

Abstract
Persisters are dormant phenotypic variants of regular cells that are tolerant to antibiotics and play an important role in recalcitrance of chronic infections to therapy. Persisters can be produced stochastically in a population untreated with antibiotics. At the same time, a deterministic component of persister formation has also been documented in a population of cells with DNA damaged by fluoroquinolone treatment. Expression of the SOS response under these conditions induces formation of persisters by increasing expression of the TisB toxin. This suggests that other stress responses may also contribute to persister formation. Of particular interest is oxidative stress that pathogens encounter during infection. Activated macrophages produce reactive oxygen and nitrogen species which induce the SoxRS and OxyR regulons. Genes controlled by these regulons deactivate the oxidants and promote repair. We examined the ability of oxidative stress induced by paraquat (PQ) to affect persister formation. Preincubation of cells with PQ produced a dramatic increase in the number of persisters surviving challenge with fluoroquinolone antibiotics. PQ did not affect killing by kanamycin or ampicillin. Persisters in a culture treated with PQ that survived a challenge with a fluoroquinolone were also highly tolerant to other antibiotics. PQ induces SoxRS, which in turn induces expression of the AcrAB-TolC multidrug-resistant (MDR) pump. Fluoroquinolones are extruded by this MDR pump, and the effect of PQ on antibiotic tolerance was largely abolished in a mutant that was defective in the pump. It appears that PQ, acting through AcrAB-TolC, reduces the concentration of fluoroquinolones in the cells. This allows a larger fraction of cells to become persisters in the presence of a fluoroquinolone. Analysis of a lexA3 mutant indeed showed a dependence of persister induction under these conditions on SOS. These findings show that induction of a classical resistance mechanism, MDR efflux, by oxidative stress leads to an increase in multidrug-tolerant persister cells.
INTRODUCTION
Persisters are dormant antibiotic tolerant cells that are produced by all bacteria tested and by the eukaryote Candida albicans (22). Persisters are phenotypic variants of regular cells that can be produced stochastically in a population. The level of persisters rises from 10−5 in an early exponential culture to 1% at stationary state, suggesting a regulatory component in their formation. The nature of the factors responsible for the rise in persisters during growth of a culture is unknown. Toxin-antitoxin (TA) modules contribute to persister formation in Escherichia coli (3, 4, 9, 28, 29, 35), and we reported that the induction of TisB toxin expression by the SOS response produces dormant persisters (12). Persisters are largely responsible for the recalcitrance of biofilms to antibiotic therapy (7, 16, 17, 19, 22, 25, 26, 37). High persister (hip) mutants of Pseudomonas aeruginosa have been isolated from patients with cystic fibrosis (30), and hip mutants of C. albicans arise in the course of treating cancer patients with oral thrush (23). These findings suggest that persister cells may be the main culprit responsible for the relapsing nature of chronic infections.
Persister formation in response to DNA damage suggests that other stresses may induce entry into a dormant state as well. We were particularly interested in the possible role of oxidative stress in drug tolerance. In the host environment, when macrophages encounter bacteria, they generate reactive oxygen species (ROS) and reactive nitrogen species (RNS), such as superoxide (O2 · −) and nitric oxide (NO) and other reactive species (31). Bacteria respond to oxidative stress by inducing a global response which eliminates ROS and repairs oxidative damage (5, 38). The sensor-regulator systems in E. coli that respond to oxidative stress are the OxyR and SoxRS regulons, which control the responses to hydrogen peroxide and superoxide, respectively (34, 43). Compounds that produce intracellular superoxide include viologens such as paraquat (PQ) and quinones such as naphthoquinone, menadione, and plumbagin (PB), a natural antimicrobial and a derivative of naphthoquinone. These compounds transfer electrons from NADPH or NADH to O2 to generate O2 · −; hence, they are commonly referred to as redox cycling agents. PQ is commonly used due to its high solubility and stability in water. The transcriptional and metabolic cascade induced by PQ is shown in Fig. 1. The soxRS regulon is induced through a two-stage process. The repressor SoxR contains a [2Fe-2S] cluster, which is inactive in the reduced form. When the [2Fe-2S] cluster is oxidized, it induces soxS expression, and SoxS in turn activates the transcription of over 100 genes in the regulon by binding to the sox box in their promoters (5, 10, 14). Superoxide is degraded to H2O2 by the superoxide dismutases SodA (Mn), SodB (Fe), and SodC (Cu, Zn). H2O2 is further degraded to water and oxygen by the enzymes KatG and AhpCF, or it can be converted to hydroxyl radical through the Fenton reaction (FR) (20, 41). OxyR is a regulator of the hydrogen peroxide response. The oxidized OxyR activates the expression of the catalase KatG and the alkyl hydroperoxide reductase AhpCF to scavenge H2O2 and the expression of proteins such as glutathione reductase GorA, glutaredoxin 1 GrxA, and thioredoxin 2 TrxC that enhance reducing capacity. PQ usage, together with the subsequent H2O2 production, also increases the expression of the IscR regulon, governing iron sulfur cluster synthesis (Fur) and cysteine synthesis (CysB) (5).
