

Abstract
Summary: The human respiratory tract is an entry point for over 200 known viruses that collectively contribute to millions of annual deaths worldwide. Consequently, the World Health Organization has designated respiratory viral infections as a priority for vaccine development. Despite enormous advances in understanding the attributes of a protective mucosal antiviral immune response, current vaccines continue to fail in effectively generating long‐lived protective CD8+ T‐cell immunity. To date, the majority of licensed human vaccines afford protection against infectious pathogens through the generation of specific immunoglobulin responses. In recent years, the selective manipulation of specific costimulatory pathways, which are critical in regulating T cell‐mediated immune responses, has generated increasing interest. Impressive results in animal models have shown that the tumor necrosis factor receptor (TNFR) family member OX40 (CD134) and its binding partner OX40L (CD252) are key costimulatory molecules involved in the generation of protective CD8+ T‐cell responses at mucosal surfaces, such as the lung. In this review, we highlight these new findings with a particular emphasis on their potential as immunological adjuvants to enhance poxvirus‐based CD8+ T‐cell vaccines.
Keywords: CD8+ T cells, vaccines, influenza, poxviruses, lung, OX40/OX40L
This article is part of a series of reviews covering TNF Receptor Family Members appearing in Volume 244 of Immunological Reviews.
Respiratory viruses: the continuing burden
The mucosal surfaces of respiratory tract provide optimal conditions for efficient gaseous exchange and, unfortunately, an ideal portal of entry for innocuous environmental antigen and human pathogens. Consequently, the body’s respiratory mucosal surfaces contain a complex array of immune regulatory mechanisms that ensures, at least in healthy individuals, a quiescent and non‐inflammatory environment that maintains optimal tissue function (1, 2, 3). However, once a pathogen establishes an infection or the regulatory mechanisms fail, a rapid cascade of event leads to the production of inflammatory cytokines that recruit immune cells in an attempt to eradicate the ensuing pathogen/antigenic stimuli while minimizing the impact on physiological function of the tissue. However, this ideal conclusion often does not occur, as many human pathogens have evolved virulence and immune modulatory mechanisms that circumvent and disrupt mucosal immune responses resulting in tissue pathology, clinical disease, and possible long‐term sequelae.
To help minimize the detrimental impact of respiratory infections, vaccines were developed against many of the most prevalent respiratory pathogens, including Bordatella pertussis (whooping cough), Corynebacterium diphtheriae (diptheria), Haemophilus influenzae type b (Hib), and Streptococcus pneumoniae (pneumococcal pneumonia and otitis media), successfully reducing infant mortality and the burden of infectious diseases worldwide (4, 5, 6). Yet, despite the continued success and widespread use of respiratory pathogen vaccines, Mycobacterium tuberculosis (TB) and a multitude of respiratory viral infections continue to cause significant morbidity and result in millions of futile deaths each year (7, 8, 9) [World Health Organization (WHO) 2004 Global Burden of Disease]. Influenza virus alone causes seasonal epidemics that can affect 10–20% of the global population (10). Recent estimates suggest that seasonal influenza viral infections are responsible for 250,000–500,000 deaths annually, which can increase during pandemics caused by the emergence of a novel reassortment viral strain (WHO 2004 Global Burden of Disease). Furthermore, the increasing number of deaths attributed to human transmission of highly pathogenic avian influenza strains (H5N1) will elevate influenza virus associated morbidity and mortality (11). In addition to influenza virus, parainfluenza virus, respiratory syncytial virus (RSV), meta‐pneumonia virus, severe acute respiratory syndrome coronavirus (SARS‐CoV), rhinovirus, measles, and adenovirus are endemic within the human population and can establish acute respiratory tract infection (11, 12, 13, 14). With a few exceptions, existing approaches have failed to develop effective vaccines against these viral pathogens. Ominously, the public health impact of respiratory infections is likely to increase in the near future due to aging global populations, increasing antibiotic resistance (in the case of TB and pneumococcus) and altering social attitudes toward vaccination (14, 15, 16). Moreover, the continuing emergence of novel respiratory viruses (through antigenic recombination events and zoonosis), such as the 2009 H1N1 influenza A virus strain, highly pathogenic avian influenza viruses, SARS coronavirus and human cases of monkeypox (11, 17, 18), taken together with the continued concern of bioterrorism (anthrax and smallpox) (19, 20) adds to the urgent need to better understand the pathogenesis of respiratory viruses and mechanisms of protection.
This review discusses the rationale for developing CD8+ T‐cell vaccines against existing and emerging human respiratory viruses, and then reviews our current understanding of antigen‐specific CD8+ T‐cell induction and memory formation in the context of respiratory viral infections. The argument will be made that applying this knowledge will be critical in future success of CD8+ T‐cell vaccines. Then, we examine how attenuated poxviruses have been developed over the past three decades as candidate vaccines for a variety of mucosal pathogens and discuss how future efforts should focus on understanding in molecular terms why live non‐attenuated vaccines result in better CD8+ T‐cell immunity. In the final section, we discuss how members of tumor necrosis factor receptor (TNFR)/TNF superfamily, specifically, OX40 (CD134) and its binding partner OX40L (CD252), are rapidly emerging as key players in the development of protective CD8+ T‐cell memory in lung. As such, it will become evident that we already have the means to develop a poxvirus‐based vaccine delivery system that establishes far better protective CD8+ T‐cell immunity in the lung than anything currently available.
The rationale for CD8+ T‐cell vaccines
Most if not all of today’s licensed vaccines work by promoting a robust and long‐lived immunoglobulin response that prevents initial pathogen infection and ensuing replication, resulting in pathogen clearance before the onset of any clinical symptoms. Apparently, this straightforward approach of safely generating neutralizing immunoglobulin via the injection of killed, subunit, or highly attenuated agents has resulted in many successful vaccines that confer life‐long protective immunity (6). However, in the context of highly pathogenic or rapidly mutating viruses that target mucosal surfaces such as the respiratory tract, this approach has proven far less successful (6, 21). The limitations of generating solely immunoglobulin‐mediated protection are highlighted by the necessity to annually develop a seasonal influenza vaccine. The protective mechanism of current subunit or inactivated influenza vaccines is through the generation of neutralizing immunoglobulins against hemagglutinin (HA) and neuraminidase (NA), two integral membrane viral proteins essential for infectivity. Although immunoglobulins against these surface proteins are long‐lived, their functional relevance diminishes rapidly due to antigenic drift in both the HA and NA surface glycoproteins (22, 23). This continual antigen drift or evolution also explains, in part, the difficulty in developing an effective vaccine against other intracellular pathogens, such as human immunodeficiency virus (HIV) and malaria (6). Both of these pathogens continually mutate, in the case of HIV, or alter, in the case of the plasmodium parasite, key surface antigens resulting in immunological escape from any induced‐specific immunoglobulin response. This continued dilemma has led to the general acceptance that to develop effective vaccines against these types of intracellular pathogens, a combination of immunoglobulin and a long‐lasting memory CD8+ T‐cell response must be generated (24, 25, 26). This approach has the added benefit of producing cross‐protective immunity mediated by CD8+ T cells that recognize conserved internal components of the specific pathogens, therefore conferring heterosubtypic immunity (27). This would be an ideal strategy in the context of a virus that rapidly mutated its external antigens while maintaining more conserved internal antigens. Strictly speaking, CD8+ T cells cannot afford protection against infection per se, but can mediate faster viral clearance and provide a substantial degree of protection against challenge with a lethal dose of virus (14). Toward this end, the scientific community has focused enormous efforts on the development of prophylactic and therapeutic vaccines that can promote CD8+ T‐cell responses with the assumption that such a strategy will enhance immunological protection through providing additional support to existing antiviral immunoglobulin response. To date, however, the development of effective T‐cell vaccines against many respiratory viruses remains elusive. In the past 10 years, immunologists have provided vaccinologists with valuable information on the regulatory mechanisms that govern not only the efficient generation of CD8+ T‐cell responses in the lung but also how different memory CD8+ T‐cell subsets are maintained over time. The current challenge is to apply this knowledge in our endeavors to develop safe and effective CD8+ T‐cell vaccines.
Overview of the CD8+ T‐cell response to respiratory viruses
Viruses are obligate intracellular parasites that upon gaining entry into a host‐cell hijack the host’s transcription and protein synthesis machinery to generate progeny virions that initiate the next cycle of infection. Consequently, the respiratory epithelium contains an array of virus detection systems, such as Toll‐like receptors (TLRs), Nod‐like receptors (NLRs), and the cytosolic RNA helicase family members, retinoic acid‐inducible gene I (RIG I) and melanoma differentiation associated gene 5 (MDA5), that act in concert to rapidly detect the presence of an invading virus (14, 28, 29). Upon viral recognition, a complex interplay between these distinct detection systems results in the activation of an array of transcription factors that culminates in the production of antiviral cytokines and inflammatory mediators (14, 28). In addition to establishing a localized inflammatory environment and recruiting innate effector cells, this initial antiviral inflammatory program is important for the activation and differentiation of lung‐resident antigen‐presenting cells (APCs), namely dendritic cells (DCs) and macrophages (30).
Much of our current understanding of CD8+ T‐cell immunity stems from studying small rodent influenza and parainfluenza infection models (14, 31, 32). These models have shown the crucial role CD8+ T cells play in controlling viral titers during primary infection and generating protection against subsequent infection. Fig. 1 depicts the events occurring in the lung parenchyma, airways, and draining lymph nodes (LNs) during the initial stages of a sublethal intranasal infection with influenza virus. Within a couple of days after infection, viral titers in the lung increase rapidly culminating in acute weight loss and cachexia. Subsequently, antigen‐loaded or infected respiratory DCs migrate to the mediastinal LNs (MLNS) within 6 h, reaching maximal numbers by 18 h postinfection (33). Intravital LN microscopy studies suggest that antigen‐bearing DCs migrate to and localize in the vicinity of the high‐endothelial venules (HEVs) (34, 35, 36, 37). This HEV localization event facilitates the DC interaction with naive antigen‐specific T cells entering the LN from peripheral circulation. The relatively rare antigen‐specific T cells are subsequently selected through T‐cell receptor (TCR)/major histocompatibility complex class I (MHC I) engagement and undergo 11–15 rounds of clonal expansion, resulting in a large population of antigen‐specific effector CD8+ T cells observed during the peak of the primary response (14, 38). During this expansion phase, CD8+ T cells simultaneously differentiate into functionally distinct effector populations based on their cytokine secretion profiles. These include T cytotoxic 1 (TC1), TC2, and TC17 cell populations, which correspond to the better described CD4+ T‐helper 1 (Th1), Th2, and Th17 cell populations. Homeostatic CD8+ T‐cell clone frequency, specific antigenic peptide abundance, and presentation kinetics can collectively influence the development and relative frequency of dominant and subdominant CD8+ T‐cell populations (39).
Figure 1.
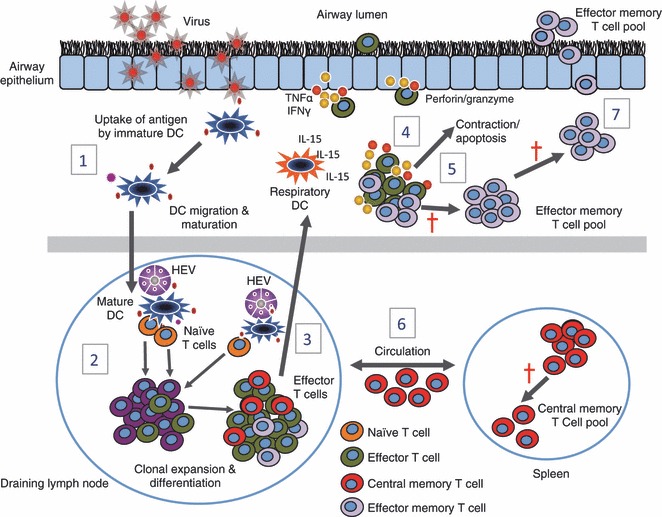
CD8+ T‐cell responses during primary respiratory virus infection. (1) Upon viral infection, lung‐resident immature dendritic cells (DC) take up viral antigen, differentiate, and migrate to the regional draining lymph nodes. (2) Antigen‐bearing DCs migrate to and localize in the vicinity of high‐endothelial venules (HEV) and interact with naive T cells that enter the lymph node (LN) via the circulation. Subsequently, viral‐specific T cells are selected through TCR/MHC I engagement and undergo rapid clonal expansion and differentiation. (3) Effector T cells migrate to the site of infection via the circulation, where they receive survival signals, in the form of IL‐15, from respiratory DCs (4) and through the release of anti‐viral cytokines and effector molecules kill virus‐infected epithelial cells. (5) After viral clearance the majority of virus‐specific effector CD8+ T cells die through apoptosis, while a hand full of surviving effector CD8+ T cells persist and seed a long‐lived memory pool (6) comprised of central memory T cells (CD62Lhi, CCR7hi), that reside in lymphoid tissue, and effector memory T cells (CD62Llo, CCR7lo) that reside at mucosal surfaces (7).
