

Abstract
Integrating high-throughput data obtained from different molecular levels is essential for understanding the mechanisms of complex diseases such as cancer. In this study, we integrated the methylation, microRNA and mRNA data from lung cancer tissues and normal lung tissues using functional gene sets. For each Gene Ontology (GO) term, three sets were defined: the methylation set, the microRNA set and the mRNA set. The discriminating ability of each gene set was represented by the Matthews correlation coefficient (MCC), as evaluated by leave-one-out cross-validation (LOOCV). Next, the MCCs in the methylation sets, the microRNA sets and the mRNA sets were ranked. By comparing the MCC ranks of methylation, microRNA and mRNA for each GO term, we classified the GO sets into six groups and identified the dysfunctional methylation, microRNA and mRNA gene sets in lung cancer. Our results provide a systematic view of the functional alterations during tumorigenesis that may help to elucidate the mechanisms of lung cancer and lead to improved treatments for patients.
Introduction
Cancer is a systems biology disease [1] that involves the dysregulation of multiple pathways at multiple levels [2]. High-throughput technologies, such as genomic sequencing and transcriptomic, proteomic and metabolomic profiling, have provided large quantities of experimental data. However, systems biology requires not only new high-throughput “-omics” data-generation technologies but also integrative analysis methods that may shed light on the potential mechanisms of complex diseases. Lung cancer is one of the leading causes of cancer death worldwide [3]. There are currently known genetic, epigenetic, transcriptomic, proteomic, metabolomic, and microRNA markers of lung cancer [4]. Because epigenetic changes occur early during tumorigenesis, methylation markers should be considered [4]. The protein is the final, functional form of the genetic information; therefore, proteomic markers are also important. Transcriptomic markers are easy to measure, and mRNA levels are frequently used as a proxy for protein abundance [5]. MicroRNA, as an important regulatory contributor, is also an excellent lung cancer biomarker [6], [7]. Whether a methylation marker, mRNA marker, or microRNA marker is considered, these markers function by affecting biological pathways or networks. The functional pathways are the common bridges between various markers and the disease.
Currently, there are several studies on multi-dimensional data integration [8]–[11]. Most of them were based on regression between different dimensions [10] and require each sample to have multiple level data [11]. The dysfunctional pathways were identified by enrichment analysis of aberrant genes [9].
In this study, we directly analyze dysfunctions of non-small-cell lung cancer (NSCLC) by comparing the functional sets of methylation, microRNA and mRNA data between lung cancer tissues and normal lung tissues. Each functional set corresponds to one Gene Ontology (GO) [12] term. Three sets of this functional unit are defined: the methylation set, the microRNA set and the mRNA set. The Matthews correlation coefficient (MCC), evaluated by leave-one-out cross-validation (LOOCV), is used to represent the discriminating ability of each gene set. The MCC ranks of each methylation set, microRNA set and mRNA set are analyzed. Six groups of GO sets are classified, and 20 dysfunctional methylation, microRNA and mRNA gene sets in lung cancer are identified. These dysfunctional sets characterize the processes of tumorigenesis. With an accurate characterization of tumorigenesis, we may better understand the mechanisms of lung cancer and improve the early diagnosis, treatment efficiency evaluation, and prognosis of lung cancer.
Materials and Methods
Data sets
We downloaded the methylation profiles of 1,413 genes in 57 NSCLC patients and 52 control samples [13] from GEO (Gene Expression Omnibus) with the accession number GSE16559. The microRNA expression profiles of 549 microRNAs in 187 NSCLC patients and 188 control samples [14] were retrieved from GEO with the accession number GSE15008. The mRNA gene expression profiles of 19,700 genes in 46 NSCLC patients and 45 control samples [15] were obtained from GEO with the accession number GSE18842.
Since the methylation data, microRNA data and mRNA data were obtained from different NSCLC studies, we compared the clinical information of patients from these three studies. The two kinds of clinical information that were given in at least two studies were age and grade of differentiation. The clinical information from these three studies is shown in
Table 1
. The average age of patients from the methylation study is 68.2 and their standard deviation is 11.4; meanwhile, the average age of patients from the microRNA study is 59.9 and the standard deviation is 9.8. The ages of patients in these two studies are similar. The percentages of well-, moderately- and poorly-differentiated cancer patients in the microRNA study and the mRNA study were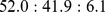 and
and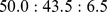 , respectively. The distributions of grades of differentiation in these two studies were very similar. Based on the available clinical information on these NSCLC patients, we think that these three data sets may represent some common dysfunctions of NSCLC.
, respectively. The distributions of grades of differentiation in these two studies were very similar. Based on the available clinical information on these NSCLC patients, we think that these three data sets may represent some common dysfunctions of NSCLC.