Fig 1.
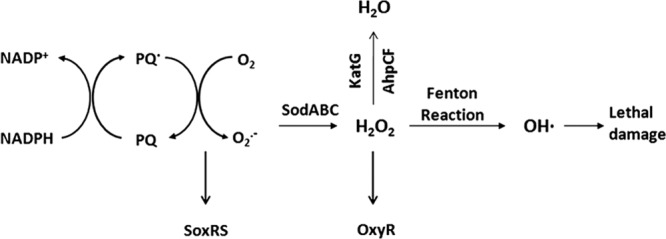
The transcriptional and metabolic cascade induced by paraquat.
In this study, we found that PQ increases the level of persisters that survive killing by fluoroquinolone antibiotics through induction of the multidrug-resistant (MDR) pump AcrAB-TolC. These surviving persisters are highly tolerant to unrelated antibiotics. This finding shows an important relationship between classical resistance (induction of an MDR pump) and drug tolerance.
MATERIALS AND METHODS
Strains and growth conditions.
E. coli K-12 BW25113, which is the parent strain for the complete single gene knockout collection (the KEIO library [2]), is the wild-type strain used in this work. Each open reading frame (ORF) in the collection is replaced by a kanamycin resistance cassette, and care was taken to choose primers for replacement that would avoid creating polar effects. The MDR pump deletion mutant acrB is from the KEIO collection. The lexA3 allele is a point mutant, deficient for SOS induction, and was moved from MV2057 into BW25113 by P1 transduction. MG1655 PsoxS-GFP (where GFP is green fluorescent protein) and MG1655 PtolC-GFP are from an E. coli library containing ∼2,000 different promoter-GFP fusions on a low-copy-number plasmid (42). Cells were cultured in Luria-Bertani (LB) broth (10 g Bacto-tryptone, 5 g yeast extract, and 10 g NaCl/liter), buffered with 0.1 M HEPES/KOH (pH 7.2), and supplemented with the appropriate antibiotic for strains carrying antibiotic markers. Overnight cultures were inoculated in 3 ml buffered LB broth in 17- by 100-mm polypropylene tubes (Fisherbrand) and incubated for 16 to 20 h at 37°C with aeration at 220 rpm.
Measurements of MICs.
The MIC was measured using 96-well plates (COSTAR; Corning Inc.). All wells in each row of the 96-well plate were filled with 50 μl of the medium, except the first one, which was filled with 100 μl of medium alone, serving as a negative control. Fifty microliters of medium containing antibiotic at 20-fold the expected MIC (1) was then added to the second well, and serial 2-fold dilutions were made from the second to the 11th well. The last well contained no antibiotics, serving as a positive control. The overnight culture of the strain to be tested was diluted 1:10 into 3 ml of fresh medium in a 17- by 100-mm polypropylene tube, which was incubated at 37°C with aeration for 1 h. The cultures were then diluted 1:2,000 in fresh medium. Fifty microliters of the cultures were added to the second to the 12th well, resulting in 105 CFU/ml. The 96-well plate was then capped and incubated at 37°C for 16 to 20 h, and the optical density (OD) was determined by a microtiter plate reader (Synergy Mx monochromator-based multimode microplate reader; BioTek). The lowest concentration at which no growth was observed was taken as the MIC. Each measurement was performed in four replicates.
Determining persister level.
