

Abstract
Astrogliosis is induced by neuronal damage and is also a pathological feature of the major aging-related neurodegenerative disorders. The mechanisms that control the cascade of astrogliosis have not been well established. In a previous study, we identified a novel androgen receptor (AR)-interacting protein, p44/WDR77, that plays a critical role in the proliferation and differentiation of prostate epithelial cells. In the present study, we found that deletion of the p44/WDR77 gene caused premature death with dramatic astrogliosis in mouse brain. We further found that p44/WDR77 is expressed in astrocytes and that loss of p44/WDR77 expression in astrocytes leads to growth arrest and astrogliosis. The astrocyte activation induced by deletion of the p44/WDR77 gene was associated with upregulation of p21Cip1 expression and NF-κB activation. Silencing p21Cip1 or NF-κB p65 expression with short hairpin RNA (shRNA) abolished astrocyte activation and rescued the astrocyte growth inhibition induced by deletion of the p44/WDR77 gene. Our results reveal a novel role for p44/WDR77 in the control of astrocyte activation through p21Cip1 and NF-κB signaling.
INTRODUCTION
The androgen receptor (AR) performs its regulatory function by acting as a ligand-activated transcription factor for androgen target genes (8, 9, 11, 25). The binding of an androgen to AR results in the release of heat shock proteins, dimerization of AR, and the binding of AR to androgen response elements located with AR target genes. During its activation by androgens, AR physically interacts with various cofactors or coregulators that modulate AR transactivation during different physiological processes (18, 21, 24). One such cofactor is p44/WDR77, which was identified by coimmunoprecipitation with AR from prostate cancer cells (22). The human p44/WDR77 protein contains 342 amino acid residues and 7 putative WD-40 repeats. Northern blot analysis showed that p44/WDR77 mRNA is expressed in multiple human tissues (22). p44/WDR77 selectively regulates a set of AR target genes in the prostate and in prostate cancer cells (17, 22, 55). In normal prostate epithelial cells, p44/WDR77 localizes to the nucleus, but in prostatic intraepithelial neoplasia and prostate cancer cells, it localizes to the cytoplasm (55). Nuclear p44/WDR77 mediates the growth inhibition (40, 55) and differentiation (17) of prostate epithelial cells by selectively modulating the expression of a set of AR target genes. The nuclear export of p44/WDR77 may be an essential step that relieves the p44/WDR77-mediated growth inhibition and enables the resumption of proliferation, which is required for prostate tumorigenesis. Studies have also implicated p44/WDR77 in the biogenesis of small nuclear ribonucleoprotein particles (14, 15, 30). However, the mechanisms by which p44/WDR77 regulates this biogenesis are not well studied.
Astrocytes are fundamental for the cellular homeostasis of the central nervous system (CNS), playing critical physiological, signaling, and immunological roles (20, 33). Insults to the CNS initiate a series of metabolic and morphological changes in astrocytes; these changes are known as reactive gliosis or astrogliosis (7). The hallmark of this phenomenon is the upregulation of glial fibrillary acidic protein (GFAP). Reactive astrocytes contribute to scar formation and neuronal death. Astrogliosis is also a pathological feature of the major aging-related neurodegenerative disorders. Under certain circumstances, astrocytes mediate dystrophic effects within the CNS and thereby contribute to a decline in neurological functions (36, 44). Many different types of intercellular signaling molecules can trigger reactive astrogliosis (45). The different molecular, morphological, and functional changes that lead to reactive astrocytes, such as GFAP upregulation, cell hypertrophy, and pro- or anti-inflammatory effects, are specifically controlled by intercellular and intracellular signaling mechanisms. However, the detailed molecular mechanisms that lead to reactive astrocytes remain incompletely characterized (46).
The brain is an important target for androgens (28). Androgens act on the brain to regulate the sexual drive and reproductive and aggressive behaviors (2, 51). Androgens also prevent neuronal death in neurodegeneration, and decreased androgen levels in plasma are therefore a risk factor for the development of neurodegenerative diseases in humans (43). During brain development, androgens affect the differentiation of GFAP-positive astrocytes (6, 31, 32). In the adult brain, androgens negatively regulate the expression of GFAP in the hippocampus (10) and inhibit astrogliosis in injured brains (3, 5, 39, 48). These studies suggest that androgens may protect neurons in the brain by inhibiting astrogliosis (34). However, the molecular mechanisms of this regulation remain unclear.
Recent studies have suggested that p21Cip1 is involved in astrocyte activation induced by lipopolysaccharide (49, 50). NF-κB acts as an inducer of reactive astrogliosis by driving the expression of inflammatory cytokines in the brain (37, 45, 54). We found that deletion of the p44/WDR77 gene led to astrogliosis with a decrease in life span in mice. The p44/WDR77 protein is expressed in astrocytes, and loss of p44/WDR77 expression leads to growth arrest and to astrocyte activation, which is accompanied by upregulation of p21Cip1 expression and NF-κB activation. The short hairpin RNA (shRNA)-mediated silencing of p21Cip1 or NF-κB p65 expression completely abolished astrocyte activation and rescued the astrocyte growth inhibition induced by the p44/WDR77 gene deletion. These results indicate a novel role for p44/WDR77 in the control of astrocyte activation through p21Cip1 and NF-κB signaling.
MATERIALS AND METHODS
Animals and dissection.
The homogeneous (p44/WDR77loxP/loxP) mice and the cross between p44/WDR77loxP/loxP and ARR2PPbi-Cre mice to generate p44/WDR77loxP/loxP; Cre mice were described previously (17). All mice were on the C57BL genetic background, and mice were handled in accordance with the guidelines published by the National Institutes of Health (33a). The M. D. Anderson Institutional Animal Care and Use Committee approved all the experimental procedures used for mice. Littermate mice of different p44/WDR77 genotypes were housed together in a pathogen-free environment and fed freely with standard mouse chow over their life spans. Mice were identified by PCR analysis of DNA from tail snips, as previously described (17, 55). Mice exhibiting extreme morbidity were killed early.
The mice were anesthetized until they no longer displayed a withdrawal reflex in the hind limbs. The animals were intracardially perfused with chilled phosphate-buffered saline (PBS) for approximately 2 min and then with Formalde-Fresh solution (Fisher Scientific) for 15 min. The animals were decapitated, the soft tissue was removed, and the skulls were postfixed in Formalde-Fresh for 16 h at 4°C. The brains were removed from the postfixed skulls and sliced into 1-mm-thick pieces. The slices were put into cassettes, which were washed with different concentrations of ethanol and then embedded in paraffin. Sections (4 μm thick) were cut and mounted on Superfrost Plus adhesion slides (Fisher) for hematoxylin and eosin (H&E) and immunohistochemical staining.
Isolation and culturing of mouse astrocytes.
A tissue culture system of primary astrocytes was derived from the cerebral cortices of newborn mice. The newborn mouse pups (n = 10) were asphyxiated in a CO2 chamber. The brain was dissected from the mouse skull and transferred to a 60-mm petri dish. The brainstem was cut off, and the hippocampus, basal nuclei, meninges, and blood vessels were removed from the two hemispheres. The hemispheres were then minced with small iris scissors and digested with 2 ml of collagenase (87.5 U/ml; Worthington Biochemical Corp.) and 40 μg/ml DNase (Sigma-Aldrich) in Dulbecco's minimum essential medium (DMEM) for 30 min. After centrifugation, the cell pellet was dispersed and resuspended in low-glucose DMEM supplemented with 10% bovine calf serum (BCS), 500 mg/liter NaHCO3, and 1% penicillin-streptomycin. Cells (1.25 × 105 cells/cm2) were placed into a flask that had been coated with 5 μg/ml mouse laminin (Sigma-Aldrich). After the astrocytes had grown to confluence (10 to 14 days), they were shaken overnight at 37°C (to remove the oligodendrocytes and neurons) and then split 1:4. Astrocytes contain characteristic intermediate filaments (GFAP), which could be readily identified in the cultured cells. The GFAP-positive cells made up >95% of the cultured-cell population.