As early as day four or five post‐infection, CD8+ T cells are present in the lung tissue and exhibit several effector functions, including the ability to secrete antiviral cytokines [interferon‐γ (IFN‐γ) and tumor necrosis factor α (TNFα)] and initiate cell killing of virus‐infected epithelial cell through Fas, TRAIL, or perforin‐mediated lysis (14). The migration of CD8+ T cells to the lung tissue and airways does not appear to be dependent on antigen, although infiltrating CD8+ T cells must acquire a sufficient level of activation to downregulate LN homing receptors and acquire lung‐homing chemokine receptors (40). Following their arrival in the lung, protective virus‐specific CD8+ T‐cell responses require additional antigen‐dependent interactions with respiratory DCs (30, 41, 42, 43). Respiratory DCs critically contribute to the sustained survival of virus‐specific effector CD8+ T cells by trans‐presentation of interleukin‐15 (IL‐15) (30, 41, 42, 43). Recent data also suggest that IL‐15 mediates the migration of effector CD8+ T cells to the lung airways (44). Together, these data support a two‐hit model for promoting effective CD8+ T‐cell responses: a first hit in the lymph node that primes T‐cell proliferation and migration to the site of infection, and a second hit that provides a survival signal to the effector T cell (42). The influx of virus‐specific CD8+ T cells into the respiratory tract coincides with decreasing viral titers, recovery of body mass and ultimately viral clearance by day eight to 10 postinfection. After viral clearance the majority of virus‐specific effector CD8+ T cells die through apoptosis, while a small population of surviving effector CD8+ T cells persist and seed a long‐lived memory pool (14). A cardinal feature of memory T cells is their ability to mediate faster, stronger, and more effective responses to secondary virus challenge than naive T cells. In the case of CD8+ T cells, increased numbers, higher activation status, more rapid induction of effector functions, and altered homing patterns contribute to their enhanced recall responses (14, 32).
In recent years, accumulating data has highlighted that memory CD8+ T cells are extremely heterogeneous in terms of their phenotype, function, and anatomical distribution (32, 45). Two major subtypes of memory CD8+ T cells have been defined based on the expression of CD62L (also known as l‐selectin), a ‘homing receptor’ for leukocytes to enter secondary lymphoid tissues via HEVs, and CCR7, a chemokine receptor that supports trafficking through secondary lymphoid tissue (46) ( Fig. 2 ). Central memory T cells (TCM) are CD62Lhi/CCR7hi cells that tend to localize in secondary lymphoid tissues, whereas effector memory T cells (TEM) are CD62Llo/CCR7lo cells that localize or traffic through peripheral tissues such as the lung. Considerable interest has been focused on elucidating functional differences between these memory T‐cell subsets and their capacity to confer protection against secondary infection (24, 32, 47, 48, 49, 50, 51, 52). After secondary encounter with antigen, TEM cells can be stimulated by respiratory DCs without the requirement for further division and can demonstrate rapid cytokine production and lytic activity. In contrast, resting TCM cells that reside in the LNs require migratory DCs for their reactivation, resulting in de novo proliferation and a ‘new’ wave of secondary effector T cells that migrate to the lung parenchyma and airways. Based on these observations, it has been proposed that TEM cells participate directly in the initiation of protective memory responses by rapidly producing effector molecules (perforin, granzyme and antiviral cytokines) at the site of antigen encounter, while TCM cells contribute to the maintenance and/or amplification of the overall secondary T‐cell response. Therefore, the specific subset of memory CD8+ T cells generated by vaccination may critically determine the ultimate effectiveness of vaccine induced‐immune protection against a natural respiratory viral infection.
Figure 2.
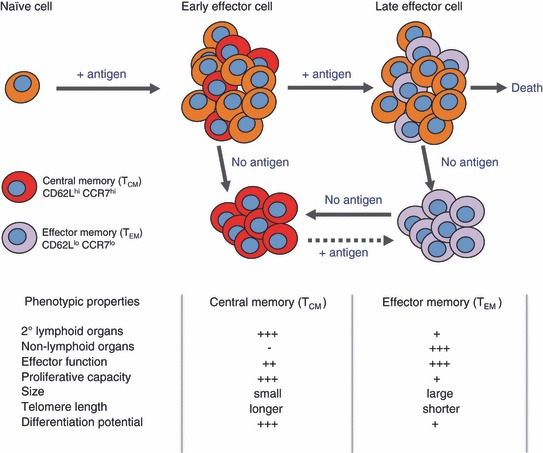
Signal strength model for memory T‐cell generation. Upon activation, naive CD8+ T cells develop into either central memory T cells (TCM) or effector memory T cells (TEM). As indicated TCM and TEM cells are distinguishable based on their phenotype, function, and anatomical location. The TCM and TEM fate decision occurs early during priming, determined by antigen access and/or dose. Brief periods of antigenic stimulation or antigen availability during priming favor the enrichment of TCM cells, whereas prolonged periods support TEM cell generation. TEM and TCM have the ability to interchange phenotype, dictated by the presence or absence of antigen. Modified from (46).
Challenges in designing CD8+ T‐cell vaccines against respiratory viruses
There are several key issues that must be taken into account to develop effective CD8+ T‐cell vaccines against respiratory viruses reviewed in detail elsewhere (24, 25, 26, 32, 53, 54, 55, 56). In this section, we will focus on the importance of memory subset (TCM versus TEM) frequency, longevity of memory subsets, and route of immunization.
Importance of TCM versus TEM frequency
There is substantial clinical evidence to suggest that protective T‐cell immunity does not last for more than a couple of years after resolution of a natural infection or vaccination, unless the response is boosted by re‐exposure to the same or cross‐reactive antigens (57, 58, 59). Similarly, in animal models of influenza and Sendai virus infection the efficacy of protection against secondary challenge wanes rapidly within 3–6 months after a primary infection (60, 61, 62). This decline in protective T‐cell immunity occurs despite the fact that the number of virus‐specific memory CD8+ T cells in the lymphoid tissues (TCM cells) remains relatively high for the life span of animal (61, 62). These studies suggest that in the context of a respiratory viral infection, TCM cells may not respond, expand, or relocate (to the lung) sufficiently quickly to provide immediate protection against disease caused by reinfection (or live‐pathogen challenge postvaccination).
More recent studies have indicated that at 1‐month postinfection, the total numbers of virus‐specific TEM cells in the lung are substantially higher than the number of TCM cells in the draining LNs (62). However, detailed kinetic studies have revealed that despite stable numbers of memory CD8+ T cells in the lymphoid organs, the numbers of lung‐ and airway‐resident memory CD8+ T cells following a primary Sendai virus or influenza infection gradually decline over the first 3–6 months before stabilizing at very low levels (62). Interestingly, this decline and stabilization in the number of memory CD8+ T cells in the lung directly correlates with a progressive decline in T cell‐mediated protection from a secondary challenge. Thus, developing strategies that elicit a sufficiently large number of long‐lived TEM cells that are not only cytolytic but also polyfunctional (defined as being able to produce high levels of antiviral cytokines IFN‐γ, TNF, and IL‐2) represents a key challenge for next generation vaccines that are able to confer protection against respiratory viruses. In this regard, it is becoming increasingly evident that the strength and duration of antigenic stimulation play important roles in determining both the frequency and long‐term persistence of TEM cells. This will be discussed below.
Role of antigen in determining the size of TCM versus TEM pools
Despite continued disagreement on the precise details of how memory T‐cell generation occurs, most people accept that the amplitude of a memory T‐cell pool generated after viral clearance is dictated by the magnitude of the primary effector T‐cell response. This phenomenon has been well studied in a number of infectious mouse models including influenza virus where between five and 10% of the CD8+ T‐cells elicited during the peak of immune response transition into memory T cells (63). The primary determinant of effector CD8+ T‐cell size appears to be a function of the initial amount of antigen present during T‐cell priming. It has been shown that brief periods of antigenic stimulation or antigen availability during priming favor the enrichment of TCM cells, whereas prolonged periods support TEM cell generation ( Fig. 2 ) (24, 46). This has important implications for vaccine development against respiratory viruses; for example, the level and time of antigen exposure that can be provided by attenuated vaccines, non‐replicating vectors, DNA vaccines, or protein subunit vaccines might be significantly less than those provided by live‐replicating vaccines. This is likely to be linked to the capacity of replicating vaccines to divide multiple times and disseminate faster, thus increasing the amount and the duration of antigen exposure. As will be discussed in more detail below, one of the key questions in inducing large effector‐cell burst size for new vaccines will be to define the precise mechanisms that regulate the formation of effector T cells in the context of high or persisting antigen load.
Role of antigen in long‐term persistence of TCM versus TEM cells
The role of antigen in determining the long‐term persistence of memory T cells has been intensely debated over the last 20 years. Initial data from mouse models suggested that T‐cell memory was short lived in the absence of periodic exposure to antigen (64). However, more recent evidence has challenged this idea and suggested that the number of TCM cells can be sustained in the absence of both specific antigen and MHC molecules (65, 66, 67). Further experimental evidence now suggests that a TCM memory pool is maintained, at least in part, through periodic slow cell division and continuous replacement of cells, a process commonly referred to as homeostatic proliferation (68, 69). This turnover of a memory TCM pool is achieved through tonic stimulation by cytokines, such as IL‐7, and IL‐15. In contrast, TEM cells are not directly maintained through IL‐15‐driven homeostatic proliferation (44).
Two different (although not mutually exclusive) models have been proposed to explain the longevity of TEM cells in the absence of re‐exposure to antigen. Initial studies suggested that TEM cell populations in the lung airways are maintained in an antigen‐independent manner by continual recruitment of new cells from the circulation (40, 70). The rate of memory cell recruitment was shown to be extremely rapid, resulting in replacement of 90% of the population every 10 days and that this can be maintained for periods over 1 year following viral clearance (70). More recent studies extended these observations by demonstrating that antigen can persist in the lung draining LNs for several weeks after influenza virus clearance and that this residual antigen contributes to the maintenance of TEM cells in the lung airways (71, 72). However, a small number of circulating memory CD8+ T‐cells generated after influenza virus infection can migrate to the airways in the absence of cognate antigen (73). Therefore, memory T‐cell recruitment to the lung airways is controlled by both antigen‐independent and antigen‐dependent mechanisms immediately following virus clearance. As residual antigen is cleared and the number of memory CD8+ T‐cells localized within the lung airways stabilizes at a low level, antigen‐independent processes are required to maintain this population over time (73). Thus, it is clear that the role of persisting antigen after vaccination is a crucial point that has to be addressed during an effective T‐cell vaccine design against respiratory viruses. This evidence clearly demonstrates that the use of conventional non‐replicating attenuated vaccine strains will fail to induce a robust and long‐lived TEM population and therefore have limited application in the design of future vaccines. This explains the need to increase the vaccine dose and/or number of booster immunizations when highly attenuated viral vaccines are utilized. However, these approaches have obvious logistical and economic limitations. Thus, to fully realize the potential benefits of attenuated vaccines or simple protein/peptide immunizations it will be of great interest to identify specific molecules and/or pathways that can be targeted to safely lower the activation threshold for antigen.
Route of immunization impacts the maintenance of memory T cells in the lung
Currently, intradermal, intramuscular, oral, intranasal, intravaginal, and even rectal routes of immunization are under active investigation. Accumulating evidence from many research groups has confirmed that the route of immunization is critical in determining both qualitative and quantitative aspects of a vaccine‐induced immune response (74, 75, 76). With regards to T‐cell vaccines against respiratory viruses, recent studies support the concept that long‐lived CD8+ T cells can be detected in the lung after mucosal, but not systemic immunization, suggesting that the maintenance of memory T cells are also dependent on the particular route of vaccination. Two recent studies provide insight into the mechanisms for this phenomenon. Verbist et al. (77) demonstrated that generation, maintenance, and function of memory CD8+ T cells after respiratory influenza infection is independent of IL‐15‐mediated survival signals. In contrast, systemically (i.v)‐induced memory T cells were dependent on IL‐15 for their long‐term survival. Thus, CD8+ T‐cells primed at a mucosal site require distinct signals for their long‐term maintenance. Shiki Takamura et al. (78) extended these results by showing that intranasally primed memory CD8+ T cells possess a unique ability to be reactivated by residual antigen in the MLN compared with intraperitoneally primed CD8+ T cells, resulting in the preferential recruitment and maintenance of intranasally primed memory CD8+ T cells in the lung airways. Furthermore, they demonstrated that the inability of intraperitoneally primed memory CD8+ T cells to access residual antigen could be corrected by a subsequent intranasal virus infection (78). Thus, initial CD8+ T‐cell priming in the MLN and prolonged presentation of residual antigen in the MLN are required to maintain large numbers of virus‐specific memory CD8+ T cells in the lung airways (78). Clearly, these findings have to be considered for the rational design of CD8+ T‐cell vaccines against respiratory pathogens.