Overnight cultures of the strains were diluted 1:100 into buffered LB broth, supplemented with appropriate antibiotics, and incubated at 37°C for 1 h (optical density at 600 nm [OD600] of ∼0.1). The culture was divided into aliquots of 5 ml per polypropylene tube (17 mm by 100 mm). The tubes were further grouped into untreated and PQ treated (paraquat dichloride; Sigma). PQ was added to the culture for 30 min (OD600 of 0.2 to ∼0.3) prior to the addition of ofloxacin, ampicillin, kanamycin, or tobramycin (all from Sigma) for an additional 24 h. The concentration of antibiotics was 10× the MIC, unless indicated otherwise. Each treatment was performed in triplicate. At designated time points, 1-ml samples were withdrawn, washed, and resuspended in 1 ml of a 1% NaCl solution. The suspension was then serially diluted and plated on LB agar plates supplemented with 20 mM MgSO4 and 2 mg/ml sodium pyruvate, an antioxidant improving plating efficiency (6). In some experiments, the entire 1-ml sample was washed and plated to increase the limit of detection.
Measurement of promoter activity.
Strains MG1655 PsoxS-GFP and MG1655 PtolC-GFP were cultured at 37°C with aeration at 220 rpm to an OD600 of ∼0.2, and the cultures were transferred into wells of a black opaque 96-well plate (Costar). The wells were divided into four groups: no PQ, no PQ with ofloxacin, PQ, and PQ with ofloxacin. PQ (0.8 mM) was added into the wells for half an hour before the addition of 1 μg/ml ofloxacin. Immediately after the addition of ofloxacin, the plate was placed into the microtiter plate reader (Synergy Mx monochromator-based multimode microplate reader; BioTek) and incubated at 37°C. The intensity of fluorescence was measured (485 nm, excitation; 585 nm, emission) every 20 min for 3 h.
RESULTS
PQ increases the persister level of a culture challenged with fluoroquinolones.
Paraquat leads to the production of ROS and was used as an oxidative stress inducer. The PQ MIC for E. coli was 1.2 mM, and a subinhibitory concentration of 0.8 mM was used to pretreat the culture in order to examine the possible effects of oxidative stress on persister levels. After incubation with PQ for 30 min, the culture was treated with ofloxacin, ampicillin, or kanamycin. PQ at this subinhibitory concentration did not affect the growth rate, but the level of persisters surviving killing by ofloxacin increased 1,000-fold (Fig. 2A). PQ had no effect on the level of cells surviving treatment by kanamycin or ampicillin (Fig. 2A). PQ pretreatment strongly increased the level of persisters in cultures exposed to other fluoroquinolones, norfloxacin, or ciprofloxacin (Fig. 2B). The different levels of persisters are apparently due to the differences in the killing ability of particular fluoroquinolones and their ability to induce the SOS response. Similarly, plumbagin, another redox cycling agent, strongly increased the level of persisters surviving ofloxacin (see Fig. S1 in the supplemental material).
Fig 2.

The influence of PQ on drug tolerance of E. coli BW25113. (A) Time-dependent killing by ampicillin (50 μg/ml), ofloxacin (1 μg/ml), and kanamycin (50 μg/ml). (B) Time-dependent killing by different fluoroquinolones: ofloxacin (1 μg/ml), norfloxacin (1 μg/ml), and ciprofloxacin (0.15 μg/ml). The growing population of BW25113 was treated with 0.8 mM PQ for 30 min before antibiotics were applied.
Persisters in a PQ-treated culture are multidrug tolerant.
PQ had a dramatic effect on the survival of persisters in a culture treated with a fluoroquinolone but not with ampicillin or kanamycin. However, it was important to determine whether these surviving persisters were tolerant to multiple antibiotics. For this reason, cells were pretreated with PQ, ofloxacin was then added, and, after another 6-h incubation, the culture was treated with high levels of ampicillin (100 μg/ml) and tobramycin (30 μg/ml) or a higher level of ofloxacin (4 μg/ml) for an additional 18 h. This exposure to additional antibiotics had little effect on survival (Fig. 3), indicating that persisters in a culture pretreated with PQ are multidrug tolerant. This experiment also suggests that the fluoroquinolone induced persisters, since a subsequent increase in the concentration of this antibiotic, or addition of a different one, had no effect on killing. Contrast this to very strong killing of cells by kanamycin, for example, when this drug was added without prior treatment with a fluoroquinolone (Fig. 2A).
Fig 3.

