

Abstract
The Ess1 prolyl isomerase in Saccharomyces cerevisiae regulates RNA polymerase II (pol II) by isomerizing peptide bonds within the pol II carboxy-terminal domain (CTD) heptapeptide repeat (YSPTSPS). Ess1 preferentially targets the Ser5-Pro6 bond when Ser5 is phosphorylated. Conformational changes in the CTD induced by Ess1 control the recruitment of essential cofactors to the pol II complex and may facilitate the ordered transition between initiation, elongation, termination, and RNA processing. Here, we show that Ess1 associates with the phospho-Ser5 form of polymerase in vivo, is present along the entire length of coding genes, and is critical for regulating the phosphorylation of Ser7 within the CTD. In addition, Ess1 represses the initiation of cryptic unstable transcripts (CUTs) and is required for efficient termination of mRNA transcription. Analysis using strains lacking nonsense-mediated decay suggests that as many as half of all yeast genes depend on Ess1 for efficient termination. Finally, we show that Ess1 is required for trimethylation of histone H3 lysine 4 (H3K4). Thus, Ess1 has direct effects on RNA polymerase transcription by controlling cofactor binding via conformationally induced changes in the CTD and indirect effects by influencing chromatin modification.
INTRODUCTION
Peptidyl-prolyl isomerases (PPIases) are enzymes that noncovalently modify target proteins by catalyzing the rotation of the peptide bond preceding proline residues. PPIases catalyze both cis→trans and trans→cis peptide bond isomerizations. The resulting changes in protein conformation can have profound functional consequences, such as altering protein-protein interactions, protein stability, or the suitability of a protein as a target for further modifications, e.g., phosphorylation/dephosphorylation (46, 47, 56).
Three classes of PPIases have been identified (5). The parvulin family of PPIases, of which Ess1 (essential 1) protein in Saccharomyces cerevisiae is the founding eukaryotic member, is distinct from the well-studied cyclophilin and FK506-binding protein (FKBP) families, which are targets of immunosuppressive drugs. In yeast, Ess1 is required for growth (20), and its human homolog, Pin1, complements yeast ess1 mutants (32). Pin1 has been linked to a number of signaling pathways in human cells and seems to target a wide variety of proteins for isomerization (33, 70). In contrast, extensive genetic studies have thus far identified only RNA polymerase II (pol II) as the target of Ess1 in yeast (21, 27, 65).
Within the RNA pol II complex, Ess1 targets the carboxy-terminal domain (CTD) of the largest subunit, Rpb1 (38, 65), which is composed of 26 repeats of the heptapeptide Y1S2P3T4S5P6S7 (62). Each repeat contains two potential Ess1 substrate binding sites (S-P), where the serines are known to be phosphorylated (24, 43). In vitro, Ess1 binds and isomerizes phosphorylated Ser5-Pro6 (pSer5-Pro6) within the heptad repeat about 5-fold better than pSer2-Pro3 (17). Ess1 stimulates pSer5-Pro6 isomerization within peptide substrates from a rate of less than 1 per minute to >15 per second (17). Genetic data also implicate pSer5-Pro6 as the in vivo target (63) where effects on the structure of the CTD, which is highly malleable (37), are likely to be significant.
The CTD is positioned near the exit site for newly synthesized RNA within the RNA polymerase II complex (3) and is thought to help recruit accessory proteins needed for RNA maturation, including cotranscriptional 5′-end capping, transcript elongation, RNA splicing, and termination/3′-end processing (8). Modifications of the CTD, whether covalent (phosphorylation) or noncovalent (isomerization), are therefore likely to be critical for cofactor recruitment during the transcription cycle (7, 14). Indeed, ess1 conditional mutants show genetic interactions with a number of genes encoding RNA pol II cofactors and exhibit defects in transcription (27, 62, 64). In the best-studied example, ess1 mutants fail to coordinate the recruitment and release of Nrd1 and Pcf11, which are required for 3′ termination of small noncoding RNAs (ncRNAs) (51). The result is transcription readthrough of snoRNAs and widespread readthrough and stabilization of cryptic unstable transcripts (CUTs) (66), stable unannotated transcripts (SUTs) (41), and other small ncRNAs transcribed by RNA pol II. One likely mechanism is that in the absence of Ess1, the CTD is an unsuitable substrate for the Ser5-Pro6-specific CTD phosphatase, Ssu72 (6, 51, 61, 67). This increases pSer5 levels, resulting in a failure to release Nrd1 and to bind Pcf11 and other factors required for termination and/or 3′-end formation (51, 59). The importance of isomerization of pSer5-Pro6 bonds at different positions along the CTD differs dramatically, with more proximal, N-terminal bonds (near the RNA pol II exit channel) being the target of Ess1 (62).
Here, we investigated three major questions prompted by previous studies. First, does Ess1 influence the recruitment of factors that function during initiation, as suggested by genetic and molecular studies (27, 63)? Second, is Ess1 important for the termination of mRNAs, as inferred genetically (21, 64)? And third, are the actions of chromatin modifiers, which may be sensitive to the phosphorylation state of the CTD (9, 52), also influenced by Ess1 (4)? These and other questions regarding the role of Ess1 during the transcription cycle are addressed. The results indicate that Ess1 functions in multiple steps of transcription; it inhibits ncRNA initiation, stimulates mRNA termination, and controls the phosphorylation state of CTD Ser7 (as well as Ser5). Finally, Ess1 is required for proper chromatin modification, specifically, the methylation of histone H3 lysine 4 (H3K4). The results support the model that Ess1 helps coordinate RNA pol II cofactor recruitment and function.
MATERIALS AND METHODS
Yeast strains, plasmids, oligonucleotides, and growth conditions.
Yeast strains are listed in Table 1. Strains YDA50 0 and YDA502 were generated using a sewing PCR strategy to insert a natMX cassette adjacent to wild-type ESS1 or ess1H164R in strain BY4742. Transformants were selected on clonNAT (nourseothricin; Werner BioAgents, Jena, Germany), and the insertions confirmed by PCR and DNA sequencing. Strain YDA511 (set1Δ) was made in the BY4741 background by replacing SET1 with kanMX, using G418 selection (Invitrogen). Plasmid p4339 (source of natMX) was a gift of Ian Willis, pUG6 (source of kanMX) was a gift of Joan Curcio, and PrADH1-HA-SET1 (pMP803) was a gift of Bernard Dichtl (19). Oligonucleotides were synthesized by Integrated DNA Technologies, and their sequences are available upon request. Yeast strains were cultured using standard conditions and media (49). Double mutants were generated by crosses and tetrad dissection using deletion strains from the EUROSCARF collection.
Table 1.
Saccharomyces cerevisiae strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| W303-1A | MATa ura3-1 trp1-1 leu2-3,112 can1-100 ade2-1 his3-11,15 [phi+] | 57 |
| W303-1B | MATα ura3-1 trp1-1 leu2-3,112 can1-100 ade2-1 his3-11,15 [phi+] | 57 |
| YGD-ts22 | MATa ura3-1 trp1-1 leu2-3,112 can1-100 ade2-1 his3-11,15 ess1164R | 64 |
| YSB2039 | MATa ura3-1 trp1-1 leu2-3,112 can1-100 ade2-1 his3-11,15 PCF11-TAP | S. Buratowski |
| YSB2040 | MATa ura3-1 trp1-1 leu2-3,112 can1-100 ade2-1 his3-11,15 PCF11-TAP ess1H164R | S. Buratowski |
| YXW137 | MATa ura3-1 trp1-1 leu2-3,112 can1-100 ade2-1 his3-11,15 ess1Δ::HIS3 (pRS315-GAL1-ESS1) | 17 |
| YXW138 | MATa ura3-1 trp1-1 leu2-3,112 can1-100 ade2-1 his3-11,15 ess1Δ::HIS3 (pRS315-GAL1-H164R) | 17 |
| YJM1 | MATa ura3-1 trp1-1 leu2-3,112 can1-100 ade2-1 his3-11,15 srb10::TRP1 | 63 |
| YJM2 | MATa ura3-1 trp1-1 leu2-3,112 can1-100 ade2-1 his3-11,15 ess1H164R srb10::TRP1 | 63 |
| CBW22 | MATa ura3-1 trp1-1 can1-100 ade2-1 his3-11,15 ess1ΔHIS3 srb10::TRP1 | 63 |
| 46a | MATa cup1Δ ura3 his3 trp1 lys2 ade2 leu2 | 53 |
| 46a nrd1-5 | MATα cup1Δ ura3 his3 trp1 lys2 ade2 leu2 nrd1-5 | 53 |
| YJC1412 | MATa ade2 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 12 |
| YJC1098 | MATa ade2 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 nab3-11 | 12 |
| NA67 | MATα ura3-1 leu2-3,112 trp1Δ his3-11,15 ade2-1 pcf11-9 | 2 |
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Open Biosystems |
| Ess1-TAP | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ESS1-TAP::HIS3 | Open Biosystems |
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Open Biosystems |
| YHD154 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] BYE1-TAP tag::HIS3MX6 | This study |
| YHD152 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] BYE1-TAP tag::HIS3MX6 ess1H164R | This study |
| YHD139 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] PAF1-TAP tag::HIS3MX6 | This study |
| YHD129 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] PAF1-TAP tag::HIS3MX6 ess1H164R | This study |
| YHD146 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] SPT4-TAP tag::HIS3MX6 | This study |
| YHD150 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] SPT4-TAP tag::HIS3MX6 ess1H164R | This study |
| YHD136 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] SEN1-TAP tag::HIS3MX6 | This study |
| YHD125 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] SEN1-TAP tag::HIS3MX6 ess1H164R | This study |
| YHD132 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] CEG1-MYC tag::TRP1 (from Kluyveromyces lactis) | This study |
| YHD119 | MATa ura3-1 leu2-3,112 trp1-1 can1-100 ade2-1 his3-11,15 [phi+] ess1H164R CEG1-MYC tag::TRP1 (from K. lactis) | This study |
| YDA500 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ESS1::natMX4 | This study |
| YDA502 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ess1H164R::natMX4 | This study |
| YDA511 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 set1Δ::kanMX4 | This study |
| YDA512 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ESS1::natMX4 set1Δ::kanMX4 | This study |
| YDA516 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ess1H164R::natMX4 set1Δ::kanMX4 | This study |
| YDA587 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ess1H164R::natMX4 jhd2Δ::kanMX4 | This study |
| YDA554 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ESS1::natMX4 nam7Δ::kanMX4 | This study |
| YDA556 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ess1H164R::natMX4 nam7Δ::kanMX4 | This study |
| YDA558 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ESS1::natMX4 rrp6Δ::kanMX4 | This study |
| YDA560 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ess1H164R::natMX4 rrp6Δ::kanMX4 | This study |
| YDA565 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ESS1::natMX4 xrn1Δ::kanMX4 | This study |
| YDA568 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ess1H164R::natMX4 xrn1Δ::kanMX4 | This study |
| Y10158 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 lcb1-4::kanMX4 | 31 |
| Y11354 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 srm1-ts::kanMX4 | 31 |
| Y07833 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 kar2-159::kanMX4 | 31 |
| Y10345 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 hts1-1::kanMX4 | 31 |
| Y04855 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 pri2-1::kanMX4 | 31 |
| Y07794 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 bos1-1::kanMX4 | 31 |
Coimmunoprecipitation (co-IP).