Table 1. Clinical information for NSCLC patients in three data sets.
| Methylation data | microRNA data | mRNA data | |
| Age: Mean (Standard Deviation) | 68.2 (11.4)s | 59.9 (9.8) | - |
| Differentiation: Well, % | - | 52.0 | 50.0 |
| Differentiation: Moderate, % | - | 41.9 | 43.5 |
| Differentiation: Poor, % | - | 6.1 | 6.5 |
The target genes of microRNAs
We define the target genes of the microRNAs to be those that were predicted by at least three out of the following six software tools: miRBase [16] (http://microrna.sanger.ac.uk/targets/v5/), TargetScan [17] (http://www.targetscan.org/), miRanda [18] (http://www.microrna.org/microrna/), TarBase [19] (http://diana.cslab.ece.ntua.gr/tarbase/), mirTarget2 [20] (http://mirdb.org/miRDB/download.html), and PicTar [21] (http://pictar.mdc-berlin.de/). Table S1 gives the microRNA - target gene pairs that are predicted by at least three tools.
The GO gene sets for methylation, microRNA and mRNA
For each GO term, we define three gene sets to represent it: first, the methylation gene set, which consists of the genes that are annotated to the GO term and for which the methylation level has been measured; second, the microRNA gene set, which consists of the microRNAs that have target genes annotated to this term; and third, the mRNA gene set, which consists of all the genes annotated to this term.
The discriminating ability of gene sets
We evaluated the discriminating ability of gene sets by constructing a prediction model. First, the Nearest Neighbor Algorithm (NNA) [5], [22]–[30] was applied to build the prediction model. Next, the prediction models were tested using LOOCV [5], [22]–[31]. Finally, the Matthews correlation coefficient (MCC) [26], [30] of LOOCV was used as the measurement of the gene set's discriminating ability.
The NNA [5], [22]–[30] is a widely used machine learning method. The NNA makes its prediction by comparing the distances between the query sample and the samples with known classes, i.e., the lung cancer samples or control samples. The query sample was predicted to have the same class as its nearest neighbor, i.e., the sample with known class that has the smallest distance. In this analysis, the distance between two samples 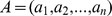 and
and 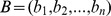 was defined as one minus the cosine similarity between the two samples [5], [23]–[27], [30], [32]–[34]:
was defined as one minus the cosine similarity between the two samples [5], [23]–[27], [30], [32]–[34]:
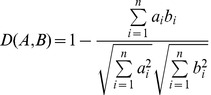 |
(1) |
The NNA program can be downloaded from http://pcal.biosino.org/NNA.html.
During LOOCV [32], [35], [36], each sample in the benchmark dataset will be chosen as the test set once and tested by the prediction model trained by the rest of the samples.
The Matthews correlation coefficient (MCC) is a balanced measurement of prediction performance that considers both sensitivity and specificity [26], [30]. It is calculated using the following formula:
| (2) |
in which TP, TN, FP and FN are the numbers of true lung cancer samples, true control samples, false lung cancer samples and false control samples, respectively.
Classification of gene sets based on their dysfunctional level: methylation, microRNA or mRNA
After we calculated the MCC of each gene set at each level, we ranked the gene sets of each level based on their MCCs and compared the ranks of the three levels, methylation, microRNA and mRNA, in each gene set. With certain values proving to be equal, their ranks were replaced by their mean ranks. As an example of a GO term, if its methylation level had changed between normal and cancer tissue, but its microRNA and mRNA levels had not changed, it was defined as a methylation dysfunctional GO gene set. Similarly, we can define other types of GO gene sets. In total, we defined six groups of gene sets, one for each possible rank ordering of methylation, microRNA and mRNA.
The work flow of dysfunctional methylation, microRNA and mRNA gene set analysis
Our strategy of dysfunctional methylation, microRNA and mRNA gene set analysis is demonstrated in Figure 1 . First, for each GO term, we defined three sets: the methylation set, the microRNA set and the mRNA set. Next, we calculated each gene set's MCC, as evaluated by LOOCV. We ranked the MCCs in the methylation sets, the microRNA sets and the mRNA sets. Next, we compared the MCC ranks of methylation, microRNA and mRNA in each GO term and classified the GO sets into six groups based on these ranks. Finally, we identified the dysfunctional methylation, microRNA and mRNA gene sets in lung cancer.
Figure 1. The work flow of dysfunctional methylation, microRNA and mRNA gene set analysis.

First, for each Gene Ontology (GO) term, we defined three gene sets: the methylation set, the microRNA set and the mRNA set. Next, we calculated the Matthews's correlation coefficient (MCC), as evaluated by leave-one-out cross-validation (LOOCV), for each gene set. Next, we ranked the MCCs in the methylation sets, the microRNA sets and the mRNA sets, and we compared the MCC ranks of methylation, microRNA and mRNA for each Gene Ontology (GO) term and classified the GO sets into six groups. Finally, we identified the dysfunctional methylation, microRNA and mRNA gene sets in lung cancer.