Multidrug tolerance of persisters induced by PQ treatment. The growing population of BW25113 was treated with 0.8 mM PQ for 30 min and then challenged with 1 μg/ml ofloxacin (the zero time point in the figure). After 6 h, the PQ treated culture was split into four aliquots: no extra antibiotic added (PQ oflo 1), 4 μg/ml ofloxacin added (PQ oflo 1 + 4), 100 μg/ml of ampicillin added (PQ oflo 1 + Amp 100), and 30 μg/ml of tobramycin added (PQ oflo 1 + Tobra 30).
The AcrAB-TolC pump is largely responsible for the survival of persisters in cultures treated with PQ.
Redox cycling agents such as PQ induce soxRS and one of the components of this regulon, AcrAB-TolC, is the major MDR pump of E. coli. Under conditions used in this study, PQ indeed induced expression of soxS and tolC (Fig. 4). AcrAB-TolC extrudes fluoroquinolones from the cell, which could contribute to persister survival. In order to test this possibility, ofloxacin was added to cultures of wild-type and acrB mutant cells pretreated with PQ at equivalent levels. Thus, the ofloxacin MIC of the wild type is 0.1 μg/ml, and cells were exposed to 1 μg/ml of ofloxacin. For the mutant, the MIC was 0.02 μg/ml, and it was exposed to 0.2 μg/ml of the antibiotic. The protective effect of PQ pretreatment on persister survival was largely eliminated in an acrB mutant strain (Fig. 5). PQ had no effect on the growth rate of the acrB strain. The MICs of colonies formed by surviving persisters were the same as for parent strains, showing that resistant mutants were not selected in the course of the experiment. These results suggest that PQ induces the AcrAB-TolC pump which lowers the concentration of the fluoroquinolone, allowing the antibiotic to induce persister formation of a larger fraction of the population. Importantly, these surviving cells become tolerant to other antibiotics, including higher levels of fluoroquinolone. This increased survival is probably due to SOS induction by the fluoroquinolone, resulting in increase of persister tolerance (11, 12). In order to test this possibility, we examined the effect of PQ pretreatment on the survival of a lexA3 mutant lacking SOS induction. A null mutation in the LexA repressor causes a derepression of all SOS-regulated genes, but the LexA3 allele fails to respond to RecA activation and is therefore unable to induce the SOS response. The fluoroquinolone MIC of the lexA3 mutant was unchanged, but the level of persisters surviving ofloxacin treatment was considerably diminished compared to the level obtained with the wild type (Fig. 6).
Fig 4.

Induction of soxS and tolC by paraquat and ofloxacin. Ofloxacin (1 μg/ml) was added to the growing cultures of MG1655 PsoxS-GFP (A) and MG1655 PtolC-GFP (B) after a 30-min incubation with 0.8 mM PQ. The level of GFP fluorescence indicates the level of promoter activity.
Fig 5.

The influence of PQ on ofloxacin tolerance of wild-type E. coli BW25113 and the MDR efflux pump mutant acrB. The growing cultures were treated with 0.8 mM PQ for 30 min before a challenge by 1 μg/ml ofloxacin to BW25113 and 0.2 μg/ml to acrB, respectively.
Fig 6.

The SOS dependence of the PQ-induced fluoroquinolone tolerance. Ofloxacin (1 μg/ml) was added to the growing populations of wild-type BW25113 and lexA3, a mutant lacking SOS induction, following a 30-min treatment of 0.8 mM PQ.
