dihydroartemisininyl 3,4,5 tri-O-methylgallate (GRB-15), dihydroartemisinin (1.0 g), trimethyl ether of gallic acid (1 g), N,N′-dicyclohexylcarbodiimide (1 g), and the catalyst 4-dimethylaminopyridine (160 mg) were stirred in dichloromethane (40 ml) for 4 h at room temperature. The reaction mixture was prepared as described above. Elution of the column with n-hexane–ethyl acetate (95:5), followed by crystallization from n-hexane–acetone (8:2), yielded GRB-15 76% (wt/wt), Rf = 0.36 (7:3 n-hexane–ethyl acetate), colorless plates (ethyl acetate), mp = 102 to 104°C; IR νmax =1,727 (ester CO), 1,591, and 1,128 cm−1; 1H NMR = δ 0.91 (3H, d, J = 6.9 Hz, H3-13), 0.99 (3H, d, J = 5.7 Hz, H3-14), 1.44 (3H, s, H3-15), 3.88, 3.91, 3.92 (3H each, s, 3×OMe), 5.53 (1H, s, H-5), 5.99 (1H, d, J = 9.6 Hz, βH-12), and 7.37 (2H, s, H-3′, and H-7′); 13C NMR = δ 12.2 (C-13), 20.2 (C-14), 22.0 (C-8), 24.5 (C-2), 25.9 (C-15), 32.0 (C-11), 34.0 (C-9), 36.2 (C-3), 37.2 (C-10), 45.3 (C-7), 51.6 (C-1), 56.3 (2×OMe), 56.9 (OMe), 80.2 (C-6), 91.6 (C-5), 92.6 (C-12), 104.4 (C-4), 107.3 (C-3′, C-7′), 124.5 (C-2′), 142.5 (C-5′), 152.8 (C-4′, C-6′), and 164.9 (C-1′). FABMS (positive) = 479 [M+H]+ (C25H34O9).
Culture of H. pylori.
Four ATCC standard strains (700392, 43504, 43629, and 49503) and six clinical isolates (80A, 121A, PCR-148, PCR-155, PCR-156, and PCR-216) were used for in vitro studies and were routinely cultured either in brain heart infusion (BHI) agar plates containing 7% fetal calf serum (FCS), 0.5% IsoVitaleX (Becton Dickinson, USA) and 0.0025% Dent (Oxoid, England) or in brucella broth (BB) medium containing 0.0025% Dent and 5% FCS under a microaerophilic environment (10% CO2, 85% N2, and 5% O2 and >95% relative humidity) at 37°C for 48 to 72 h (11) in a double-gas CO2 incubator (Heraeus, model HERAcell 240 or BB6020). For working purposes, either a 3-day-old culture in BB containing 5% FCS and 0.0025% Dent (CFU at ca. 5 × 107/ml) or freshly grown cells in BHI agar plates, scraped, and diluted in BB (CFU at ca. 107 to 108/ml) was used. The strains were routinely stored in BHI medium containing 20% glycerol (vol/vol) in −70°C for further use. The Sydney strain SS1, used for in vivo studies, was grown and maintained as described above; however, for large-scale production, we used a shake-culture method (detailed below). One ATCC standard strain, 700392, and all of the clinical strains were kindly provided by A. K. Mukhopadhyay, National Institute of Cholera and Enteric Diseases (NICED), Kolkata, India. The Sydney strain SS1 was procured from J. O'Rourke, University of New South Wales, Sydney, Australia. Standard strain ATCC 49503 was kindly provided by M. Sitaram Kumar, Dr. Reddy's Laboratory, Hyderabad, India.
Disc diffusion sensitivity assay.
For H. pylori susceptibility testing, 0.5-ml portions of the inoculum (∼108 CFU/ml) for each of the strains tested were flooded on freshly prepared BHI agar plates. Sterilized discs (5 mm in diameter), each containing either a test compound or a standard antibiotic, were placed on the agar surface. The plates were incubated for 3 days in a microaerophilic atmosphere at 37°C. At the end of the incubation period, the inhibition zone diameter (IZD) was measured in centimeters (31). General antibacterial and antifungal activities were determined using 12 bacterial strains, including seven Gram-positive and five Gram-negative aerobic bacteria and eight fungal species. A petri dish containing 20 ml of either Mueller-Hinton (MH) agar (for bacteria) or Sabouraud dextrose agar (for fungi) was spread plated with 100 μl of a 0.5 McFarland standard inoculum of the strain. Sterile discs (6 mm in diameter) impregnated with the test compounds were placed on the plate, followed by incubation at 37°C for 24 h for bacteria or at 28°C for 48 h for fungi (2). The IZDs were measured, and samples were graded accordingly.
Broth microdilution assay.
Twofold serial dilutions of the test compounds were prepared in a 96-well microtiter plate containing 100 μl of MH broth supplemented with 5% FCS and 0.0025% Dent. A 3-day-old H. pylori liquid culture was diluted 10 times in MH broth and 100 μl of this was inoculated into each well to give a final concentration of ca. 5 × 105 to 1 × 106 CFU/well. The plates were incubated for 3 days in a microaerophilic atmosphere at 37°C. After incubation, the plates were examined visually, and the lowest concentration showing complete inhibition of growth was recorded as the MIC of the respective material (13). Aliquots (10 μl) of a 72-h-old culture, in which no growth had been detected, were taken from the wells of the above microtiter plates and streaked onto fresh BHI agar plates. The MBCs were determined by visual inspection of such plates after further incubation for 72 h at 37°C. The titer point at which no growth (fewer than 10 colonies) appeared was considered the MBC. Samples pretreated in acidic pH were also examined for MIC and MBC values vis-à-vis the chemical stability. Briefly, a 1-ml stock (4 to 8 mg/ml) of any sample was treated with 200 to 300 μl of 0.12 N HCl to bring the pH of the solution to ∼2.0 and then kept for 24 h at 25°C. After 2 h, a part of such acid-treated samples was serially diluted (2-fold) with medium in 96-well microtiter plates to determine the MIC and MBC values (10), while the other part was analyzed for acid stability at different time points (0, 1, 2, and 24 h) by reversed-phase high-performance liquid chromatography (HPLC) using the following operating conditions: a X Bridge C18 column (250 by 4.6 mm [inner diameter], 5 μm), acetonitrile–water–1,4-dioxane (65:32:3 [vol/vol/vol]), a flow rate of 1 ml/min, and a detector wavelength of 235 nm (44).
Bactericidal kinetics.
The rate and extent of killing of H. pylori by the test compounds were assessed in liquid culture with constant shaking (22). In 50-ml flasks, 5 ml of BB containing 5% FCS in the presence or absence (control) of samples at 0 to 32 times the MIC was inoculated with bacteria obtained from an overnight culture to yield an initial cell density of ∼106 CFU/ml. The culture was shaken at 150 rpm inside the incubator in an otherwise identical growth condition as stated before. At different time points (0, 3, 6, 9, 12, 24, and 36 h), aliquots (100 μl) of culture were taken out to monitor the growth by direct microscopic examination, and the viable cells were counted by inoculating 10-fold serially diluted suspensions onto BHI agar plates. Colonies were counted after 4 days of incubation, and the bactericidal activities were assessed in terms of decrease in cell count (CFU/ml). The rate and extent of killing were determined by plotting viable counts (log10 CFU/ml) against time (h).
Drug resistance study.
Samples were prepared as 2-fold serial dilutions in MH broth containing 5% FCS and 0.0025% Dent in 96-well microtiter plates. H. pylori strains (ATCC 43504 and clinical isolate 80A) were inoculated into each dilution series at an inoculum size of ca. 106 to 107 CFU/ml. After 3 days of incubation, the culture from each series with the highest concentration of test compound or antibiotic and also showing turbidity was subcultured in a fresh series of the same sample. This procedure was repeated for up to 10 cycles, and alterations in MICs during the course of continued exposure were determined (17).
Combination effect of antibiotics with test compounds.
Checkerboard titration was carried out using test samples and standard antibiotics (CLR, AMX, and MNZ), alone or in combination, to determine fractional inhibitory concentration (FIC) index (24, 49). The inoculum size and the media and culture conditions were the same as those described for the MIC determination by broth microdilution assay. The FIC indices were interpreted as follows: ≤0.5 = synergy, >0.5 to ≤4.0 = nonsynergistic, and >4.0 = antagonistic.
Ultrastructural alteration study.
Sample processing was performed essentially according to a published procedure (38). Briefly, H. pylori cells (ATCC 43504), after exposure to test compound GRB-1 (1 μg/ml) for 24 h under microaerophilic condition at 37°C, were harvested by centrifugation, resuspended, washed in 0.1 M potassium phosphate buffer (pH 7.0), and fixed at 4°C for 24 h by the addition of glutaraldehyde to a final concentration of 2.5%. The samples were then rinsed with 0.1 M potassium phosphate buffer, centrifuged at 9,000 rpm for 20 min (twice), and postfixed with 1.0% osmium tetroxide in 0.1 M potassium phosphate buffer overnight at room temperature. After a wash with the same buffer, the samples were dehydrated multiple times in increasing concentrations of ethanol and embedded in Spurr resin. Sections (30 to 50 nm thick) were cut with a diamond knife on an ultramicrotome (Leica EM FC6) and applied to copper grids. The grids were contrasted with uranyl acetate and lead citrate. The sections were examined with a transmission electron microscope (FEI Tecnai Spirit; FEI, Eindhoven, Netherlands) at an accelerating voltage of 60 kV.