Immortalization of mouse astrocytes.
The Phoenix-A packaging cell line was maintained in DMEM supplemented with 10% fetal bovine serum and transfected with pLXSP:HPV16-E6/E7 virus using Lipofectamine 2000 (Invitrogen). The culture medium was replaced with fresh DMEM supplemented with 10% BCS 48 h after transfection. The virus was collected 12 h later and used to infect the primary astrocytes in the presence of 8 μg/ml Polybrene (Sigma-Aldrich). Two days after infection, the astrocytes were split 1:4, and cells were then selected with G418 (0.5 mg/ml) for 2 weeks. The G418-resistant cell colonies were selected and expanded. A total of 8 stable astrocyte lines were obtained.
Cre-mediated deletion of the p44/WDR77 gene in mouse astrocytes.
Immortalized astrocytes (2.5 × 105) were plated in a 100-mm plate in low-glucose DMEM with 10% BCS and supplements; 18 h later, the cells were infected with 1.25 × 109 particles of adenovirus Ad-GFP or Ad-Cre-GFP (Vector Development Laboratory, Baylor College of Medicine). Fresh medium was added 3 days after infection. Green fluorescent protein (GFP) expression in the infected astrocytes was examined under a fluorescence microscope, and 100% infection efficiency was achieved. The two adenoviruses used were replication deficient, and expression of GFP and Cre was under the control of the cytomegalovirus (CMV) promoter.
Cell growth assay.
Cells (1 × 104/well) were plated in 6-well plates in low-glucose DMEM with 10% BCS. Cells were counted every day with a hemacytometer.
Immunohistochemistry.
The antigen-purified anti-p44/WDR77 (1:100) antibody was described previously (22, 53). Anti-GFAP (1:1,000) was purchased from Chemicon, and anti-S100B (1:200) was obtained from Santa Cruz Biotechnology. Tissue sections were blocked with 1% fish gel, and antibodies were applied to the tissue sections and incubated overnight. A streptavidin-biotin peroxidase detection system (Dako) was used according to the manufacturer's instructions. For double-immunofluorescence staining, the Alexa Fluor 488-labeled anti-rabbit immunoglobulin G (IgG) antibody (1:1,000; Dako) and Alexa Fluor 594-labeled anti-chicken IgG were used. The fluorescence signals were observed under a confocal microscope with a red filter (to detect p44/WDR77 protein) or a blue filter (to detect GFAP protein). The primary antibodies were omitted in negative controls.
Northern blot assay.
Northern blot analysis was performed as previously described (16). mRNA was isolated from whole brains of 2-month-old wild-type (WT)(n = 5) or mutant (MT)(n = 5) male mice and transferred to a Hybond-N+ membrane (Amersham Biosciences). The membrane was hybridized with cDNA probes of the p44/WDR77 and β-actin genes.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation was performed as described previously (16). Briefly, astrocytes (p44/WDR77loxP/loxP) were infected with adenovirus (Ad)-GFP or Ad-Cre-GFP and grown for 6 days. The cells were cross-linked with 10% formalin for 10 min at room temperature. The DNA-protein complexes were purified with 2 μg of antigen-purified anti-p44/WDR77 antibody or rabbit IgG (as a negative control). The purified DNA was amplified by a PCR with two specific primers derived from the promoter region of p21 (bp −93 to −215). The same set of PCRs was performed with the chromatin DNA used for chromatin immunoprecipitation.
RNA interference.
p21Cip1 shRNA (target sequence, 5′-TTCTCCGAACGTGTCACGT-3′), p65 shRNA (target sequence, 5′-TTAGGACTCAACCGTAATA-3′), and a nontargeting (NT) shRNA (target sequence, 5′-TTCTCCGAACGTGTCACGT-3′) were designed with a hairpin and sticky ends (ClaI and MluI). The oligonucleotides were annealed into the lentiviral gene transfer vector, pLVTHM, using the ClaI and MluI restriction enzyme sites. The DNA constructs were sequenced to test for proper insertion and length of the inserts. The lentivirus was then produced by transfecting human embryonic kidney cells (293FT; Invitrogen) with the sequence-verified pLVTHM vector, the packaging plasmid MD2G, and the envelope plasmid PAX2, which are required for viral production. The viral supernatant was collected 3 days later and filtered to remove cellular debris. Astrocytes (1 × 105/well) were plated in 6-well plates and transduced with the virus. After 16 h, the virus-containing medium was removed and replaced with normal growth medium. Three days after infection, cells were split 1:6, and the cells were then grown for 3 days. Whole-cell lysates prepared from the infected cells were subjected to Western blot analysis.
Exogenous expression of p21Cip1 and p65.
Human cDNA encoding p21Cip1 or p65 protein was subcloned into a lentiviral expression vector (dsRed-OG2). The recombinant lentivirus was produced with 293FT cells as described above. Astrocytes (1 × 105/well) were plated in 6-well plates and transduced with lentivirus containing p21Cip1 or p65 expression vector or empty vector. After 48 h, the cells were replated, and the expression of p21Cip1 or p65 was confirmed by Western blotting.
Quantification and statistical analysis.
Data were analyzed by using the unpaired Student's t test. Differences between experimental groups were considered significant at a P value of <0.05. All values represent means and standard errors of the mean (SEM).
RESULTS
Loss of the p44/wdr77 gene decreases life span in mice.
We crossed p44/WDR77loxP/loxP mice with the ARR2PPbi-Cre transgenic line (17). A previous study indicated that Cre recombinase was highly expressed in all lobes of the mouse prostate (26). We noticed an unexpected change in the longevity of p44/WDR77loxP/loxP; Cre (MT) mice (Fig. 1A). The median life span of MT mice (120 days) was dramatically reduced compared with that of p44/WDR77loxP/loxP mice. The observed reductions in life span were similar in male and female mice.
Fig 1.

Deletion of the p44/WDR77 gene leads to decreased life span in mice. (A) Survival data are presented for p44/WDR77loxP/loxP; Cre (MT) and p44/WDR77loxP/loxP (WT) mice. The median life span was 120 days for MT mice. (B) Comparison of mean body weights of WT and MT mice. The error bars indicate SEM.
Careful examinations of the MT mice by a veterinary pathologist at The University of Texas M. D. Anderson Cancer Center failed to reveal any obvious pathological defects. The MT mice appeared morphologically identical to their p44/WDR77loxP/loxP (WT) littermates when they were born. However, the mean body weight of MT mice was significantly less than that of their WT littermates by the age of 60 days and did not increase after that (Fig. 1B). By 90 days, virtually all of the MT mice exhibited signs of a lack of vigor.
Cre-mediated deletion of the p44/wdr77 gene in the mouse brain.