Wild-type yeast (W303-1A) was grown in 30 ml yeast extract-peptone-dextrose (YEPD) medium to an optical density at 600 nm (OD600) of 0.6, and cells were pelleted, washed, and resuspended in FA lysis buffer (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, pH 8, 1% Triton X-100, 0.1% sodium deoxycholate with 1× protease, and phosphatase inhibitors) and broken with an equal volume of glass beads using a cell disruptor at 4°C. The extracts were clarified by microcentrifugation at 14,000 rpm. The protein concentration was determined using a Bio-Rad protein assay. About 2 mg of total protein lysate was incubated with 5 μl affinity-purified anti-Ess1 antibody overnight at 4°C. Thirty microliters of protein A-agarose bead slurry was added for 2 h, followed by 2 washes with low-salt (140 mM NaCl) and high-salt (250 mM NaCl) FA lysis buffers. Protein A-agarose beads were resuspended in 30 μl FA lysis buffer and 30 μl 2× sample buffer (62.5 mM Tris-HCl, pH 6.8, 25% glycerol, 2% SDS, 0.01% bromophenol blue, 5% beta-mercaptoethanol), boiled for 10 min, and clarified by centrifugation. Supernatants were loaded on 8% SDS-PAGE gels for Western blot detection of hypophospho-CTD, pSer5, and pSer2, as well as Ess1, with antibodies described below.
Western analysis.
Western analysis was carried out as described previously (51). The primary antibodies were H14 (pSer5) and 8WG16 (hypophosphorylated CTD) from Covance, H5 (pSer2) from Bethyl, 4E12 (pSer7) (11), anti-H3K4me3 (trimethylated H3K4) from Upstate, anti-H3K36me3 and anti-H3 from Abcam, and anti-Ess1 (65). Antitubulin antibody (Abcam) was used for loading controls. Secondary monoclonal antibodies (MAbs; anti-mouse, anti-rat, or anti-rabbit immunoglobulin, IgG, or anti-mouse IgM) conjugated to horseradish peroxidase (Amersham) were used as appropriate.
ChIP.
Chromatin immunoprecipitation (ChIP) was performed as described previously (51). The antibodies used were as follows: anti-Ess1 (65), anti-Rpb3 (1:100 dilution; Neoclone), anti-pSer2 (1:100 dilution), anti-pSer5 (1:100 dilution), anti-TATA binding protein (1:100 dilution; Santa Cruz), anti-TFIIB (1:100 dilution; a gift of Anthony Weil), anti-Ssu72 (1:100 dilution; a gift of Michael Hampsey), anti-TFIIS (1:100 dilution; a gift of Caroline Kane), anti-Nrd1 (1:100 dilution; a gift of D. Brow), anti-H3K4me3 (1 μl; Upstate), anti-H3K36me3 (1 μl; Abcam), and anti-H3 (1 μl; Abcam) antibodies. Tandem affinity purification (TAP)-tagged proteins (Pcf11, Sen1, Paf1, Spt4, and Bye1) were immunoprecipitated using anti-protein A and then with protein A-agarose (Santa Cruz). Myc-tagged protein (Ceg1) was precipitated using anti-Myc antibody (Santa Cruz) and protein A-agarose. For each antibody, the immunoprecipitated DNA fragments from at least three biological replicates were isolated. The relative proportion was then analyzed by quantitative real-time PCR (qRT-PCR). For normalization across a set of samples, quantitative real-time PCR values (normalized to inputs and a chromosome V control, except that for H3K4me3, a tRNA gene was used for internal control) were summed for each experiment and the sums set to the same arbitrary value. The normalized values thus obtained for each ChIP sample were then used to obtain averages and standard deviations (71). For the experiment whose results are shown in Fig. 4, ChIP samples were analyzed using standard PCR (26 cycles) and products resolved by gel electrophoresis. Ethidium bromide-stained gel scans were quantitated using ImageJ software. Representative results from one of three replicates (for PYK1, PTC1, and ARD1) or two replicates (for BUD3) are shown.
RNA isolation, cDNA synthesis, qRT-PCR, and Northern analysis.
Cells were grown at 30°C to an OD600 of 0.5 to 0.8. For Northern analysis, cells were shifted to 37°C for 2 h. RNA was purified using hot phenol as described previously (48). The RNA was treated with DNase (Turbo DNA-free DNase; Ambion) and quantitated spectrophotometrically. An aliquot was used for qRT-PCR (described below) to test the efficacy of the DNase treatment. cDNA synthesis was carried out in a 10-μl reaction mixture containing 1 μg of RNA, random hexamers at 3.75 μM or gene-specific primers at 500 nM, and deoxynucleoside triphosphates (dNTPs) at 500 μM. This was incubated at 65°C for 10 min to denature the RNA. Two units of RNase inhibitor (USB), 1 U Moloney murine leukemia virus (M-MLV) reverse transcriptase (USB),10× buffer (supplied), and water were added to bring the reaction mixture to 1×, followed by incubation at 44°C for 1 h. Amounts of 0.5 μl of this cDNA and 1 μl of a primer mixture at 10 μM each were added to 6.25 μl of HotStart-IT SYBR green qPCR master mix (USB) and 4.75 μl H2O. qRT-PCR was carried out in an Eppendorf Realplex Mastercycler Epgradient S as follows: 95°C for 2 min, followed by 40 cycles of 95°C 15 s, 60°C 15 s, and 68°C for 20 s. All PCR primers were tested for efficiency on DNA templates using standard reaction mixtures, and the products examined on ethidium bromide-stained gels. The comparative CT (cycle threshold) method (Applied Biosystems), in which the amount of target gene amplification is normalized to the amplification of an NADPH internal control, was used for quantitation. The relative (fold) enrichment is calculated as follows: 2−ΔΔCT = 2[ΔCT (control) − ΔCT (sample)], where ΔCT = CT (sample) − CT (control). Error bars in figures show standard deviations from the mean log values of at least three biological replicates. The internal control was the SNR6 gene (a pol III product), and all values were normalized to the transcript levels in the upstream coding region as follows: [(intergenic region/SNR6)/(upstream control/SNR6)]mutant/[(intergenic region/SNR6)/(upstream control/SNR6)]wild type. Northern analysis was carried out by standard methods, using formaldehyde gel electrophoresis, GeneScreen+ membranes (Perkin-Elmer), and 32P-labeled probes ([α-32P]dATP, 3,000 Ci/mmol; Perkin-Elmer) generated by random priming (Sequenase; USB-Affymetrix) of glass powder-and-gel-purified PCR-generated gene fragments.
RESULTS
Ess1 targets pSer5-CTD and controls Ser7 phosphorylation levels.
To test whether Ess1 associates with the pSer5 form of RNA pol II in vivo, we performed coimmunoprecipitation (co-IP) experiments. Rabbit anti-Ess1 antibodies were used against yeast whole-cell extracts, and the precipitates analyzed by Western blotting with CTD phospho-specific antibodies. The results indicate that Ess1 preferentially associates with the pSer5 form or the pSer2-pSer5 doubly phosphorylated form, which is also recognized by the H14 MAb (11), over the pSer2 alone or hypophosphorylated forms of pol II (Fig. 1A). Thus, the specificity of Ess1 in vivo appears to be similar to that defined in vitro and by genetic inference (17, 63).
Fig 1.