Infection of mice with H. pylori strain SS1.
Female C57BL/6J mice (procured from National Institute of Nutrition, Hyderabad, India) were used as the host, and the H. pylori SS1 strain was used as the infecting agent. Experiments approved by the Animal Ethics committee of the Institute were carried out according to the guidelines provided to use the minimum number of animals for valid evaluation. Each mouse (body weight, 20 to 24 g) was acclimatized in laboratory conditions under a 12-h light-dark schedule for at least 2 weeks prior to the experiment in a clean setup and fed standard pellets and water ad libitum. Freshly cultured H. pylori SS1 from about four BHI agar plates was scraped, inoculated into four 100-ml flasks containing 20 ml of BHI broth supplemented with 5% FCS, 0.5% IsoVitaleX, and 0.0025% Dent, and then incubated at 37°C in a microaerophilic environment for 36 to 48 h with shaking at 60 rpm. After incubation, the cultures were centrifuged at 5,000 rpm for 5 min, and the cell pellets were pooled and resuspended in 2 ml of sterile BHI (∼109 CFU/ml). Prior to infection, the cultures were examined microscopically to confirm purity and motility (mostly spiral rather than coccoid form). Animals were inoculated orogastrically three times over a 5-day period with a 0.1 ml of suspension containing ∼108 organisms. At 2 weeks after inoculation, the mice were randomly divided into four groups and treated as follows: group 1 (eight animals) was the infection control with no treatment, group 2 (six animals) received treatment with a standard drug combination (triple-therapy omeprazole-CLR-MNZ [OCM]), group 3 (six animals) received treatment with dual therapy (CLR-MNZ [CM]), and group 4 (six animals) received treatment with the test compound. The standard triple therapy was administered perorally twice daily for 7 days: omeprazole (400 μmol/kg/day) was fed 30 to 60 min before the administration of combined CLR (7.15 mg/kg/day) and MNZ (14.2 mg/kg/day). The test sample (50 mg/kg/day) and the antibiotics were suspended in 0.5% hydroxypropylmethylcellulose. Each animal received the corresponding treatment twice daily for 7 days (46). Animals were sacrificed 36 h and 29 days, respectively, after the cessation of treatment for the assessment of H. pylori colonization for clearance and eradication profiles. Assessment of remission of infection was carried out as follows. One-half of each stomach was dipped in urease reagent containing 10% urea and 0.05% phenol red in phosphate-buffered saline (PBS) for qualitative assessment of rapid urease activity. The other half of the stomach was used for the determination of viable counts. Weighed stomach halves were placed in 1 ml of BHI broth and homogenized gently in a hand homogenizer using a Teflon pestle (10 strokes). Serial dilutions (both 10- and 100-fold) were prepared in PBS. Aliquots containing various dilutions were streaked onto fresh BHI agar plates and incubated for 3 to 5 days under microaerophilic conditions at 37°C. Viable counts were expressed as the log10 CFU/g of stomach tissue (wet weight).
Cytotoxicity assay using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] method.
HepG2 (human hepatocellular carcinoma), MCF-7 (human breast adenocarcinoma), AGS (human gastric adenocarcinoma), and RAW 264.7 (mouse leukemic monocytic macrophage) cells, all procured from Infectious Diseases and Immunology division of the Indian Institute of Chemical Biology (IICB), were grown as monolayer cultures in Dulbecco modified Eagle medium supplemented with 10% FCS and 1% penicillin-streptomycin by incubation in a humidified atmosphere of 5% CO2 and 95% air at 37°C. Mouse peritoneal macrophages (MPM) were isolated from 6- to 8-week-old female BALB/c mice by peritoneal lavage with ice-cold PBS at 48 h after intraperitoneal injection of 2.5 ml of sterile 4% starch suspension (9). The cells were washed in cold PBS, resuspended in the medium described above, and incubated at 37°C for 24 h. The nonadhering cells were then removed by washing with PBS and prewarmed to 37°C. More than 95% of the adherent cells were macrophages.
For evaluation of the cytotoxicity, cells were seeded in a 96-well microplate at 104 cells per well (105 cells/well for MPM) and cultured at 37°C for 24 h for all of the cell lines except for MPM, which were incubated for 48 h. The test compounds were then added at the indicated concentrations, using the solvent dimethyl sulfoxide (DMSO) as an untreated control, and further incubated for 48 h (24 h for MPM). At the end of the treatment, MTT was added into each well at 0.2 mg/ml, followed by incubation at 37°C for 4 h in dark (35). The culture medium containing MTT was aspirated off, and dye crystals were dissolved in 100 μl of DMSO. The viable cells were detected by reading the absorbance of formazan at 570 nm using a Bio-Rad microplate reader (model 680 XR; Bio-Rad Laboratories, CA). The results are expressed as the 50% inhibitory concentration (IC50), the dose capable of killing 50% of the cells compared to the control.
Predictive values of biological properties.
Physicochemical values such as lipophilicity (log P) and plasma protein binding (PPB) values were calculated by predicted mathematical methods using Cerius2 software, version 4.10 (Accelrys, Inc., San Diego, CA), wherein the PPB levels are defined as 0 (binding < 90%), 1 (binding ≥ 90%), and 2 (binding ≥ 95%). The log S values were calculated based on atom contribution approach (16).
RESULTS
Synthesis and screening of artemisinin derivatives against H. pylori.
During large-scale random screening of natural product based molecules for anti-H. pylori potential, we had observed artemisinin to exhibit a ca. 1.5- to 2.5-cm inhibition zone diameter (IZD) at a dose of 1 to 5 μg/disc during antibacterial susceptibility testing against both clinical and standard strains. The compound also exhibited both potent bacteriostatic and bactericidal activity (MIC ∼ 1 μg/ml, MBC ∼ 4 μg/ml). It did not show any antibacterial activity against a few Gram-positive and Gram-negative bacteria (MIC > 8 μg/ml). All such primary observations, made for the first time, tempted us to investigate further about the therapeutic prospect of such sesquiterpene lactone endoperoxides. Since many synthetic analogues of artemisinin have been prepared for the sake of designing an appropriate antimalarial drug (25, 27, 42), it was of interest to examine such compounds for the putative potential of anti-H. pylori therapeutics. Further, looking at the primary results with known artemisinin analogues, we attempted to synthesize a few more derivatives with a view to generating more potent and useful anti-H. pylori molecules.
Artemisinin (GRB-17) was reduced with sodium borohydride to prepare dihydroartemisinin (GRB-8). In this reduction step, the lactone or C-12 carbonyl function of artemisinin was converted to lactol (hemiacetal) function. Since presence of the lactol function in dihydroartemisinin was reported to make the compound a more potent antimalarial than artemisinin, we synthesized a number of dihydroartemisinin ether/ester/12-dehydrated derivatives focusing changes at the C-12 position (Fig. 1B). Alkylation of dihydroartemisinin with different alcohols gave rise to 10 new or known ether derivatives, among which artecyclopropylmethers (GRB-1 and GRB-16), artepipernoylether (GRB-9), artecyclopentanylmether (GRB-10), artefurfurylether (GRB-5), and arte-2,4-dichlorobenzylether (GRB-6) are new compounds. Next, dihydroartemisinin was acylated to obtain three C-12 esters, of which dihydroartemisininyl-4-chlorobenzoate (GRB-7) and dihydroartemisininyl-3,4,5-tri-O-methylgallate (GRB-15) are new. The alkylated products obtained were predominantly β-isomers, whereas the α-isomers were found to be the major products during acylation process. The minor α-ether (except α-artemether) and β-ester derivatives could not, however, be isolated. Dehydration of dihydroartemisinin afforded anhydrodihydroartemisinin (GRB-13). All of the semisynthetic compounds were identified by NMR (1H, 13C, DEPT, and COSY) and MS and by comparison with data in the literature. The 1H and 13C NMR data of the eight newly synthesized compounds (GRB-1, GRB-5, GRB-6, GRB-7, GRB-9, GRB-10, GRB-15, and GRB-16) are reported here for the first time.
The 17 test molecules (abbreviated as GRB-1 to GRB-17), which included the two naturally isolated compounds artemisinin (GRB-17) and artemisinic acid (GRB-12, not an endoperoxide, however), were examined against six clinical isolates and four ATCC standard strains using disc diffusion susceptibility assay. Based on the experimental outcome with majority of the strains at a 1-μg/disc dose, the samples were initially grouped as “strongly active” with an IZD > 2.0 cm (GRB-1, GRB-2, GRB-4, and GRB-5), “moderately active” with an IZD ∼ 1.5 to 2.0 cm (GRB-3, GRB-7, GRB-8, GRB-9, and GRB-10), and “active” with an IZD ∼ 1.0 to 1.5 cm (GRB-6, GRB-11, GRB-12, GRB-13, GRB-14, GRB-15, GRB-16, and GRB-17). Under identical conditions, clarithromycin showed IZD values of 1.8 cm for the strains 80A and 121A at 0.01 μg/disc, 1.0 to 1.5 cm for PCR-148, PCR-155, PCR-156, and PCR-216 at 0.02 μg/disc, and 2.8, 2.0, 1.7, and 1.5 cm, respectively, with strains 700392 (0.4 μg/disc), 43504 (0.04 μg/disc), 43629 (0.1 μg/disc), and 49503 (0.005 μg/disc). Evaluation of the results in terms of range and average values revealed the rank order to remain similar at both doses of 0.5 and 1.0 μg/ml (Table 1). Interestingly, we observed standard strains to exhibit better susceptibility compared to clinical strains, particularly in the case of less active molecules. The newly synthesized molecules GRB-1 and GRB-5 appeared more promising.