Results and Discussion
The GO gene sets of methylation, microRNA and mRNA
We cross-referenced the three data sets that measured the methylation, microRNA and mRNA of lung cancer tissues and control tissues with GO and found 4,381 GO gene sets that have methylation, microRNA and mRNA data. The three levels of gene sets for these 4,381 GO terms were compiled as follows: the methylation set for each GO term consists of the genes that had methylation data and were annotated to this term, the microRNA set consists of the microRNAs that had target genes annotated to this term, and the mRNA set consists of all of the genes that were annotated to this term. The 4,381 GO sets of mRNA, microRNA and methylation can be found in Dataset S1, Dataset S2 and Dataset S3, respectively.
The discriminating ability of the methylation, microRNA and mRNA gene sets
We measured the ability of the gene sets to discriminate between cancer and normal tissue using the Matthews correlation coefficient (MCC) of the NNA prediction model evaluated by LOOCV. We compared the MCCs of methylation, microRNA and mRNA. Figure 2 shows the MCC distributions of the methylation, microRNA and mRNA gene sets. The mean MCCs of the mRNA, microRNA and methylation gene sets are 0.897, 0.702 and 0.561, respectively. The one-side-greater t-test p-value for the mRNA and microRNA sets is less than 2.2e-16. The one-side-greater t-test p-value for the microRNA and methylation sets is also less than 2.2e-16. These results indicate that the MCCs of the mRNA sets are significantly greater than the MCCs of the microRNA sets, which are, in turn, significantly greater than the MCCs of the methylation sets.
Figure 2. The MCC boxplot of methylation, microRNA and mRNA gene sets.

The mean MCCs of the mRNA, microRNA and methylation gene sets were 0.897, 0.702 and 0.561, respectively. The MCCs of the mRNA sets were significantly greater than the MCCs of the microRNA sets with a one-sided t-test p-value of less than 2.2e-16, and the MCCs of the microRNA sets were, in turn, significantly greater than the MCCs of the methylation sets with a one-sided t-test p-value of less than 2.2e-16.
Classification of gene sets based on their dysfunctional level: methylation, microRNA or mRNA
By comparing the MCC ranks of the gene sets at the methylation, microRNA or mRNA level, we defined six groups of gene sets. There are 960 gene sets in which methylation rank < microRNA rank < mRNA rank; 638 gene sets in which methylation rank < mRNA rank < microRNA rank; 721 gene sets in which microRNA rank < methylation rank < mRNA rank; 684 gene sets in which microRNA rank < mRNA rank < methylation rank; 584 gene sets in which mRNA rank < methylation rank < microRNA rank; and 794 gene sets in which mRNA rank < microRNA rank < methylation rank. Table S2 shows the methylation, microRNA and mRNA dysfunction groups of the 4,381 GO gene sets.
The dysfunctional gene sets in lung cancer
We ranked the dysfunctional gene sets in lung cancer based on the summed MCC ranks of methylation, microRNA and mRNA. The top 20 dysfunctional gene sets in lung cancer shown in Table S3 were analyzed. These 20 dysfunctional gene sets in lung cancer are GO:0048585 (negative regulation of response to stimulus), GO:0007517 (muscle organ development), GO:0048514 (blood vessel morphogenesis), GO:0051146 (striated muscle cell differentiation), GO:0001525 (angiogenesis), GO:0045595 (regulation of cell differentiation), GO:0007162 (negative regulation of cell adhesion), GO:0060191 (regulation of lipase activity), GO:0006275 (regulation of DNA replication), GO:0061061 (muscle structure development), GO:0022008 (neurogenesis), GO:0008543 (fibroblast growth factor receptor signaling pathway), GO:0035107 (appendage morphogenesis), GO:0035108 (limb morphogenesis), GO:0001568 (blood vessel development), GO:0005576 (extracellular region), GO:0050793( regulation of developmental processes), GO:0010648 (negative regulation of cell communication), GO:0023057 (negative regulation of signaling), and GO:0019216 (regulation of lipid metabolic processes). Many of these GO terms have been reported to be associated with lung cancer. We analyze several GO sets as examples.
GO:0045595 (regulation of cell differentiation, ranked 6th) and GO:0050793 (regulation of developmental processes, ranked 17th)
Developmental processes and cell differentiation are regulated by a series of similar genes in normal tissues. Therefore, changes in these genes are frequently associated with carcinogenesis. Naveen Babbar et al. reported that TNFα can activate NFκB signaling in NSCLC cells [37], which results in decreased cell growth and increased apoptosis [37]. A role for FGF/FGFR family members has also been indicated in lung cancer. For example, frequent amplification of FGFR1 was identified in human squamous cell lung cancer [38]. Additionally, somatic mutations in several of these genes were identified in lung carcinomas, including FGFR1, FGFR2, and FGF2/10 [39]–[42]. Usually, tumor suppressor genes, such as P53, CDKN2A/B, and STK11, are downregulated, and oncogenes (such as KRAS and ERBB2/4) are upregulated in lung cancer [40]. MicroRNAs are involved in lung cancer due to the epigenetic changes that occur in cancer cells. The low expression of miR-200 and miR-205 is associated with the epithelial-mesenchymal transition (EMT) and stem-cell-like properties of cancer cells and promotes invasion and translocation [43]–[45]. The enforced expression of miR-29 family members in lung cancer cells can restore normal patterns of DNA methylation, induce the re-expression of methylation-silenced tumor suppressor genes, such as FHIT and WWOX, and inhibit tumorigenicity [46].