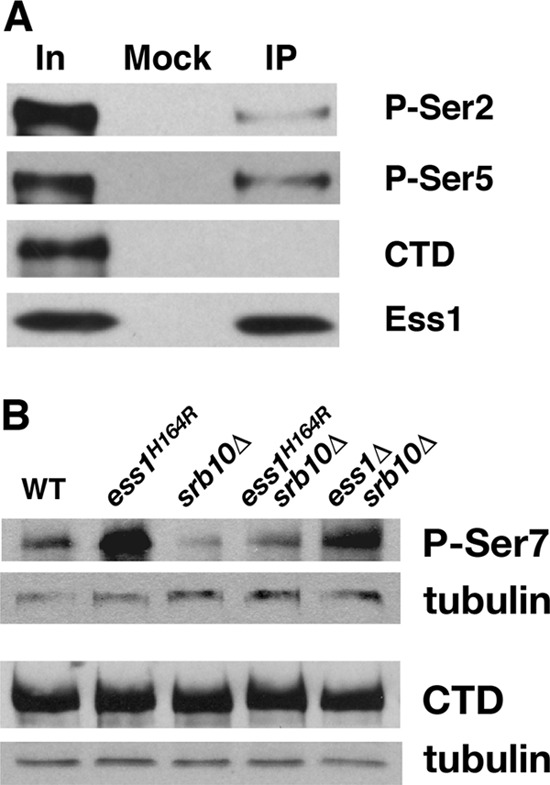
Ess1 preferentially associates with pSer5 CTD in vivo and promotes dephosphorylation of pSer7 CTD. (A) Wild-type (WT) cells were subjected to immunoprecipitation (IP) with anti-Ess1 polyclonal antibodies, followed by Western analysis with monoclonal antibodies H5 (P-Ser2), H14 (P-Ser5), and 8WG16 (hypophosphorylated CTD). Sample inputs (In) are indicated. Note that the H14 antibody actually recognizes doubly phosphorylated pSer2/pSer5 (11). (B) Western analysis of protein (10 μg) from whole-cell extracts of the indicated strains grown at 30°C. The 4E12 monoclonal antibody was used to detect levels of pSer7 CTD, and the 8WG16 monoclonal antibody (hypophosphorylated CTD) as a control for overall levels of RNA pol II CTD. Antitubulin antibodies were used for detection of the loading control. Note that ESS1 is an essential gene but can be suppressed by deletion of SRB10 (62).
We and others showed that the levels of pSer5 become elevated in ess1 mutant cells, whereas the levels of pSer2 remain unchanged (6, 27, 51). This is probably due to loss of Ess1-directed prolyl bond isomerization at pSer5-Pro6 and consequent reduction of the affinity of the Ssu72 phosphatase, which targets pSer5 (61). Ser7 phosphorylation in the CTD seems to be linked to that of Ser5 (15, 26, 36, 58). We therefore examined whether the levels of Ser7 phosphorylation are affected in ess1 mutant cells. Using two different mutants (ess1H164R and ess1Δ srb10Δ), Western blotting of whole-cell extracts with pSer7-specific antibodies showed that pSer7 levels were indeed elevated (Fig. 1B), mirroring those of pSer5 (51). Note that pSer7 is followed not by a proline residue in the CTD (Y-S-P-T-S-P-S)n but, rather, by a tyrosine. Therefore, the effect of Ess1 on Ser7 phosphorylation is likely to occur by an indirect mechanism, perhaps involving a processive coupling of dephosphorylation by Ssu72 that initiates at pSer5-Pro6. Genome-wide analysis has also implicated Ess1 in control of pSer7 levels (6). One potential caveat to these experiments is the possibility that loss of Ess1 activity alters the isomeric state of the CTD in such a way as to increase recognition by the monoclonal antibodies used for detecting the phosphorylated form of Ser7. We do not favor this explanation, however, as the increase in Ser7 phosphorylation appears to be greater than stoichiometric, suggesting a catalytic effect, more consistent with a loss of Ess1's known stimulation of protein phosphatase activity (61).
Ess1 represses initiation of small noncoding RNAs.
A genome-wide tiling microarray analysis of ess1 mutants led to the discovery of defects in Nrd1-dependent transcription termination of small noncoding RNAs (51). A large number of CUTs were also identified in ess1 mutants. To study their production further, we used chromatin immunoprecipitation (ChIP) to compare the pol II CTD phosphorylation profiles and localization of various transcription cofactors across representative loci in wild-type and ess1 mutant cells.
Three loci were chosen for study. First, we chose the TEF2-MUD2 locus, where we found a prominent previously annotated CUT (51, 66) to be upregulated in ess1 mutant cells, as well as the RNQ1-FUS1 locus that contains a putative CUT. Second, we examined the SRG1-SER3 locus, where SRG1 is a SUT-like noncoding RNA that represses the expression of SER3 in response to serine levels via a mechanism of transcriptional interference (35). Finally, the PYK1 gene served as a control for a midsized protein-coding gene (∼1.5 kb) that contained no obvious CUTs in ess1 mutants (51).
In the first set of experiments, we carried out ChIP across these loci using pSer2-specific (H5) or pSer5-specific (H14) antibodies. Antibodies to the Rpb3 subunit of RNA pol II were used as a control for the total amount of pol II complex present. The results revealed an increase in the total amount of pol II complex recruited to the position of the NBR024W CUT, the putative RNQ1-FUS1 intergenic CUT, and SRG1 in the ess1 mutant (Fig. 2A to C). Most of the increases are due to enrichment of the pSer5 form of the pol II CTD, consistent with elevated pSer5 levels at the 5′ ends of actively transcribed genes. The control gene, PYK1, did not show either any substantive changes in pSer5 or pSer2 phosphorylation (Fig. 2D) or changes in expression in ess1 mutant cells (51).
Fig 2.

Ess1 mutants show increased recruitment to ncRNA loci of the initiation form (pSer5) of RNA pol II. Wild-type and ess1H164R mutant cells were grown to mid-logarithmic phase at 30°C. Chromatin immunoprecipitation was used to monitor recruitment of total RNA pol II complex (α-Rpb3) and the pSer2 or pSer5 forms of Rpb1. (A to C) Results of ChIP across three intergenic regions containing known CUTs (NBR024W and SRG1) or a suspected CUT (between RNQ1 and FUS1). Increased recruitment of the pSer5 but not the pSer2 form of pol II is detected in all three. (D) Results of control ChIP across the PYK1-coding gene locus. No increase in recruitment of either form of pol II is detected. Fold changes are relative to the results for a chromosome V control. Numbered horizontal bars represent approximate locations of qRT-PCR products (also in Fig. 3, 4, and 9). Error bars show standard deviations of three biological replicates.
In the second set of experiments, we examined recruitment to three loci of selected cofactors important for initiation, elongation, and termination of transcription. Consistent with the strong induction of the NBR024W CUT, the recruitment of TATA-binding protein (TBP), the TFIIB initiation factor, and capping enzyme Ceg1 all increased (Fig. 3A). Similar results were observed at the SRG1 locus. For both loci, the majority of the increase in recruitment occurred at a position encompassing the beginning of the CUT transcripts. In contrast, there was no enhancement of initiation factors on the PYK1 gene in ess1 mutant cells. These results suggest that in ess1 mutants, increased production of small noncoding RNAs is due, at least in part, to increased transcription initiation. Thus, one role of Ess1 in wild-type cells is to repress CUT/small ncRNA initiation.
Fig 3.

Ess1 mutants show increased recruitment of the initiation factors but not general elongation factors to ncRNA loci. (A) Results of ChIP to detect recruitment of Ceg1 (capping enzyme), and initiation factors TBP and TFIIB to ncRNA loci in ess1H164R mutant cells. Increases are detected across intergenic regions of TEF2-MUD1 and SRG1 but not on the coding gene PYK1. (B) ChIP to monitor recruitment of elongation factors. No significant changes are detected in ess1H164R cells for TFIIS and Paf1. Spt4 shows a modest increase on SRG1. A larger increase in recruitment of the ess1-specific suppressor Bye1 (bypass of Ess1) is detected on ncRNA loci in ess1H164R cells.
Similar experiments were performed to examine recruitment of elongation (Fig. 3B). For elongation factors TFIIS and Paf1, we found no alteration in recruitment, and for Spt4, the changes did not appear significant (Fig. 3B). In contrast, we observed a strong increase in recruitment of Bye1 in ess1 mutants (Fig. 3B). The latter is not entirely surprising given that Bye1 was discovered as a high-copy-number suppressor of Ess1, and it is possible that in the absence of Ess1, high levels of Bye1 play a substitute role in controlling elongation (63). Given that TFIIS, Paf1, and Spt4 function to promote elongation, whereas Bye1 acts to inhibit elongation (64), it seems unlikely that stimulation of elongation is a decisive event in CUT/small ncRNA induction in ess1 mutants.
Finally, we examined termination factor recruitment. We chose two factors important for small ncRNA termination, Nrd1 and Sen1, and two factors that work universally in termination, Pcf11 (which binds the CTD and nascent RNA) and Ssu72, the Ser5 phosphatase that is required for termination and 3′-end processing. No change was observed for Ssu72 or Sen1 recruitment on any of the loci examined (Fig. 4). In the case of Ssu72, this enzyme might bind normally but not be able to dephosphorylate the CTD because it is not in the preferred isomerization state (cis) (61). In contrast, in the ess1 mutant, recruitment of Nrd1 is increased and Pcf11 is decreased on the NBR024W, SRG1, and RNQ1-FUS1 loci (Fig. 4), similar to earlier studies on snoRNA termination sites (51). This aberrant recruitment pattern might lead to transcription readthrough, as previously observed (51). In summary, the CTD phosphorylation and ChIP data are consistent with the idea that increased initiation and aberrant termination contribute to CUT and small ncRNA overexpression/stabilization in ess1 mutants. Ess1's normal function, therefore, is to repress aberrant initiation of CUTs/SUTs, as well as to promote their proper termination.
Fig 4.

Recruitment of some (Nrd1 and Pcf11) but not other termination factors (Sen1 and Ssu72) is altered in ess1 mutants. ChIP analysis of four termination factors was performed. No significant effects were detected on recruitment of termination factors (Sen1 and Ssu72) to the intergenic region of TEF2-MUD1 or to the SRG1 ncRNA gene or the PYK1 control gene. An increase in Nrd1 and a decrease in Pcf11 are observed at the 3′ regions of TEF2, SRG1, and RNQ1, similar to previous results for snoRNA loci (51). The inability to bind or release these factors in ess1 mutants may contribute to defective termination of these genes. Data in panels indicated by stars are reprinted from Molecular Cell (51) with permission of the publisher and are reproduced here for completeness.
Ess1 is localized along mRNA coding genes.