To investigate whether tissues other than the prostate also express Cre recombinase in the ARR2PPbi-Cre transgenic mouse, we examined the status of p44/WDR77 gene deletion by sensitive PCR analysis. PCR analysis showed that the p44/WDR77 gene was deleted, as indicated by excision of exons 2 to 5 of the p44/WDR77 gene, in the MT prostate but not in the WT prostate (Fig. 2A, lanes 2 and 3). This observation is consistent with expression of Cre recombinase in the prostate, as previously reported (26). Cre-mediated deletion of the p44/WDR77 gene was also detected in the penis, eye, and brain of MT mice (lanes 4, 8, and 10). The p44/WDR77 alleles were not deleted in the heart, smooth muscle (lane 6), skeletal muscle, liver, spleen, lung, kidney, skin, testis (lane 12), small intestine, large intestine, pancreas, tongue, stomach, bone marrow, or lymph node in MT mice.
Fig 2.
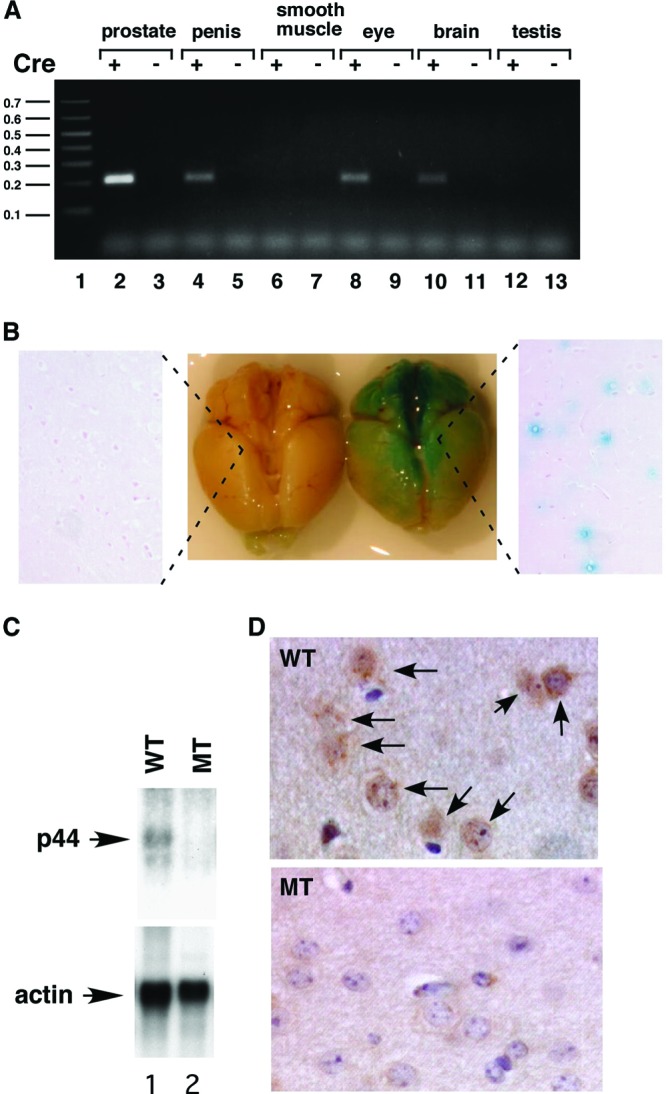
The p44/WDR77 gene was deleted in the mouse brain. (A) PCR analysis of deletion of the p44/WDR77 gene in various organs in the MT mouse. Genomic DNA was isolated from organs of WT and MT mice and subjected to PCR analysis. Excision of exons 2 to 5 of the p44/WDR77 gene generated the 220-bp DNA fragment in the PCR. Lane 1, 0.1-kb DNA ladder. (B) Expression of Cre recombinase in the brains of ARR2PPbi-Cre transgenic mice. The ARR2PPbi-Cre transgenic mouse was crossed with the B6; 129-Gt(ROSA)26Sor/J mouse. Brains from 2-month-old B6; 129-Gt(ROSA)26Sor/J (left) or B6; 129-Gt(ROSA)26Sor/J-ARR2PPbi-Cre (right) mice were stained with β-Gal (middle) and then formalin fixed and paraffin embedded. The slides were counterstained with eosin (left and right). (C) Loss of p44/WDR77 gene expression in the MT mouse brain. Shown is Northern blot analysis of p44/WDR77 and β-actin mRNAs in the mouse brain. mRNA was isolated from the brains of 2-month-old WT (n = 5; lane 1) or MT (n = 5; lane 2) mice, fractionated by electrophoresis, and transferred to a Hybond N+ membrane. The membrane was hybridized with probes as indicated. (D) Loss of the p44/WDR77 protein in the brain of the MT mouse. Tissues from the cerebral cortices of WT (top) and MT (bottom) mouse brains were immunostained with anti-p44/WDR77 antibody. Representative photomicrographs are shown. The p44/WDR77 staining seen in WT mouse brain cells (indicated by the arrows) was dramatically decreased in MT mouse brain cells.
To investigate whether Cre recombinase is expressed in the brain in the ARR2PPbi-Cre transgenic mouse, we crossed this mouse line with the B6; 129-Gt(ROSA)26Sor/J mouse (Jackson Laboratory; 003309). Mice heterozygous or homozygous for the B6; 129-Gt(ROSA)26Sor/J targeted mutation were used to test the tissue or cellular expression pattern of the Cre transgene in all transgenic strains in which Cre expression was under the control of a promoter. Cre expression results in the removal of a loxP-flanked DNA segment that prevents expression of a lacZ gene. When crossed with a Cre transgenic strain, LacZ is expressed in cells or tissues where Cre is expressed. Tissue sections from F1 heterozygous mice were subjected to β-galactosidase (β-Gal) staining to indicate Cre recombinase expression. As expected, the prostate stained strongly for β-Gal. The brain also showed clear β-Gal staining (Fig. 2B, middle). Sections of the stained brain revealed that some cells stained positively for β-Gal (Fig. 2B, right), indicating that Cre is expressed in the brain in the ARR2PPbi-Cre transgenic line. Consistent with the PCR analysis, sections of the eye and penis were also found to stain positively for β-Gal.
To further confirm p44/WDR77 gene deletion in the brain, we prepared mRNA from brains of 2-month-old WT and MT mice. Northern blot analysis showed the Cre-mediated loss of p44/WDR77 mRNA expression in the MT mouse brain (Fig. 2C, top). Immunostaining analysis demonstrated that p44/WDR77 protein levels were high in the nuclei of brain cells from WT mice (Fig. 2D, top, arrows), whereas these signals were dramatically reduced in brain cells from MT mice (Fig. 2D, bottom). These findings indicate that the p44/WDR77 gene is deleted in the brains of MT mice.
Astrocytes express p44/WDR77 protein.
To determine which cells in the brain express p44/WDR77 protein, we used mouse brain tissues double-immunostained for p44/WDR77 and GFAP (astrocyte markers) or for NeuN and MAP2 (neuronal markers). We found that p44/WDR77 staining colocalized with GFAP staining in some cells (indicated by red arrowheads in Fig. 3a), indicating that p44/WDR77 is expressed in astrocytes. However, in other cells (indicated by the arrowheads in Fig. 4A), p44/WDR77 staining did not colocalize with GFAP staining; immunostaining analysis with anti-NeuN and anti-MAP2 antibodies indicated that those cells were neurons (data not shown). Similar analysis of human brain tissue microarrays (Chemicon International; TMA3001-4) indicated that p44/WDR77 is also expressed in astrocytes of human brain tissues (data not shown).
Fig 3.

p44/WDR77 expression in astrocytes of the mouse brain. (a and b) Tissues from cerebral cortices of WT mouse brains were double immunostained for p44/WDR77 (a) and GFAP (b). (c) p44/WDR77 staining merged with GFAP staining.
Fig 4.