GO:0022008 (neurogenesis, ranked 11th)
Several genes annotated to this GO term are associated with acantha and brain metastases; for example, mutations in activating epidermal growth factor receptor (EGFR) were found in many lung cancer patients [47]. Human lung cancer features extensive alterations of microRNA expression that may deregulate cancer-related genes; for example, hsa-miR-125a-5p silencing unregulated ROCK1, miR-34b methylation caused c-Met overexpression, and miR-200c was silenced by methylation and downregulated TCF8 and E-cadherin, which resulted in cancer invasion and deterioration [48]–[50]. Demethylation and mutation of genes (ERBB2, KRAS) can also cause carcinogenesis [51], [52]. Methylation of the Death-associated protein kinase (DAPK) promoter and the opioid binding protein/cell adhesion molecule-like gene (OPCML) has been found in both adenocarcinoma and squamous-cell carcinoma [53], [54].
GO:0005576 (extracellular region, ranked 16th)
Epithelial Mesenchymal Transition (EMT) is the main process required for tumor invasion and translocation. Mutations in TIMP3, LAMA/B/C, TMEFF2, CDH13 and other genes are involved in lung cancer deterioration [55]. IL-8 can initiate an airway epithelial signaling pathway, and deregulation of this gene may cause tobacco-related lung cancer [56]. Five microRNAs (hsa-miR-155, hsa-miR-17-3p, hsa-let-7a-2, hsa-miR-145, and hsa-miR-21) are seen to be expressed differently in lung cancer tissues versus the corresponding noncancerous lung tissues. Among these microRNAs, let-7a can regulate RAS activity [57]. Epigenetic activation of human kallikrein 13 (KLK13) enhances the malignancy of lung adenocarcinoma by promoting N-cadherin expression and laminin degradation [58]. Recently, MMP1 was reported to be associated with lung cancer. The -16071G-2G polymorphism of MMP1 results in transcriptional up regulation [58]. X Xiang et al. reported that the stable expression of miR-155 significantly reduces the aggressiveness of tumor cell dissemination by preventing the EMT of tumor cells in vivo [59]. Furthermore, miR-155 directly suppresses the expression of the transcription factor TCF4, which is an important regulator of EMT [59].
The high frequency genes and microRNAs in the top dysfunctional gene sets
We calculated the frequency of genes or microRNAs in the top 300 dysfunctional gene sets. The genes in either mRNA or methylation gene sets with frequency higher than 50 were defined as high frequency genes. Similarly, the high frequency microRNAs were defined as microRNAs that have frequency higher than 50 in the top 300 dysfunctional gene sets. The high frequency genes and microRNAs are given in Table S4.
We tested the discriminating ability of these high frequency genes in an independent data set which includes 58 lung cancer samples and 58 adjacent normal samples. The independent data set was downloaded from GEO with the accession number GSE32863. It was found that the high frequency genes can perfectly differentiate the lung cancer tissues from adjacent normal tissues. The prediction MCC was 1, which means that all samples were correctly classified in their actual group, tumor or normal. The heatmap of the high frequency genes and the tumor/normal samples is shown in Figure 3 . The tumor and normal samples were clearly differentiated by the high frequency genes.
Figure 3. The heatmap of the high frequency genes and the tumor/normal samples.

The green bars indicate the tumor samples and the blue bars indicate the normal samples. The tumor and normal samples were clearly differentiated by the high frequency genes.
We did a hypergeometric test [5], [24], [25], [32], [36] to investigate whether the high frequency genes are significantly overlapped with the KEGG pathway “hsa05223 Non-small cell lung cancer”. The hypergeometric test p value was a highly significant 1.61E-26. This result suggests that many higher frequency genes are known “hsa05223 Non-small cell lung cancer” genes.