The anti-H. pylori potential of all of the compounds was further assessed in terms of bacteriostatic and bactericidal potential using a broth microdilution assay against all 10 strains (Table 2). The molecules showing MIC50 values ranging from ca. 0.5 to 1.0 μg/ml and MBC50 values ranging from ca. 1 to 2 μg/ml were separated out (GRB-1 to GRB-5 and GRB-14 to GRB-16). The next best group comprised GRB-8 and GRB-13, which exhibited MIC50 values from ca. 1 to 2 μg/ml and MBC50 values from ca. 2 to 4 μg/ml. A third group comprising GRB-7, GRB-9, GRB-11, and GRB-17 showed MIC50s of 2 to 8 μg/ml and MBC50s of 4 to 8 μg/ml. Among the 17 compounds, GRB-6 and GRB-12 appeared to be the least active molecules (both MIC50 and MBC50 ≥ 8 μg/ml). However, if one considers the MIC range, the ester derivatives GRB-14 and GRB-15 appear to be as potent as the ether derivatives (GRB-1 to GRB-5). An assessment of anti-H. pylori spectrum in terms of MIC90 values tends to indicate that ether derivatives GRB-1 to GRB-5 have significant potential (MIC90 = 0.5 to 2 μg/ml). However, based on evaluation of the MBC range and mean MBC values, GRB-5 appeared to be the most potent, followed by GRB-1, GRB-3, GRB-4, and GRB-14. The mean MBC value (in μg/ml), if extrapolated in terms of the micromolar concentration, exhibited a remarkable similarity in rank order for all 17 molecules. The relatively higher sensitivity of the less active molecules toward standard strains, particularly ATCC 43629, was pronounced in terms of the MIC and MBC also. Further, the ether derivatives were found to be more active than esters. Summarizing, we were successful in synthesizing few new compounds that proved to be highly potent, such as GRB-1 and GRB-5.
Table 2.
Anti-H. pylori spectrum of artemisinin and its derivatives as determined by broth microdilution assaya
| Compound | MIC (μg/ml) |
MBC (μg/ml) |
||||
|---|---|---|---|---|---|---|
| Range | MIC50 | MIC90 | Range | MBC50 | Meanb | |
| GRB-1 | 0.25–1 | 1 | 1 | 1–16 | 2 | 3.7 (10.95) |
| GRB-2 | 0.5–4 | 1 | 2 | 0.5–≥16 | 4 | 5.25 (16.83) |
| GRB-3 | 0.5–1 | 0.5 | 0.5 | 1–>16 | 1 | 4.1 (13.76) |
| GRB-4 | 0.5–2 | 0.5 | 1 | 0.5–>16 | 1 | 3.0 (10.07) |
| GRB-5 | 0.5–1 | 0.5 | 1 | 0.5–4 | 1 | 1.85 (5.08) |
| GRB-6 | 4–>128 | ≥8 | >128 | >8–>128 | ≥32 | 37.6 (84.90) |
| GRB-7 | 2–8 | 2 | 4 | 2–16 | 8 | 6.8 (16.08) |
| GRB-8 | 0.5–8 | 2 | 4 | 1–≥8 | 4 | 4.5 (15.85) |
| GRB-9 | 1–8 | 4 | 8 | 2–≥8 | 8 | 7.0 (16.75) |
| GRB-10 | 2–>128 | ≥8 | >128 | 4–>128 | ≥16 | 33.2 (90.70) |
| GRB-11 | 4–>128 | 8 | >128 | 8–>128 | ≥8 | 36.8 (77.64) |
| GRB-12 | 32–>128 | 64 | >128 | 128–>128 | ≥128 | 128 (547) |
| GRB-13 | 1–8 | 2 | 8 | 1–≥8 | 4 | 4.7 (17.60) |
| GRB-14 | 0.25–4 | 0.5 | 4 | 1–16 | 2 | 4.1 (10.68) |
| GRB-15 | 0.5–4 | 1 | 4 | 1–32 | 4 | 8.8 (18.41) |
| GRB-16 | 0.5–8 | 1 | 8 | 0.5–32 | 2 | 8.05 (23.82) |
| GRB-17 | 0.5–≥8 | 2 | 8 | 1–≥32 | 8 | 13.3 (47.16) |
| CLR | 0.016–0.064 | 0.032 | 0.064 | 0.032–0.128 | 0.064 | 0.080 (0.107) |
| AMX | 0.016–0.064 | 0.032 | 0.064 | 0.032–1 | 0.128 | 0.225 (0.616) |
| MNZ | 1.56–100 | 25 | 100 | 6.25–>100 | 50 | 50.375 (294.6) |
For the clinical strains 80A and 121A and the reference strains 700392, 43504, and 49503, the compounds were examined in the range from 8 to 0.125 μg/ml (except for GRB-12, for which a dose range of 128 to 2 μg/ml was used), and for strains PCR-148, PCR-155, PCR-156, PCR-216, and ATCC 43629, for which a dose range of 128 to 0.125 μg/ml was used.
Data in parentheses represent mean MBCs in micromolar (μM) concentrations.
Antimicrobial spectrum of artemisinin derivatives.
With a view to understanding the nature of specificity of such compounds against H. pylori, a general antimicrobial pattern of all 17 molecules was evaluated using 12 bacterial strains and 8 fungal species (Table 3). Between the two natural product molecules, artemisinin (GRB-17) did not exhibit any significant zone of inhibition with the bacterial strains used, whereas artemisinic acid (GRB-12) showed a somewhat strong inhibition of S. mutans, S. aureus, methicillin-resistant S. aureus (MRSA), and S. epidermidis, besides exerting mild inhibitory effect on three other bacterial strains. Of the most potent anti-H. pylori derivatives (GRB-1 to GRB-5), GRB-2 and GRB-4 did not show sensitivity to any of the bacterial strains and therefore may be considered as specific anti-H. pylori molecules. The compound GRB-3, however, showed mild activity against E. coli, E. aerogenes, and S. aureus (J) (IZD = 1.2 to 1.6 cm). While GRB-5 exhibited high activity against S. aureus (IZD > 1.8 cm) and mild activity against E. coli, S. mutans, S. aureus (J), MRSA, S. epidermidis, and B. subtilis (IZD = 1.2 to 1.6 cm), GRB-1 showed relatively strong inhibition of S. aureus (J) (IZD > 1.8 cm) and mild activity against E. coli, S. aureus and MRSA (IZD ∼ 1.6 cm). Standard antibiotics amikacin (30 μg/disc) and ampicillin (10 μg/disc) showed IZDs of 1.2 to 2.4 cm and 1.0 to 1.6 cm, respectively, against all of the bacterial strains. Further, when GRB-1, GRB-3, and GRB-5 were examined for MIC values using a broth microdilution assay, none of the samples showed activity below 100 μg/ml, where standard antibiotics amikacin and ampicillin exhibited MIC ranges of 1 to 4 and 2 to 4 μg/ml, respectively.
Table 3.
Antibacterial and antifungal activities of artemisinin and its derivativesa
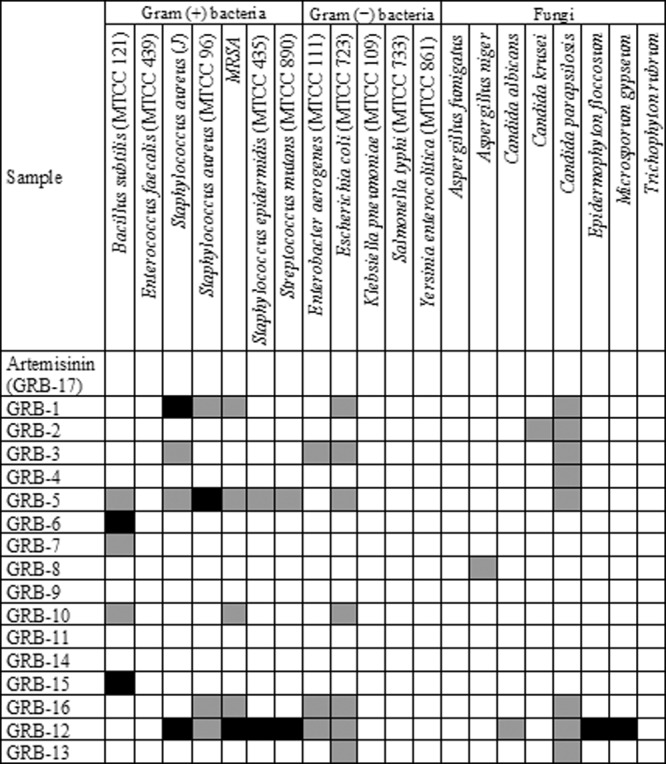
Discs were impregnated with 80 μg and 160 μg of the compounds to assess antibacterial and antifungal activities, respectively. The values for IZD were graded, and the results have been presented in a spectrum format as follows: IZD < 1.2 cm, not active (white); IZD = 1.2–1.6 cm, intermediate active (grey); and IZD > 1.6 cm, active (black).