DISCUSSION
Understanding the molecular basis of persister formation and their drug tolerance is necessary to devise countermeasures against these specialized survivor cells, which are largely responsible for recalcitrance of chronic infections (23, 30). The study of persisters, however, presents a formidable challenge, since these cells are genetically identical to their propagating kin and form a small and temporary subpopulation, and the mechanisms of their formation are highly redundant (24). In E. coli, the transcriptome of persisters pointed to overexpression of chromosomal toxin-antitoxin modules which were originally discovered as a plasmid maintenance mechanism (17, 36). Overexpression of toxins such as RelE or MazF, which inhibit protein synthesis by degrading mRNA, stopped cell division and led to drug tolerance (8, 33, 40). Similarly, overexpression of the toxin HipA, which inhibits protein synthesis by the phosphorylating elongation factor EF-Tu, leads to multidrug tolerance (21). In E. coli, there are over 10 TA modules that degrade mRNA, and the total number of toxins-antitoxins in this species is over 20 (12, 32). This high level of redundancy is problematic when trying to link a particular persister candidate gene to function. In E. coli, only after knocking out 6 TA modules is there an observable decrease in persister levels (27). In M. tuberculosis, there are over 65 TA modules (18), making analysis particularly daunting. One possible reason for this redundancy is that each persister gene is largely responsible for persister formation under a given condition. In E. coli, the TisAB TA module is controlled by the SOS response, and we found that treating cells with DNA-damaging agents such as fluoroquinolones induces expression of TisB (12). Under these conditions, most of the persisters are formed in a TisB-dependent manner. The TisB protein is a typical antimicrobial peptide (13, 39), which acts by forming anion channels in the membrane (15). A decrease in proton motive force (PMF) and ATP then shuts down the antibiotic targets, leading to dormancy and drug tolerance.
We reasoned that other stresses may be linked to persister formation, similar to the SOS response (11). In this study, we examined the effects of oxidative stress on persister formation. Incubating cells with an oxidative stress inducer, PQ, sharply increased the level of persisters in a population treated with fluoroquinolones. Unexpectedly, this effect was limited to this class of antibiotics: pretreatment with PQ had no effect on the action of other bactericidal compounds, ampicillin, and kanamycin. Further investigation showed that the increased persister levels were due to the PQ-induced expression of the MDR pump AcrAB-TolC, which extrudes fluoroquinolones from the cell. We also found that persisters surviving a challenge with fluoroquinolone under these conditions are tolerant to other antibiotics. This suggests that the fluoroquinolone was actually inducing their formation. Indeed, tolerance of persisters was reduced in a lexA3 mutant lacking induction of the SOS response.
The findings of the current study suggest an interesting and complex interplay between classical resistance (MDR efflux) and tolerance. During infection, a pathogen will encounter activated macrophages that produce ROS and RNS compounds, triggering oxidative stress. E. coli infection is typically treated with fluoroquinolones, and the AcrAB-TolC MDR will lower the concentration of these compounds in the cell, increasing the level of surviving persisters and their tolerance to this and other antibiotics, exacerbating recalcitrance. It appears that one of the main immune defenses, the production of ROS and RNS, has a side effect of activating both classical resistance and multidrug tolerance in pathogen cells.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by grant T-RO1AI085585 from the NIAID.
Footnotes
Published ahead of print 9 July 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Andrews JM. 2001. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 48(Suppl 1):5–16 [DOI] [PubMed] [Google Scholar]
- 2. Baba T, et al. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Black DS, Irwin B, Moyed HS. 1994. Autoregulation of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J. Bacteriol. 176:4081–4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Black DS, Kelly AJ, Mardis MJ, Moyed HS. 1991. Structure and organization of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J. Bacteriol. 173:5732–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blanchard JL, Wholey WY, Conlon EM, Pomposiello PJ. 2007. Rapid changes in gene expression dynamics in response to superoxide reveal SoxRS-dependent and independent transcriptional networks. PLoS One 2:e1186 doi:10.1371/journal.pone.0001186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boaretti M, Lleo MM, Bonato B, Signoretto C, Canepari P. 2003. Involvement of rpoS in the survival of Escherichia coli in the viable but non-culturable state. Environ. Microbiol. 5:986–996 [DOI] [PubMed] [Google Scholar]
- 7. Brooun A, Liu S, Lewis K. 2000. A dose-response study of antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 44:640–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Christensen SK, Pedersen K, Hansen FG, Gerdes K. 2003. Toxin-antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J. Mol. Biol. 332:809–819 [DOI] [PubMed] [Google Scholar]