In Figure 4 , we highlighted the high frequency genes we discovered in the KEGG pathway “hsa05223 Non-small cell lung cancer”. Many hub genes of the KEGG pathway “hsa05223 Non-small cell lung cancer” were high frequency dysfunctional genes, such as KRAS, EGFR, ERBB2, CDKN2A and RB1. And the hub high frequency genes tend to be dysfunctional at both the methylation and mRNA levels. It is known that KRAS can initiate tumorgenesis by affecting the endodermal progenitor [60]. The copy number alterations of KRAS are strongly associated with clinical outcomes of lung cancer patients [61]. EGFR is a receptor of the epidermal growth factor family. Binding of EGFR to a ligand will induce cell proliferation [62]. EGFR mutations are very common in lung cancer [63] and are associated with prognosis of NSCLC [64]. They can alter the signaling cascades of NSCLC [65]. ERBB2 is mutated in 4% of NSCLC [66] and its polymorphisms increase the risk of lung cancer [67]. Methylation of CDKN2A occurs more frequently in NSCLC tissues than in non-tumor tissues [68]. CDKN2A is involved in the p16/pRb/cyclin-D1 pathway [69]. RB1 can regulate cell proliferation, differentiation, and apoptosis in human NSCLC [70]. In advanced NSCLC patients, the frequency of Rb loss is high [71].
Figure 4. The high frequency genes and microRNAs of the KEGG pathway “hsa05223 Non-small cell lung cancer”.

The green nodes denote high frequency microRNAs. The red nodes denote high frequency genes in both methylation and mRNA dysfunctional sets. The yellow nodes indicate high frequency genes in mRNA dysfunctional sets only. There is no specific high frequency gene in methylation dysfunctional sets. The white nodes indicate non-high frequency genes. The black edges show interactions from the KEGG pathway “hsa05223 Non-small cell lung cancer”. The green edges show regulation by high frequency microRNAs on their target genes.
In Figure 4 , there are some high frequency microRNAs, such as hsa-miR-495, hsa-miR-96, has-miR-106a, has-miR-137, has-miR-372, hsa-miR-183, hsa-miR-182, hsa-miR-203, hsa-miR-15a, hsa-miR-15b and hsa-miR-7. hsa-miR-495 regulates two high frequency dysfunctional genes, STK4 and PRKCB. It was reported that miR-495 is upregulated in KRAS-positive NSCLC [72]. hsa-miR-96 is downregulated in NSCLC [73]. has-miR-106a is related to lung cancer patient survival [56]. Patients with high expression of has-miR-106a tend to have a worse prognosis [56]. has-miR-137 and has-miR-372 are both upregulated in NSCLC and their expression levels are associated with survival and relapse in NSCLC patients [74]. has-miR-183 is a potential metastasis-inhibitor of lung cancer and can regulate migration and invasion genes [75]. hsa-miR-183 and hsa-miR-182 were reported as the most differentially expressed microRNAs between lung cancer tissues with adjacent normal tissues [76]. hsa-miR-203 is upregulated in lung cancer tissues [56]. hsa-miR-15a is frequently deleted or down-regulated in NSCLC [77] and its expression inversely correlates with the expression of cyclin D1[77]. hsa-miR-15 b is differentially expressed in tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) resistant NSCLC cells [73]. hsa-miR-7 is downregulated in lung cancer and it can regulate epidermal growth factor receptor signaling [78].
The advantages and limitations of our methods
Obtaining a systematic understanding of pathological change is an essential problem in medical and pharmaceutical studies. Tumorigenesis involves alterations to many proteins, molecules and pathways. Eventually, however, all these changes cause cancer through functional effects. In this study, we used GO to describe biological functions and stratified the functions into three levels: methylation, microRNA and mRNA. In each level, we calculated and ranked the discriminating ability of the functional set for this level that was measured by the MCC correctly classifying cancer and normal tissues. For each functional set, we compared the MCC rank of each level, and we subsequently grouped the functional sets into six patterns based on the relationships of the MCC ranks of the different levels. Some functional sets may function at the methylation level; others may function at the microRNA level. Taking all three levels into consideration, we ranked the functional sets based on their overall ranks on the three levels. The overall ranking of the functional sets appears reasonable and is consistent with several published studies.
There are still several limitations to this research. Firstly, the methylation, microRNA and mRNA data for lung cancer and normal tissues are obtained from different studies, which may affect the results. Ideally, all of the data would be derived from the same study. To partially overcome this problem, we used the MCC rank, instead of the MCC itself, when comparing among the different levels. Secondly, the links between microRNAs and their target genes are based on predictions. Due to the low proportion of experimentally confirmed microRNA and target gene pairs, we used the microRNA and target gene pairs that were predicted by at least three popular microRNA target-gene predictors. Thirdly, not all functional sets were analyzed. The methylation, microRNA and mRNA data we used were generated with microarray technology. Certain genes or microRNAs were not measured, especially with respect to the methylation status of genes. With the development of sequencing technology and sequence capture technology, increasing numbers of genes can be measured, allowing us to analyze more functional sets and obtain a more comprehensive view of tumorigenesis.
Overall, our methods provide a means of performing “multi-omics” dysfunctional set analysis, which could be useful in the study of complex diseases. Our results yield a systematic view of tumorigenesis that may shed light on the diagnosis and prognosis of lung cancer.