Against a panel of eight fungal species, artemisinin and some of its synthetic analogues (GRB-6, GRB-7, GRB-9 to GRB-11, GRB-14, and GRB-15) were found to be inactive against all of the fungi. However, potent anti-H. pylori derivatives such as GRB-1 to GRB-5 exhibited mild activity against one strain, C. parapsilosis. Standard antifungal fluconazole at 25 μg/disc exhibited IZDs of 1.0 to 2.0 cm against all of the fungal strains. An assessment of MIC values of such five compounds against C. parapsilosis indicated values of 62.5 to 125 μg/ml, where fluconazole exhibited an MIC of 0.976 μg/ml. In contrast, the least anti-H. pylori-specific compound, GRB-12, showed strong inhibition of Microsporum gypseum and Epidermophyton floccosum and mild activity against Candida albicans and C. parapsilosis. Therefore, the five active anti-H. pylori analogues (GRB-1 to GRB-5) besides the parent molecule (GRB-17) may be considered to have insignificant activity against such fungi.
Acid stability of artemisinin and some analogues.
To be an effective anti-H. pylori agent, the molecule is required to be stable in a stomach acidic pH environment, because inactivation by low pH may be a factor that contributes to the limited clinical efficacies of antimicrobial agents active in vitro against H. pylori. This is particularly important in view of the report (21) that the acetal (C-O) type artemisinin derivatives have shorter half-life in simulated stomach acidic environment. Therefore, the acid stability of a few potent compounds such as GRB-1 to GRB-5 and also artemisinin (GRB-17) was assessed in terms of functional competency (MIC/MBC determination) vis-à-vis chemical degradation (HPLC analysis). Upon acid treatment, there was no perceptible increase in the MIC and MBC values of such molecules in general when examined against both the ATCC standard strain 43504 and the clinical isolate 80A (Table 4). Only a 2-fold increase in MIC value was observed with GRB-4, GRB-5, and artemisinin against both strains, whereas the molecule GRB-1 showed a 2-fold increase in MIC against ATCC 43504 alone. The bactericidal activity, on the other hand, did not vary with any of the tested compounds when the standard strain was used. However, a 2-fold increase in MBC value against clinical strain was noted in the case of GRB-1, GRB-2, and GRB-4.
Table 4.
Determination of acid stability of artemisinin and its five potent derivativesa
| Sample | MIC (μg/ml) |
MBC (μg/ml) |
% Decomposition during acidic exposure for: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal |
Acid treated |
Normal |
Acid treated |
|||||||||
| 80A | 43504 | 80A | 43504 | 80A | 43504 | 80A | 43504 | 0 h | 1 h | 2 h | 24 h | |
| GRB-1 | 2 | 0.5 | 2 | 1 | 2 | 1 | 4 | 1 | 8 | 5 | 5 | 18 |
| GRB-2 | 1 | 0.5 | 1 | 0.5 | 2 | 0.5 | 4 | 0.5 | 5 | 24 | 25 | 77 |
| GRB-3 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 4 | 6 | 5 | 2 |
| GRB-4 | 1 | 0.5 | 2 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 18 |
| GRB-5 | 2 | 2 | 4 | 4 | 8 | 4 | 8 | 4 | 6 | 0 | 4 | 31 |
| GRB-17 | 2 | 2 | 4 | 4 | 8 | 4 | 8 | 4 | 4 | 5 | 0 | 6 |
Acid treatment (pH 2.0) of the compounds was carried out for 24 h at 25°C. For MIC/MBC determination by broth microdilution assay, acid-treated (2 h) and untreated samples were serially diluted (2-fold) in 96-well microtiter plates in duplicate containing a total volume of 0.1 ml of Mueller-Hinton broth supplemented with 5% FCS and 0.0025% Dent. The chemical decomposition was assessed using HPLC by estimating the reduction in peak areas at the indicated time points (“0 h” means just upon addition of HCl) with respect to untreated controls.
HPLC analysis of the molecules, as determined before and after the addition of HCl at different time points (i.e., after 0, 1, 2, and 24 h of exposure), indicated ca. 1 to 10% decomposition at up to 2 h of exposure at pH 2.0 in 0.1 N HCl at 25°C (Table 4). However, some of these compounds do get degraded to higher extents after 24 h of exposure under identical acidic conditions (GRB-2 exhibited ca. 80% degradation; GRB-1, GRB-4, and GRB-5 showed ca. 20 to 30%), as was observed by others (21). It seems that the decomposition of the molecules under simulated acidic conditions is negligible up to 2 h, and this corroborates well with the insignificant changes in MICs and MBCs observed upon acid treatment. This implies that such potent molecules are expected to be functionally active in a stomach acidic environment except for GRB-2. It should be remembered that under acidic conditions, acetal-type derivatives are expected to generate dihydroartemisinin (GRB-8), which is an intermediate-type anti-H. pylori molecule. The equilibrium mixture of the putative drug and the compound GRB-8 could thus act as a functionally effective molecule as the pH of the stomach oscillates between 2.0 and 6.8.
Killing kinetics.
The rate and extent of killing of H. pylori by artemisinin along with its five potent analogues, namely, GRB-1 to GRB-5, were assessed against the reference strain ATCC 43504 with a view to assessing their effectiveness as bactericidal agents (Fig. 2). The experiments were initiated using 106 to 107 CFU/ml. The control growth pattern was as follows: an initial slow-growth period for up to 12 h, followed by exponential growth over 12 to 24 h. After this, the growth remained stationary for another 12 h or so, and then a decline phase slowly started. With compound GRB-1 (MIC = 1 μg/ml), a 1-log decrease in cell count was observed within 12 h, which culminated in complete killing at 24 h at 0.5× MIC. At 1× MIC and higher, total killing was attained well before 20 h (Fig. 2A). With the sample GRB-2 (MIC 0.5 μg/ml), only marginal killing effect was observed at 24 h at 1× MIC and 2× MIC doses, but complete killing was achieved at ∼36 h. At 4× MIC and above, a 1-log decrease in cell count was observed within 12 h, and complete killing was ensured within 24 h (Fig. 2B). With another ether derivative GRB-3 (MIC 1 μg/ml), bacteriostatic activity was observed till 12 h at 0.5× MIC, followed by some increase in growth till 48 h, indicating no effect at this half-MIC dose. At 1× MIC or 2× MIC, no inhibition was observed till 24 h. However, dose-dependent decrease in cell count was observed at or near 48 h. At 4× MIC, a 1-log decrease in cell count was observed after 24 h, with total killing at 48 h (Fig. 2C). The β-isomer GRB-4 (MIC 1 μg/ml) exhibited an almost identical kill kinetics profile (Fig. 2D) as its α-isomer GRB-3. With the sample GRB-5 (MIC 0.5 μg/ml), dose-dependent inhibition was observed within 6 h; a 1-log decrease in cell count was evident with 4× MIC. At 0.5× MIC, 1- and 2-log decreases in cell count were observed at 12 and 24 h, respectively, which remained static till 48 h. Complete killing of bacteria was observed at 1× MIC to 4× MIC at 36 h (Fig. 2E). The natural molecule artemisinin at the MIC dose or double the MIC dose could not kill the bacteria over the entire period of incubation (data not shown). However, at a 4× MIC dose (4 μg/ml), it could reduce the bacterial count to some extent at 24 h and drastically kill the bacteria at the end of 48 h. Using still higher doses of 8× MIC, 16× MIC, and 32× MIC, only a 2-log decrease in cell count was observed after 24 h, although total killing was attained only at 48 h (Fig. 2F). Summarizing, GRB-1 appeared to be the strongest anti-H. pylori molecule among the potent compounds tested, as far as the killing efficacy is concerned. At a dose range of 1 to 4 μg/ml, it induced complete killing of the bacteria within 20 h, whereas at 0.5 μg/ml complete killing was observed only after 24 h. GRB-5 appeared to be the next most effective, exhibiting complete killing at 0.5 to 2 μg/ml after 30 h. The third promising candidate is GRB-2, which at 0.5 to 1 μg/ml killed the bacteria within 36 h, while at the higher dose of 2 to 8 μg/ml the same was achieved at ∼24 h. The compounds GRB-3 and GRB-4, on the other hand, appeared to be less active among these potent molecules in exhibiting their bactericidal potential. To summarize, all of the compounds exhibited true bactericidal activity (>3-log decrease in viable cell count) after 24 h and are more effective than the natural molecule artemisinin (GRB-17) as far as killing efficacy is concerned.
Fig 2.

Kill kinetics of H. pylori ATCC 43504 by artemisinin and its five potent derivatives. Time- and dose-dependent killing of the bacteria by artemisinin derivatives GRB-1, GRB-2, GRB-3, GRB-4, and GRB-5 (panels A to E, respectively) and artemisinin (GRB-17) (F) were evaluated. The MICs of GRB-17, GRB-1, GRB-3, and GRB-4 were considered as 1 μg/ml and that of GRB-2 and GRB-5 was considered as 0.5 μg/ml. Symbols: ●, control; ○, 0.5× MIC; □, 1× MIC; △, 2× MIC; ▽, 4× MIC; ♢, 8× MIC; ⎔, 16× MIC; ✚, 32 × MIC.