Loss of the p44/WDR77 gene leads to reactive astrocytes in the mouse brain. (A) Reactive astrocytes were observed in the MT mouse brain. Sections at the 2.00-mm lateral plane of cerebral cortices from WT (left) or MT (right) brains were stained with anti-p44/WDR77 (top) or anti-GFAP (middle) antibodies. (Bottom) p44/WDR77 staining merged with GFAP staining. Some reactive astrocytes are indicated by arrowheads. (B) Counts of reactive astrocytes in WT and MT brains. The error bars indicate SEM. (C) Loss of p44/WDR77 expression in astrocytes isolated from MT mouse brains. Whole-cell lysates (20 μg of protein) were prepared from WT (lanes 1, 3, and 5) and MT (lanes 2, 4, and 6) astrocytes. Western blot analysis was performed with the indicated antibodies.
Loss of p44/WDR77 expression leads to astrogliosis.
To determine whether loss of p44/WDR77 expression increases the number of reactive astrocytes, we used sections at the 2.00-mm lateral plane of brain tissues from WT and MT mice double immunostained for p44/WDR77 and GFAP (Fig. 4A). p44/WDR77-positive signals were observed in many astrocytes in brains from WT mice but in only a few astrocytes in brains of MT mice (Fig. 4Ab versus a). In brains from WT mice, only a few reactive astrocytes (those with strong GFAP staining and spiny shapes) were observed (Fig. 4Ac, indicated by the arrowheads). In contrast, in brains from MT mice, more reactive astrocytes were observed throughout the cerebral cortex (Fig. 4Ad, some indicated by arrowheads). The cerebral cortex at the 2.00-mm lateral plane contained significantly more reactive astrocytes in MT mice (1,560 ± 166) than WT mice (280 ± 31) (Fig. 4B). None of the reactive astrocytes were observed to express p44/WDR77 (Fig. 4Ae and f), suggesting that loss of p44/WDR77 expression in the brain leads to the activation of astrocytes. Astrocytes were isolated from brains of WT and MT mice, and whole-cell lysates were prepared. Western blot assays demonstrated p44/WDR77 expression in isolated WT astrocytes (Fig. 4C, lane 1); p44/WDR77 expression was markedly less in isolated MT astrocytes (Fig. 4B, lane 2). Consistent with astrocytes in the mouse brain, the MT astrocytes expressed higher levels of GFAP than the WT astrocytes (Fig. 4B, lane 4 versus lane 3).
p44/WDR77 is essential for growth of astrocytes.
To further study the role of p44/WDR77, we isolated astrocytes from brains of WT (p44/WDR77loxP/loxP) mice and immortalized them with retrovirus expressing E6/E7 (47). The E6 protein interacts with p53, resulting in its degradation via the ubiquitin pathway, and E7 associates with Rb, leading to disruption of the interaction between the Rb and E2F transcription factors, efficiently immortalizing cells (52). We obtained 8 cell lines from 10 newborn mice. Two immortalized astrocyte lines (E3.1 and E3.2) were infected with adenovirus expressing either GFP (Ad-GFP) or Cre recombinase (Ad-Cre) to generate 4 cell lines (WT, E3.1 GFP and E3.2 GFP; MT, E3.1 Cre and E3.2 Cre). PCR analysis confirmed the deletion of the p44/WDR77 gene in MT astrocytes (data not shown), and Western blot analysis showed loss of p44/WDR77 expression in MT astrocytes (Fig. 5A, top, lane 2). Nine days after infection, the MT and WT astrocytes were immunostained for p44/WDR77, and the p44/WDR77 signals in MT astrocytes decreased to the level of the negative control (Fig. 5B, top). In contrast, p44/WDR77 expression was evident in WT astrocytes (Fig. 5B, bottom).
Fig 5.

p44/WDR77 is essential for growth of astrocytes. (A and B) Loss of p44/WDR77 expression in p44/WDR77-null astrocytes. (A) Whole-cell lysates (10 μg of protein) were isolated from WT (lane 1) and MT (lane 2) astrocytes. Western blot analysis was performed with the indicated antibodies. (B) Astrocytes were infected with Ad-GFP (WT) or Ad-Cre-GFP (MT) and stained for the nucleus (a and d) and p44/WDR77 expression (b and e). (c and f) p44/WDR77 staining merged with nuclear staining. The insets (a to c) depict staining of WT astrocytes without the p44/WDR77 antibody. (C) Growth curves of WT and MT astrocytes after infection with adenovirus. The error bars indicate SEM.
We then examined the growth of astrocytes from day 9 to day 13 after adenovirus infection. The numbers of WT (E3.1 GFP and E3.2 GFP) astrocytes continued to increase until day 12, when they reached 100% confluence (Fig. 5C). The doubling times of E3.1 GFP and E3.2 GFP astrocytes were 24 h and 22 h, respectively. In contrast, the numbers of MT (E3.1 Cre and E3.2 Cre) astrocytes did not increase over the course of 4 days (Fig. 5C). Thus, p44/WDR77 is essential for growth of astrocytes.
Deletion of the p44/WDR77 gene leads to reactive astrocytes.
Previous studies demonstrated that immortalized astrocytes express low levels of GFAP and S100B and have the cellular structure of nonreactive astrocytes (41). Consistent with the results of previous studies (41), we found that immortalized WT astrocytes expressed low levels of GFAP and S100B and exhibited the cellular structure of nonreactive astrocytes (Fig. 6e and k). Upon loss of p44/WDR77 expression (12 days after infection), MT astrocytes expressed much higher levels of GFAP and S100B (Fig. 6b and h), suggestive of astrocyte activation. In MT astrocytes, hypertrophy of cell nuclei (Fig. 6a, indicated by the arrowheads) and thin processes protruding from the cell body (Fig. 6c, indicated by the arrowheads) were evident; both are characteristics of astrocyte activation (50).
Fig 6.

Deletion of the p44/WDR77 gene led to reactive astrocytes. Astrocytes were infected with Ad-GFP (WT) or Ad-Cre-GFP (MT) and stained for the nucleus (a, d, g, and j) and GFAP (b and e) or S100B (h and k). (c, f, i, and l) GFAP or S100B staining merged with nuclear staining. The arrowheads in panel a indicate hypertrophy of cell nuclei, and those in panel c indicate thin processes protruding from the cell body.
Upregulation of p21cip1 expression is essential for astrocyte activation induced by deletion of the p44/WDR77 gene.
Recent studies have suggested that p21Cip1 is involved in astrocyte activation induced by lipopolysaccharide (49, 50). To examine p21Cip1 expression in the mouse brain, we isolated mRNA from the brains of 2-month-old WT and MT mice and subjected the mRNA to Northern blot analysis. We found that Cre-mediated deletion the p44/WDR77 gene led to a 5-fold increase in p21Cip1 mRNA levels (Fig. 7A, lane 3 versus lane 4). This upregulation of p21Cip1 gene expression is not likely to occur via the p53 signaling pathway, because expression of other p53 target genes (Gadd45A and RGS16) did not change (Fig. 7A, lanes 5 to 8). To assess p21Cip1 expression in astrocytes, we isolated total RNA from immortalized astrocytes and subjected the RNA to quantitative reverse transcription (qRT)-PCR with primers for p21Cip1. The qRT-PCR analysis revealed a 4.5-fold increase in p21Cip1 mRNA levels in MT astrocytes compared with WT astrocytes, and Western blot analysis showed a 4-fold increase in p21Cip1 protein levels associated with the loss of p44/WDR77 expression (Fig. 7B, top, lane MT versus lane WT). Thus, deletion of the p44/WDR77 gene upregulated p21Cip1 expression in astrocytes. Chromatin immunoprecipitation analysis revealed that the p44/WDR77 protein was recruited onto the p21Cip1 promoter (Fig. 7C, lane 3), suggesting that p44/WDR77 directly target the p21Cip1 promoter in astrocytes.