- 9. Correia FF, et al. 2006. Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. J. Bacteriol. 188:8360–8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding H, Hidalgo E, Demple B. 1996. The redox state of the [2Fe-2S] clusters in SoxR protein regulates its activity as a transcription factor. J. Biol. Chem. 271:33173–33175 [DOI] [PubMed] [Google Scholar]
- 11. Dörr T, Lewis K, Vulic M. 2009. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5:e1000760 doi:10.1371/journal.pgen.1000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dörr T, Vulic M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8:e1000317 doi:10.1371/journal.pbio.1000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evans ME, Feola DJ, Rapp RP. 1999. Polymyxin B sulfate and colistin: old antibiotics for emerging multiresistant gram-negative bacteria. Ann. Pharmacother. 33:960–967 [DOI] [PubMed] [Google Scholar]
- 14. Gaudu P, Weiss B. 1996. SoxR, a [2Fe-2S] transcription factor, is active only in its oxidized form. Proc. Natl. Acad. Sci. U. S. A. 93:10094–10098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gurnev PA, Ortenberg R, Dörr T, Lewis K, Bezrukov SM. 21 June 2012. Persister-promoting bacterial toxin TisB produces anion-selective pores in planar lipid bilayers. FEBS Lett. [Epub ahead of print.] doi:10.1016/j.febslet.2012.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaldalu N, Mei R, Lewis K. 2004. Killing by ampicillin and ofloxacin induces overlapping changes in Escherichia coli transcription profile. Antimicrob. Agents Chemother. 48:890–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230:13–18 [DOI] [PubMed] [Google Scholar]
- 18. Keren I, Minami S, Rubin E, Lewis K. 2011. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2:e00100–11 doi:10.1128/mBio.00100–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186:8172–8180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810 [DOI] [PubMed] [Google Scholar]
- 21. Korch SB, Henderson TA, Hill TM. 2003. Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol. 50:1199–1213 [DOI] [PubMed] [Google Scholar]
- 22. LaFleur MD, Kumamoto CA, Lewis K. 2006. Candida albicans biofilms produce antifungal-tolerant persister cells. Antimicrob. Agents Chemother. 50:3839–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. LaFleur MD, Qi Q, Lewis K. 2010. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob. Agents Chemother. 54:39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lewis K. 2010. Persister cells. Annu. Rev. Microbiol. 64:357–372 [DOI] [PubMed] [Google Scholar]
- 25. Lewis K. 2005. Persister cells and the riddle of biofilm survival. Biochemistry (Mosc.) 70:267–274 [DOI] [PubMed] [Google Scholar]
- 26. Lewis K. 2000. Programmed death in bacteria. Microbiol. Mol. Biol. Rev. 64:503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maisonneuve E, Shakespeare LJ, Jorgensen MG, Gerdes K. 2011. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. U. S. A. 108:13206–13211 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28. Moyed HS, Bertrand KP. 1983. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155:768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moyed HS, Broderick SH. 1986. Molecular cloning and expression of hipA, a gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 166:399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mulcahy LR, Burns JL, Lory S, Lewis K. 2010. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 192:6191–6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nunoshiba T, DeRojas-Walker T, Tannenbaum SR, Demple B. 1995. Roles of nitric oxide in inducible resistance of Escherichia coli to activated murine macrophages. Infect. Immun. 63:794–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pandey DP, Gerdes K. 2005. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33:966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pedersen K, Christensen SK, Gerdes K. 2002. Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol. Microbiol. 45:501–510 [DOI] [PubMed] [Google Scholar]
- 34. Pomposiello PJ, Demple B. 2002. Global adjustment of microbial physiology during free radical stress. Adv. Microb. Physiol. 46:319–341 [DOI] [PubMed] [Google Scholar]
- 35. Schumacher MA, et al. 2009. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323:396–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shah D, Zhang Z, Khodursky AB, Kaldalu N, Kurg K, Lewis K. 2006. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spoering AL, Lewis K. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183:6746–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Touati D. 2000. Sensing and protecting against superoxide stress in Escherichia coli–how many ways are there to trigger soxRS response? Redox Rep. 5:287–293 [DOI] [PubMed] [Google Scholar]
- 39. Unoson C, Wagner EG. 2008. A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Mol. Microbiol. 70:258–270 [DOI] [PubMed] [Google Scholar]
- 40. Vázquez-Laslop N, Lee H, Neyfakh AA. 2006. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J. Bacteriol. 188:3494–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang X, Zhao X. 2009. Contribution of oxidative damage to antimicrobial lethality. Antimicrob. Agents Chemother. 53:1395–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zaslaver A, et al. 2006. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods 3:623–628 [DOI] [PubMed] [Google Scholar]
- 43. Zheng M, Storz G. 2000. Redox sensing by prokaryotic transcription factors. Biochem. Pharmacol. 59:1–6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.