Our tiling array of ess1 mutants revealed readthrough of mRNA genes only in rare instances (51). We wondered whether this was because Ess1 does not bind to RNA pol II engaged on longer transcription units and therefore, plays no role in termination of mRNAs. To address this question, we carried out ChIP to localize Ess1 protein across mRNA genes of various lengths. The genes analyzed were PYK1 (1.5 kb), PTC1 (0.8 kb), BUD3 (6.4 kb), and ARD1 (0.7 kb) (Fig. 5A). Quantitation of these results indicates that Ess1 was present at 5′, middle, and 3′ locations, although the amount of Ess1 varied between loci (Fig. 5B). As expected, a control ChIP shows localization of TBP to the 5′ end of the PYK1 gene. These results suggest that Ess1 associates with RNA pol II across the entire length of protein-coding genes. Thus, our failure to detect an effect on mRNA termination was not due to the lack of Ess1 on protein-coding genes.
Fig 5.

Ess1 is present along transcribed protein-coding genes of all sizes. (A) Results of ChIP with antibodies to Ess1 to monitor the presence of Ess1 at 5′, coding, and 3′ regions of four protein-coding genes. No-antibody and input chromatin (in) controls are included. Shown are representative ethidium bromide-stained gels (image inverted) of PCR products for PYK1, PTC1, BUD3, and ARD1 genes, as well as an untranscribed region of chromosome V (ChrV). Results of a ChIP for TBP are also included. (B) Quantitation of the results in panel A, with ChIP signals normalized to input and ChrV signals [(IP/input)/(IP/inputChrV)]. Similar results were obtained when ChIP signals were normalized to those of no-antibody controls (not shown).
Ess1 is required for efficient mRNA termination.
We next considered the possibility that Ess1 might be important for mRNA gene termination but, in ess1 mutants, readthrough transcripts were degraded by an RNA surveillance mechanism (1). This would have precluded their detection using a tiling microarray approach (51). First, we carried out genetic interaction tests to determine whether nonsense-mediated decay (NMD) was involved (39). We generated double-mutant cells that were defective for both ess1 and genes required for NMD, including UPF1 (also known as NAM7), which encodes an RNA helicase that plays a key role in nonsense recognition and targeting of aberrant RNAs (29). In addition, we made double mutants with XRN1 (cytoplasmic 5′→3′ exonuclease) and RRP6 (nuclear 3′→5′ exonuclease), which encode P-body and exosome components required for degrading aberrant mRNAs. The prediction is that the severity of the growth defect of an ess1ts conditional mutant would be enhanced in a background that failed to degrade readthrough transcripts. This is exactly what we observed (Fig. 6), with severe growth defects in the double mutants apparent even at 30°C, which is normally permissive for the growth of ess1H164R cells.
Fig 6.
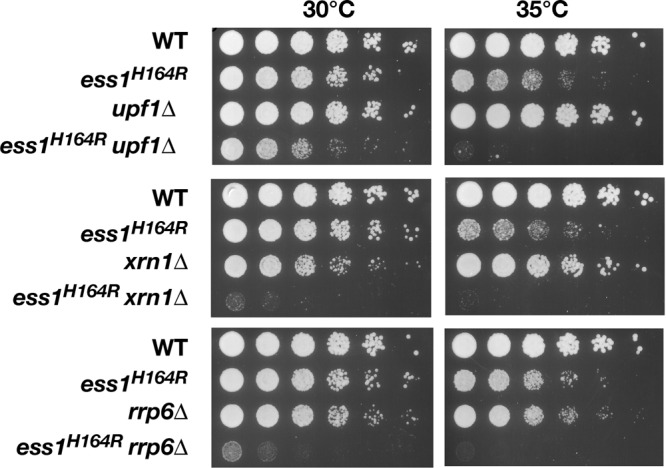
Ess1 mutants are synthetic lethal with NMD and RNA decay mutants. Serial dilutions (1:5, starting with cells at an OD600 of 0.5) of the wild type or the indicated mutant cells were grown at 30°C or 35°C on YEPD for 3 days. At both permissive (30°C) and semipermissive (35°C) temperatures, the growth defect of the ess1H164R mutant is strongly enhanced by upf1 (NMD pathway), xrn1 (cytoplasmic nuclease), and rrp6 (nuclear exonuclease) mutations. At 35°C, growth of the double mutants was negligible. Single upf1 mutants have no discernible growth defects, while xrn1 and rrp6 mutants have only minor growth defects at 30°C or 35°C, respectively.
To determine whether loss of NMD stabilized mRNA readthrough transcripts in ess1 mutants, we looked directly for readthrough products using reverse transcription-PCR. To simplify the analysis, we examined loci that met the following criteria: (i) mRNA genes that did not show obvious readthrough in our prior tiling array analysis (51), (ii) genes that were oriented in the same direction so as to avoid potential transcriptional interference by converging polymerases, and (iii) loci that lacked obvious CUTs in ess1 mutants (based on prior tiling array data).
The initial experiments were done using random hexamers as primers for reverse transcription, followed by PCR to detect intergenic transcripts. Nearly all 14 loci examined displayed intergenic transcription in ess1 mutants. Representative data for three loci are shown (Fig. 7A) and reveal strong intergenic transcription in ess1 mutants for two of the three loci (TAE1-RGD1 and SMK1-SEC8). For the third locus (HMG2-LEU3), only a small increase in transcript levels relative to the levels in wild-type cells was detected. Importantly, for TAE1-RGD1, the intergenic transcripts are dramatically increased in the NMD-deficient background (ess1H164R upf1Δ) (Fig. 7A), suggesting that they are normally degraded by NMD. The intergenic transcription could result from mRNA readthrough but could also be due to CUTs or other small ncRNAs templated from either DNA strand (see schematic).
Fig 7.

Strong mRNA termination defects were detected in ess1 mutants using cells that lack nonsense-mediated decay. (A) Intergenic transcription is detected in ess1 mutants. Quantitative reverse transcription–RT-PCR was used to detect intergenic transcription. Results are shown for three representative loci, HMG2-LEU3, TAE1-RGD1, and SMK1-SEC8. Total RNA from the indicated strains was reverse transcribed using random hexamer primers, and the cDNA products were amplified by PCR for 30, 30, and 34 cycles, respectively, and visualized on an agarose gel. Intergenic primers were used for PCR as indicated in the schematic. (B) Ess1 mutants fail to terminate at mRNA genes, and the readthrough transcripts are degraded by NMD. In contrast to the experiment whose results are shown in panel A, gene-specific primers were used for first-strand cDNA synthesis (see schematic) to allow strand-specific detection of intergenic transcripts (primer set 1), as well as the longer fusion transcripts (primer set 2). The presence of long products at the TAE1-RGD1 locus in ess1H164R mutant cells indicates mRNA readthrough. Thirty cycles were used for PCR. Genomic DNA controls show the efficacy of the primers. Mixtures for control reactions without reverse transcription (no RT) contained 4× as much input template as the experimental samples. (C) Northern analysis of ess1 and ess1 upf1 double mutants and controls showing aberrantly long (readthrough) transcripts. Fifteen micrograms of total RNA was used. The 32P-labeled probes (specific activity, >5 × 108/μg DNA) were generated from the open reading frames of the genes indicated. The numbers refer to the sizes of the open reading frames in base pairs. Both the upstream and downstream gene probes at each locus were used. (D) Additional loci are analyzed as described for panel C, except that only the upstream gene probe for each locus was used. Ten micrograms of total RNA was used for these samples.
To help distinguish CUTs from readthrough transcripts, we used gene-specific (and strand-specific) primers for reverse transcription of RNA that would only allow the detection of forward transcripts (Fig. 7B, schematic). We also used an additional primer set at each locus that allowed the detection of longer fusion transcripts (potential readthroughs) composed of both coding and downstream intergenic sequences. As shown in Fig. 7B, both short intergenic (primer set 1) and long fusion transcripts (primer set 2) are detected at the TAE1-RGD1 locus, and both are stabilized in the NMD-deficient background. In contrast, at the SMK1-SEC8 locus, only the short intergenic transcripts and not the long fusion transcripts are detected, suggesting the presence of a forward-oriented CUT.
The gel data (Fig. 7A and B) illustrate key points, but are only qualitative. We also took a quantitative approach on more than a dozen individual loci using reverse transcription–real-time PCR (Table 2). The results are expressed relative to the results for the wild type and as a ratio of the amount of intergenic transcript relative to the amount of upstream mRNA to normalize for any changes that might occur in the transcription of the gene in mutant backgrounds. For the 14 loci examined, changes in upstream gene expression (data not shown) were minor if any compared with changes in the levels of intergenic transcripts (Table 2). Each sample was also normalized to an internal control gene (SNR6, a pol III product) to control for RNA integrity and real-time amplification efficiency, and the data are the averages of three biological replicates. This approach was very sensitive and detected intergenic transcription in ess1 mutants even when NMD was operational. However, when NMD was inactivated, the levels of intragenic/readthrough transcription detected in ess1 mutants increased dramatically (Table 2).
Table 2.