Supporting Information
The 4,381 Gene Ontology (GO) sets of mRNA. Each line describes a gene set. The first field contains the Gene Ontology (GO) term name, the second field contains the number of mRNAs in the set, and the remaining fields list the mRNAs in the set.
(TXT)
The 4,381 Gene Ontology (GO) sets of microRNA. Each line describes a microRNA set. The first field contains the Gene Ontology (GO) term name, the second field contains the number of microRNAs in the set, and the remaining fields list the microRNAs in the set.
(TXT)
The 4,381 Gene Ontology (GO) sets of methylation. Each line describes a gene set. The first field contains the Gene Ontology (GO) term name, the second field contains the number of genes in the set, and the remaining fields list the genes in the set.
(TXT)
The microRNA - target gene pairs that were predicted by at least three tools.
(XLSX)
The methylation, microRNA and mRNA dysfunction groups of the 4,381 Gene Ontology (GO) gene sets.
(XLSX)
The top 20 dysfunctional gene sets in lung cancer.
(PDF)
The high frequency genes and microRNAs.
(XLSX)
Funding Statement
This work was supported by grants from the National Basic Research Program of China (2011CB510102, 2011CB510101, 2011CB910200 and 2010CB912702), the National Natural Science Foundation of China (90913009), Research Program of the Chinese Academy of Sciences (KSCX2-EW-R-04) and the Innovation Program of the Shanghai Municipal Education Commission (12ZZ087). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hornberg JJ, Bruggeman FJ, Westerhoff HV, Lankelma J (2006) Cancer: a Systems Biology disease. Biosystems 83: 81–90. [DOI] [PubMed] [Google Scholar]
- 2. Kreeger PK, Lauffenburger DA (2010) Cancer systems biology: a network modeling perspective. Carcinogenesis 31: 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, et al. (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131: 1190–1203. [DOI] [PubMed] [Google Scholar]
- 4. Ponomareva AA, Rykova E, Cherdyntseva NV, Choinzonov EL, Laktionov PP, et al. (2011) [Molecular-genetic markers in lung cancer diagnostics]. Mol Biol (Mosk) 45: 203–217. [PubMed] [Google Scholar]
- 5. Huang T, Wan S, Xu Z, Zheng Y, Feng KY, et al. (2011) Analysis and prediction of translation rate based on sequence and functional features of the mRNA. PLoS ONE 6: e16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee JH, Voortman J, Dingemans AM, Voeller DM, Pham T, et al. (2011) MicroRNA expression and clinical outcome of small cell lung cancer. PLoS ONE 6: e21300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Voortman J, Goto A, Mendiboure J, Sohn JJ, Schetter AJ, et al. (2010) MicroRNA expression and clinical outcomes in patients treated with adjuvant chemotherapy after complete resection of non-small cell lung carcinoma. Cancer Res 70: 8288–8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chari R, Coe BP, Wedseltoft C, Benetti M, Wilson IM, et al. (2008) SIGMA2: a system for the integrative genomic multi-dimensional analysis of cancer genomes, epigenomes, and transcriptomes. BMC Bioinformatics 9: 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chari R, Coe BP, Vucic EA, Lockwood WW, Lam WL (2010) An integrative multi-dimensional genetic and epigenetic strategy to identify aberrant genes and pathways in cancer. BMC Syst Biol 4: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peng J, Zhu J, Bergamaschi A, Han W, Noh D-Y, et al. (2010) Regularized multivariate regression for identifying master predictors with application to integrative genomics study of breast cancer. Annals Of Applied Statistics 4: 53–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akavia UD, Litvin O, Kim J, Sanchez-Garcia F, Kotliar D, et al. (2010) An integrated approach to uncover drivers of cancer. Cell 143: 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Christensen BC, Marsit CJ, Houseman EA, Godleski JJ, Longacker JL, et al. (2009) Differentiation of lung adenocarcinoma, pleural mesothelioma, and nonmalignant pulmonary tissues using DNA methylation profiles. Cancer Res 69: 6315–6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan X, Qin W, Zhang L, Hang J, Li B, et al.. (2011) A Five-microRNA Signature for Squamous Cell Lung Carcinoma (SCC) Diagnosis and Hsa-miR-31 for SCC Prognosis. Clin Cancer Res. [DOI] [PubMed]
- 15.Sanchez-Palencia A, Gomez-Morales M, Gomez-Capilla JA, Pedraza V, Boyero L, et al.. (2010) Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. Int J Cancer. [DOI] [PubMed]
- 16. Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34: D140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Friedman RC, Farh KK, Burge CB, Bartel DP (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Betel D, Wilson M, Gabow A, Marks DS, Sander C (2008) The microRNA.org resource: targets and expression. Nucleic Acids Res 36: D149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sethupathy P, Corda B, Hatzigeorgiou AG (2006) TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA 12: 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X, El Naqa IM (2008) Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics 24: 325–332. [DOI] [PubMed] [Google Scholar]