Resistance development.
Considering drug resistance as the primary cause of failure of H. pylori treatment, artemisinin (GRB-17) and its five potent derivatives (GRB-1 to GRB-5) were examined for the possible development of insensitivity toward the bacteria upon repeated subculture (Fig. 3). Employing one reference strain, ATCC 43504, and one clinical isolate, 80A (both MNZ resistant), 10 repeated transfers of the bacteria were carried out. No change in MIC value of the derivative GRB-2 was observed with the standard strain ATCC 43504, whereas only a marginal increase (2-fold) was noted with artemisinin, GRB-1, GRB-3, GRB-4, and GRB-5 (Fig. 3A). Against clinical strain 80A, the derivative GRB-3 did not show any alteration in its MIC value, but others showed 2-fold increase (Fig. 3B). With the standard antibiotic CLR, the MIC was observed to increase 4-fold in both strains. Although the AMX MIC did not change for the reference strain, it increased 4-fold for the clinical strain. With MNZ, the MIC increased in both the strains, but more particularly with the clinical strain (8-fold). It therefore appears that artemisinin and its five potent analogues, particularly GRB-2 and GRB-3, would not exhibit any significant development of resistance in strains that are resistant to the standard antibiotic MNZ.
Fig 3.

Development of resistance to artemisinin, its five potent derivatives, and standard antibiotics in ATCC 43504 (A) and clinical isolate 80A (B). The results for artemisinin (⎔), GRB-1 (○), GRB-2 (■), GRB-3 (◆), GRB-4 (▲), GRB-5 (△), CLR (●), AMX (□), and MNZ (♢) are presented.
Combination effect of antibiotics with artemisinin and GRB-1.
To assess whether such molecules can manifest a synergistic effect with antibiotics used to treat H. pylori infection, a checkerboard titration analysis was carried out against a panel of 10 strains using the most potent derivative GRB-1 and the natural compound artemisinin (GRB-17). Of the 10 strains used for this investigation, all six clinical isolates and one standard strain (ATCC 43504) are MNZ resistant (MIC > 12.5 μg/ml), whereas all of the strains are CLR and AMX sensitive. As evident from Table 5, artemisinin and its semisynthetic derivative GRB-1 exhibited a nonsynergistic effect when combined with CLR, AMX, or MNZ. Interestingly, GRB-1 showed synergism with AMX in the case of 10% strains (FIC index 0.345). Upon analyzing the data, it was found that the artemisinin-MNZ combination exhibited an FIC index of 0.75 that is close to synergy in 40% of the strains, all of them being MNZ resistant. On the other hand, the GRB-1 and MNZ combination seemed to be approaching synergy for displaying an FIC index of 0.56 to 0.75 in 70% strains where as many as five of seven strains are MNZ resistant. Further, the combination of GRB-1 and CLR exhibited similar effects in half of the strains with an FIC index of 0.75. This may be taken to mean that the addition of artemisinin or its derivative GRB-1 with MNZ under in vitro conditions has resulted in the development of sensitivity in MNZ-resistant strains. For example, the standard strain ATCC 43504 is MNZ resistant (MIC 50 μg/ml) but, in combination with 1 μg of GRB-1/ml, the MIC value of MNZ was reduced to 6.25 μg/ml, resulting in an FIC index of 0.625. The MIC value of MNZ thus decreased 8-fold, indicating that GRB-1 in combination with MNZ can enhance the activity of the antibiotic against resistant strains. The same observation was noted against clinical strain 80A, where MNZ alone showed an MIC of 50 μg/ml, while in combination with GRB-1 (1 μg/ml), the MIC value decreased to 6.25 μg/ml. In the case of MNZ-sensitive H. pylori, the MIC values of two of three strains were found to decrease 8- to 16-fold, whereas in all 10 CLR-sensitive strains a 4-fold decrease in MIC values of CLR was observed with half of the strains. Taking all of these results into account, it is evident that both GRB-1 and GRB-17 lack the potential to exhibit synergism with the three antibiotics tested.
Table 5.
Checkerboard titration assay of artemisinin and GRB-1 in combination with antibioticsa
| FIC index (interpretation) | No. (%) of isolates |
|||||
|---|---|---|---|---|---|---|
| GRB-17 (artemisinin) |
GRB-1 |
|||||
| CLR | AMX | MNZ | CLR | AMX | MNZ | |
| ≤0.5 (synergy) | 0 | 0 | 0 | 0 | 1 (10) | 0 |
| >0.5 to ≤4.0 (nonsynergistic) | 10 (10) | 10 (10) | 10 (10) | 10 (10) | 9 (10) | 10 (10) |
| >4.0 (antagonism) | 0 | 0 | 0 | 0 | 0 | 0 |
Checkerboard titration for determining synergism between artemisinin (GRB-17) or GRB-1 with standard antibiotics against 10 H. pylori strains was performed using a broth microdilution assay.
Effect on morphology.
The morphological alterations of H. pylori were assessed after exposing the standard strain ATCC 43504 to 1 μg of GRB-1/ml for 24 h under shake-culture conditions. Transmission electron micrographs demonstrated that GRB-1 treatment induced swelling and vacuole-like structures in the cell cytoplasm (Fig. 4B and C), whereas the control cells (i.e., without any drug [Fig. 4A]) remained rod-shaped (Fig. 4Aa and Ab) with intact flagella (Fig. 4Ac). Exposure to GRB-1 transformed the shape and size of the organism from a bacillus- to a doughnut-shaped form. The majority of the bacteria became curved (Fig. 4Bb) and some even full coccoid (Fig. 4Bc) shaped. The detachment of the outer cell membrane was evident in a few of the population, and other features, such as the ingrowth of periplasmic membranes (Fig. 4Ba), an increase in the periplasmic cylinder (Fig. 4Ca and Cb), and the invagination of membranes, were typically observed. The ultrastructural changes associated with GRB-1 treatment also includes detachment of the outer membrane from the inner membrane and an increase in the periplasmic space filled with the electron-rich region. Moreover, the outer envelop became detached from the inner side of the bend, leading to extensive deformity.
Fig 4.

Transmission electron micrograph of H. pylori exposed to GRB-1. H. pylori ATCC 43504 cells were incubated with 1 μg of GRB-1/ml (shown at 11,500× magnification [B and C]) or in the absence (at 8,200× magnification [A]) for 24 h under microaerophilic condition at 37°C. The typical ingrowth of periplasmic membranes (Ba), swelling and conversion to U shape (Bb), invagination of membranes to form full coccoid (Bc), increase in periplasmic cylinder (Ca and Cb), and bilayer coccoid (Cc) are shown (arrow) as signs of structural deformity in the marked field (bar, 2 μm). The control cells exhibit rod shape (Aa) and polar membranes (Ab) attached to flagella (Ac) (arrow) (bar, 2 μm).
In vivo effect.
We finally wanted to probe the potent molecule GRB-1 for its in vivo potential in the H. pylori-induced infection model in terms of clearance and eradication. Using C57BL/6J mice in four treatment groups—infection controls (8 mice), triple-therapy omeprazole-CLR-MNZ (6 mice), double therapy CLR-MNZ (6 mice), and monotherapy with GRB-1 (6 mice)—we wanted to compare the standalone efficacy of GRB-1 with triple or double therapy. In terms of clearance as measured 36 h after the last treatment (Fig. 5A), the OCM- and the CM-treated groups exhibited significant attenuation of bacterial load (means of 1.20 × 104 CFU/g and 1.40 × 104 CFU/g tissue, respectively) as determined by a negative rapid urease test (one animal in the OCM group died during the treatment period), whereas the control mice were found to get substantially infected (mean of 1.18 × 107 CFU/g tissue) and showed a urease-positive reaction. In three GRB-1-treated animals sacrificed for the clearance study, the bacterial counts were 3.70 × 105, 9.22 × 106, and 5.69 × 106 CFU/g of tissue (average, 5.09 × 106 CFU/g tissue). Although two stomach tissues showing higher count were urease negative, one that exhibited a lower count was, however, found to be urease positive. For the eradication experiment (Fig. 5B), the remaining mice were sacrificed 29 days after last treatment (one mouse in the CM group, however, died during the intervening period). The bacterial load in the infection controls reached still-higher levels (mean of 1.39 × 108 CFU/g tissue; all urease positive), indicating successful colonization by H. pylori under these experimental conditions. Substantial eradication of the H. pylori burden was perceptible in OCM-treated animals (mean cell count of 1.17 × 104 CFU/g tissue), wherein the corresponding tissues were also urease negative. In the CM-treated group, although almost total eradication (1.50 × 104 CFU/g) was observed in one mice (urease negative), the other one exhibited a higher bacterial count (1.00 × 107 CFU/g of tissue) with positive urease tests. It was reported earlier that omission of omeprazole in the OCM triple therapy is not efficient enough to prevent the relapse of bacterial colonization at longer durations (46). In the three GRB-1-treated mice, the bacterial counts were 9.30 × 106, 5.60 × 105, and 9.50 × 105 CFU/g of tissue (average of 3.60 × 106 CFU/g of tissue). All three tissues however remained urease positive. It seems that treatment with GRB-1 alone at 50 mg/kg/day (single therapy) could reduce the H. pylori load in vivo to some extent during clearance stage and perhaps a little more at the eradication stage. Such indications using monotherapy are encouraging, especially since this novel artemisinin derivative did not elicit any toxicity at this dose, whereas we typically observed diarrhea-like symptom in most of the animals treated with OCM and CM.