Results of quantitative reverse transcription–real-time PCR across selected intergenic regions
| Locus | cDNA primera | Primer set | Normalized ΔCT value ± SDb |
Interpretation | ||
|---|---|---|---|---|---|---|
| ess1H164R | upf1Δ | ess1H164R upf1Δ | ||||
| PTP1-SSB1 | RH | 1 | 0.34 ± 0.56 | 2.45 ± 5.82 | 0.58 ± 0.88 | No aberrant transcripts |
| GUS1-RTF1 | RH | 1 | 0.40 ± 0.99 | 6.59 ± 3.93 | 3.68 ± 1.24 | |
| TAE1-RGD1 | RH | 1 | 11.79 ± 1.05 | 9.85 ± 0.39 | 68.95 ± 1.02 | Readthrough |
| GSS | 1 | 6.37 ± 0.75 | 3.74 ± 1.12 | 226.76 ± 0.66 | ||
| GSS | 2 | 24.20 ± 1.18 | 12.01 ± 1.56 | 1,166.80 ± 0.51 | ||
| SMK1-SEC8 | RH | 1 | 18.77 ± 1.03 | 4.54 ± 1.30 | 28.80 ± 1.03 | CUT |
| GSS | 1 | 2.36 ± 0.38 | ND | 9.63 ± 0.39 | ||
| GSS | 2 | ND | ND | ND | ||
| KTR3-FTH1 | RH | 1 | 27.54 ± 0.54 | 8.37 ± 1.63 | 58.42 ± 0.55 | CUT (reverse) and readthrough |
| GSS | 1 | 3.63 ± 0.76 | 6.61 ± 1.89 | 42.23 ± 0.89 | ||
| GSS | 2 | 2.64 ± 0.79 | 4.75 ± 1.65 | 24.30 ± 0.77 | ||
| RNQ1-FUS1 | RH | 1 | 92.41 ± 1.10 | 10.57 ± 2.79 | 192.45 ± 0.66 | CUT (reverse) and readthrough |
| GSS | 1 | 9.49 ± 0.60 | 5.19 ± 1.73 | 191.00 ± 0.69 | ||
| GSS | 2 | 77.35 ± 0.53 | 18.16 ± 1.30 | 1,103.86 ± 0.34 | ||
| TEF2-MUD1 | RH | 1 | 594.97 ± 1.08 | 3.69 ± 1.42 | 7,152.18 ± 4.05 | CUT (forward) and readthrough |
| GSS | 1 | 168.90 ± 0.93 | 5.17 ± 1.46 | 5,887.07 ± 1.18 | ||
| GSS | 2 | 435.54 ± 1.51 | 9.17 ± 1.66 | 9,079.10 ± 1.57 | ||
| AIM5-TAE1 | RH | 1 | 1.23 ± 1.42 | 2.59 ± 2.26 | 3.44 ± 0.95 | Readthrough |
| GSS | 1 | 0.37 ± 1.18 | 1.84 ± 4.36 | 12.29 ± 2.20 | ||
| GSS | 2 | 1.50 ± 0.97 | 6.53 ± 4.88 | 55.66 ± 2.07 | ||
| YPR114W-RGC1 | RH | 1 | 91.35 ± 0.33 | 4.07 ± 0.77 | 12,677.66 ± 1.31 | CUT (reverse) |
| GSS | 1 | 1.27 ± 0.56 | 2.19 ± 4.13 | 9.84 ± 0.84 | ||
| GSS | 2 | 1.05 ± 0.92 | 1.57 ± 6.66 | 1.26 ± 0.42 | ||
| HMG2-LEU3 | RH | 1 | 3.47 ± 0.41 | 5.68 ± 2.22 | 15.21 ± 1.20 | Possible (minor) readthrough |
| GSS | 1 | 0.84 ± 0.49 | 0.92 ± 0.67 | 3.18 ± 1.25 | ||
| GSS | 2 | 0.33 ± 0.49 | 1.85 ± 1.01 | 11.24 ± 0.14 | ||
| FIG4-PFA3 | RH | 1 | 1.84 ± 1.72 | 2.65 ± 1.13 | 9.18 ± 2.60 | CUT (forward) |
| GSS | 1 | 255.41 ± 0.80 | 142.35 ± 1.44 | 5,395.35 ± 0.94 | ||
| GSS | 2 | ND | ND | ND | ||
| SLM1-SHQ1 | RH | 1 | 34.86 ± 1.02 | 2.90 ± 4.49 | 66.48 ± 1.13 | CUT (forward) |
| GSS | 1 | 17.63 ± 1.34 | 3.25 ± 4.90 | 218.40 ± 1.58 | ||
| GSS | 2 | ND | ND | ND | ||
| YLR173W-IDP2 | RH | 1 | 1.69 ± 0.38 | 4.17 ± 3.13 | 5.00 ± 0.77 | Readthrough |
| GSS | 1 | 0.75 ± 0.58 | 3.39 ± 4.08 | 10.60 ± 0.96 | ||
| GSS | 2 | 1.85 ± 0.16 | 25.05 ± 3.96 | 105.85 ± 0.37 | ||
| SKM1-MSB4 | RH | 1 | 1.11 ± 1.02 | 21.61 ± 5.69 | 13.67 ± 0.52 | CUT (forward) revealed by upf1Δ |
| GSS | 1 | 0.81 ± 1.02 | 29.45 ± 3.36 | 13.79 ± 0.78 | ||
| GSS | 2 | ND | ND | ND | ||
RH, random hexamers; GSS, gene and strand specific.
All data are expressed as the ratio over the data for the wild type. Data have been normalized to SNR6 expression (to control for RNA and cDNA integrity and qRT-PCR efficiency) and to a region within the 5′ coding region of the upstream gene (to control for any potential changes in mRNA levels in the mutant backgrounds). See Materials and Methods for details. Primer set locations are approximately as depicted in Fig. 7. cDNA synthesis was primed using either random hexamers or gene-specific primers oriented so as to template only forward transcripts, as indicated. Random-hexamer-primed cDNAs typically resulted in stronger signals than cDNA made using a single gene-specific primer (i.e., using the same PCR primer sets). N.B., for the FIG4-PFA3 GSS primer set 1 samples, since WT readthrough was undetectable, we set the ΔCT(control) value to 37 cycles, which is average for samples with no readthrough. ND, none detected.
Of the genes analyzed, almost half (6 or 7 of 14) had transcript profiles indicative of readthrough transcription (e.g., HMG2, TAE1, TEF2, KTR3, RNQ1, AIM5, and YLR173W), suggesting that Ess1 is required for efficient termination of a substantial number of mRNA genes. These results cannot be the result of overall changes in the expression of these genes in the mutant strains, as this was accounted for by the normalization to upstream products. Interestingly, in ESS1+ cells in which NMD is inactivated, some readthrough is also detected (e.g., TAE1 and KTR3), indicating that a basal level of readthrough occurs even in wild-type cells but these transcripts are normally degraded. CUTs were also prevalent in ess1 backgrounds (8 of 14 loci). Some occurred at loci that were also exhibiting hallmarks of mRNA readthrough (3 of 8). CUTs also fell into two groups, those that are stabilized in the NMD-deficient background (e.g., YPR114W-RGC1) and those that are not (e.g., SLM1-SHQ1). The data indicate that some CUTs are subject to degradation by NMD. For both qualitative and quantitative experiments, controls in which no reverse transcriptase was added to the RNA samples ruled out potential DNA contamination (Fig. 7B and data not shown).
To confirm the qRT-PCR results and to investigate the nature of the readthrough products, we carried out Northern analysis on a subset of the above-mentioned genes. Consistent with the qRT-PCR results, we detected aberrantly long transcripts in ess1ts mutants whose abundance increased in the NMD mutant background (ess1ts upf1) (Fig. 7C). These transcripts were more prominent at the restrictive temperature (37°C) (Fig. 7C and D) than at 30°C (data not shown), as expected. To confirm that the longer transcripts were indeed readthrough products (i.e., fusion transcripts), duplicate blots were probed with the downstream genes (RGD1 and FTH1) (Fig. 7C). The same aberrant products were detected using the downstream probes, and their sizes were consistent with termination occurring after the downstream gene. Longer exposures of these gels revealed upward smears of signal from the readthrough bands, suggesting the presence of additional readthrough transcripts with a range of extended 3′ ends (data not shown). Readthrough transcripts were also observed for three additional loci that were examined (Fig. 7D).
Finally, to ensure that transcription readthrough was not simply a consequence of slow growth at 30°C of the ess1 upf1 double mutants, we also analyzed six other slow-growing strains, obtained from the C. Boone ts collection (University of Toronto), by qRT-PCR. Five of the six mutants showed no evidence of readthrough defects on seven different loci examined (data not shown). Mutations in these strains (lcb1-1, kar2-159, hts1-1, pri2-1, and bos1-1 mutants) are unrelated to transcription or mRNA processing. The sixth mutant (srm1-ts) did show increased intergenic transcription relative to the amount in the wild type. However, this was not entirely surprising given that SRM1, also known as PRP20 (for pre-mRNA processing), is required for mRNA biogenesis and nucleocytoplasmic trafficking and, like ESS1, interacts genetically with the NMD pathway (UPF2) (13).
On the basis of these results, we suggest that Ess1 is required for efficient termination of the transcription of a sizable fraction (perhaps half) of mRNA genes in vivo and that the bulk of aberrant transcripts produced in ess1 mutants are degraded by NMD. Genome-wide double-mutant analysis would give us a better indication of the total number of genes whose transcription termination is Ess1 dependent.
Ess1 promotes H3K4 trimethylation.
The results described above show that Ess1 targets the phosphorylated Ser5-CTD form of RNA pol II in vivo. pSer5-CTD levels are highest near the 5′ ends of active genes, and a number of components of the histone modification machinery seem to require pSer5-CTD for binding (18, 42). We therefore asked whether Ess1 might play a role in histone modification, for example, by altering the conformation of the CTD and/or influencing its phosphorylation state, thereby effecting the recruitment or activity of histone-modifying enzymes. We first took a genetic approach, using mutants with mutations of the Set1 and Set2 histone methyltransferases. Set1, as part of the COMPASS complex, generates a trimethylation mark, H3K4me3, which is associated with the 5′ ends of active genes (reviewed in reference 50). Importantly, this mark has been also associated with Nrd1-dependent termination (55). Set2 trimethylates H3K36, and the modified histone helps recruit the Rpd3S deacetylase complex in the wake of elongating polymerase to help reform repressive chromatin to prevent cryptic transcription (10, 25).