Poxvirus‐based vectors as vaccine‐delivery systems: past, present, and future
Several approaches have been used to elicit protective CD8+ T‐cell responses (25, 79, 80). These include the delivery of proteins in replication‐competent attenuated recombinant viruses (live‐attenuated vaccines), in replication‐defective attenuated recombinant viruses, in DNA vaccines, protein/peptide subunit vaccines, and through combining one or more of these modalities in a heterologous prime‐boost strategy. Currently, the two most extensively utilized recombinant viral vectors in preclinical and clinical studies are derived from poxviruses and adenoviruses (25, 79, 80, 81). Detailed discussion on adenovirus vectors, DNA vaccines, and protein subunit vaccines will not be provided here, and for further information the reader is referred to the following reviews (25, 79, 80). The focus of remaining sections of this review center around the poxvirus‐based vectors listed in Table 1 , and more specifically how the pursuit of safer vaccines has paradoxically resulted in largely ineffective CD8+ T‐cell vaccines against mucosal pathogens.
Table 1.
Current pox‐virus based vaccine vectors.
| Original virus strain | Derived virus | Derivation approach | Virulence |
|---|---|---|---|
| NYCBOH | ACAM1000, ACAM2000, ACAM3000, CCSV | Tissue culture VACV | Replication‐competent, attenuated |
| Lister | Lc16m8 | >50 serial passages in primary rabbit kidney cells | Replication‐competent, highly attenuated |
| Ankara | MVA | >570 serial passages on primary chick embryo fibroblasts | Replication defective, highly attenuated |
| Copenhagen | NYVAC | Precise deletion of 18 open reading frames | Replication defective, highly attenuated |
| Canary pox | ALVAC | >200 serial passages in canary embryonic fibroblasts | Replication defective, highly attenuated |
A summary of the live‐attenuated VACV strains used during the global smallpox eradication program and their corresponding replication‐competent attenuated and replication‐defective attenuated VACV‐vaccine strains. Abbreviations: MVA, modified vaccinia virus Ankara; VACV, vaccinia virus
Replication‐competent (live) attenuated poxviruses as vaccines against mucosal viruses
Poxviruses are a family of large enveloped viruses that contain a linear double‐stranded DNA genome, ranging in size between 130 and 300 kb pairs, that is capped by a hairpin loop at each end (82). They are also unique in that they encode their own transcription machinery enabling poxviruses to replicate in the cytoplasm of an infected cell (82, 83). The concept of utilizing live‐attenuated poxviruses as an immunizing agent against respiratory viruses can be traced back to the global‐smallpox eradication program (82, 83). The strategy for this program involved mass vaccination with VACV, an antigenically similar virus to Variola major, the etiological agent of smallpox disease. Many live‐attenuated VACV strains (first generation vaccines) were used in different parts of the world (83): Lister or Lister/Elstree strain was the most widely used vaccine globally; New York City Board of Health (NYCBOH; later produced by Wyeth Laboratories as Dryvax) strain was used in the Americas and West Africa; Copenhagen strain (Denmark), and the Ankara strain (Turkey) were also widely used. These VACV variants are known to differ in the expression of several virulence factors that determine their replicative capacity and safety profiles (83, 84). It was the ability of these VACV strains to induce robust humoral and cell‐mediated immunity (82, 83, 85) that led to the World Health Organization declaring the eradication of smallpox in 1980 and shortly thereafter routine vaccination with VACV could be discontinued in most countries.
Serendipitously, at the same time as the global VACV vaccination program was terminated, novel molecular biological tools had identified VACV as an extremely versatile eukaryotic expression vector (86, 87). One of the main properties of VACV, and poxviruses in general, that provides an advantage over other viral vectors is the relative ease by which large inserts of foreign DNA can be cloned into them. This allows for the insertion of multiple genes and the creation of multivalent vaccines. Furthermore, poxviruses have the potential to be administered by different routes, such as intradermal, intramuscular, oral, intranasal, intravaginal, and intrarectal routes to generate immunoglobulin and T‐cell responses. Over the last 30 years, a growing number of reports have described the utility of recombinant vaccinia viruses (rVACVs) expressing relevant antigens from multiple viral, bacterial, and parasitic pathogens to confer protection to immunized animals (81, 88, 89, 90, 91, 92, 93).
The VACV strain most extensively utilized in animal models has been the highly virulent Western Reserve (WR) strain, which was derived from the NYCBOH strain by multiple passages in mice. For obvious safety reasons, the WR strain does not represent a viable strain for use in human vaccine applications. Moreover, the well‐documented severe, albeit rare, complications following vaccination with first generation VACV strains, and the increasing number of immune‐compromised individuals have raised legitimate concerns about their widespread use as a vaccine‐delivery system in humans (84). Thus, during the past three decades, a major focus of research has been on developing novel, highly attenuated, recombinant VACV strains (rVACV) that demonstrate significantly improved safety profiles and can be used in humans as potential vaccines candidates (83, 84) ( Table 1 ).
Replication‐defective highly attenuated poxviruses as vaccines against mucosal viruses
As briefly mentioned above, the safety concerns surrounding the WR and first generation VACV‐based vaccine candidates have been addressed through the development of highly attenuated VACV strains (83, 84) ( Table 1 ). These include, attenuated modified vaccinia virus Ankara (MVA) (94) and the Copenhagen‐derived NYVAC strain (95). MVA is replication deficient in mammalian cells but importantly retains the capacity to synthesize viral proteins, while NYVAC is a highly attenuated virus that is unable to produce infectious particles in humans. Both virus vectors are extremely safe and can be used in young children and immuno‐compromised individuals (84, 96). In addition, an attenuated Avipoxvirus‐vaccine vector, the canary pox like ALVAC, was also developed for use in humans (97, 98). Due to its restricted tissue tropism to avian cells, ALVAC can be safely used in immune compromised humans (84).
In recent years, MVA, NYVAC, and ALVAC have all been utilized, with varying degrees of success, as promising vaccine vectors for many infectious diseases, including influenza, SARS, HIV, HSV, TB, and malaria (81, 84, 99). Furthermore, these novel vaccine vectors have been increasingly used, again with varying degrees of success, in the development of therapeutic vaccines against several common cancer antigens (81, 100). The large number of clinical trials has produced a complicated picture with regards to using these vaccine vectors to generate a robust CD8+ T‐cell response. Despite a small number of clinical trials providing promising preliminary evidence for CD8+ T‐cell induction (101, 102), unfortunately, many trials demonstrate a limited induction of antigen‐specific CD8+ T cell against the infectious disease or cancer target of interest. For example, the immunogenicity of an attenuated ALVAC HIV‐1 vaccine in humans was shown to be low, eliciting HIV‐1‐specific CD8+ T‐cell responses in less than 25% of normal volunteers (103, 104, 105, 106). Similarly, the immunogenicity of MVA expressing the GAG protein (consensus of HIV‐1 clade A) and several immunodominant CD8+ T‐cell epitopes was also shown to be unsatisfactory (17% response) (107). A smaller study using the same vaccines at a higher dose showed enhanced immunogenicity (40% response), but the T‐cell responses induced were exclusively CD4+ T‐cell driven (108). With regards to respiratory viruses, a small phase I clinical trial in healthy adults investigating a novel MVA‐based heterosubtypic influenza A vaccine (MVA – NP + M1) demonstrated that all volunteers had a significant increase in the number of IFN‐γ producing CD8+ T cells in their blood at weeks one, three and eight after intradermal or intramuscular vaccination (109). This response, however, rapidly declined over time, reaching prevaccination levels by week 52 postvaccination (109). This is consistent with an earlier preclinical study that demonstrated MVA‐induced T cell responses are followed several weeks later by a dramatic (20‐ to 40‐fold) contraction, a phenomenon that also occurred with DNA‐poxvirus regimens in which NYVAC and ALVAC were used for boosting (110). Another interesting characteristic of MVA vaccines was revealed by Pillai et al. 2011, in which MVA‐primed HIV‐1 Gag‐specific memory cells were found to be predominantly of the TCM phenotype (CD62L and IL‐2 positive) (111). Similarly, after a single immunization with MVA vector expressing pre‐erythrocytic malaria antigens, Reyes‐Sandoval et al. (112) found that MVA‐induced accelerated TCM rather than TEM generation and consequently failed to protect against malaria. These studies suggests that MVA in its current form may not be the most effective poxvirus vector for induction of strong and long‐lasting protective CD8+ T‐cell responses in peripheral tissues, such as the lung.
If one looks back in the literature it becomes evident that these somewhat disappointing clinical results with non‐replicating highly attenuated poxviruses could have been predicted based on studies carried out more than 20‐years ago. A seminal study by Morgan et al. (113) demonstrated that the virulence of a VACV vector critically influences the protective efficacy of the recombinant vaccine. Cottontop tamarins vaccinated with a VACV‐WR‐based recombinant expressing the gp340 envelope antigen of Epstein‐Barr virus (EBV) were protected against EBV‐induced lymphoma, but animals inoculated with the attenuated Wyeth (VACV‐NYCBOH)‐based recombinant, were not. The precise mechanism for this was not investigated in detail, but it appeared to be independent of humoral immunity.
In summary, 30 years of increasing safety and regulatory concerns surrounding replication‐competent recombinant poxviruses has unfortunately resulted in the widespread use of vaccine‐delivery vectors that are ineffective at inducing robust and long‐lived memory CD8+ T cells necessary to confer protection against highly virulent respiratory viruses.
Lessons learnt from the past: focusing on defining the mechanisms that link virulence and immunogenecity
At the dawn of recombinant poxvirus‐vaccine research, relatively little was known about the precise molecular mechanisms that govern the quantity, phenotype, and quality of memory T‐cells following infection with a live (replicating) virus. In the interim, we have gained a number of important insights, which must be considered in our pursuit of safe and effective poxvirus‐based T‐cell vaccines. First, replication‐competent or highly virulent poxviruses, in general, promote a more potent primary and memory CD8+ T‐cell response compared with that of replication‐defective or highly attenuated vaccine strains. Second, replication‐defective poxviruses predominantly drive the induction of TCM cells over TEM cells. Third, in many instances, multiple dosing, or higher dosing (107–109 pfu), with replication‐defective or highly attenuated poxvirus‐vaccine strains are necessary to achieve comparable levels of CD8+ T cells to those produced by live poxvirus‐vaccine strains. Finally, while attenuated poxvirus strains provide good short term T‐cell protection, they are not effective at conferring long term T‐cell‐mediated protection. As discussed previously, these limitations are logically related to antigen load, antigen persistence and the ability to effectively stimulate CD8+ T‐cell responses. Thus, a convincing argument can be made that a key challenge in utilizing attenuated poxvirus vectors to develop efficacious T‐cell vaccines is to define the molecular mechanisms that link virulence and immunogenicity. Toward this end, several recent studies have indicated that key events during initial T‐cell priming, including inflammatory stimuli (24, 54), co‐stimulation (114), CD4+ T‐cell help (115), and the cytokine mileu (54), have long‐lasting programing effects on the quantity, phenotype, and quality of the memory T cells generated in response to a live viral infection. In the following sections, we will discuss the importance of T‐cell costimulatory receptors, specifically, we will focus on the importance of TNF/TNFR family members, OX40 and OX40L, in eliciting protective T‐cell memory and their potential use as immunological adjuvants to enhance poxvirus‐based CD8+ T‐cell vaccines.
Significance of T‐cell stimulatory receptors to antiviral immunity: an evolutionary argument
It has been hypothesized that the coevolution of pathogens and their hosts has been a major driving force behind the development of a highly sophisticated and extremely adaptable vertebrate immune system. This coevolution is clearly demonstrated by the fact that many large DNA viruses, such as poxviruses and herpesviruses, possess multiple homologues of cytokines, chemokines, or the associated receptors of these molecules, which can neutralize or modulate host immunity to favor the virus (116, 117, 118, 119). It is assumed that the main goal of these immune modulatory strategies is to enhance viral replication and survival; however, it is highly plausible that there exists unknown consequences of viral immune evasion strategies that may impact the host’s immune response. Could it be possible that certain molecular pathways that regulate alternative immune responses, such as those involved in autoimmunity, allergic or even anti‐tumor responses, might have evolved or be differentially utilized as a consequence of viral evasion tactics. An obvious example is the large number of costimulatory receptors, especially in the immunoglobulin (Ig) and TNF/TNFR superfamilies that are expressed on T cells, and known to be critical for their activity. CD28‐B7, ICOS‐ICOSL, and CD2‐LFA‐3 typify costimulatory molecules of the Ig superfamily (120, 121), whereas TNFR/TNF family members are typified by, but are not exclusive to, OX40‐OX40L, 4‐1BB‐4‐1BBL, CD27‐CD70, GITR‐GITRL, CD30‐CD30L, and HVEM‐LIGHT (114, 122, 123, 124). Members of the TNF/TNFR superfamily are recognized for their ability to stimulate T cells and provide signals to promote sustained T‐cell clonal expansion and long‐term survival (114, 122, 123, 124). This is achieved through the selective targeting of intracellular signaling molecules that promote effector cytokine production, cell division and suppress apoptosis (114, 122, 123). Interestingly, there are a number of receptor‐ligand pairs within the TNF/TNFR superfamily that are not constitutively expressed, but are induced during specific inflammatory scenarios. They are thought to provide ‘late’ signals that subtly regulate T‐cell responses in both a quantitative and qualitative manner (114, 120, 122). Why so many costimulatory molecules exist, with often apparently similar and overlapping functional attributes, remains to be determined. Several pox and herpes viruses encode TNFR superfamily homologues (TNFR itself, CD30, and HVEM) that act as decoy receptors to modulate host immune responses to favor viral replication and persistence (125, 126, 127, 128). It is of interest, that recent analysis of the teleost genome, an organism in which the first fully functional adaptive immune system was thought to have developed, has revealed the presence of many human TNF superfamily orthologs. This provides evidence to suggest that many of the TNF superfamily members co‐emerged around the same time as did antigen receptors (129). Interestingly, however, four ligands were not found to exist within the teleost genome, namely OX40L, CD70 (the ligand for CD27), GITRL, and CD30L (129). As briefly discussed above, these are important regulators of the later phases of T‐cell expansion and survival, leading to the hypothesis that these ligands may have arisen more recently to allow immune adaptation, plasticity and effective development of protective immunity against pathogens. To test this hypothesis, we recently utilized several poxvirus infection models and investigated the use and role of select TNF/TNFR and Ig family members in initiating and sustaining protective CD8+ T‐cell responses.