- 21. Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, et al. (2005) Combinatorial microRNA target predictions. Nat Genet 37: 495–500. [DOI] [PubMed] [Google Scholar]
- 22.Huang T, Zhang J, Xie L, Dong X, Zhang L, et al.. (2011) Crosstissue Coexpression Network of Aging. OMICS. [DOI] [PubMed]
- 23. Huang T, Xu Z, Chen L, Cai YD, Kong X (2011) Computational Analysis of HIV-1 Resistance Based on Gene Expression Profiles and the Virus-Host Interaction Network. PLoS ONE 6: e17291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang T, Wang P, Ye ZQ, Xu H, He Z, et al. (2010) Prediction of Deleterious Non-Synonymous SNPs Based on Protein Interaction Network and Hybrid Properties. PLoS ONE 5: e11900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang T, Shi XH, Wang P, He Z, Feng KY, et al. (2010) Analysis and prediction of the metabolic stability of proteins based on their sequential features, subcellular locations and interaction networks. PLoS ONE 5: e10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang T, Niu S, Xu Z, Huang Y, Kong X, et al. (2011) Predicting Transcriptional Activity of Multiple Site p53 Mutants Based on Hybrid Properties. PLoS ONE 6: e22940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang T, Cui W, Hu L, Feng K, Li YX, et al. (2009) Prediction of pharmacological and xenobiotic responses to drugs based on time course gene expression profiles. PLoS ONE 4: e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang T, Chen L, Liu X-J, Cai Y-D (2011) Predicting triplet of transcription factor - mediating enzyme - target gene by functional profiles. Neurocomputing 74: 3677–3681. [Google Scholar]
- 29. Huang T, Chen L, Cai Y-D, Chou K-C (2011) Classification and analysis of regulatory pathways using graph property, biochemical and physicochemical property, and functional property. PLoS ONE 6: e25297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai Y, Huang T, Hu L, Shi X, Xie L, et al.. (2011) Prediction of lysine ubiquitination with mRMR feature selection and analysis. Amino Acids. [DOI] [PubMed]
- 31. Huang T, Tu K, Shyr Y, Wei CC, Xie L, et al. (2008) The prediction of interferon treatment effects based on time series microarray gene expression profiles. J Transl Med 6: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang T, Zhang J, Xu ZP, Hu LL, Chen L, et al. (2012) Deciphering the effects of gene deletion on yeast longevity using network and machine learning approaches. Biochimie 94: 1017–1025. [DOI] [PubMed] [Google Scholar]
- 33. Huang T, Zhang J, Xie L, Dong X, Zhang L, et al. (2011) Crosstissue coexpression network of aging. OMICS 15: 665–671. [DOI] [PubMed] [Google Scholar]
- 34. Huang T, Chen L, Cai YD, Chou KC (2011) Classification and analysis of regulatory pathways using graph property, biochemical and physicochemical property, and functional property. PLoS ONE 6: e25297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang T, Wang J, Cai Y-D, Yu H, Chou K-C (2012) Hepatitis C Virus Network Based Classification of Hepatocellular Cirrhosis and Carcinoma. PLoS ONE 7: e34460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang T, Wang C, Zhang G, Xie L, Li Y (2012) SySAP: a system-level predictor of deleterious single amino acid polymorphisms. Protein Cell 3: 38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Babbar N HA, Huang Y, Casero RA Jr (2006) Tumor necrosis factor alpha induces spermidine/spermine N1-acetyltransferase through nuclear factor kappaB in non-small cell lung cancer cells. J Biol Chem 281(34): 24182–24192. [DOI] [PubMed] [Google Scholar]
- 38. Wesche J, Haglund K, Haugsten EM (2011) Fibroblast growth factors and their receptors in cancer. Biochem J 437: 199–213. [DOI] [PubMed] [Google Scholar]
- 39. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, et al. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, et al. (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455: 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weiss J, Sos ML, Seidel D, Peifer M, Zander T, et al. (2010) Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2: 62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, et al. (2010) Lung organogenesis. Curr Top Dev Biol 90: 73–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ceppi P, Mudduluru G, Kumarswamy R, Rapa I, Scagliotti GV, et al. (2010) Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol Cancer Res 8: 1207–1216. [DOI] [PubMed] [Google Scholar]
- 44. Hurteau GJ, Carlson JA, Spivack SD, Brock GJ (2007) Overexpression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res 67: 7972–7976. [DOI] [PubMed] [Google Scholar]