We found that ess1H164R set1Δ double mutants showed synthetic slow growth/lethality, while high-copy-number expression of Set1 rescued the growth defect of ess1H164R cells at 37°C (Fig. 8). These results lead to the inference that Ess1 positively affects H3K4 methylation. If true, then in ess1 mutant cells, H3K4 methylation would decrease, leading to slow growth, and this growth defect should be reversed by mutation of the H3K4 demethylase, Jhd2. Indeed, jhd2Δ partially suppressed the ess1 growth defect at 37°C (Fig. 8). In contrast, the ess1H164R set2Δ double mutants showed genetic suppression whereby set2Δ rescued the growth defect of ess1 mutants (Fig. 8, bottom).
Fig 8.
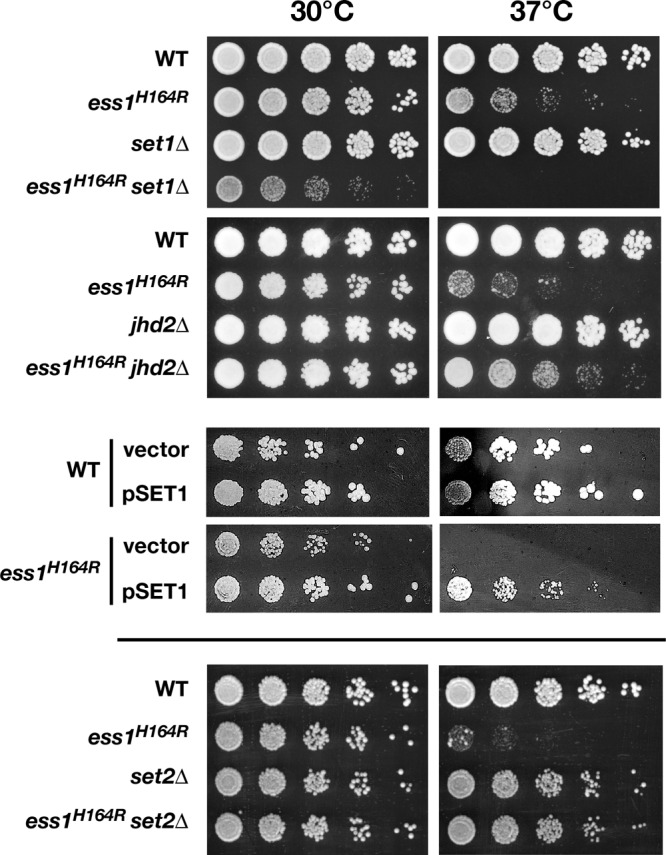
Genetic interactions between ESS1 and SET1/JHD2 suggest that Ess1 is required for histone H3K4 methylation. Serial dilutions (1:5) starting at a 1:5 dilution (except for set1 mutant cells shown in the top panels, which started at 1:1) of wild-type or mutant cells at an OD600 of 0.5 were grown at 30°C or 37°C on YEPD for 2 to 3 days or, for the experiments using a plasmid overexpressing SET1 (pSET1), on CSM minus uracil (49) for 4 days. Deletion of the gene encoding the Set1 histone methyltransferase is synthetic lethal with ess1H164R, whereas deletion of JHD2, the gene encoding the H3K4 demethylase, rescues ess1H164R cells. The plasmid that overexpresses SET1 (pMP803-ADH/SET1) suppresses the growth defects of ess1H164R cells at permissive (30°C) and nonpermissive (37°C) temperatures, but the control vector (pRS416) does not. A set2Δ mutant rescues growth of ess1H164 cells at 37°C.
To determine whether Ess1 affects H3K4 methylation, we used Western analysis to compare the bulk H3K4me3 levels in wild-type and ess1 mutant cells (Fig. 9A). In ess1H164R and ess1Δ srb10Δ mutants, H3K4me3 levels were reduced relative to the levels in their respective control strains (the wild type and srb10Δ). No change occurred in the overall levels of either H3K36me3 or H3 and tubulin in the controls. We also monitored H3K4me3 and H3K36me3 levels across individual genes using ChIP and found significant reductions in H3K4me3 in ess1 mutants on two out of three loci examined (TEF2-MUD1 and SRG1-SER3 but not PYK1), suggesting that Ess1 promotes H3K4 trimethylation (Fig. 9B). No changes were observed for H3K36me3. Together, the genetic and molecular analyses indicate that Ess1 plays a role in histone modification and that it affects some modifications more than others.
Fig 9.

Levels of H3K4me3 are reduced in ess1 mutants. (A) Western analysis using protein from whole-cell extracts probed with the indicated antibodies (see Materials and Methods for details). Cells were grown at 30°C. Bulk levels of H3K4me3 are reduced in ess1 mutants. Levels of H3K36 methylation were unchanged, as were overall levels of histone H3. Tubulin was used as a loading control to assess protein integrity and concentration. Equal amounts of protein (10 μg) were loaded in all wells. Blots were generated individually and not reprobed. (B) ChIP analysis to monitor levels of H3K4me3 in ess1 mutants along individual gene loci TEF2-MUD1 and SRG1-SER3. Levels of H3K4me3 but not H3K36me3 are reduced across ncRNA loci. No effect was observed at PYK1. Fold changes are relative to the results for H3 after normalization to a chromosome V control (H3K36me3) or a tRNA gene control (H3K4me3). Error bars show standard deviations of three biological replicates.
DISCUSSION
Covalent and noncovalent modifications of the CTD.
In this work, we monitored the impact of Ess1 mutations on transcription, CTD phosphorylation, and the recruitment of cofactors to transcribed genes. Although the isomerization state of the CTD cannot be measured directly in vivo, we detected widespread transcription defects in cells in which the catalytic activity of the Ess1 enzyme is compromised. The most commonly used allele, ess1H164R, which is temperature sensitive for growth, has less than 0.01% of the activity of the wild type on phospho-CTD peptides in vitro (17) and is likely to cause substantial alterations in CTD structure and function in vivo. Consistent with a role for Ess1 in regulating RNA pol II activity, we could immunoprecipitate pol II (mostly the pSer5 form) with Ess1 (Fig. 1), and using ChIP, we showed that Ess1 is recruited to the 5′, middle, and 3′ regions of the protein-coding genes examined (Fig. 5).
Ess1 is required to keep CUTs silent.
A previous tiling array study showed a dramatic increase in the number of CUTs in ess1 mutants, due in part to faulty transcription termination (and stabilization) of small ncRNAs (51). In this work, ChIP analysis of ess1 mutants at selected CUT/SUT loci (Fig. 2 and 3) showed increased recruitment of the Ser5-phosphorylated form of RNA pol II, as well as initiation factors TBP and TFIIB and the 5′-capping enzyme Ceg1. This result suggests that, in addition to being required for CUT termination, Ess1 represses the initiation of CUTs and, similarly, keeps SUTs from being overexpressed. The mechanism is not yet clear, but it could be due to direct effects of Ess1 on CTD structure that affect the binding of initiation factors or to indirect effects through changes in CTD phosphorylation or chromatin structure (see below).
A role for Ess1 in initiation was previously suggested by Krishnamurthy et al. (27), based on its strong genetic interaction with SUA7, which encodes TFIIB. These authors found ess1 mutants to be defective for the induction of GAL1, PHO5, and INO1 reporters (although they did not formally demonstrate that this was due to initiation defects). We found no effect of Ess1 on the recruitment of initiation factors to a control mRNA gene, PYK1. One difference is that PYK1 is constitutively expressed, whereas the genes studied by Krishnamurthy et al. are highly inducible. Thus, the function of Ess1 during initiation may vary depending on the mRNA gene, probably because of the requirements for different combinations of regulators at individual promoters.
Ess1 is important for mRNA termination.
Early studies recovered Ess1 (Ptf1) in a genetic screen for defective 3′-end processing/termination (21, 22). Later work showed readthrough of reporter constructs in ess1 mutant cells (27, 64). However, none of these studies clearly demonstrated readthrough of chromosomal genes. Even genome-wide tiling array analysis revealed only a small number of mRNAs exhibiting readthrough in ess1 mutants (51). Perhaps the nuclear and/or cytoplasmic RNA quality control mechanisms (reviewed in references 16 and 39) were degrading the readthrough transcripts in ess1 mutants, explaining why they eluded detection. Indeed, inactivating NMD, the major RNA surveillance system, revealed strong readthrough in ess1 mutants in almost half of the chromosomal mRNA genes tested (Fig. 7 and Table 2). Although the results were complicated by the induction and stabilization of intergenic CUTs (in ess1 mutants), we could distinguish these from transcription readthrough and showed that Ess1 is required for efficient mRNA termination in yeast. It is likely that some mRNA readthrough products are subject to degradation by pathways other than NMD (e.g., Rrp6/nuclear exosome), and thus, our estimate of about half of mRNA genes being dependent on Ess1 for efficient termination might be an underestimate.
While we do not know how readthrough transcripts in ess1 mutants reach the cytoplasm, the site of NMD-mediated RNA decay, there are several possibilities. Termination and polyadenylation may occur in the downstream gene at cryptic or normal poly(A) sites. In this case, the transcripts would be exported from the nucleus and would trigger NMD due to premature termination codons encoded in the intergenic regions. Indeed, our Northern analysis, which detected discrete-length readthrough transcripts, was consistent with this idea. Second, it is possible that in ess1 mutants, the normal RNA quality control mechanisms for export are defective or are saturated by widespread mRNA readthrough products, thereby allowing the defective transcripts to enter the cytoplasm for degradation. There was also evidence for transcripts with heterogeneous 3′ ends in ess1 upf1 double mutants, as detected by Northern analysis. It is possible that these are incorrectly processed mRNAs.