Fig 7.
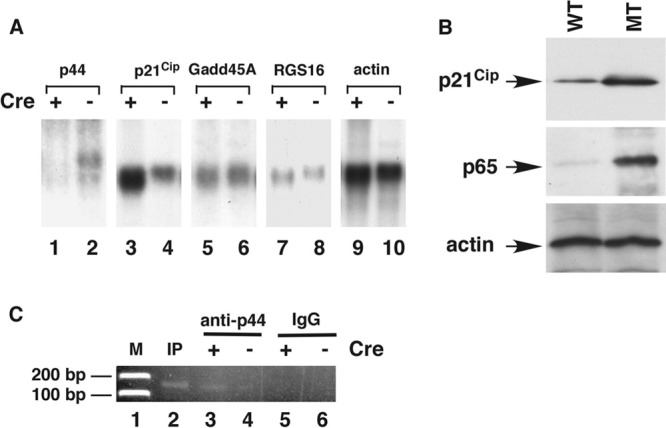
Loss of the p44/WDR77 gene induced p21Cip1 expression and NF-κB activation. (A) Loss of the p44/WDR77 gene induced p21Cip1 expression in mouse brains. Shown is Northern blot analysis of p44/WDR77, p21Cip1, Gadd45A, RGS16, and β-actin mRNAs in mouse brains. mRNA was isolated from the brains of 2-month-old WT (n = 5; lanes 2, 4, 6, 8, and 10) or MT (n = 5; lanes 1, 3, 5, 7, and 9) mice, fractionated by electrophoresis, and transferred to a Hybond N+ membrane. The membrane was hybridized with probes as indicated. (B) Loss of the p44/WDR77 gene induced p21Cip1 and p65 expression in astrocytes. Whole-cell lysates (20 μg of protein) were prepared from WT (lane WT) and MT (lane MT) astrocytes. Western blot analysis was performed with anti-p21Cip1 (top), anti-p65 (middle), or antiactin (bottom) antibody. (C) Chromatin immunoprecipitation assay to identify p21Cip1 as a direct target gene of p44. Immunoprecipitation was performed with antigen-purified anti-p44 (lanes 3 and 4) antibodies or rabbit IgG (lanes 5 and 6). The purified DNA was amplified by PCR with two specific primers derived from the promoter region (bp −93 to −215) of p21Cip1. The same set of PCRs was performed with chromatin DNA used for immunoprecipitation (IP) (lane 2).
To assess the role of p21Cip1 in astrocyte activation induced by the loss of p44/WDR77, we silenced p21Cip1 expression in astrocytes via lentivirus expressing NT or p21Cip1 shRNA and then infected astrocytes with Ad-Cre-GFP to delete the p44/WDR77 gene. The p21Cip1 shRNA abolished the upregulation of both p21Cip1 and GFAP expression in astrocytes induced by p44/WDR77 deletion (Fig. 8A, top and third rows, lanes 1, 5, and 6). Similarly to our earlier findings, WT (p44/WDR77loxP/loxP; NT shRNA) astrocytes expressed low levels of GFAP and exhibited the cellular structure of nonreactive astrocytes (Fig. 8Ba). Upon loss of p44/WDR77 (12 days after Ad-Cre-GFP infection), MT (p44/WDR77−/−; NT shRNA) astrocytes expressed high levels of GFAP and exhibited cellular and nuclear hypertrophy (Fig. 8Bb), indicative of astrocyte activation. However, p21Cip1-silenced MT (p44/WDR77−/−; p21Cip1 shRNA) astrocytes showed GFAP expression levels and cellular structure similar to those of nonreactive astrocytes (Fig. 8Bd), suggesting that upregulation of p21Cip1 expression is essential for astrocyte activation induced by p44/WDR77 deletion.
Fig 8.

p21Cip1 mediated the astrocyte activation induced by loss of p44/WDR77. (A) Silencing p21Cip1 and/or p65 expression in WT and MT astrocytes. Whole-cell lysates (20 μg of protein) were prepared from WT (lanes 1 to 4) and MT (lanes 5 to 8) astrocytes expressing NT (lanes 1 and 5), p21Cip1 (lanes 2 and 6), p65 (lanes 3 and 7), or p21Cip1 plus p65 (lanes 4 and 8) shRNA. Western blot analysis was performed with the indicated antibodies. (B) Silencing p21Cip1 expression abolished the astrocyte activation induced by loss of p44/WDR77. WT (left) and MT (right) astrocytes expressing NT (top) or p21Cip1 (bottom) shRNA were stained for GFAP. (C) Growth curves of WT and MT astrocytes expressing NT or p21Cip1 shRNA. The error bars indicate SEM. (D) Shown is overexpression of p21Cip1 and/or p65 in astrocytes. Whole-cell lysates (20 μg of protein) were prepared from control astrocytes (lane 1) or astrocytes expressing p21Cip1 (lane 2), p65 (lane 3), or p21Cip1 plus p65 (lane 4). Western blot analysis was performed with the indicated antibodies. Endogenous (mouse) and exogenous (human) p21Cip1 are indicated by the arrows on the right. (E) Overexpression of p21Cip1 induced astrocyte activation. Control astrocytes (a) and astrocytes expressing p21Cip1 (b) were stained for GFAP. (F) Growth curves of control astrocytes or astrocytes expressing p21Cip1.
We examined the growth rate of these cells from day 9 to day 13 after adenovirus infection. The numbers of WT (NT shRNA or p21Cip1 shRNA) astrocytes continued to increase until day 12, when they reached 100% confluence (Fig. 8C). The doubling times of NT shRNA and p21Cip1 shRNA astrocytes were 26 h and 22 h, respectively (Fig. 8C). The p21Cip1 shRNA MT astrocytes had a doubling time of 31 h and never reached 100% confluence during the 4 days assayed. In contrast, the number of NT shRNA MT astrocytes increased only slightly over the course of 4 days (Fig. 8C). Thus, silencing p21Cip1 expression partially rescued the astrocyte growth inhibition induced by deletion of the p44/WDR77 gene.
To further explore the role of p21Cip1 in astrocytes, we infected astrocytes with lentivirus expressing exogenous human p21Cip1. Western blot analysis indicated expression of high levels of human p21Cip1 protein in mouse astrocytes (Fig. 8D, top, lane 2 versus lane 1). Consistent with previous studies (49, 50) suggesting that p21Cip1 is involved in astrocyte activation, we found that p21Cip1 overexpression moderately increased GFAP expression and induced the cellular structure of reactive astrocytes (Fig. 8Eb). Human p21Cip1 expression decreased the growth of astrocytes (Fig. 8F) and increased the doubling time of astrocytes from 24 h to 30 h. However, p21Cip1 overexpression did not inhibit cell growth to the extent induced by loss of p44/WDR77 expression (Fig. 8F).
NF-κB mediates p44/WDR77 function in astrocytes.
NF-κB acts as an inducer of reactive astrogliosis by driving the expression of inflammatory cytokines in the brain (37, 45, 54). Expression of p65 protein was induced in MT astrocytes (Fig. 7B, middle; Fig. 8A, second gel from top, lane 5 versus lane 1). Immunostaining of WT (p44/WDR77loxP/loxP) astrocytes revealed low cytoplasmic levels of the p65 subunit of NF-κB (data not shown), indicating the lack of NF-κB activation in astrocytes. Nine days after infection, we observed high levels of p65 in the nuclei of MT astrocytes (data not shown), indicative of NF-κB activation upon p44/WDR77 deletion.