Poxvirus vaccines and protective CD8+ T‐cell responses against respiratory virus challenge: virulence matters
Utilizing a panel of VACV strains with varying degrees of virulence in mice, our laboratory recently demonstrated that the relative virulence of a virus/vaccine dictates the quantity and protective capacity of the resulting memory CD8+ T‐cells generated (76, 130). The three VACV strains used in our study included the highly virulent WR strain and two clinically relevant VACV‐vaccine strains, Lister, and NYCBOH. All VACV strains are known to differ in their expression of several virulence factors that impact on their replicative capacity in vivo. Consequently, we found that after intranasal or intraperitoneal infection only the WR VACV strain replicated rapidly, reaching high titers across multiple tissues (76 and Salek‐Ardakani unpublished observations), and resulting in severe and sustained inflammation. In contrast, the Lister and NYCBOH strains were cleared more rapidly and were unable to disseminate to the same extent as WR.
Mice infected with an attenuated VACV‐vaccine strain (Lister or NYCBOH) via the intranasal or intraperitoneal route generated five to 10‐fold fewer lung resident virus‐specific memory CD8+ T cells compared to mice infected with the virulent, replication sufficient, WR strain (76). Similar differences were observed at the peak (days seven and eight) of the initial effector CD8+ T‐cell response, implying that altered molecular regulation at this phase of the response could explain the difference in size of the memory pool generated. To highlight the importance of CD8+ T cells in protecting against subsequent viral encounter, MHC class‐II‐deficient mice (MHC II−/−), that lack CD4+ T cells and therefore can not generate VACV‐specific humoral immunity, were vaccinated with WR, Lister, and NYCBOH and challenged 70 days later with a lethal intranasal dose of VACV‐WR (76). All mice vaccinated with the WR strain, the most virulent virus, survived the lethal infection and presented mild disease symptoms. In contrast, no protection was evident in mice vaccinated with either attenuated strain (Lister or NYCBOH), resulting in comparable mortality and disease severity to that observed in naïve (unvaccinated) mice. This demonstrated that a VACV strain capable of replicating to greater titers, in multiple tissues and over a longer period of time promotes greater number of persisting antigen‐reactive CD8+ T cells that afford protection against a highly lethal respiratory virus challenge. In humans, immunization with live‐attenuated VACV (Lister or NYCBOH) elicits what was previously thought to be a good memory CD8+ T‐cell response (85, 131, 132). The VACV‐specific memory CD8+ T‐cell pool has been shown to persist for many years in the majority of vaccine recipients; however, recent observations have highlighted a rapid decline in memory CD8+ T‐cell numbers does occur in a significant group of individuals over time (133, 134, 135). This raised concerns as to whether optimal long‐lived CD8+ T‐cell immunity can in fact be generated using first generation VACV strains. Our data provides considerable evidence to warrant these concerns as vaccination using the virulent VACV‐WR promoted far superior numbers of protective memory CD8+ T‐cells. This result raised the question as to whether a specific molecular mechanism exists that is engaged during an infection with the virulent WR strain, but not the attenuated Lister or NYCBOH VACV strains. If alternate molecular control mechanisms are evident, they could potentially be harnessed to enhance CD8+ T‐cell responses generated by attenuated poxvirus vaccines.
OX40 and OX40L link viral virulence to protective CD8+ T‐cell memory
OX40 is a member of the TNFR family and is not constitutively expressed on naïve T cells, but is induced hours to days after TCR engagement (114, 122, 124). Maximal expression is typically observed between two and five days following T‐cell activation after which OX40 is downregulated, implying a delayed mode of action in primary immune responses (114, 122, 124). Consistent with this, we recently reported that OX40 was seen on a proportion of VACV‐specific CD8+ T cells as early as day four and peaked on day five postinfection with VACV‐WR (136). In our initial studies, we found that the development of high numbers of effector CD8+ T cells capable of producing both IFNγ and TNFα was strongly impaired when OX40 was lacking on a CD8+ T‐cell responding to VACV (124, 136). Analysis of the main immunodominant CD8+ T‐cell populations (B8R, A3L, A8R, A23R, and B2R), which account for close to 70% of the whole VACV‐specific response, demonstrated a global impairment in CD8+ T‐cell priming in the absence of OX40 such that the response was reduced by between 70% and 80% (136). We further demonstrated that the engagement and activity of OX40 during a virulent viral infection ensured the generation of a high frequency of persisting and functional memory CD8+ T cells that located to the lung (136). Most interestingly, OX40 driven induction of lung‐resident memory CD8+ T cells correlated directly with a robust protection against a highly lethal intranasal infection with VV‐WR (76, 136).
Subsequently, we assessed the requirement for OX40 after infection with the Lister and NYCBOH VACV strains (76). In striking contrast to VACV‐WR, little difference in the frequency of virus‐specific effector and memory CD8+ T cells was observed in the absence of OX40 signaling. Thus, virulence and host evasion mechanisms of VACV‐WR result in persistent viral replication and the engagement of costimulatory receptor OX40. Subsequently, this results in enhanced induction of memory CD8+ T cells that afford protection against a highly lethal respiratory virus challenge.
In light of these studies, we postulated whether the differential engagement of OX40 applies to other T‐cell stimulatory receptors (76). In this regard, recent data from our group and others have shown that in addition to OX40, CD28 (a co‐stimulatory receptor in the Ig superfamily) (137, 138), and CD27 (another TNFR family member) (139), can be utilized during a VACV‐WR infection and each can contribute to the generation of effector CD8+ T‐cell responses. CD28 and CD27 are distinguishable from OX40 in that they are constitutively present on resting CD8+ T cells, but their respective ligands, B7.2 and CD70, are similar to OX40L in that they are induced once an APC receives certain inflammatory stimuli (120, 137, 140, 141). In contrast to our OX40 studies, we found that CD28 was indispensible for optimal effector CD8+ T‐cell generation regardless of whether mice were infected with Lister or NYCBOH (76). However, similar to OX40, deficiency in CD27 resulted in impaired CD8+ T‐cell responses to WR, but little or no defect to Lister or NYCBOH (76).
Another interesting distinction between infection with the virulent WR and the attenuated viruses was only evident when VACV‐specific T‐cell responses were assessed during the memory phase. CD28‐deficient mice exhibited defective memory CD8+ T‐cell generation in response to Lister and NYCBOH, but not interestingly to WR, even though CD28 was required for an optimal effector response to this virus (76). This indicates that in response to VACV‐WR infection, there exists a CD28‐dependent phase of CD8+ T‐cell priming followed by a CD28‐independent phase. CD27 signaling has previously been shown to prolong CD8+ T‐cell survival beyond the effector phase of the immune response and thus serves to increase the number of memory cells by inhibiting effector T‐cell death (124, 142, 143, 144). Consistent with these results, blocking the CD27 and CD70 interactions in CD28‐deficient mice late in the primary response (between five and eight days) completely abrogated the CD28‐independent CD8+ T‐cell responses to VACV‐WR (76). These results support a model in which a temporal sequence of events is necessary for optimal virus‐specific CD8+ T‐cell proliferation, survival and memory generation, brought about by a specific sequential engagement of costimulatory molecules CD28, OX40, and CD27.
In summary, we show that viral virulence and evasion strategies that impact viral replication, dissemination and invariably the inflammatory milieu, can lead to differential use of costimulatory receptors for T cells, which dictate the magnitude and effectiveness of the CD8+ T‐cell response ( Fig. 3 ). Furthermore, these data are of high potential significance to vaccination, and promote the notion that the use of attenuated viruses may not elicit the best long‐term T‐cell memory because they do not allow molecules, such as OX40 and CD27 or their ligands, that may have evolved for the purpose of promoting T‐cell memory, to be engaged. This provides a hypothetical model that might in part explain the abundance of such stimulatory receptors for T cells, and demonstrate that molecular plasticity can occur during antiviral responses, where certain immune mechanisms thought non‐essential could become highly relevant.
Figure 3.
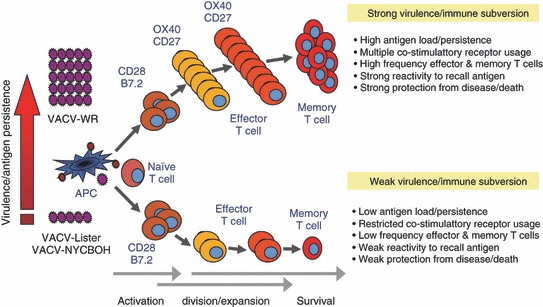
TNFR family members OX40 and CD27 link viral virulence to protective T‐cell vaccines. The model explains why the strongly replicating (live) vaccinia virus (VACV) Western Resrve strain (VACV‐WR) results in better CD8+ T‐cell immunity as compared with attenuated VACV strains (VACV‐Lister or VACV‐NYCBOH). The level of virus replication, brought about by virulence and immune evasion tactics, can lead to differential use by a CD8+ T cell of stimulatory receptors in the TNFR and Ig superfamilies, and that this dictates the magnitude of the T‐cell response. Two TNFR family members, OX40 and CD27, drive the generation of memory CD8+ T cells with virus that replicates strongly, or when higher doses of attenuated virus are used for inoculation. In contrast, CD28/B7 interactions, but not OX40/OX40L or CD27/CD70 interactions, are used to generate memory responses to attenuated viruses that are rapidly cleared, and this corresponds with strongly reduced T‐cell memory. This differential molecular use and altered CD8+ T‐cell memory determines the ability of the host to protect against subsequent respiratory infection.
Role of OX40 in other virus infection models
Contrary to our VACV‐WR studies, but similar to the data with VACV‐Lister and VACV‐NYCBOH discussed above, the regulation of CD8+ T‐cell responses in other viral settings is variably dependent on OX40 and OX40L signaling. Initial studies on OX40−/− and OX40L−/− mice independently infected with lymphocytic choriomeningitis virus (LCMV), vesicular stomatitis virus (VSV), Theiler’s murine encephalomyelitis (TMEV), and influenza virus demonstrated no defect in control of viral replication or the generation of effector CD8+ T‐cell populations (124, 145, 146, 147, 148). However, recent studies investigating the impact of OX40/OX40L signaling on memory and recall CD8+ T‐cell responses demonstrated a reduction in the number and secondary expansion of memory CD8+ T cells in the lung, despite not influencing initial CD8+ T‐cell priming (148). Similarly, OX40 signaling regulates the accumulation of CD8+ T cells reactive with the persistent‐phase epitopes during a mouse cytomegalovirus (MCMV) infection (149).
Collectively, these data suggest that no two viruses initiate the same combination of costimulatory pathways. This raises many questions surrounding the specific signals that initiate the ‘program’ of costimulatory pathways required for induction, maintenance and recall of memory CD8+ T‐cell populations. As discussed above, the level of virus replication and persistence, brought about by virulence and immune evasion strategies, is one parameter that dictates the differential use of costimulatory receptors and subsequently the magnitude and efficacy of the ensuing CD8+ T‐cell response. Furthermore, the inflammatory signature initiated by a specific virus strain or the presence of virus‐specific virulence factors may also authorize particular costimulatory interactions. APC derived cytokines, such as IL‐1, IL‐2, IL‐12, and TNFα have been shown to influence the extent and length of costimulatory receptors/ligand expression (114, 124). While a recent LCMV study revealed a differential requirement for type I IFNs in controlling the initial expansion and generation of CD8+ T cells that is not used in response to a VACV infection (150). Furthermore, the CD40 engagement, TLR ligation and IL‐18 derived signals can also regulate the expression of OX40L on APCs (114, 140).
In summary, the extent of viral replication, in terms of peak titer and tissue tropism, and the unique inflammatory signature of a specific viral infection is likely to influence the use of a specific combination of costimulatory molecules. Further studies to investigate the spatial and temporal expression profiles of both costimulatory ligands and receptors in different viral settings will almost certainly add to our understanding and help in the development of safe yet effective vaccines against viral infections.