The defect in mRNA termination in ess1 mutants is probably due to a failure to coordinate the recruitment and release of key termination factors. Pcf11 recruitment to the 3′ end of several genes was reduced in ess1 mutants (Fig. 4), while the levels of Nrd1, which is not known to function in mRNA termination, increased, perhaps inhibiting the process. Both proteins are sensitive to the phosphorylation state of the CTD, with Nrd1 preferring the pSer5 form and Pcf11 preferring the pSer2 form (59). Ess1's effect on their recruitment may therefore be due to changes in CTD phosphorylation, with ess1 mutants showing increased levels of pSer5 (27, 51) and pSer7 (Fig. 1). Bataille et al. showed by genome-wide ChIP with microarray technology (ChIP-chip) analysis that both pSer5 and pSer7 levels increased significantly at the 3′ ends of nearly all protein-coding genes in ess1 mutants (6). Interestingly, pSer7 in human cells is important for the recruitment of the RPAP2 Ser5-specific phosphatase (Rtr1 in yeast) to small nuclear RNA genes, also suggesting a functional linkage between these two marks that may also be critical for termination, although a distinct mechanism seems to operate at mRNA genes (15).
Ess1 probably increases the activity of the Ssu72 phosphatase, which prefers the cis form of CTD peptide substrates (61) and targets both pSer5 and pSer7 in the CTD (6). We found no significant changes in the levels of Ssu72 at several loci in ess1 mutants (Fig. 4 and data not shown), suggesting that Ess1 affects its activity on its substrate(s) but not its recruitment. Many other factors are required for mRNA termination and 3′-end processing in yeast (28), and genetic studies suggest that at least some are influenced by Ess1 (C. Barnes, J. Alafifi, and S. Hanes, unpublished observations).
Ess1 is important for H3K4 methylation.
Prompted by close linkages between histone modifiers and the CTD (e.g., see references 18 and 42; reviewed in reference 8) and prior studies linking ess1 to the rpd3 histone deacetylase and Gcn5 histone acetyltransferase (4), we investigated the link between Ess1 and two additional histone modifiers, Set1 and Set2. The Set1 H3K4 histone methyltransferase specifically targets the pSer5 form of RNA polymerase and is associated with active transcription (42). The Set2 H3K36 methyltransferase, which contains a WW domain, binds the doubly phosphorylated form (pSer2/pSer5) and is associated with elongating polymerases (30, 44). Our results indicate a functional link between Ess1 and histone H3K4 methylation by Set1 (Fig. 8 and 9). In contrast, although we found strong genetic interactions between ESS1 and SET2 (Fig. 8), we did not detect changes in H3K36 trimethylation (Fig. 9).
Ess1 may help recruit Set1 to the early form of elongating polymerase. This may occur directly, by conformation-induced changes in CTD structure, or indirectly, through Ess1's effect on Ser5 CTD phosphorylation or, potentially, on other histone modifications. For example, ess1 is synthetic lethal with a deletion of RAD6 (data not shown), which encodes a ubiquitin-conjugating enzyme (E2) that monoubiquitylates histone H2B at lysine 123 (45). H2B K123 ubiquitylation is coupled to H3K4 trimethylation by Set1 (40, 54). If indeed Ess1 is important for Rad6 activity, then loss of Ess1 would indirectly reduce H3K4 methylation. This would be consistent with genetic results that show the opposite genetic relationship between ess1H164R and set1Δ (synthetic lethality) versus that of ess1H164R and set2Δ (rescue), since Rad6-dependent ubiquitylation of H2B is positively associated with Set1-dependent H3K4 trimethylation but negatively associated with Set2-dependent H3K36 activity (23). Finally, it is possible that Ess1 acts directly on Set1 protein or other subunits of the COMPASS complex to stimulate their activity. Localization of Set1p by ChIP in ess1 mutants may help distinguish between these models.
Two recent studies are of special interest. First, Wang et al. (60) found that H3K4me3 can prevent promoter activation by recruitment of the Rpd3L deacetylase complex. When H3K4me3 levels are reduced, Rpd3L is not recruited and basal transcription of the PHO5 promoter is derepressed (i.e., in under high-Pi conditions), probably due to increased H3 and H4 acetylation and a more open chromatin structure. A similar mechanism could occur at CUT loci in ess1 mutants, which show decreased H3K4me3 levels (Fig. 9) and increased recruitment of pol II and initiation factors (Fig. 2 and 3). Thus, loss of Ess1 function would reduce H3K4me3 and would lower histone deacetylase (Rpd3L) recruitment at cryptic promoters and derepress their transcription.
In the second study, Terzi et al. (55) showed that deletion of SET1 reduced Nrd1 recruitment, resulting in transcription readthrough of snoRNAs and CUTs. Their results suggested a model in which Set1 H3K4 trimethylation recruits RpdL, as well as the NuA3 histone acetyltransferase, and that the proper balance of acetylation at these promoters is necessary to load the Nrd1-Nab3-Sen1 termination complex onto the elongating polymerase. We also see readthrough of snoRNAs and CUTs in ess1 mutants (51), as well as decreased H3K4me3 (Fig. 9). However, we do not see reduced Nrd1 recruitment but, rather, results consistent with a failure of exchange of Nrd1 (increased levels) and Pcf11 (decreased levels) (Fig. 4) (51). Perhaps this indicates that the rate of exchange of termination factors, facilitated by CTD isomerization, is as important as the absolute levels of recruitment. Interestingly, Terzi et al. (55) found no effect of set1Δ on mRNA termination, suggesting that the mRNA readthrough defects we detect in ess1 mutants are probably not mediated through changes in levels of H3K4 trimethylation.
Ess1 function in termination might be conserved.
The biochemical activities of Ess1 and Pin1 are similar (72), and the fact that PIN1 genes from Drosophila (34), Xenopus (C. B. Wilcox and S. D. Hanes, unpublished), and humans (32) rescue ess1Δ mutants indicates a conserved in vivo function. Preliminary experiments in human (HeLa) cells suggest that small interfering RNA knockdown of Pin1 may cause readthrough of snoRNAs (A. Burch, Z. Ma, S. Hanes, data not shown). Prior studies showed Pin1-dependent inhibition of transcription in vitro (69), and overexpression of Pin1 increased CTD phosphorylation levels on Ser5 (68). This later effect was opposite to that observed in yeast (overexpression of Ess1 decreased pSer5 [51]); however, it nonetheless supports the idea that Ess1 controls RNA pol II function in all eukaryotes. It remains to be determined whether Pin1 is important for mRNA termination. Further studies should provide the answers.
ACKNOWLEDGMENTS
We are grateful to David Brow, Joan Curcio, Steve Buratowski, Brian Dichtl, Dirk Eick, Mike Hampsey, Caroline Kane, Ian Willis, and Anthony Weil for plasmids, strains, or antibodies, to the Wadsworth Center Molecular Genetics and Media facilities, and to Dave Amberg, Randy Morse, Navjot Singh, and Joe Wade for helpful discussions. We thank Taylor Moon (SUNY—Upstate Summer Undergraduate Fellow) for pilot qRT-PCR experiments and April Burch (Wadsworth Center) for Pin1 experiments.
S.D.H. dedicates this paper to the memory of his mother, Helen Hanes, who first introduced him to research at the famed Bell Telephone Laboratories in Murray Hill, NJ.
This work was funded by grants to S.D.H. from the NIH (grants 3R01-GM055108 and ARRA-GM055108-S1), the NSF (grant MCB-0613001), and SUNY—Upstate Medical University.