Expression of p65 was silenced by p65 shRNA (Fig. 8A, second gel from top, lanes 3 and 7). To assess the role of NF-κB in astrocyte activation induced by the loss of p44/WDR77, we infected WT (p44/WDR77loxP/loxP) astrocytes with lentivirus expressing NT shRNA or p65 shRNA. Four days later, we infected the astrocytes with either Ad-GFP or Ad-Cre-GFP. Silencing p65 expression inhibited the GFAP expression induced by p44/WDR77 deletion and reversed the induction of the cellular structure of reactive astrocytes induced by p44/WDR77 deletion (Fig. 9A, top, versus Fig. 8B, top). Thus, NF-κB activation is also required for astrocyte activation induced by p44/WDR77 deletion.
Fig 9.

NF-κB mediated the astrocyte activation induced by loss of p44/WDR77. (A) Silencing p65 expression abolished the astrocyte activation induced by loss of p44/WDR77. WT (left) and MT (right) astrocytes expressing p65 (top) or p21Cip1 plus p65 (bottom) shRNA were stained for GFAP. (B) Growth curves of WT and MT astrocytes expressing NT, p65, or p21Cip1 plus p65 shRNA. The error bars indicate SEM. (C) Overexpression of p65 or p21Cip1 plus p65 induced astrocyte activation. Astrocytes expressing p65 (a) or p21Cip1 plus p65 (b) were stained for GFAP. (D) Growth curves of control astrocytes or astrocytes expressing p65 or p21Cip1 plus p65. (E) Model of how p44/WDR77 induces astrocyte activation by inhibiting p21Cip1, NF-κB, and other (unknown [?]) signals.
In the growth rate analysis, the numbers of WT (NT shRNA or p65 shRNA) and MT (p65 shRNA) astrocytes continued to increase until day 12, when they reached 100% confluence (Fig. 9B). The doubling times of NT shRNA WT, p65 shRNA WT, and p65 shRNA MT astrocytes were 26 h, 23 h, and 23 h, respectively. In contrast, the number of MT (NT shRNA) astrocytes increased only slightly over the course of 4 days (Fig. 9B). Thus, silencing p65 expression fully rescued the astrocyte growth inhibition induced by p44/WDR77 deletion.
To further explore the role of NF-κB in astrocytes, we infected WT astrocytes with control lentivirus or lentivirus expressing human p65 (Fig. 8D, middle, lane 3). Nine days after viral infection, astrocytes were immunostained for GFAP. We found that p65 overexpression marginally increased GFAP expression and led to the cellular structure of reactive astrocytes (Fig. 9Ca versus Fig. 8Ba). The doubling time of the WT control astrocytes was 24 h. However, the number of p65-overexpressing astrocytes increased only slightly over the course of 4 days (Fig. 9D).
Silencing p65 expression in astrocytes also inhibited the p21Cip1 expression induced by p44/WDR77 deletion (Fig. 8A, top, lane 7 versus lane 5), indicating that NF-κB signaling may mediate the upregulation of p21Cip1 expression in p44/WDR77-null astrocytes. However, silencing p21Cip1 expression did not inhibit the increase in p65 expression induced by p44/WDR77 deletion (Fig. 8A, second gel from top, lane 6 versus lane 5), suggesting that p44/WDR77 regulates p65 expression independent of p21Cip1. Silencing both p21Cip1 and p65 completely abolished astrocyte activation (Fig. 8A, third gel, lane 8, and 9Ad) and the growth inhibition induced by p44/WDR77 deletion (Fig. 9B). However, overexpression of both p21Cip1 and p65 still failed to activate astrocytes to the levels that resulted from p44/WDR77 deletion (Fig. 9Cb versus 6b and 8Bb), indicating that additional signals mediate the astrocyte activation induced by p44/WDR77 deletion.
DISCUSSION
In this study, we demonstrated that loss of the p44/WDR77 gene leads to a decrease in mouse longevity and results in growth arrest and astrocyte activation. NF-κB and p21Cip1 mediate the growth arrest and astrocyte activation induced by deletion of the p44/WDR77 gene. This study reveals a novel role for p44/WDR77 in the control of astrocyte activation.
Role of p44/WDR77 in astrocyte activation.
The statistically significant (P < 0.0001) increase in astrogliosis in brains of MT mice led us to investigate the role of p44/WDR77 in astrocytes. p44/WDR77 expression in astrocytes, as revealed by immunostaining, was markedly decreased in the brains of MT mice. p44/WDR77 expression was confirmed in cultured astrocytes by immunostaining and Western blotting. Astrogliosis was observed only in astrocytes that had lost p44/WDR77 expression. Furthermore, we demonstrated that p44/WRD77 is required for the growth of astrocytes and that loss of p44/WDR77 expression led to dramatic changes in the cellular structure and increased the expression of GFAP and S100B; these are characteristics of astrogliosis. Thus, p44/WDR77 prevents astrogliosis in the brain.
We previously showed that p44/WDR77 plays important roles in the mouse prostate by regulating expression of AR target genes (18, 19). Decreased levels of glucocorticoids and androgens have been associated with an increased risk for neurodegenerative diseases (35, 42). Androgens and glucocorticoids downregulate reactive gliosis after a neural injury (3, 13). p44/WDR77 can act as a cofactor to modulate AR-dependent or glucocorticoid receptor-dependent gene expression (22). On the basis of these observations, we anticipate that p44/WDR77 may mediate the suppressive effect of androgens and/or glucocorticoids on astrocyte activation. p44/WDR77 may also interact and function with other unknown factors to regulate astrocyte activation.
p21cip1 expression is essential for astrocyte activation induced by loss of the p44/WDR77 gene.
Recent studies have suggested that p21Cip1 plays a role in astrocyte activation (49, 50). The statistically significant (P < 0.0001) increase in astrogliosis in brains and astrocytes from p44/WDR77-null mice, accompanied by increased p21Cip1 expression, led us to investigate the role of p21Cip1 in astrocyte activation. p21Cip1 shRNA was effective in silencing p21Cip1 expression and blocking the astrocyte activation induced by p44/WDR77 deletion. Furthermore, we used overexpression of human p21Cip1 to confirm the ability of p21Cip1 to induce astrocyte activation (49, 50). These results suggest that p44/WDR77 influences astrocyte activation by regulating p21Cip1 expression. However, p21Cip1 overexpression did not induce GFAP expression in astrocytes to the extent induced by p44/WDR77 deletion. A possible explanation for this finding is that p44/WDR77 controls astrocyte activation, not only through p21Cip1 upregulation, but also via other, unknown factors. Although there is growing evidence that cell cycle control systems perform regulatory functions in reactive gliosis, the mechanistic actions of p21Cip1 in reactive gliosis remain incompletely characterized (12, 27, 56).
Cell growth and progression through the cell cycle are orchestrated by the interaction of cyclin–cyclin-dependent kinase (CDK) complexes. p21Cip1 is a powerful CDK inhibitor. Through protein-protein interactions with cyclin-CDK2 or cyclin-CDK4 complexes, p21Cip1 directly inhibits their activity, and in doing so acts as a regulator of cell cycle progression at G1 (4, 19). We found that p21Cip1 expression was upregulated in activated astrocytes induced by deletion of the p44/WDR77 gene. We demonstrated that p21Cip1 upregulation is partially responsible for the dramatic inhibition of the growth of MT astrocytes induced by p44/WDR77 deletion. Furthermore, we confirmed the ability of p21Cip1 to directly inhibit astrocyte growth by overexpressing human p21Cip1 in mouse astrocytes, which is functionally conserved in mice (4, 19). These findings suggest that p44/WDR77 regulates astrocyte proliferation by decreasing p21Cip1 expression.