Targeting OX40 to boost poxvirus‐based CD8+ T‐cell vaccines against respiratory viruses
Accumulating evidence from our laboratory and others support the notion that a critical component of any future CD8+ T‐cell vaccine against highly virulent respiratory viruses must be the capacity to: (i) promote robust expansion of naïve precursor T cells reactive with viral antigenic peptides; (ii) allow a high frequency of these virus‐specific effector T cells to survive over time as memory T cells; (iii) allow high numbers of these memory cells to persist in the lung parenchyma and airways in the absence of persisting antigen; and (iv) to retain high effector function to provide optimal surveillance against subsequent infections. As alluded to earlier, replication‐defective highly attenuated poxvirus vectors, although extremely safe, are unlikely to satisfy a number of these important requirements. Consequently, a number of innovative strategies have been developed to facilitate and enhance the immunogenicity of attenuated poxvirus vectors, including the use of heterologous prime‐boost regiments (151), coadministration of TLR agonists (152), cytokines (153, 154, 155, 156, 157), and targeting T‐cell costimulatory receptors (158, 159, 160).
Our studies, highlighted above, suggest that attenuated VACV‐vaccine strains will not elicit the most effective CD8+ T‐cell memory responses in the lungs due to the lack of OX40 and CD27 engagement. Therefore, we investigated whether engagement of OX40 would boost the response to attenuated VACV‐vaccine strains by treating with an agonist antibody (76). Agonistic OX40 treatment during the initial priming of CD8+ T‐cells markedly enhanced the numbers of effector and memory cells not only in the spleen, but also in the lungs. This was observed regardless of whether vaccination was via the intraperitoneal or dermal scarification route. Most significantly, agonistic OX40 provided strong protection against a lethal respiratory virus challenge in MHC II−/− mice vaccinated with Lister or NYCBOH, which alone were ineffective at preventing lethality. The extent of protection when mice were vaccinated with the combination of attenuated vaccine strains and agonistic OX40 was similar to mice vaccinated with the virulent WR strain.
To extend these results, we focused on priming CD8+ T cells by subcutaneous immunization with the immunodominant VACV peptide epitope, B8R20‐27, given in IFA (161). This strategy allowed us to assess the role of memory CD8+ T cells in the absence of any pre‐existing VACV‐specific immunoglobulin and in the presence of an intact naïve CD4+ T‐cell population. Several weeks later, once memory cells had developed, mice were challenged intranasally with a lethal dose of VACV‐WR. Correlating with our prior data, agonistic OX40 provided strong protection in a CD8+‐dependent CD4+‐independent manner in mice vaccinated with a high dose of B8R20‐27 peptide, in that no disease, as measured by weight loss, was observed. It should be stressed that prevention of weight loss during the first seven days of infection is extremely difficult to achieve, especially with such high‐challenge doses (x 300 LD50) that were used in our experiments, and therefore an important demonstration of the utility of agonistically targeting OX40 in antiviral protection. More interestingly, targeting OX40 induced almost complete protection in mice immunized with a low dose of B8R20‐27 that alone was ineffective at preventing lethality. This was reflected in both minimal loss of weight and mortality, as well as lung immunopathology (161). As before, the extent of protection directly correlated with the number of IFN‐γ producing B8R‐specific memory CD8+ T‐cells that were generated in the lung after OX40 engagement, and the number that accumulated after intranasal infection. These findings are reminiscent of those obtained with secondary influenza or Sendai virus infection, in which the presence of significant numbers of CD8+ T cells in the lung before challenge correlated with immunity (162, 163).
The development of polyfunctional CD8+ T cells, which are considered the most potent memory cells for antiviral immunity, is a powerful metric for potency of adjuvanticity (55). Agonist OX40 treatment at the time of vaccination elicits highly polyfunctional B8R‐specific CD8+ T cells that persisted, specifically, in the lung tissue in high numbers for at least 18‐months after secondary challenge with VACV and importantly, was independent of CD4+ T‐cell help. As previously discussed, to effectively promote lung tropic memory CD8+ T cells both the route of vaccination and the virulence/immunogenicity of the vaccine vector is paramount. Collectively, our studies demonstrate that intraperitoneal infection with attenuated VACV‐vaccine strains or subcutaneous vaccination of a viral peptide in the presence of an agonistic OX40 antibody can generate long‐lasting memory CD8+ T cell that persist in the lung. Therefore, targeting OX40 during priming of VACV‐specific CD8+ T cells elicits fully protective and long‐lived antiviral CD8+ T‐cell responses in the lung irrespective of the site of vaccination and the context of viral antigen.
Several other groups have raised the possibility that triggering OX40 could enhance vaccination efficacy against other viral (and non‐viral) pathogens. The use of pox‐viral vectors that encode an antigen of interest in combination with OX40L has demonstrated enhanced splenic CD4+ and CD8+ T‐cell responses to a hepatitis B surface antigen (HBsAg) (164) and improved protective efficacy of a foot‐and‐mouth disease (FMD) vaccine (165). Furthermore, a recombinant poxvirus vector encoding OX40L in combination with B7‐1, ICAM‐1 and LFA‐3 enhanced both CD4+ and CD8+ T‐cell proliferative capacity and cytokine production after antigen re‐stimulation in vitro (166). A similar approach, in which a HIV‐1 canary pox vaccine vector was co‐administered intramuscularly with an OX40L‐expressing canarypox vector resulted in the expansion of HIV‐specific CD8+ T cells detected in the spleen six‐weeks postinfection (167). The use of an agonistic OX40 antibody and fusion proteins has also produced very promising results. Coadministration of an agonistic OX40 antibody with SIV‐gp130 in rhesus monkeys resulted in enhanced virus‐specific T cell and immunoglobulin recall responses (168). While the targeting of OX40 using an agonistic monoclonal antibody elicited strong antiviral CD8+ T cells during a MCMV infection, resulting in enhanced clearance of the virus (149). Furthermore, stimulation of OX40, in cooperation with 4‐1BB, during vaccination with an OVA‐expressing poxvirus vector also enhanced OVA‐specific memory responses (169). This approach also promoted fungal clearance and enhanced T‐cell responses against the respiratory fungal pathogen Cryptococcus neoformans (170). A summary of these agonistic and vector‐based adjuvant strategies is shown in Table 2 .
Table 2.
Targeting OX40/X40L in different infectious disease models.
| Therapy and route of administration | Target antigen/infection | Therapy effect | Infection/immunization route | Study number |
|---|---|---|---|---|
| OX40 agonist (i.p.) | VACV‐Lister and NYCBOH | Enhanced effector and memory CD8+ T cell response. Protected against lethal VACV‐WR challenge | i.p. and d.s. | 1 |
| OX40 agonist (i.p.) | B8R20‐27 VACV peptide | Protected against lethal VACV‐WR challenge. Enhanced IFN‐y B8R specific long‐term mucosal CD8+ T cells | s.c. in IFA | 1 |
| OX40 agonist (i.v.) | SIV‐gp130 | Enhanced virus‐specific T cell and immunoglobulin recall responses | s.c. | 2 |
| OX40 agonist (i.p.) | MCMV | Increased expansion of protective CD8+ T cells (CD4+ dependent) | i.p. | 3 |
| OX40 and 41BB agonist | OVA‐VACV | Enhanced OVA‐specific memory response | i.p. | 4 |
| OX40 agonist (i.p.) | C. neoformans | Promoted fungal clearance from the lung | i.n. | 5 |
| OX40L encoding vector | HBsAg | Boosted CD4+, CD8+ and immunoglobulin responses against HBsAg | – | 6 |
| OX40L and 4‐1BBL encoding vector (i.m.) | Foot‐and‐mouth disease virus (FMDV) – VP1 | Enhanced antigen‐specific CD4+, CD8+, immunoglobulin responses. Protected against live infection with FMDV | i.m. | 7 |
| OX40L encoding vector | vCP1452‐HIVgp120/41 | Enhanced antigen‐specific CD8+ T cells detected in the spleen six weeks postinfection | i.m. | 8 |
| OX40L, B7‐1, ICAM‐1 and LFA‐3 encoding vector | In vitro stimulation assay | Enhanced CD4+ and CD8+ T cell activation, proliferation, cytokine production and re‐stimulation responses in vitro | – | 9 |
A summary of an agonistic and vector‐based adjuvant strategies used in viral and non viral infectious disease models. Abbreviations: i.p., intraperitoneal; i.v., intravenous; i.m., intramuscular; i.n., intranasal; s.c., sub‐cutaneous; IFA, incomplete freund’s adjuvant; d.s., dermal scarification; VACV, vaccinia virus; VACV‐WR, vaccinia virus western reserve; HBsAg, Hepatitis B surface antigen; SIV, simian immunodeficiency virus; MCMV, murine cytomegalovirus; HIV, human immunodeficiency virus. Corresponding references; study number reference; 1 (161), 2 (168), 3 (149), 4 (169), 5 (170), 6 (164), 7 (165), 8 (167), 9 (166)
Collectively, these studies suggest that OX40 is a promising target for enhancing T‐cell responses against a variety of antigens, and that agonist reagents, to OX40, or OX40L encoded within a vaccine vector, might be useful in vaccination regimens. This strategy could enable the highly desirable ability to retain the use of attenuated vaccines or simple peptide or protein immunization while promote both immunoglobulin and T‐cell components of the immune response. For a more comprehensive insight into the molecular mechanisms of agonistic OX40 therapies reviewed in (171, 172, 173, 174, 175, 176, 177, 178).
Caveats and limitations to OX40 adjuvant strategies
Without doubt the greatest concern with enhancing CD8+ T‐cell responses through the use of agonistic OX40 antibodies or vector encoded OX40L is a hyper‐inflammatory immune response. A clinical trial investigating the propensity of inducing regulatory T cell development through the use of an anti‐CD28 super‐agonist antibody (TGN1412) resulted in six human volunteers experiencing life‐threatening complications (179). Consequently, future clinical trials targeting T‐cell stimulatory receptors will undoubtedly face more stringent safety requirements by regulatory authorities. To this end, toxicity trials in non‐human primates have already demonstrated that agonistic OX40 antibody therapy is well tolerated and safe, therefore providing a strong rationale to pursue clinical tests in humans (168).
Direct interaction between viral pathogens and OX40 itself also raise concerns surrounding the targeting of OX40 to enhance T‐cell responses. Recently, OX40 was identified as cell entry receptor for both feline (FIV) (180, 181) and HIV (182). By replicating in and subsequently killing OX40 expressing cells, both FIV and HIV would preferentially deplete virus‐specific T cells that would otherwise mediate antiviral immune responses. T‐cell tropic viruses may also exploit the OX40‐OX40L axis to aid their dissemination. For example, infection with Human T‐cell leukemia virus type 1 (HTLV‐1), the causative agent of adult T‐cell leukemia (ATL), enhances OX40 expression in a co‐operative process involving NF‐κB and the viral oncoprotein, Tax (183). Intriguingly, a small number of ATL patients displayed OX40 dependent adhesion of leukemic cells to endothelial cells (184), suggesting that virus‐induced OX40 expression could enhance leukemic cell infiltration and viral dissemination.
Viruses can hijack transcriptional activation events triggered by OX40 to induce the expression of their own genes. A number of viruses including cytomegalovirus (185, 186) and HIV (187) have NF‐κB response elements incorporated into their genomes, which can enhance viral reactivation, in the case of CMV, and viral replication (182, 188). The possible influence of OX40 on viral replication, survival and/or reactivation from latency represents an interesting area for future research. Finally, research into the potential pathogenicity of enhanced T‐cell responses upon natural virus re‐exposure is necessary. An inappropriate memory T‐cell response upon re‐exposure to RSV after vaccination with a formalin‐inactivated vaccine resulted in several infant deaths and countless hospitalizations (189). Consequently, it would be necessary to determine the safe upper limit for the number of memory CD8+ T‐cells desired and also whether maintaining large numbers of tissue resident (lung) memory CD8+ T cells is detrimental over time. In summary, although OX40 is a promising target for enhancing protection against infection and reducing viral‐induced immunopathology, the positive effect OX40 may have on virus replication and survival in infected host cells raises new considerations for its manipulation, making the infectious status of the patient of paramount importance.
Conclusions and future directions
Accumulating evidence now indicates that targeting human OX40 and OX40L holds great promise for future vaccine strategies against highly virulent respiratory viruses and other unmet clinical infectious diseases, such as HIV, malaria and TB. The literature discussed above provide compelling evidence to suggests the targeting of costimulatory pathways, such as OX40/OX40L, or other similar molecules in this family, can promote robust expansion of antigen‐specific effector T cells in response to safe attenuated VACV‐vaccine strains and peptide antigen. Furthermore, OX40 stimulation generates long‐lived memory effector T cells that are capable of surviving at mucosal sites in the absence of persisting antigen while maintaining their effector function to provide protection against natural re‐exposure. There is no doubt that increasing our understanding of basic CD8+ T‐cell biology and the dynamic regulatory influences the many costimulatory pathways have on the generation, phenotype, maintenance and reactivation of CD8+ T cells will facilitate the development of novel CD8+ T‐cell vaccines. A key area of research that will provide important insight into the plasticity and adaptation of costimulatory regulation of T‐cell responses is identifying the differences in inflammatory signature between viruses that elicit potent CD8+ T‐cell responses and those that do not. This will enable us to recapitulate the signature of a virulent virus while using a safe attenuated vaccine virus to promote life‐long CD8+ T cell‐mediated immunity.