Footnotes
Published ahead of print 9 July 2012
REFERENCES
- 1. Amrani N, et al. 2006. Aberrant termination triggers nonsense-mediated mRNA decay. Biochem. Soc. Trans. 34: 39–42 [DOI] [PubMed] [Google Scholar]
- 2. Amrani N, et al. 1997. PCF11 encodes a third protein component of yeast cleavage and polyadenylation factor I. Mol. Cell. Biol. 17: 1102–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrecka J, et al. 2008. Single-molecule tracking of mRNA exiting from RNA polymerase II. Proc. Natl. Acad. Sci. U. S. A. 105: 135–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arevalo-Rodriguez M, Cardenas ME, Wu X, Hanes SD, Heitman J. 2000. Cyclophilin A and Ess1 interact with and regulate silencing by the Sin3-Rpd3 histone deacetylase. EMBO J. 19: 3739–3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arevalo-Rodriguez M, Wu X, Hanes SD, Heitman J. 2004. Prolyl isomerases in yeast. Front. Biosci. 9: 2420–2446 [DOI] [PubMed] [Google Scholar]
- 6. Bataille AR, et al. 2012. A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase and isomerase enzymes along genes. Mol. Cell 45: 158–170 [DOI] [PubMed] [Google Scholar]
- 7. Buratowski S. 2003. The CTD code. Nat. Struct. Biol. 10: 679–680 [DOI] [PubMed] [Google Scholar]
- 8. Buratowski S. 2009. Progression through the RNA polymerase II CTD cycle. Mol. Cell 36: 541–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buratowski S, Kim T. 2010. The role of cotranscriptional histone methylations. Cold Spring Harb. Symp. Quant. Biol. 75: 95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carrozza MJ, et al. 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123: 581–592 [DOI] [PubMed] [Google Scholar]
- 11. Chapman RD, et al. 2007. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 318: 1780–1782 [DOI] [PubMed] [Google Scholar]
- 12. Conrad NK, et al. 2000. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics 154: 557–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Costanzo M, et al. 2010. The genetic landscape of a cell. Science 327: 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Egloff S, Murphy S. 2008. Cracking the RNA polymerase II CTD code. Trends Genet. 24: 280–288 [DOI] [PubMed] [Google Scholar]
- 15. Egloff S, Zaborowska J, Laitem C, Kiss T, Murphy S. 2012. Ser7 phosphorylation of the CTD recruits the RPAP2 Ser5 phosphatase to snRNA genes. Mol. Cell 45: 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fasken MB, Corbett AH. 2009. Mechanisms of nuclear mRNA quality control. RNA Biol. 6: 237–241 [DOI] [PubMed] [Google Scholar]
- 17. Gemmill TR, Wu X, Hanes SD. 2005. Vanishingly low levels of Ess1 prolyl-isomerase activity are sufficient for growth in Saccharomyces cerevisiae. J. Biol. Chem. 280: 15510–15517 [DOI] [PubMed] [Google Scholar]
- 18. Govind CK, et al. 2010. Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol. Cell 39: 234–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Halbach A, et al. 2009. Cotranslational assembly of the yeast SET1C histone methyltransferase complex. EMBO J. 28: 2959–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hanes SD, Shank PR, Bostian KA. 1989. Sequence and mutational analysis of ESS1, a gene essential for growth in Saccharomyces cerevisiae. Yeast 5: 55–72 [DOI] [PubMed] [Google Scholar]
- 21. Hani J, et al. 1999. Mutations in a peptidylprolyl-cis/trans-isomerase gene lead to a defect in 3′-end formation of a pre-mRNA in Saccharomyces cerevisiae. J. Biol. Chem. 274: 108–116 [DOI] [PubMed] [Google Scholar]
- 22. Hani J, Stumpf G, Domdey H. 1995. PTF1 encodes an essential protein in Saccharomyces cerevisiae, which shows strong homology with a new putative family of PPIases. FEBS Lett. 365: 198–202 [DOI] [PubMed] [Google Scholar]
- 23. Henry KW, et al. 2003. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 17: 2648–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hirose Y, Ohkuma Y. 2007. Phosphorylation of the C-terminal domain of RNA polymerase II plays central roles in the integrated events of eucaryotic gene expression. J. Biochem. 141: 601–608 [DOI] [PubMed] [Google Scholar]
- 25. Keogh MC, et al. 2005. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123: 593–605 [DOI] [PubMed] [Google Scholar]
- 26. Kim H, et al. 2010. Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat. Struct. Mol. Biol. 17: 1279–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krishnamurthy S, Ghazy MA, Moore C, Hampsey M. 2009. Functional interaction of the Ess1 prolyl isomerase with components of the RNA polymerase II initiation and termination machineries. Mol. Cell. Biol. 29: 2925–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuehner JN, Pearson EL, Moore C. 2011. Unravelling the means to an end: RNA polymerase II transcription termination. Nat. Rev. Mol. Cell. Biol. 12: 283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leeds P, Peltz SW, Jacobson A, Culbertson MR. 1991. The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev. 5: 2303–2314 [DOI] [PubMed] [Google Scholar]
- 30. Li J, Moazed D, Gygi SP. 2002. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J. Biol. Chem. 277: 49383–49388 [DOI] [PubMed] [Google Scholar]
- 31. Li Z, et al. 2011. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat. Biotechnol. 29: 361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu KP, Hanes SD, Hunter T. 1996. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380: 544–547 [DOI] [PubMed] [Google Scholar]
- 33. Lu KP, Zhou XZ. 2007. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8: 904–916 [DOI] [PubMed] [Google Scholar]
- 34. Maleszka R, Hanes SD, Hackett RL, de Couet HG, Miklos GL. 1996. The Drosophila melanogaster dodo (dod) gene, conserved in humans, is functionally interchangeable with the ESS1 cell division gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 93: 447–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martens JA, Wu PY, Winston F. 2005. Regulation of an intergenic transcript controls adjacent gene transcription in Saccharomyces cerevisiae. Genes Dev. 19: 2695–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mayer A, et al. 2010. Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 17: 1272–1278 [DOI] [PubMed] [Google Scholar]
- 37. Meinhart A, Kamenski T, Hoeppner S, Baumli S, Cramer P. 2005. A structural perspective of CTD function. Genes Dev. 19: 1401–1415 [DOI] [PubMed] [Google Scholar]
- 38. Morris DP, Phatnani HP, Greenleaf AL. 1999. Phospho-carboxyl-terminal domain binding and the role of a prolyl isomerase in pre-mRNA 3′-end formation. J. Biol. Chem. 274: 31583–31587 [DOI] [PubMed] [Google Scholar]
- 39. Muhlemann O, Eberle AB, Stalder L, Zamudio Orozco R. 2008. Recognition and elimination of nonsense mRNA. Biochim. Biophys. Acta 1779: 538–549 [DOI] [PubMed] [Google Scholar]
- 40. Nakanishi S, et al. 2009. Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J. Cell Biol. 186: 371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neil H, et al. 2009. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457: 1038–1042 [DOI] [PubMed] [Google Scholar]
- 42. Ng HH, Robert F, Young RA, Struhl K. 2003. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell 11: 709–719 [DOI] [PubMed] [Google Scholar]
- 43. Phatnani HP, Greenleaf AL. 2006. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20: 2922–2936 [DOI] [PubMed] [Google Scholar]
- 44. Phatnani HP, Jones JC, Greenleaf AL. 2004. Expanding the functional repertoire of CTD kinase I and RNA polymerase II: novel phosphoCTD-associating proteins in the yeast proteome. Biochemistry 43: 15702–15719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Robzyk K, Recht J, Osley MA. 2000. Rad6-dependent ubiquitination of histone H2B in yeast. Science 287: 501–504 [DOI] [PubMed] [Google Scholar]
- 46. Schiene C, Fischer G. 2000. Enzymes that catalyse the restructuring of proteins. Curr. Opin. Struct. Biol. 10: 40–45 [DOI] [PubMed] [Google Scholar]
- 47. Schiene-Fischer C, Aumuller T, Fischer G. 20 May2011. Peptide bond cis/trans isomerases: a biocatalysis perspective of conformational dynamics in proteins. Top. Curr. Chem. [Epub ahead of print.] doi:10.1007/128_2011_151 [DOI] [PubMed] [Google Scholar]
- 48. Schmitt ME, Brown TA, Trumpower BL. 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 18: 3091–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sherman F. 1991. Getting started with yeast. Methods Enzymol. 194: 3–21 [DOI] [PubMed] [Google Scholar]
- 50. Shilatifard A. 2008. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 20: 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Singh N, et al. 2009. The Ess1 prolyl isomerase is required for transcription termination of small non-coding regulatory RNAs via the Nrd1 pathway. Mol. Cell 36: 255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Spain MM, Govind CK. 2011. A role for phosphorylated Pol II CTD in modulating transcription coupled histone dynamics. Transcription 2: 78–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steinmetz EJ, Brow DA. 1996. Repression of gene expression by an exogenous sequence element acting in concert with a heterogeneous nuclear ribonucleoprotein-like protein, Nrd1, and the putative helicase Sen1. Mol. Cell. Biol. 16: 6993–7003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun ZW, Allis CD. 2002. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418: 104–108 [DOI] [PubMed] [Google Scholar]
- 55. Terzi N, Churchman LS, Vasiljeva L, Weissman J, Buratowski S. 2011. H3K4 trimethylation by Set1 promotes efficient termination by the Nrd1-Nab3-Sen1 pathway. Mol. Cell. Biol. 31: 3569–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Theuerkorn M, Fischer G, Schiene-Fischer C. 2011. Prolyl cis/trans isomerase signalling pathways in cancer. Curr. Opin. Pharmacol. 11: 281–287 [DOI] [PubMed] [Google Scholar]
- 57. Thomas BJ, Rothstein R. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56: 619–630 [DOI] [PubMed] [Google Scholar]
- 58. Tietjen JR, et al. 2010. Chemical-genomic dissection of the CTD code. Nat. Struct. Mol. Biol. 17: 1154–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vasiljeva L, Kim M, Mutschler H, Buratowski S, Meinhart A. 2008. The Nrd1-Nab3-Sen1 termination complex interacts with the Ser5-phosphorylated RNA polymerase II C-terminal domain. Nat. Struct. Mol. Biol. 15: 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang SS, Zhou BO, Zhou JQ. 2011. Histone H3 lysine 4 hypermethylation prevents aberrant nucleosome remodeling at the PHO5 promoter. Mol. Cell. Biol. 31: 3171–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Werner-Allen JW, et al. 2011. cis-Proline-mediated Ser(P)5 dephosphorylation by the RNA polymerase II C-terminal domain phosphatase Ssu72. J. Biol. Chem. 286: 5717–5726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. West ML, Corden JL. 1995. Construction and analysis of yeast RNA polymerase II CTD deletion and substitution mutations. Genetics 140: 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilcox CB, Rossettini A, Hanes SD. 2004. Genetic interactions with C-terminal domain (CTD) kinases and the CTD of RNA Pol II suggest a role for ESS1 in transcription initiation and elongation in Saccharomyces cerevisiae. Genetics 167: 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wu X, Rossettini A, Hanes SD. 2003. The ESS1 prolyl isomerase and its suppressor BYE1 interact with RNA pol II to inhibit transcription elongation in Saccharomyces cerevisiae. Genetics 165: 1687–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu X, et al. 2000. The Ess1 prolyl isomerase is linked to chromatin remodeling complexes and the general transcription machinery. EMBO J. 19: 3727–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wyers F, et al. 2005. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121: 725–737 [DOI] [PubMed] [Google Scholar]
- 67. Xiang K, et al. 2010. Crystal structure of the human symplekin-Ssu72-CTD phosphopeptide complex. Nature 467: 729–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu YX, Hirose Y, Zhou XZ, Lu KP, Manley JL. 2003. Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 17: 2765–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu YX, Manley JL. 2007. Pin1 modulates RNA polymerase II activity during the transcription cycle. Genes Dev. 21: 2950–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yeh ES, Means AR. 2007. PIN1, the cell cycle and cancer. Nat. Rev. Cancer 7: 381–388 [DOI] [PubMed] [Google Scholar]
- 71. Yu C, Palumbo MJ, Lawrence CE, Morse RH. 2006. Contribution of the histone H3 and H4 amino termini to Gcn4p- and Gcn5p-mediated transcription in yeast. J. Biol. Chem. 281: 9755–9764 [DOI] [PubMed] [Google Scholar]
- 72. Zhang Y, Fussel S, Reimer U, Schutkowski M, Fischer G. 2002. Substrate-based design of reversible Pin1 inhibitors. Biochemistry 41: 11868–11877 [DOI] [PubMed] [Google Scholar]