Fig 5.

In vivo effect of GRB-1, dual therapy (CM [clarithromycin and metronidazole]), and triple therapy (OCM [omeprazole, clarithromycin, and metronidazole]) on H. pylori infection in C57BL/6J mice. Clearance (A) indicates reduction in bacterial load 36 h after the cessation of treatment, whereas eradication (B) indicates the same 29 days after the treatment (reduction in bacterial load in terms of 8 mice for the infection control, five each for OCM and CM, and six for GRB-1, 50 mg/kg/day). The animals were sacrificed after 36 h (4 mice for the infection control, 2 mice for OCM, 3 mice with CM and GRB-1 each) and 29 days (4 mice for the infection control, 3 mice for OCM, 2 mice for CM, and 3 mice GRB-1) after the last treatment. Each point represents the viable count obtained from each animal in the particular group.
In vitro cytotoxicity evaluation.
In order to assess the cytotoxicity, all 17 molecules were tested against five different cell lines. Except for GRB-10 and GRB-11, most of the compounds, such as GRB-1 to GRB-5, GRB-12, GRB-13, GRB-16, and GRB-17 (artemisinin), appeared to be safe since none of them showed any potential cytotoxic effect around their MBC50 values against H. pylori (Table 6). The cytotoxicity values obtained with GRB-8, GRB-14, and GRB-17 correlate well with the experimental findings of other groups (see, for example, reference 5), and hence the activity of eight new molecules, including GRB-1 to GRB-5, could be accepted as valid. The compounds GRB-6 to GRB-9, GRB-14, and GRB-15 can be considered intermediate cytotoxic. Nevertheless, in the tumor macrophage cell line (RAW 264.7), but not in the normal macrophage primary culture, the majority of the molecules exhibited cytotoxicity. The potential lethal effects of most of the compounds against RAW 264.7 cell lines indicate that all of the compounds probably have dramatic effect on tumor-associated macrophages where the cells automatically develop anti-inflammatory cytokines such as interleukin-10 and show signs of tissue repair functions (14). In contrast, none of the compounds exerted cytotoxic effects against normal mouse macrophage cell lines (which generate huge amounts of proinflammatory cytokines, express elevated levels of major histocompatibility complex molecules, and are powerful killers of pathogens and tumor cells).
Table 6.
Cytotoxic activity and pharmacokinetic parameters of the test compoundsa
| Sample code (mol wt) | H. pylori activity (MBC50 [μg/ml]) | Cytotoxicity (IC50 [μg/ml]) |
Pharmacokinetic parameters |
||||||
|---|---|---|---|---|---|---|---|---|---|
| HepG2 | MCF-7 | AGS | RAW 264.7 | MPM | log P | log S | PPBb (%) | ||
| GRB-1 (338) | 2 | >100 | 50 | 50 | 10 | >100 | 4.0407 | –4.17808 | <90 |
| GRB-2 (312) | 4 | >100 | 75 | 75 | 10 | 100 | 3.6733 | –3.87658 | <90 |
| GRB-3 (298) | 1 | >100 | >100 | >100 | 75 | >100 | 3.3308 | –3.54937 | <90 |
| GRB-4 (298) | 1 | >100 | 100 | 100 | 75 | >100 | 3.3308 | –3.54937 | <90 |
| GRB-5 (364) | 1 | >100 | 50 | 75 | 10 | 75 | 4.0558 | –5.06884 | <90 |
| GRB-6 (443) | >32 | 75 | 100 | 75 | 5 | 50 | 6.1434 | –6.78585 | ≥95 |
| GRB-7 (423) | 8 | >50 | 100 | 100 | 5 | 75 | 5.6125 | –6.31709 | ≥95 |
| GRB-8 (284) | 4 | ∼50 | 50 | 100 | 5 | 100 | 3.0525 | –3.20419 | <90 |
| GRB-9 (418) | 8 | 75 | 50 | 50 | 5 | >100 | 4.4419 | –5.27237 | ≥95 |
| GRB-10 (366) | >16 | 75 | 50 | 30 | 0.5 | 75 | 4.8333 | –5.20852 | ≥90 |
| GRB-11 (474) | >8 | >100 | 0.5 | 0.5 | 0.5 | 75 | 7.1118 | –9.07303 | ≥95 |
| GRB-12 (234) | >128 | 100 | >100 | >100 | 100 | >100 | 3.4474 | –4.21376 | <90 |
| GRB-13 (267) | 4 | >100 | 100 | 100 | 100 | >100 | 3.2401 | –2.97035 | <90 |
| GRB-14 (384) | 2 | 50 | 100 | 100 | 1 | 10 | 2.9735 | –3.57458 | <90 |
| GRB-15 (478) | 4 | 50 | 100 | 100 | 1 | 50 | 4.3364 | –5.73394 | ≥95 |
| GRB-16 (338) | 2 | >100 | 100 | 100 | 10 | 100 | 4.0407 | –4.17808 | <90 |
| GRB-17 (282) | 8 | >100 | >100 | >100 | >100 | >100 | 3.0853 | –3.47168 | <90 |
| Mitomycin C | 1.1 | 6.8 | 2.5 | 3.3 | |||||
For evaluation of cytotoxicity by an MTT assay, test compounds and the standard mitomycin C were examined over dose ranges of 100 to 0.5 μg/ml and 16.7 to 0.17 μg/ml, respectively. The pharmacokinetic parameters were calculated by using Cerius2 software, version 4.10.
PPB, plasma protein binding.
Pharmacokinetic property predictions.
The compounds showed lipophilicity, with log P values in the range of 2.9735 to 7.1118, and aqueous solubility, with log S values in the range of −2.97035 to −6.78585. GRB-6, GRB-7, and GRB-11 were the most lipophilic compounds of the series and thus considered not appropriate for drug therapy. Interestingly, the five potent anti-H. pylori molecules (GRB-1 to GRB-5), along with artemisinin (GRB-17) and dihydroartemisinin (GRB-8), exhibited log P values in the range of 3.0853 to 4.0558 and log S values in the range of −3.20419 to −5.06884, indicating their potential as candidate drugs (Table 6). Plasma protein binding (PPB) levels determine the extent to which a compound is likely to be bound with the protein fraction of blood. This influences the half-life of the compound in the body and hence is an important parameter. Most of the molecules exhibited acceptable PPB levels with a binding efficiency of <90% (PPB level 0), including artemisinin, dihydroartemisinin, and the five potent compounds. GRB-10 had an efficiency of ≥90% (PPB level 1) and GRB-6, GRB-7, GRB-9, GRB-11, and GRB-15 had an efficiency of ≥95% (PPB level 2), indicating a lower probability of free drug concentration at the site of action and hence were considered less suitable for drug therapy.
DISCUSSION
Considering the widespread and commensal nature of H. pylori and the rate at which resistance to some of the more common antibiotics used in H. pylori treatments manifests jeopardizing their effectiveness in managing other infections (7, 26, 32, 34), it is necessary to look for chemotherapeutic agents that should be both effective and safe in eradicating both antibiotic-susceptible and -resistant strains. A novel anti-H. pylori drug should (i) exert potent and selective antibacterial action against both clinical and standard H. pylori strains, (ii) be stable at the acidic pH of the stomach, (iii) show antibacterial action against CLR-resistant and MNZ-resistant strains, (iv) not lead to the development of drug resistance, (v) possess wide safety, and (vi) be easily affordable. The present investigation was therefore designed to find potent anti-H. pylori molecules and also to consider their functional usefulness in identifying prospective candidate leads.
Based on our serendipitous observation that artemisinin could be effective against H. pylori, we evaluated a series of semisynthetic analogues, including a few newly synthesized molecules, with a view to profiling their anti-H. pylori features. When screened for their potential in terms of disc diffusion sensitivity, the MIC and MBC values against a panel of clinical and standard strains of H. pylori, 5 of 17 compounds, namely, β-artecyclopropylmether, β-arteether, α-artemether, β-artemether, and β-artefurfurylether (GRB-1 to GRB-5) proved to be reasonably prospective (Tables 1 and 2). Such strongly active compounds can be considered specific anti-H. pylori agents because all of them demonstrated a lack of sensitivity against the majority of other bacterial and fungal strains tested, barring perhaps C. parapsilosis; β-artefurfurylether, however, exhibited the highest antimicrobial activity of the five molecules (Table 3). Looking at the structural features of the compounds, it seems that a small, nonpolar, β-oriented C-12 substituent besides the endoperoxide linkage confers higher activity. Presumably, the α-substituent, having the same orientation as the peroxo bridge, offers steric hindrance and interferes with the activity.