Acknowledgements
This work was supported by NIH grants AI77079 and AI087734 to S.S.‐A., and by a fellowship from the Center for Infectious Disease at the La Jolla Institute for Allergy and Immunology to S.S.‐A. This is publication 1420 from the La Jolla Institute for Allergy and Immunology. None of the authors have any conflicts of interest.
References
- 1. Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol 2008;8:142–152. [DOI] [PubMed] [Google Scholar]
- 2. Snelgrove RJ, Godlee A, Hussell T. Airway immune homeostasis and implications for influenza‐induced inflammation. Trends Immunol 2011;32:328–334. [DOI] [PubMed] [Google Scholar]
- 3. Wissinger E, Goulding J, Hussell T. Immune homeostasis in the respiratory tract and its impact on heterologous infection. Semin Immunol 2009;21:147–155. [DOI] [PubMed] [Google Scholar]
- 4. Pollard AJ, Perrett KP, Beverley PC. Maintaining protection against invasive bacteria with protein‐polysaccharide conjugate vaccines. Nat Rev Immunol 2009;9:213–220. [DOI] [PubMed] [Google Scholar]
- 5. Plotkin SA. Vaccines: past, present and future. Nat Med 2005;11:S5–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rappuoli R. Bridging the knowledge gaps in vaccine design. Nat Biotechnol 2007;25:1361–1366. [DOI] [PubMed] [Google Scholar]
- 7. Mizgerd JP. Lung infection – a public health priority. PLoS Med 2006;3:e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michaud CM, Murray CJ, Bloom BR. Burden of disease – implications for future research. JAMA 2001;285:535–539. [DOI] [PubMed] [Google Scholar]
- 9. Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997;349:1436–1442. [DOI] [PubMed] [Google Scholar]
- 10. Horimoto T, Kawaoka Y. Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol 2005;3:591–600. [DOI] [PubMed] [Google Scholar]
- 11. Beigel JH, et al. Avian influenza A (H5N1) infection in humans. N Engl J Med 2005;353:1374–1385. [DOI] [PubMed] [Google Scholar]
- 12. Lipscomb MF, Hutt J, Lovchik J, Wu T, Lyons CR. The pathogenesis of acute pulmonary viral and bacterial infections: investigations in animal models. Annu Rev Pathol 2010;5:223–252. [DOI] [PubMed] [Google Scholar]
- 13. Wroblewska M. New developments in vaccines against respiratory viruses. Curr Opin Investig Drugs 2008;9:846–855. [PubMed] [Google Scholar]
- 14. Kohlmeier JE, Woodland DL. Immunity to respiratory viruses. Annu Rev Immunol 2009;27:61–82. [DOI] [PubMed] [Google Scholar]
- 15. Hanage WP, Fraser C, Tang J, Connor TR, Corander J. Hyper‐recombination, diversity, and antibiotic resistance in pneumococcus . Science 2009;324:1454–1457. [DOI] [PubMed] [Google Scholar]
- 16. Brown KF, et al. Factors underlying parental decisions about combination childhood vaccinations including MMR: a systematic review. Vaccine 2010;28:4235–4248. [DOI] [PubMed] [Google Scholar]
- 17. Girard MP, Tam JS, Assossou OM, Kieny MP. The 2009 A (H1N1) influenza virus pandemic: a review. Vaccine 2010;28:4895–4902. [DOI] [PubMed] [Google Scholar]
- 18. Donnelly CA, et al. Epidemiological and genetic analysis of severe acute respiratory syndrome. Lancet Infect Dis 2004;4:672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reed KD, et al. The detection of monkeypox in humans in the Western Hemisphere. N Engl J Med 2004;350:342–350. [DOI] [PubMed] [Google Scholar]
- 20. Parker S, Nuara A, Buller RM, Schultz DA. Human monkeypox: an emerging zoonotic disease. Future Microbiol 2007;2:17–34. [DOI] [PubMed] [Google Scholar]
- 21. Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med 2005;11:S45–S53. [DOI] [PubMed] [Google Scholar]
- 22. Yu X, et al. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 2008;455:532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dormitzer PR, et al. Influenza vaccine immunology. Immunol Rev 2011;239:167–177. [DOI] [PubMed] [Google Scholar]
- 24. Ahlers JD, Belyakov IM. Memories that last forever: strategies for optimizing vaccine T‐cell memory. Blood 2010;115:1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robinson HL, Amara RR. T cell vaccines for microbial infections. Nat Med 2005;11:S25–S32. [DOI] [PubMed] [Google Scholar]
- 26. Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity 2010;33:451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grebe KM, Yewdell JW, Bennink JR. Heterosubtypic immunity to influenza A virus: where do we stand? Microbes Infect 2008;10:1024–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ehrhardt C, Seyer R, Hrincius ER, Eierhoff T, Wolff T, Ludwig S. Interplay between influenza A virus and the innate immune signaling. Microbes Infect 2010;12:81–87. [DOI] [PubMed] [Google Scholar]
- 29. Sanders CJ, Doherty PC, Thomas PG. Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res 2011;343:13–21. [DOI] [PubMed] [Google Scholar]
- 30. McGill J, Heusel JW, Legge KL. Innate immune control and regulation of influenza virus infections. J Leukoc Biol 2009;86:803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woodland DL, Scott I. T cell memory in the lung airways. Proc Am Thorac Soc 2005;2:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol 2009;9:153–161. [DOI] [PubMed] [Google Scholar]
- 33. Legge KL, Braciale TJ. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity 2003;18:265–277. [DOI] [PubMed] [Google Scholar]
- 34. Bajenoff M, Granjeaud S, Guerder S. The strategy of T cell antigen‐presenting cell encounter in antigen‐draining lymph nodes revealed by imaging of initial T cell activation. J Exp Med 2003;198:715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mempel TR, Henrickson SE, Von Andrian UH. T‐cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004;427:154–159. [DOI] [PubMed] [Google Scholar]
- 36. Mempel TR, Scimone ML, Mora JR, von Andrian UH. In vivo imaging of leukocyte trafficking in blood vessels and tissues. Curr Opin Immunol 2004;16:406–417. [DOI] [PubMed] [Google Scholar]
- 37. Hugues S, Fetler L, Bonifaz L, Helft J, Amblard F, Amigorena S. Distinct T cell dynamics in lymph nodes during the induction of tolerance and immunity. Nat Immunol 2004;5:1235–1242. [DOI] [PubMed] [Google Scholar]
- 38. Butz EA, Bevan MJ. Massive expansion of antigen‐specific CD8+ T cells during an acute virus infection. Immunity 1998;8:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crowe SR, Miller SC, Woodland DL. Identification of protective and non‐protective T cell epitopes in influenza. Vaccine 2006;24:452–456. [DOI] [PubMed] [Google Scholar]
- 40. Topham DJ, Castrucci MR, Wingo FS, Belz GT, Doherty PC. The role of antigen in the localization of naive, acutely activated, and memory CD8(+) T cells to the lung during influenza pneumonia. J Immunol 2001;167:6983–6990. [DOI] [PubMed] [Google Scholar]
- 41. McGill J, Legge KL. Cutting edge: contribution of lung‐resident T cell proliferation to the overall magnitude of the antigen‐specific CD8+ T cell response in the lungs following murine influenza virus infection. J Immunol 2009;183:4177–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McGill J, Van Rooijen N, Legge KL. IL‐15 trans‐presentation by pulmonary dendritic cells promotes effector CD8+ T cell survival during influenza virus infection. J Exp Med 2010;207:521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McGill J, Van Rooijen N, Legge KL. Protective influenza‐specific CD8+ T cell responses require interactions with dendritic cells in the lungs. J Exp Med 2008;205:1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Verbist KC, Cole CJ, Field MB, Klonowski KD. A role for IL‐15 in the migration of effector CD8+ T cells to the lung airways following influenza infection. J Immunol 2011;186:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Woodland DL, Dutton RW. Heterogeneity of CD4(+) and CD8(+) T cells. Curr Opin Immunol 2003;15:336–342. [DOI] [PubMed] [Google Scholar]
- 46. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol 2004;22:745–763. [DOI] [PubMed] [Google Scholar]
- 47. Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, Woodland DL. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity 2010;33:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hikono H, et al. T‐cell memory and recall responses to respiratory virus infections. Immunol Rev 2006;211:119–132. [DOI] [PubMed] [Google Scholar]
- 49. Hikono H, Kohlmeier JE, Takamura S, Wittmer ST, Roberts AD, Woodland DL. Activation phenotype, rather than central‐ or effector‐memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J Exp Med 2007;204:1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roberts AD, Ely KH, Woodland DL. Differential contributions of central and effector memory T cells to recall responses. J Exp Med 2005;202:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roberts AD, Woodland DL. Cutting edge: effector memory CD8+ T cells play a prominent role in recall responses to secondary viral infection in the lung. J Immunol 2004;172:6533–6537. [DOI] [PubMed] [Google Scholar]
- 52. Bachmann MF, Wolint P, Schwarz K, Oxenius A. Recall proliferation potential of memory CD8+ T cells and antiviral protection. J Immunol 2005;175:4677–4685. [DOI] [PubMed] [Google Scholar]
- 53. Esser MT, et al. Memory T cells and vaccines. Vaccine 2003;21:419–430. [DOI] [PubMed] [Google Scholar]
- 54. Harty JT, Badovinac VP. Shaping and reshaping CD8+ T‐cell memory. Nat Rev Immunol 2008;8:107–119. [DOI] [PubMed] [Google Scholar]
- 55. Seder RA, Darrah PA, Roederer M. T‐cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol 2008;8:247–258. [DOI] [PubMed] [Google Scholar]
- 56. Appay V, Douek DC, Price DA. CD8+ T cell efficacy in vaccination and disease. Nat Med 2008;14:623–628. [DOI] [PubMed] [Google Scholar]
- 57. Sonoguchi T, Naito H, Hara M, Takeuchi Y, Fukumi H. Cross‐subtype protection in humans during sequential, overlapping, and/or concurrent epidemics caused by H3N2 and H1N1 influenza viruses. J Infect Dis 1985;151:81–88. [DOI] [PubMed] [Google Scholar]
- 58. Frank AL, Taber LH, Wells JM. Individuals infected with two subtypes of influenza A virus in the same season. J Infect Dis 1983;147:120–124. [DOI] [PubMed] [Google Scholar]
- 59. Woodland DL, et al. Antiviral memory T‐cell responses in the lung. Microbes Infect 2002;4:1091–1098. [DOI] [PubMed] [Google Scholar]
- 60. Liang S, Mozdzanowska K, Palladino G, Gerhard W. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J Immunol 1994;152:1653–1661. [PubMed] [Google Scholar]
- 61. Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC. Virus‐specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity 1998;8:683–691. [DOI] [PubMed] [Google Scholar]
- 62. Hogan RJ, et al. Activated antigen‐specific CD8+ cells persist in the lungs following recovery from respiratory virus infections. J Immunol 2001;166:1813–1822. [DOI] [PubMed] [Google Scholar]
- 63. Hou S, Hyland L, Ryan KW, Portner A, Doherty PC. Virus‐specific CD8+ T‐cell memory determined by clonal burst size. Nature 1994;369:652–654. [DOI] [PubMed] [Google Scholar]
- 64. Gray D, Matzinger P. T cell memory is short‐lived in the absence of antigen. J Exp Med 1991;174:969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science 1997;276:2057–2062. [DOI] [PubMed] [Google Scholar]
- 66. Murali‐Krishna K, Lau LL, Sambhara S, Lemonnier F, Altman J, Ahmed R. Persistence of memory CD8+ T cells in MHC class I‐deficient mice. Science 1999;286:1377–1381. [DOI] [PubMed] [Google Scholar]
- 67. Gray D. A role for antigen in the maintenance of immunological memory. Nat Rev Immunol 2002;2:60–65. [DOI] [PubMed] [Google Scholar]
- 68. Surh CD, Boyman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev 2006;211:154–163. [DOI] [PubMed] [Google Scholar]
- 69. Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity 2008;29:848–862. [DOI] [PubMed] [Google Scholar]
- 70. Ely KH, Cauley LS, Roberts AD, Brennan JW, Cookenham T, Woodland DL. Nonspecific recruitment of memory CD8+ T cells to the lung airways during respiratory virus infections. J Immunol 2003;170:1423–1429. [DOI] [PubMed] [Google Scholar]