Role of NF-κΒ in astrocyte activation induced by loss of the p44/WDR77 gene.
Activation of NF-κB signaling has been implicated in the regulation of astrocyte activation (37, 45, 54). A previous study with transgenic mice expressing a dominant-negative form of the inhibitor of κBα driven by the GFAP promoter demonstrated that the loss of NF-κB activity after spinal cord injury resulted in decreased GFAP expression and in scar formation (23). This alteration in astrocyte activation was accompanied by a reduction in astrocytic production of proinflammatory chemokines and cytokines, such as CXCL10 and CCL2 (23). That study, along with other studies, has suggested that astrocytic NF-κB plays a role in reactive astrogliosis and neuropathology (45). Here, immunostaining revealed NF-κB activation upon induction of astrogliosis by p44/WDR77 gene deletion. p65 shRNA was effective in silencing p65 expression and blocking the astrocyte activation induced by p44/WDR77 gene deletion, indicating an essential role for NF-κB in astrocyte activation induced by loss of the p44/WDR77 gene.
We demonstrated that inhibition of NF-κB pathway activity by silencing p65 expression completely rescued the dramatic inhibition of astrocyte growth induced by loss of p44/WDR77 expression. Furthermore, we demonstrated that NF-κB overexpression could decrease astrocyte growth to the levels seen in p44/WDR77-null astrocytes. These findings suggest that p44/WDR77 regulates astrocyte proliferation by repressing NF-κB signaling. However, the molecular mechanisms by which NF-κB influences astrocyte proliferation remain unclear.
It has previously been demonstrated that AR and p44/WDR77 target the p21Cip1 promoter in prostate cancer cells (40, 55). Also, previous work has suggested that AR and NF-κB physically interact, resulting in mutual antagonism (1, 29, 38). Further study is needed to investigate whether p44/WDR77 regulates p21Cip1 and NF-κB through AR in astrocytes. Studies using p21Cip1-null astrocytes have suggested that p21Cip1 expression can increase NF-κB activity in astrocytes (49, 50). We confirmed this observation in astrocytes. We also observed that exogenous expression of p65 increased p21Cip1 expression and that silencing p65 expression decreased p21Cip1 expression induced by p44/WDR77 deletion. These studies allude to a possible bidirectional mechanism in which p21Cip1 and NF-κB activity are interconnected. However, the molecular mechanisms governing the interaction between p21Cip1 and NF-κB in astrocytes remain to be elucidated. The in vitro and in vivo studies presented here clearly demonstrate an important role for p44/WDR77 in astrocyte activation through p21Cip1 and NF-κB signaling. However, overexpression of p65, p21Cip1, or p65 plus p21Cip1 did not induce astrocyte activation to the extent induced by p44/WDR77 deletion. This finding indicates that other changes are induced by the loss of p44/WDR77 in astrocytes, thus prompting further study of the role of p44/WDR77 in astrocyte activation.
ACKNOWLEDGMENTS
We thank Karen Muller for critical editorial review of the manuscript.
This work was supported in part by the National Institutes of Health through M. D. Anderson Cancer Center support grant CA016672. No additional external funding was received for this study. B.V. was supported in part by a Smith Research Fellowship.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We declare that no competing interests exist.
Footnotes
Published ahead of print 2 July 2012
REFERENCES
- 1. Altuwaijri S, et al. 2003. Interruption of nuclear factor kappaB signaling by the androgen receptor facilitates 12-O-tetradecanoylphorbolacetate-induced apoptosis in androgen-sensitive prostate cancer LNCaP cells. Cancer Res. 63:7106–7112 [PubMed] [Google Scholar]
- 2. Bancroft J. 2005. The endocrinology of sexual arousal. J. Endocrinol. 186:411–427 [DOI] [PubMed] [Google Scholar]
- 3. Barreto G, Veiga S, Azcoitia I, Garcia-Segura LM, Garcia-Ovejero D. 2007. Testosterone decreases reactive astroglia and reactive microglia after brain injury in male rats: role of its metabolites, oestradiol and dihydrotestosterone. Eur. J. Neurosci. 25:3039–3046 [DOI] [PubMed] [Google Scholar]
- 4. Besson A, Dowdy SF, Roberts JM. 2008. CDK inhibitors: cell cycle regulators and beyond. Dev. Cell 14:159–169 [DOI] [PubMed] [Google Scholar]
- 5. Coers S, Tanzer L, Jones KJ. 2002. Testosterone treatment attenuates the effects of facial nerve transection on glial fibrillary acidic protein (GFAP) levels in the hamster facial motor nucleus. Metab. Brain Dis. 17:55–63 [DOI] [PubMed] [Google Scholar]
- 6. Conejo NM, et al. 2005. Influence of gonadal steroids on the glial fibrillary acidic protein-immunoreactive astrocyte population in young rat hippocampus. J. Neurosci. Res. 79:488–494 [DOI] [PubMed] [Google Scholar]
- 7. Correa-Cerro LS, Mandell JW. 2007. Molecular mechanisms of astrogliosis: new approaches with mouse genetics. J. Neuropathol. Exp. Neurol. 66:169–176 [DOI] [PubMed] [Google Scholar]
- 8. Craft N, et al. 1999. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res. 59:5030–5036 [PubMed] [Google Scholar]
- 9. Craft N, Sawyers CL. 1998. Mechanistic concepts in androgen-dependence of prostate cancer. Cancer Metastasis Rev. 17:421–427 [DOI] [PubMed] [Google Scholar]
- 10. Day JR, et al. 1993. Gonadal steroids regulate the expression of glial fibrillary acidic protein in the adult male rat hippocampus. Neuroscience 55:435–443 [DOI] [PubMed] [Google Scholar]
- 11. Denmeade SR, Lin XS, Isaacs JT. 1996. Role of programmed (apoptotic) cell death during the progression and therapy for prostate cancer. Prostate 28:251–265 [DOI] [PubMed] [Google Scholar]
- 12. Di Giovanni S, et al. 2005. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. U. S. A. 102:8333–8338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Felszeghy K, Banisadr G, Rostene W, Nyakas C, Haour F. 2004. Dexamethasone downregulates chemokine receptor CXCR4 and exerts neuroprotection against hypoxia/ischemia-induced brain injury in neonatal rats. Neuroimmunomodulation 11:404–413 [DOI] [PubMed] [Google Scholar]
- 14. Friesen WJ, et al. 2001. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol. Cell. Biol. 21:8289–8300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Friesen WJ, et al. 2002. A novel WD repeat protein component of the methylosome binds Sm proteins. J. Biol. Chem. 277:8243–8247 [DOI] [PubMed] [Google Scholar]
- 16. Gao S, et al. 2005. The androgen receptor directly targets the cellular Fas/FasL-associated death domain protein-like inhibitory protein gene to promote the androgen-independent growth of prostate cancer cells. Mol. Endocrinol. 19:1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao S, Wu H, Wang F, Wang Z. 2010. Altered differentiation and proliferation of prostate epithelium in mice lacking the androgen receptor cofactor p44/WDR77. Endocrinology 151:3941–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gelmann EP. 2002. Molecular biology of the androgen receptor. J. Clin. Oncol. 20:3001–3015 [DOI] [PubMed] [Google Scholar]