We performed a series of experiments with these five alkoxy derivatives to probe their functional efficacy. The derivatives showed no significant changes in their MIC and MBC values even after exposure to acidic pH (Table 4). This finding is in contrast to CLR, for which the MIC/MBC increases with a decrease in pH (8), necessitating the enhancement of the stomach pH by gastric antisecretory agents such as H2 receptor blockers or proton pump inhibitors for the effective clearance and eradication of H. pylori-induced gastroduodenal ulcers by the antibiotics (46). Thus, functional competency at a stomach acidic pH would augur well for such smaller alkoxy derivatives of artemisinin. Second, all five molecules showed potential activity even against resistant strains, a desirable attribute for any putative anti-H. pylori agent. This is more important because of the indiscriminate use of antibiotics such as CLR, AMX, tetracycline, and particularly MNZ for curing H. pylori-induced gastric ulcer, leading to the development of resistance to this organism (7, 12, 26, 32). Third, the killing efficacy of artemisinin and its five derivatives indicated β-artecyclopropylmether as the most potent molecule, which killed bacteria within 24 h at 0.5 μg/ml and within 20 h at ≥1 μg/ml (Fig. 2A). The next effective molecule is β-artefurfurylether, which showed killing within 36 h at doses of 0.5 to 2 μg/ml (Fig. 2E). Interestingly, α-artecyclopropylmether was found to be much less active than its β-isomer (data not shown). Fourth, the five potent compounds did not show any tendency to develop resistance against either the clinical isolate or standard strain (Fig. 3). Thus, such molecules would not be expected to develop any resistance and, therefore, could prove useful in treating H. pylori infection successfully, particularly in low socioeconomic settings where majority of the isolated strains were found to be antibiotic resistant (7, 33, 34). It was of further interest to see whether such activity is due to the manifestation of urease inhibition as H. pylori surface is richly coated with urease and is considered as a putative target in designing potential anti-H. pylori principles (28). However, artemisinin and the potent five analogues did not inhibit H. pylori urease in vitro (data not shown).
A comparative viewing of these five derivatives in terms of IZD, MIC, and MBC values, as well as the four experimental outcomes described above, indicated that in majority of the experiments, both β-artecyclopropylmether and β-artefurfurylether proved to be superior compared to β-arteether, α-artemether, and β-artemether. However, in terms of killing efficacy, β-artecyclopropylmether proved to be the most potent. We therefore further examined this lead molecule for its capacity to induce morphological deformity, show synergism with anti-H. pylori antibiotics, and reduce the H. pylori burden in vivo. The phenotypic changes of the bacteria in terms of morphological transformation from rod to spiral to coccoid form, and the cytological alteration, leading to the formation of swelling and vacuole-like structures in the cytoplasm with detachment of outer cell membranes (Fig. 4) have been discernible in the presence of β-artecyclopropylmether. Next, a checkerboard titration assay indicated that β-artecyclopropylmether in combination with MNZ under in vitro condition exhibited the development of sensitivity in MNZ-resistant strains, suggesting its potential as a therapeutic agent against resistant strains, although the compound could not qualify as synergistic with any of the antibiotics tested (Table 5). The molecule, however, exhibits a somewhat mild in vivo potential as a single therapeutic agent in the chronic H. pylori infection model at a dose of 50 mg/kg/day (Fig. 5). It remains to be seen whether it can substitute one or both antibiotics in triple OCM therapy or at least reduce the dose requirement of such gastric irritant antibiotics. Since artemisinin and other derivatives, including β-artecyclopropylmether, did not exhibit gastric anti-H+,K+-ATPase activity (data not shown), we do not anticipate the molecule to replace omeprazole in triple therapy. Further experiments are required to be carried out with β-artecyclopropylmether and CLR-MNZ in a combination format and also as a function of dosage. Although the majority of the in vivo validation experiments with newly discovered anti-H. pylori principle(s) tend to indicate the necessity of simultaneous use of one or two conventional antibiotics, albeit with lower doses (24), success with monotherapy has also been reported (22).
In an attempt to see the role of antimalarial drugs devoid of peroxide linkage, we have examined seven commonly used antimalarials, namely, quinine, chloroquine, primaquine, pyrimethamine, lumefantrine, piperaquine, and mefloquine, against one clinical strain (80A) and one standard strain (ATCC 49503) of H. pylori. Barring mefloquine (MIC = 16 to 32 μg/ml) and pyrimethamine (MIC = 32 to 64 μg/ml), none could exert any significant anti-H. pylori potential. It may not be out of place to mention here that a recent epidemiological study in child refugees in Western Australia indicated that premigration antimalarial treatment significantly reduced the odds of H. pylori infection after adjusting for age and sex and therefore predicted a protective effect of antimalarials against H. pylori infection (6). An earlier in vitro study with backpackers from developed countries after travel to tropical countries indicated that mefloquine, a traveler's choice antimalarial, has anti-H. pylori activity (39). Whereas the latter study corroborates the findings described above, the former study indirectly supports our serendipitous observation that artemisinin has strong anti-H. pylori effect. We therefore anticipate observing the absence of H. pylori seroprevalence among malaria patients treated with artemisinin combination drugs as opposed to antimalarials devoid of artemisinin.
Whatever may be the mechanism(s) of action of artemisinin analogues in manifesting strong and potentially useful anti-H. pylori features, it is worth reiterating that artemisinin is a cheap and relatively safe drug with few and minor side effects even at high doses. Because much of the pharmacokinetic and pharmacodynamic information is available, it can very well take a faster route in developing an appropriate candidate drug against H. pylori infections. Our efforts in evaluating physicochemical parameters (log P, aqueous solubility, and PPB) and the cytotoxicity profiles, particularly of the eight newly synthesized molecules, revealed that the majority of the compounds, including GRB-1 to GRB-5, lie well within acceptable ranges to qualify as therapeutic candidate (Table 6). Further investigation using quantitative structure-activity relationship studies would help predict which among such potent molecules would be best for the development of a candidate drug for H. pylori treatment. Such preclinical findings must, however, be validated in a clinical setting to judge the efficacy of the molecule as a useful anti-H. pylori agent and hence as a gastric anti-ulcer principle.
ACKNOWLEDGMENTS
This study was supported by an interlaboratory network project (COR-0023) of the Council of Scientific and Industrial Research (CSIR). We also thank the CSIR for the award of Senior Research Fellowships to S.G., A.C., and S.K.K. during the course of this investigation.
We thank A. K. Mukhopadhyay, NICED, Kolkata, India, for providing clinical strains and one standard strain ATCC 700392 and M. Sitaram Kumar of Dr. Reddy's Laboratory, Hyderabad, India, for the standard strain ATCC 49503 of H. pylori. The other bacterial strains were kindly provided by Anil Tyagi, University of Delhi South Campus, Delhi, India (all MTCC strains); Amita Jain, King George Medical University, Lucknow, India (MRSA strain); and A. K. Saxena, Indian Institute of Integrative Medicine, Jammu, India [S. aureus (J)]. Uma Banerjee of the Department of Microbiology, All India Institute of Medical Sciences, New Delhi, India, kindly provided all of the clinical fungal strains. We also thank S. K. Puri of Central Drug Research Institute (CSIR-CDRI), Lucknow, India, for kindly providing the seven antimalarial compounds. We acknowledge the interpretation of EM pictures by J. Sengupta, the calculation of physicochemical values (log P, log S, and PPB) by M. Prabu, and the HPLC study by B. Chakraborty of CSIR-IICB. We are indebted to B. Achari (CSIR-IICB) for valuable comments and critical review of the manuscript.