- 71. Jelley‐Gibbs DM, Brown DM, Dibble JP, Haynes L, Eaton SM, Swain SL. Unexpected prolonged presentation of influenza antigens promotes CD4+ T cell memory generation. J Exp Med 2005;202:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zammit DJ, Turner DL, Klonowski KD, Lefrancois L, Cauley LS. Residual antigen presentation after influenza virus infection affects CD8+ T cell activation and migration. Immunity 2006;24:439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kohlmeier JE, Miller SC, Woodland DL. Cutting edge: antigen is not required for the activation and maintenance of virus‐specific memory CD8+ T cells in the lung airways. J Immunol 2007;178:4721–4725. [DOI] [PubMed] [Google Scholar]
- 74. Belyakov IM, Ahlers JD. What role does the route of immunization play in the generation of protective immunity against mucosal pathogens? J Immunol 2009;183:6883–6892. [DOI] [PubMed] [Google Scholar]
- 75. Liu L, Zhong Q, Tian T, Dubin K, Athale SK, Kupper TS. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell‐mediated immunity. Nat Med 2010;16:224–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Salek‐Ardakani S, et al. The TNFR family members OX40 and CD27 link viral virulence to protective T cell vaccines in mice. J Clin Invest 2011;121:296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Verbist KC, Field MB, Klonowski KD. Cutting edge: IL‐15‐independent maintenance of mucosally generated memory CD8+ T cells. J Immunol 2011;186:6667–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Takamura S, Roberts AD, Jelley‐Gibbs DM, Wittmer ST, Kohlmeier JE, Woodland DL. The route of priming influences the ability of respiratory virus‐specific memory CD8+ T cells to be activated by residual antigen. J Exp Med 2010;207:1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu MA. Immunologic basis of vaccine vectors. Immunity 2010;33:504–515. [DOI] [PubMed] [Google Scholar]
- 80. Rollier CS, Reyes‐Sandoval A, Cottingham MG, Ewer K, Hill AV. Viral vectors as vaccine platforms: deployment in sight. Curr Opin Immunol 2011;23:377–382. [DOI] [PubMed] [Google Scholar]
- 81. Perkus ME, Tartaglia J, Paoletti E. Poxvirus‐based vaccine candidates for cancer, AIDS, and other infectious diseases. J Leukoc Biol 1995;58:1–13. [DOI] [PubMed] [Google Scholar]
- 82. Moss B. Smallpox vaccines: targets of protective immunity. Immunol Rev 2011;239:8–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jacobs BL, et al. Vaccinia virus vaccines: past, present and future. Antiviral Res 2009;84:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Artenstein AW. New generation smallpox vaccines: a review of preclinical and clinical data. Rev Med Virol 2008;18:217–231. [DOI] [PubMed] [Google Scholar]
- 85. Amanna IJ, Slifka MK, Crotty S. Immunity and immunological memory following smallpox vaccination. Immunol Rev 2006;211:320–337. [DOI] [PubMed] [Google Scholar]
- 86. Panicali D, Paoletti E. Construction of poxviruses as cloning vectors: insertion of the thymidine kinase gene from herpes simplex virus into the DNA of infectious vaccinia virus. Proc Natl Acad Sci USA 1982;79:4927–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mackett M, Smith GL, Moss B. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proc Natl Acad Sci USA 1982;79:7415–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Smith GL, Mackett M, Moss B. Infectious vaccinia virus recombinants that express hepatitis B virus surface antigen. Nature 1983;302:490–495. [DOI] [PubMed] [Google Scholar]
- 89. Wiktor TJ, et al. Protection from rabies by a vaccinia virus recombinant containing the rabies virus glycoprotein gene. Proc Natl Acad Sci USA 1984;81:7194–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mackett M, Yilma T, Rose JK, Moss B. Vaccinia virus recombinants: expression of VSV genes and protective immunization of mice and cattle. Science 1985;227:433–435. [DOI] [PubMed] [Google Scholar]
- 91. Cremer KJ, Mackett M, Wohlenberg C, Notkins AL, Moss B. Vaccinia virus recombinant expressing herpes simplex virus type 1 glycoprotein D prevents latent herpes in mice. Science 1985;228:737–740. [DOI] [PubMed] [Google Scholar]
- 92. Mackett M, Arrand JR. Recombinant vaccinia virus induces neutralising antibodies in rabbits against Epstein‐Barr virus membrane antigen gp340. EMBO J 1985;4:3229–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Perkus ME, et al. Live attenuated vaccinia and other poxviruses as delivery systems: public health issues. Ann N Y Acad Sci 1995;754:222–233. [DOI] [PubMed] [Google Scholar]
- 94. Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol 1991;72:1031–1038. [DOI] [PubMed] [Google Scholar]
- 95. Tartaglia J, et al. NYVAC: a highly attenuated strain of vaccinia virus. Virology 1992;188:217–232. [DOI] [PubMed] [Google Scholar]
- 96. Paoletti E, Tartaglia J, Taylor J. Safe and effective poxvirus vectors – NYVAC and ALVAC. Dev Biol Stand 1994;82:65–69. [PubMed] [Google Scholar]
- 97. Taylor J, et al. Nonreplicating viral vectors as potential vaccines: recombinant canarypox virus expressing measles virus fusion (F) and hemagglutinin (HA) glycoproteins. Virology 1992;187:321–328. [DOI] [PubMed] [Google Scholar]
- 98. Taylor J, et al. Vaccinia virus recombinants expressing either the measles virus fusion or hemagglutinin glycoprotein protect dogs against canine distemper virus challenge. J Virol 1991;65:4263–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gomez CE, Najera JL, Krupa M, Esteban M. The poxvirus vectors MVA and NYVAC as gene delivery systems for vaccination against infectious diseases and cancer. Curr Gene Ther 2008;8:97–120. [DOI] [PubMed] [Google Scholar]
- 100. Kwak H, Horig H, Kaufman HL. Poxviruses as vectors for cancer immunotherapy. Curr Opin Drug Discov Devel 2003;6:161–168. [PubMed] [Google Scholar]
- 101. Marthas ML, et al. Partial efficacy of a VSV‐SIV/MVA‐SIV vaccine regimen against oral SIV challenge in infant macaques. Vaccine 2011;29:3124–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rerks‐Ngarm S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV‐1 infection in Thailand. N Engl J Med 2009;361:2209–2220. [DOI] [PubMed] [Google Scholar]
- 103. Cleghorn F, et al. Lessons from a multisite international trial in the Caribbean and South America of an HIV‐1 Canarypox vaccine (ALVAC‐HIV vCP1452) with or without boosting with MN rgp120. J Acquir Immune Defic Syndr 2007;46:222–230. [DOI] [PubMed] [Google Scholar]
- 104. Goepfert PA, et al. High‐dose recombinant Canarypox vaccine expressing HIV‐1 protein, in seronegative human subjects. J Infect Dis 2005;192:1249–1259. [DOI] [PubMed] [Google Scholar]
- 105. Goepfert PA, et al. Phase 1 safety and immunogenicity testing of DNA and recombinant modified vaccinia Ankara vaccines expressing HIV‐1 virus‐like particles. J Infect Dis 2011;203:610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Thongcharoen P, et al. A phase 1/2 comparative vaccine trial of the safety and immunogenicity of a CRF01_AE (subtype E) candidate vaccine: ALVAC‐HIV (vCP1521) prime with oligomeric gp160 (92TH023/LAI‐DID) or bivalent gp120 (CM235/SF2) boost. J Acquir Immune Defic Syndr 2007;46:48–55. [DOI] [PubMed] [Google Scholar]
- 107. Cebere I, et al. Phase I clinical trial safety of DNA‐ and modified virus Ankara‐vectored human immunodeficiency virus type 1 (HIV‐1) vaccines administered alone and in a prime‐boost regime to healthy HIV‐1‐uninfected volunteers. Vaccine 2006;24:417–425. [DOI] [PubMed] [Google Scholar]
- 108. Peters BS, et al. Studies of a prophylactic HIV‐1 vaccine candidate based on modified vaccinia virus Ankara (MVA) with and without DNA priming: effects of dosage and route on safety and immunogenicity. Vaccine 2007;25:2120–2127. [DOI] [PubMed] [Google Scholar]
- 109. Berthoud TK, et al. Potent CD8+ T‐cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA‐NP+M1. Clin Infect Dis 2011;52:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Santra S, et al. Recombinant poxvirus boosting of DNA‐primed rhesus monkeys augments peak but not memory T lymphocyte responses. Proc Natl Acad Sci USA 2004;101:11088–11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pillai VK, et al. Different patterns of expansion, contraction and memory differentiation of HIV‐1 Gag‐specific CD8+ T cells elicited by adenovirus type 5 and modified vaccinia Ankara vaccines. Vaccine 2011;29:5399–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Reyes‐Sandoval A, et al. CD8+ T effector memory cells protect against liver‐stage malaria. J Immunol 2011;187:1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Morgan AJ, Mackett M, Finerty S, Arrand JR, Scullion FT, Epstein MA. Recombinant vaccinia virus expressing Epstein‐Barr virus glycoprotein gp340 protects cottontop tamarins against EB virus‐induced malignant lymphomas. J Med Virol 1988;25:189–195. [DOI] [PubMed] [Google Scholar]
- 114. Croft M. Co‐stimulatory members of the TNFR family: keys to effective T‐cell immunity? Nat Rev Immunol 2003;3:609–620. [DOI] [PubMed] [Google Scholar]
- 115. Bevan MJ. Helping the CD8(+) T‐cell response. Nat Rev Immunol 2004;4:595–602. [DOI] [PubMed] [Google Scholar]
- 116. Smith GL, Symons JA, Khanna A, Vanderplasschen A, Alcami A. Vaccinia virus immune evasion. Immunol Rev 1997;159:137–154. [DOI] [PubMed] [Google Scholar]
- 117. Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. Viral subversion of the immune system. Annu Rev Immunol 2000;18:861–926. [DOI] [PubMed] [Google Scholar]
- 118. Alcami A. Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol 2003;3:36–50. [DOI] [PubMed] [Google Scholar]
- 119. Finlay BB, McFadden G. Anti‐immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 2006;124:767–782. [DOI] [PubMed] [Google Scholar]
- 120. Sharpe AH, Freeman GJ. The B7‐CD28 superfamily. Nat Rev Immunol 2002;2:116–126. [DOI] [PubMed] [Google Scholar]
- 121. Chen L. Co‐inhibitory molecules of the B7‐CD28 family in the control of T‐cell immunity. Nat Rev Immunol 2004;4:336–347. [DOI] [PubMed] [Google Scholar]
- 122. Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol 2005;23:23–68. [DOI] [PubMed] [Google Scholar]
- 123. Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol 2005;23:787–819. [DOI] [PubMed] [Google Scholar]
- 124. Salek‐Ardakani S, Croft M. Tumor necrosis factor receptor/tumor necrosis factor family members in antiviral CD8+ T‐cell immunity. J Interferon Cytokine Res 2010;30:205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Sedger LM, et al. Poxvirus tumor necrosis factor receptor (TNFR)‐like T2 proteins contain a conserved preligand assembly domain that inhibits cellular TNFR1‐induced cell death. J Virol 2006;80:9300–9309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sedy JR, Spear PG, Ware CF. Cross‐regulation between herpesviruses and the TNF superfamily members. Nat Rev Immunol 2008;8:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Schneider K, Potter KG, Ware CF. Lymphotoxin and LIGHT signaling pathways and target genes. Immunol Rev 2004;202:49–66. [DOI] [PubMed] [Google Scholar]
- 128. Saraiva M, Smith P, Fallon PG, Alcami A. Inhibition of type 1 cytokine‐mediated inflammation by a soluble CD30 homologue encoded by ectromelia (mousepox) virus. J Exp Med 2002;196:829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Glenney GW, Wiens GD. Early diversification of the TNF superfamily in teleosts: genomic characterization and expression analysis. J Immunol 2007;178:7955–7973. [DOI] [PubMed] [Google Scholar]
- 130. Isaacs SN. A stimulating way to improve T cell responses to poxvirus‐vectored vaccines. J Clin Invest 2011;121:19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Amara RR, Nigam P, Sharma S, Liu J, Bostik V. Long‐lived poxvirus immunity, robust CD4 help, and better persistence of CD4 than CD8+ T cells. J Virol 2004;78:3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Hammarlund E, Lewis MW, Hanifin JM, Mori M, Koudelka CW, Slifka MK. Duration of antiviral immunity after smallpox vaccination. Nat Med 2003;9:1131–1137. [DOI] [PubMed] [Google Scholar]