- 19. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805–816 [DOI] [PubMed] [Google Scholar]
- 20. He F, Sun YE. 2007. Glial cells more than support cells? Int. J. Biochem. Cell Biol. 39:661–665 [DOI] [PubMed] [Google Scholar]
- 21. Heinlein CA, Chang C. 2002. Androgen receptor (AR) coregulators: an overview. Endocr. Rev. 23:175–200 [DOI] [PubMed] [Google Scholar]
- 22. Hosohata K, et al. 2003. Purification and identification of a novel complex which is involved in androgen receptor-dependent transcription. Mol. Cell. Biol. 23:7019–7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu WH, et al. 2005. NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J. Biol. Chem. 280:29233–29241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Janne OA, et al. 2000. Androgen-receptor-interacting nuclear proteins. Biochem. Soc. Trans. 28:401–405 [PubMed] [Google Scholar]
- 25. Jenster G. 1999. The role of the androgen receptor in the development and progression of prostate cancer. Semin. Oncol. 26:407–421 [PubMed] [Google Scholar]
- 26. Jin C, McKeehan K, Wang F. 2003. Transgenic mouse with high Cre recombinase activity in all prostate lobes, seminal vesicle, and ductus deferens. Prostate 57:160–164 [DOI] [PubMed] [Google Scholar]
- 27. Koguchi K, Nakatsuji Y, Okuno T, Sawada M, Sakoda S. 2003. Microglial cell cycle-associated proteins control microglial proliferation in vivo and in vitro and are regulated by GM-CSF and density-dependent inhibition. J. Neurosci. Res. 74:898–905 [DOI] [PubMed] [Google Scholar]
- 28. McCarthy MM, Amateau SK, Mong JA. 2002. Steroid modulation of astrocytes in the neonatal brain: implications for adult reproductive function. Biol. Reprod. 67:691–698 [DOI] [PubMed] [Google Scholar]
- 29. McKay LI, Cidlowski JA. 1998. Cross-talk between nuclear factor-kappa B and the steroid hormone receptors: mechanisms of mutual antagonism. Mol. Endocrinol. 12:45–56 [DOI] [PubMed] [Google Scholar]
- 30. Meister G, et al. 2001. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 11:1990–1994 [DOI] [PubMed] [Google Scholar]
- 31. Mong JA, Glaser E, McCarthy MM. 1999. Gonadal steroids promote glial differentiation and alter neuronal morphology in the developing hypothalamus in a regionally specific manner. J. Neurosci. 19:1464–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mong JA, McCarthy MM. 1999. Steroid-induced developmental plasticity in hypothalamic astrocytes: implications for synaptic patterning. J. Neurobiol. 40:602–619 [DOI] [PubMed] [Google Scholar]
- 33. Mori T, Buffo A, Gotz M. 2005. The novel roles of glial cells revisited: the contribution of radial glia and astrocytes to neurogenesis. Curr. Top. Dev. Biol. 69:67–99. [DOI] [PubMed] [Google Scholar]
- 33a. National Institutes of Health 1996. Guide for the care and use of laboratory animals. National Institutes of Health, Washington, DC [Google Scholar]
- 34. Nichols NR. 1999. Glial responses to steroids as markers of brain aging. J. Neurobiol. 40:585–601 [PubMed] [Google Scholar]
- 35. Nichols NR, Agolley D, Zieba M, Bye N. 2005. Glucocorticoid regulation of glial responses during hippocampal neurodegeneration and regeneration. Brain Res. Brain Res. Rev. 48:287–301 [DOI] [PubMed] [Google Scholar]
- 36. Nichols NR, Day JR, Laping NJ, Johnson SA, Finch CE. 1993. GFAP mRNA increases with age in rat and human brain. Neurobiol Aging 14:421–429 [DOI] [PubMed] [Google Scholar]
- 37. O'Neill LA, Kaltschmidt C. 1997. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 20:252–258 [DOI] [PubMed] [Google Scholar]
- 38. Palvimo JJ, et al. 1996. Mutual transcriptional interference between RelA and androgen receptor. J. Biol. Chem. 271:24151–24156 [DOI] [PubMed] [Google Scholar]
- 39. Pan Y, et al. 2005. Effect of testosterone on functional recovery in a castrate male rat stroke model. Brain Res. 1043:195–204 [DOI] [PubMed] [Google Scholar]
- 40. Peng Y, et al. 2008. Distinct nuclear and cytoplasmic functions of androgen receptor cofactor p44 and association with androgen-independent prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 105:5236–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perng MD, et al. 2008. Glial fibrillary acidic protein filaments can tolerate the incorporation of assembly-compromised GFAP-delta, but with consequences for filament organization and alphaB-crystallin association. Mol. Biol. Cell 19:4521–4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pike CJ, et al. 2008. Androgen cell signaling pathways involved in neuroprotective actions. Horm. Behav. 53:693–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pike CJ, Rosario ER, Nguyen TV. 2006. Androgens, aging, and Alzheimer's disease. Endocrine 29:233–241 [DOI] [PubMed] [Google Scholar]
- 44. Schipper HM. 1996. Astrocytes, brain aging, and neurodegeneration. Neurobiol. Aging 17:467–480 [DOI] [PubMed] [Google Scholar]
- 45. Sofroniew MV. 2009. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32:638–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sofroniew MV, Vinters HV. 2010. Astrocytes: biology and pathology. Acta Neuropathol. 119:7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sonoda Y, et al. 2001. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 61:4956–4960 [PubMed] [Google Scholar]
- 48. Storer PD, Jones KJ. 2003. Glial fibrillary acidic protein expression in the hamster red nucleus: effects of axotomy and testosterone treatment. Exp. Neurol. 184:939–946 [DOI] [PubMed] [Google Scholar]
- 49. Tusell JM, et al. 2009. Upregulation of p21Cip1 in activated glial cells. Glia 57:524–534 [DOI] [PubMed] [Google Scholar]
- 50. Tusell JM, Saura J, Serratosa J. 2005. Absence of the cell cycle inhibitor p21Cip1 reduces LPS-induced NO release and activation of the transcription factor NF-kappaB in mixed glial cultures. Glia 49:52–58 [DOI] [PubMed] [Google Scholar]
- 51. Wilson JD. 2001. Androgens, androgen receptors, and male gender role behavior. Horm. Behav. 40:358–366 [DOI] [PubMed] [Google Scholar]
- 52. Wise-Draper TM, Wells SI. 2008. Papillomavirus E6 and E7 proteins and their cellular targets. Front. Biosci. 13:1003–1017 [DOI] [PubMed] [Google Scholar]
- 53. Yu X, Li P, Roeder RG, Wang Z. 2001. Inhibition of androgen receptor-mediated transcription by amino-terminal enhancer of split. Mol. Cell. Biol. 21:4614–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zaheer A, Yorek MA, Lim R. 2001. Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion. Neurochem. Res. 26:1293–1299 [DOI] [PubMed] [Google Scholar]
- 55. Zhou L, Wu H, Lee P, Wang Z. 2006. Roles of the androgen receptor cofactor p44 in the growth of prostate epithelial cells. J. Mol. Endocrinol. 37:283–300 [DOI] [PubMed] [Google Scholar]
- 56. Zhu Z, et al. 2007. Inhibiting cell cycle progression reduces reactive astrogliosis initiated by scratch injury in vitro and by cerebral ischemia in vivo. Glia 55:546–558 [DOI] [PubMed] [Google Scholar]