Footnotes
Published ahead of print 11 June 2012
REFERENCES
- 1. Andersen LP. 2007. Colonization and infection by Helicobacter pylori in humans. Helicobacter 12(Suppl 2):12–15 [DOI] [PubMed] [Google Scholar]
- 2. Bauer AW, Kirby WM, Sherris JC, Turck M. 1966. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 45:493–496 [PubMed] [Google Scholar]
- 3. Bhakuni RS, Jain DC, Sharma RP. 1995. An improved procedure for the synthesis of ether of dihydroartemisinin. Ind. J. Chem. 34B:529–530 [Google Scholar]
- 4. Bhakuni RS, et al. January 2004. Single pot conversion of artemisinin into artemether. U.S. patent 6,683,193, MY-129031-A(2007)
- 5. Chaijaroenkul W, Viyanant V, Mahavorasirikul W, Na-Bangchang K. 2011. Cytotoxic activity of artemisinin derivatives against cholangiocarcinoma (CL-6) and hepatocarcinoma (Hep-G2) cell lines. Asian Pac. J. Cancer Prev. 12:55–59 [PubMed] [Google Scholar]
- 6. Cherian S, Forbes D, Sanfilippo F, Cook A, Burgner D. 2008. The epidemiology of Helicobacter pylori infection in African refugee children resettled in Australia. Med. J. Aust. 189:438–441 [DOI] [PubMed] [Google Scholar]
- 7. Datta S, et al. 2005. Most Helicobacter pylori strains of Kolkata in India are resistant to metronidazole but susceptible to other drugs commonly used for eradication and ulcer therapy. Aliment. Pharmacol. Ther. 22:51–57 [DOI] [PubMed] [Google Scholar]
- 8. Erah PO, Goddard AF, Barrett DA, Shaw PN, Spiller RC. 1997. The stability of amoxicillin, clarithromycin, and metronidazole in gastric juice: relevance to the treatment of Helicobacter pylori infection. J. Antimicrob. Chemother. 39:5–12 [DOI] [PubMed] [Google Scholar]
- 9. Fahey TJ, et al. 1992. Macrophage inflammatory protein 1 modulates macrophage function. J. Immunol. 148:2764–2769 [PubMed] [Google Scholar]
- 10. Funatogawa K, et al. 2004. Antibacterial activity of hydrolyzable tannins derived from medicinal plants against Helicobacter pylori. Microbiol. Immunol. 48:251–261 [DOI] [PubMed] [Google Scholar]
- 11. Glupczynski Y. 1996. Culture of Helicobacter pylori from gastric biopsies and antimicrobial susceptibility testing, p 17–28 In Lee A, Megraud F. (ed), Helicobacter pylori: techniques for clinical diagnosis and basic research. WB Saunders Co, Ltd, London, England [Google Scholar]
- 12. Graham DY, Qureshi WA. 2000. Antibiotic-resistant Helicobacter pylori infection and its treatment. Curr. Pharm. Des. 6:1537–1544 [DOI] [PubMed] [Google Scholar]
- 13. Hachem CY, et al. 1996. Antimicrobial susceptibility testing of Helicobacter pylori: comparison of E-test, broth microdilution, and disc diffusion for ampicillin, clarithromycin, and metronidazole. Diagn. Microbiol. Infect. Dis. 24:37–41 [DOI] [PubMed] [Google Scholar]
- 14. Hagemann T, et al. 2008. “Re-educating” tumor-associated macrophages by targeting NF-κB. J. Exp. Med. 205:1261–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haynes RK, et al. 2002. C-10 ester and ether derivatives of dihydroartemisinin–10-α artesunate, preparation of authentic 10-β artesunate, and of other ester and ether derivatives bearing potential aromatic intercalating groups at C-10. Eur. J. Org. Chem. 2002:113–132 [Google Scholar]
- 16. Hou TJ, Xia K, Zhang W, Xu XJ. 2004. ADME evaluation in drug discovery. 4. prediction of aqueous solubility based on atom contribution approach. J. Chem. Infect. Comput. Sci. 44:266–275 [DOI] [PubMed] [Google Scholar]
- 17. Iwao E, Yamamoto K, Yokoyama Y, Hirayama F, Haga K. 2004. Potent antibacterial activity of Y-754, a novel benzimidazole compound with selective action against Helicobacter pylori. J. Infect. Chemother. 10:90–96 [DOI] [PubMed] [Google Scholar]
- 18. Jain DC, et al. September 1999. Process for the simultaneous production of artemisinin and essential oil from plant Artemisia annua. U.S. patent 5,955,084
- 19. Jayaraman KS. 2003. Technology, tradition unite in India's drug discovery scheme. Nat. Med. 9:982. [DOI] [PubMed] [Google Scholar]
- 20. Jung M, Lee K, Kim H, Park M. 2004. Recent advances in artemisinin and its derivatives as antimalarial and antitumor agents. Curr. Med. Chem. 11:1265–1284 [DOI] [PubMed] [Google Scholar]
- 21. Jung M, Lee S. 1998. Stability of acetal and non-acetal-type analogs of artemisinin in simulated stomach acid. Bioorg. Med. Chem. Lett. 8:1003–1006 [DOI] [PubMed] [Google Scholar]
- 22. Kanamaru T, et al. 2001. In vitro and in vivo antibacterial activities of TAK-083, an agent for treatment of Helicobacter pylori infection. Antimicrob. Agents Chemother. 45:2455–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kawase M, Motohashi N. 2004. Plant-derived leading compound for eradication of Helicobacter pylori. Curr. Med. Chem. 3:89–100 [Google Scholar]
- 24. Koga T, et al. 2002. Effect of plaunotol in combination with clarithromycin or amoxicillin on Helicobacter pylori in vitro and in vivo. J. Antimicrob. Chemother. 50:133–136 [DOI] [PubMed] [Google Scholar]
- 25. Kuhn T, Wang Y. 2008. Artemisinin: an innovative cornerstone for anti-malaria therapy. Prog. Drug Res. 383:385–422 [DOI] [PubMed] [Google Scholar]
- 26. Kwon DH, et al. 2000. Isolation and characterization of tetracycline-resistant clinical isolates of Helicobacter pylori. Antimicrob. Agents Chemother. 44:3203–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li J, Zhou B. 2010. Biological actions of artemisinin: insights from medicinal chemistry studies. Molecules 15:1378–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin YT, Kwon YI, Labbe RG, Shetty K. 2005. Inhibition of Helicobacter pylori and associated urease by oregano and cranberry phytochemical synergies. Appl. Environ. Microbiol. 71:8558–8564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malfertheiner P, et al. 2005. Helicobacter pylori eradication has the potential to prevent gastric cancer: a state-of-the-art critique. Am. J. Gastroenterol. 100:2100–2115 [DOI] [PubMed] [Google Scholar]
- 30. Marshall BJ. 2001. One hundred years of discovery and rediscovery of Helicobacter pylori and its association with peptic ulcer disease, p 19–24 In Mobley HLT, Mendz GL, Hazell SL. (ed), Helicobacter pylori: physiology and genetics. ASM Press, Washington, DC: [PubMed] [Google Scholar]
- 31. McNulty C, et al. 2002. Helicobacter pylori susceptibility testing by disc diffusion. J. Antimicrob. Chemother. 49:601–609 [DOI] [PubMed] [Google Scholar]
- 32. Megraud F. 2004. Basis for the management of drug-resistant Helicobacter pylori infection. Drugs 64:1893–1904 [DOI] [PubMed] [Google Scholar]
- 33. Megraud F, Lehn N, Lind T. 1997. The MACH 2 Study: Helicobacter pylori resistance to antimicrobial agents and its influence on clinical outcome. Gastroenterology 112:A216 [Google Scholar]
- 34. Mendonça S, et al. 2000. Prevalence of Helicobacter pylori resistance to metronidazole, clarithromycin, amoxicillin, tetracycline, and furazolidone in Brazil. Helicobacter 5:79–83 [DOI] [PubMed] [Google Scholar]
- 35. Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65:55–63 [DOI] [PubMed] [Google Scholar]
- 36. Nostro A, et al. 2005. Antibacterial effect of plant extracts against Helicobacter pylori. Phytother. Res. 19:198–202 [DOI] [PubMed] [Google Scholar]
- 37. O'Neill PM, et al. 2001. Optimization of the allylsilane approach to C-10 deoxo-carba analogues of dihydroartemisinin: synthesis and in vitro antimalarial activity of new, metabolically stable C-10 analogues. J. Chem. Soc. Perkin Trans. 1:2682–2689 [Google Scholar]
- 38. O'Rourke J, Bode G. 2001. Morphology and ultrastructure, p 53–67 In Mobley HLT, Mendz GL, Hazell SL. (ed), Helicobacter pylori: physiology and genetics. ASM Press, Washington, DC: [PubMed] [Google Scholar]
- 39. Potasman I, Yitzhak A. 1998. Helicobacter pylori serostatus in backpackers following travel to tropical countries. Am. J. Trop. Med. Hyg. 58:305–308 [DOI] [PubMed] [Google Scholar]
- 40. Posner GH, et al. 1999. Antimalarial, antiproliferative, and antitumor activities of artemisinin-derived, chemically robust, trioxane dimers. J. Med. Chem. 42:4275–4280 [DOI] [PubMed] [Google Scholar]
- 41. Schuster BG. 2001. Demonstrating the validity of natural products as anti-infective drugs. J. Altern. Complement. Med. 7(Suppl 1):73–82 [DOI] [PubMed] [Google Scholar]
- 42. Shrimali M, Bhattacharya AK, Bhakuni RS, Jain DC, Sharma RP. 2001. A process for the preparation of sodium artelinate. Indian patent 185198
- 43. Simon JE, Chadwick AF, Craker LE. 1984. Herbs: an indexed bibliography, p 1971–1980 In The scientific literature on selected herbs and aromatic and medicinal plants of the temperate zone. Archon Books, Hamden, CT [Google Scholar]
- 44. Singh BL, et al. 2001. Simultaneous determination of antimalarial artemisinin, dihydroartemisinin and arteether using reversed phase high performance liquid chromatography. J. Ind. Chem. Soc. 78:489–491 [Google Scholar]
- 45. Tawfik AF, Bishop SJ, Ayalp A, el-Feraly FS. 1990. Effects of artemisinin, dihydroartemisinin and arteether on immune responses of normal mice. Int. J. Immunopharmacol. 12:385–389 [DOI] [PubMed] [Google Scholar]
- 46. van Zanten SJOV, Kolesnikow T, Leung V, O'Rourke JL, Lee A. 2003. Gastric transitional zones, areas where Helicobacter treatment fails: results of a treatment trial using the Sydney strain mouse model. Antimicrob. Agents Chemother. 47:2249–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang J, Guo Y, Zhang BC, Chen ZT, Gao JF. 2007. Induction of apoptosis and inhibition of cell migration and tube-like formation by dihydortemisinin in murine lymphatic endothelial cells. Pharmacology 80:207–218 [DOI] [PubMed] [Google Scholar]
- 48. Williamson JS. 2001. Helicobacter pylori: current chemotherapy and new targets for drug design. Curr. Pharm. Des. 7:355–392 [DOI] [PubMed] [Google Scholar]
- 49. Yanagawa Y, Yamamoto Y, Hara Y, Shimamura T. 2003. A combination effect of epigallocatechin gallate, a major compound of green tea catechins, with antibiotics on Helicobacter pylori growth in vitro. Curr. Microbiol. 47:244–249 [DOI] [PubMed] [Google Scholar]