- 133. Combadiere B, et al. Distinct time effects of vaccination on long‐term proliferative and IFN‐gamma‐producing T cell memory to smallpox in humans. J Exp Med 2004;199:1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hsieh SM, Pan SC, Chen SY, Huang PF, Chang SC. Age distribution for T cell reactivity to vaccinia virus in a healthy population. Clin Infect Dis 2004;38:86–89. [DOI] [PubMed] [Google Scholar]
- 135. Kim SH, Oh MD. Persistence of cell‐mediated immunity to vaccinia virus. J Infect Dis 2007;196:805–806. [DOI] [PubMed] [Google Scholar]
- 136. Salek‐Ardakani S, Moutaftsi M, Crotty S, Sette A, Croft M. OX40 drives protective vaccinia virus‐specific CD8+ T cells. J Immunol 2008;181:7969–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Salek‐Ardakani S, Arens R, Flynn R, Sette A, Schoenberger SP, Croft M. Preferential use of B7.2 and not B7.1 in priming of vaccinia virus‐specific CD8+ T cells. J Immunol 2009;182:2909–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Fang M, Sigal LJ. Direct CD28 costimulation is required for CD8+ T cell‐mediated resistance to an acute viral disease in a natural host. J Immunol 2006;177:8027–8036. [DOI] [PubMed] [Google Scholar]
- 139. Schildknecht A, Miescher I, Yagita H, van den Broek M. Priming of CD8+ T cell responses by pathogens typically depends on CD70‐mediated interactions with dendritic cells. Eur J Immunol 2007;37:716–728. [DOI] [PubMed] [Google Scholar]
- 140. Croft M. The role of TNF superfamily members in T‐cell function and diseases. Nat Rev Immunol 2009;9:271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Borst J, Hendriks J, Xiao Y. CD27 and CD70 in T cell and B cell activation. Curr Opin Immunol 2005;17:275–281. [DOI] [PubMed] [Google Scholar]
- 142. Peperzak V, Xiao Y, Veraar EA, Borst J. CD27 sustains survival of CTLs in virus‐infected nonlymphoid tissue in mice by inducing autocrine IL‐2 production. J Clin Invest 2010;120:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Peperzak V, Veraar EA, Keller AM, Xiao Y, Borst J. The Pim kinase pathway contributes to survival signaling in primed CD8+ T cells upon CD27 costimulation. J Immunol 2010;185:6670–6678. [DOI] [PubMed] [Google Scholar]
- 144. Dolfi DV, Boesteanu AC, Petrovas C, Xia D, Butz EA, Katsikis PD. Late signals from CD27 prevent Fas‐dependent apoptosis of primary CD8+ T cells. J Immunol 2008;180:2912–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Pippig SD, et al. Robust B cell immunity but impaired T cell proliferation in the absence of CD134 (OX40). J Immunol 1999;163:6520–6529. [PubMed] [Google Scholar]
- 146. Kopf M, et al. OX40‐deficient mice are defective in Th cell proliferation but are competent in generating B cell and CTL responses after virus infection. Immunity 1999;11:699–708. [DOI] [PubMed] [Google Scholar]
- 147. Dawicki W, Bertram EM, Sharpe AH, Watts TH. 4‐1BB and OX40 act independently to facilitate robust CD8 and CD4 recall responses. J Immunol 2004;173:5944–5951. [DOI] [PubMed] [Google Scholar]
- 148. Hendriks J, et al. During viral infection of the respiratory tract, CD27, 4‐1BB, and OX40 collectively determine formation of CD8+ memory T cells and their capacity for secondary expansion. J Immunol 2005;175:1665–1676. [DOI] [PubMed] [Google Scholar]
- 149. Humphreys IR, et al. OX40 costimulation promotes persistence of cytomegalovirus‐specific CD8+ T Cells: a CD4‐dependent mechanism. J Immunol 2007;179:2195–2202. [DOI] [PubMed] [Google Scholar]
- 150. Thompson LJ, Kolumam GA, Thomas S, Murali‐Krishna K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN‐I dependence of CD8+ T cells for clonal expansion and memory formation. J Immunol 2006;177:1746–1754. [DOI] [PubMed] [Google Scholar]
- 151. Ranasinghe C, et al. Evaluation of fowlpox‐vaccinia virus prime‐boost vaccine strategies for high‐level mucosal and systemic immunity against HIV‐1. Vaccine 2006;24:5881–5895. [DOI] [PubMed] [Google Scholar]
- 152. Belyakov IM, Isakov D, Zhu Q, Dzutsev A, Klinman D, Berzofsky JA. Enhancement of CD8+ T cell immunity in the lung by CpG oligodeoxynucleotides increases protective efficacy of a modified vaccinia Ankara vaccine against lethal poxvirus infection even in a CD4‐deficient host. J Immunol 2006;177:6336–6343. [DOI] [PubMed] [Google Scholar]
- 153. Kaufman HL, Flanagan K, Lee CS, Perretta DJ, Horig H. Insertion of interleukin‐2 (IL‐2) and interleukin‐12 (IL‐12) genes into vaccinia virus results in effective anti‐tumor responses without toxicity. Vaccine 2002;20:1862–1869. [DOI] [PubMed] [Google Scholar]
- 154. Li S, et al. IL‐15 increases the frequency of effector memory CD8+ T cells in rhesus monkeys immunized with HIV vaccine. Cell Mol Immunol 2010;7:491–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Zielinski RJ, et al. Smallpox vaccine with integrated IL‐15 demonstrates enhanced in vivo viral clearance in immunodeficient mice and confers long term protection against a lethal monkeypox challenge in cynomolgus monkeys. Vaccine 2010;28:7081–7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Poon LL, et al. Vaccinia virus‐based multivalent H5N1 avian influenza vaccines adjuvanted with IL‐15 confer sterile cross‐clade protection in mice. J Immunol 2009;182:3063–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Oh S, Berzofsky JA, Burke DS, Waldmann TA, Perera LP. Coadministration of HIV vaccine vectors with vaccinia viruses expressing IL‐15 but not IL‐2 induces long‐lasting cellular immunity. Proc Natl Acad Sci USA 2003;100:3392–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Feder‐Mengus C, et al. Nonreplicating recombinant vaccinia virus expressing CD40 ligand enhances APC capacity to stimulate specific CD4+ and CD8+ T cell responses. Hum Gene Ther 2005;16:348–360. [DOI] [PubMed] [Google Scholar]
- 159. Barr TA, Carlring J, Heath AW. Co‐stimulatory agonists as immunological adjuvants. Vaccine 2006;24:3399–3407. [DOI] [PubMed] [Google Scholar]
- 160. Kudo‐Saito C, Hodge JW, Kwak H, Kim‐Schulze S, Schlom J, Kaufman HL. 4‐1BB ligand enhances tumor‐specific immunity of poxvirus vaccines. Vaccine 2006;24:4975–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Salek‐Ardakani S, Moutaftsi M, Sette A, Croft M. Targeting OX40 promotes lung resident memory CD8+ T‐cell populations that protect against respiratory poxvirus infection. J Virol 2011;85:9051–9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ely KH, Roberts AD, Woodland DL. Cutting edge: effector memory CD8+ T cells in the lung airways retain the potential to mediate recall responses. J Immunol 2003;171:3338–3342. [DOI] [PubMed] [Google Scholar]
- 163. Cerwenka A, Morgan TM, Harmsen AG, Dutton RW. Migration kinetics and final destination of type 1 and type 2 CD8 effector cells predict protection against pulmonary virus infection. J Exp Med 1999;189:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Du X, et al. The adjuvant effects of co‐stimulatory molecules on cellular and memory responses to HBsAg DNA vaccination. J Gene Med 2007;9:136–146. [DOI] [PubMed] [Google Scholar]
- 165. Xiao C, et al. Enhanced protective efficacy and reduced viral load of foot‐and‐mouth disease DNA vaccine with co‐stimulatory molecules as the molecular adjuvants. Antiviral Res 2007;76:11–20. [DOI] [PubMed] [Google Scholar]
- 166. Grosenbach DW, Schlom J, Gritz L, Gomez Yafal A, Hodge JW. A recombinant vector expressing transgenes for four T‐cell costimulatory molecules (OX40L, B7‐1, ICAM‐1, LFA‐3) induces sustained CD4+ and CD8+ T‐cell activation, protection from apoptosis, and enhanced cytokine production. Cell Immunol 2003;222:45–57. [DOI] [PubMed] [Google Scholar]
- 167. Liu J, Ngai N, Stone GW, Yue FY, Ostrowski MA. The adjuvancy of OX40 ligand (CD252) on an HIV‐1 canarypox vaccine. Vaccine 2009;27:5077–5084. [DOI] [PubMed] [Google Scholar]
- 168. Weinberg AD, et al. Anti‐OX40 (CD134) administration to nonhuman primates: immunostimulatory effects and toxicokinetic study. J Immunother 2006;29:575–585. [DOI] [PubMed] [Google Scholar]
- 169. Munks MW, Mourich DV, Mittler RS, Weinberg AD, Hill AB. 4‐1BB and OX40 stimulation enhance CD8 and CD4+ T‐cell responses to a DNA prime, poxvirus boost vaccine. Immunology 2004;112:559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Humphreys IR, et al. OX40 ligation on activated T cells enhances the control of Cryptococcus neoformans and reduces pulmonary eosinophilia. J Immunol 2003;170:6125–6132. [DOI] [PubMed] [Google Scholar]
- 171. Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T‐cell co‐stimulatory molecule OX40. Nat Rev Immunol 2004;4:420–431. [DOI] [PubMed] [Google Scholar]
- 172. Croft M. Control of immunity by the TNFR‐related molecule OX40 (CD134). Annu Rev Immunol 2010;28:57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Vasilevsky NA, Ruby CE, Hurlin PJ, Weinberg AD. OX40 engagement stabilizes Mxd4 and Mnt protein levels in antigen‐stimulated T cells leading to an increase in cell survival. Eur J Immunol 2011;41:1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Ruby CE, Montler R, Zheng R, Shu S, Weinberg AD. IL‐12 is required for anti‐OX40‐mediated CD4+ T cell survival. J Immunol 2008;180:2140–2148. [DOI] [PubMed] [Google Scholar]
- 175. Ruby CE, Redmond WL, Haley D, Weinberg AD. Anti‐OX40 stimulation in vivo enhances CD8+ memory T cell survival and significantly increases recall responses. Eur J Immunol 2007;37:157–166. [DOI] [PubMed] [Google Scholar]
- 176. Williams CA, Murray SE, Weinberg AD, Parker DC. OX40‐mediated differentiation to effector function requires IL‐2 receptor signaling but not CD28, CD40, IL‐12Rbeta2, or T‐bet. J Immunol 2007;178:7694–7702. [DOI] [PubMed] [Google Scholar]
- 177. Lee SJ, et al. CD134 costimulation couples the CD137 pathway to induce production of supereffector CD8+ T cells that become IL‐7 dependent. J Immunol 2007;179:2203–2214. [DOI] [PubMed] [Google Scholar]
- 178. Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood 2005;105:2845–2851. [DOI] [PubMed] [Google Scholar]
- 179. Stebbings R, Poole S, Thorpe R. Safety of biologics, lessons learnt from TGN1412. Curr Opin Biotechnol 2009;20:673–677. [DOI] [PubMed] [Google Scholar]
- 180. Shiao SL, McNiff JM, Pober JS. Memory T cells and their costimulators in human allograft injury. J Immunol 2005;175:4886–4896. [DOI] [PubMed] [Google Scholar]
- 181. de Parseval A, Chatterji U, Morris G, Sun P, Olson AJ, Elder JH. Structural mapping of CD134 residues critical for interaction with feline immunodeficiency virus. Nat Struct Mol Biol 2005;12:60–66. [DOI] [PubMed] [Google Scholar]
- 182. Takahashi Y, Tanaka Y, Yamashita A, Koyanagi Y, Nakamura M, Yamamoto N. OX40 stimulation by gp34/OX40 ligand enhances productive human immunodeficiency virus type 1 infection. J Virol 2001;75:6748–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Pankow R, et al. The HTLV‐I tax protein transcriptionally modulates OX40 antigen expression. J Immunol 2000;165:263–270. [DOI] [PubMed] [Google Scholar]
- 184. Imura A, et al. OX40 expressed on fresh leukemic cells from adult T‐cell leukemia patients mediates cell adhesion to vascular endothelial cells: implication for the possible involvement of OX40 in leukemic cell infiltration. Blood 1997;89:2951–2958. [PubMed] [Google Scholar]
- 185. Sambucetti LC, Cherrington JM, Wilkinson GW, Mocarski ES. NF‐kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J 1989;8:4251–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Yurochko AD, Kowalik TF, Huong SM, Huang ES. Human cytomegalovirus upregulates NF‐kappa B activity by transactivating the NF‐kappa B p105/p50 and p65 promoters. J Virol 1995;69:5391–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. DeLuca C, Petropoulos L, Zmeureanu D, Hiscott J. Nuclear IkappaBbeta maintains persistent NF‐kappaB activation in HIV‐1‐infected myeloid cells. J Biol Chem 1999;274:13010–13016. [DOI] [PubMed] [Google Scholar]
- 188. Benedict CA, et al. Neutrality of the canonical NF‐kappaB‐dependent pathway for human and murine cytomegalovirus transcription and replication in vitro . J Virol 2004;78:741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Openshaw PJ, Culley FJ, Olszewska W. Immunopathogenesis of vaccine‐enhanced RSV disease. Vaccine 2001;20(Suppl):S27–S31. [DOI] [PubMed] [Google Scholar]