- 45. Tellez CS, Juri DE, Do K, Bernauer AM, Thomas CL, et al. (2011) EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res 71: 3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, et al. (2007) MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A 104: 15805–15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fruh M (2011) The search for improved systemic therapy of non-small cell lung cancer–what are today′s options? Lung Cancer 72: 265.–270. [DOI] [PubMed] [Google Scholar]
- 48. Jiang L, Zhang Q, Chang J, Qiu X, Wang E (2009) [hsa-miR-125a-5p Enhances Invasion Ability in Non-Small Lung Carcinoma Cell Lines.]. Zhongguo Fei Ai Za Zhi 12: 951–955. [DOI] [PubMed] [Google Scholar]
- 49. Jiang L, Zhang Q, Chang H, Qiu X, Wang E (2009) [hsa-miR-125a-5p Enhances Invasion in Non-small Cell Lung Carcinoma Cell Lines by Upregulating Rock-1.]. Zhongguo Fei Ai Za Zhi 12: 1069–1073. [DOI] [PubMed] [Google Scholar]
- 50. Watanabe K, Emoto N, Hamano E, Sunohara M, Kawakami M, et al. (2012) Genome structure-based screening identified epigenetically silenced microRNA associated with invasiveness in non-small-cell lung cancer. Int J Cancer 130: 2580–2590. [DOI] [PubMed] [Google Scholar]
- 51. Davies H, Hunter C, Smith R, Stephens P, Greenman C, et al. (2005) Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res 65: 7591–7595. [DOI] [PubMed] [Google Scholar]
- 52. Toyooka S, Mitsudomi T, Soh J, Aokage K, Yamane M, et al. (2011) Molecular oncology of lung cancer. Gen Thorac Cardiovasc Surg 59: 527–537. [DOI] [PubMed] [Google Scholar]
- 53. Belinsky SA (2004) Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer 4: 707–717. [DOI] [PubMed] [Google Scholar]
- 54. Selamat SA, Galler JS, Joshi AD, Fyfe MN, Campan M, et al. (2011) DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS ONE 6: e21443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Belinsky SA (2004) Gene-promoter hypermethylation as a biomarker in lung cancer. Nature Reviews Cancer 4: 707–717. [DOI] [PubMed] [Google Scholar]
- 56. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, et al. (2006) Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9: 189–198. [DOI] [PubMed] [Google Scholar]
- 57. Chou RH, Lin SC, Wen HC, Wu CW, Chang WS (2011) Epigenetic activation of human kallikrein 13 enhances malignancy of lung adenocarcinoma by promoting N-cadherin expression and laminin degradation. Biochem Biophys Res Commun 409: 442–447. [DOI] [PubMed] [Google Scholar]
- 58. Liu L, Wu J, Wu C, Wang Y, Zhong R, et al. (2011) A functional polymorphism (-1607 1G–>2G) in the matrix metalloproteinase-1 promoter is associated with development and progression of lung cancer. Cancer 117: 5172–5181. [DOI] [PubMed] [Google Scholar]
- 59. Xiang X, Zhuang X, Ju S, Zhang S, Jiang H, et al. (2011) miR-155 promotes macroscopic tumor formation yet inhibits tumor dissemination from mammary fat pads to the lung by preventing EMT. Oncogene 30: 3440–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, et al. (2008) A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res 68: 8535–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chitale D, Gong Y, Taylor BS, Broderick S, Brennan C, et al. (2009) An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene 28: 2773–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oda K, Matsuoka Y, Funahashi A, Kitano H (2005) A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol 1: 2005 0010. [DOI] [PMC free article] [PubMed]
- 63. Reinmuth N, Jauch A, Xu EC, Muley T, Granzow M, et al. (2008) Correlation of EGFR mutations with chromosomal alterations and expression of EGFR, ErbB3 and VEGF in tumor samples of lung adenocarcinoma patients. Lung Cancer 62: 193–201. [DOI] [PubMed] [Google Scholar]
- 64. Sasaki H, Okuda K, Shimizu S, Takada M, Kawahara M, et al. (2009) EGFR R497K polymorphism is a favorable prognostic factor for advanced lung cancer. J Cancer Res Clin Oncol 135: 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zimmer S, Kahl P, Buhl TM, Steiner S, Wardelmann E, et al. (2009) Epidermal growth factor receptor mutations in non-small cell lung cancer influence downstream Akt, MAPK and Stat3 signaling. J Cancer Res Clin Oncol 135: 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, et al. (2006) HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 10: 25–38. [DOI] [PubMed] [Google Scholar]
- 67. Jo UH, Han SG, Seo JH, Park KH, Lee JW, et al. (2008) The genetic polymorphisms of HER-2 and the risk of lung cancer in a Korean population. BMC Cancer 8: 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. De Jong WK, Verpooten GF, Kramer H, Louwagie J, Groen HJ (2009) Promoter methylation primarily occurs in tumor cells of patients with non-small cell lung cancer. Anticancer Res 29: 363–369. [PubMed] [Google Scholar]
- 69. Bastide K, Guilly MN, Bernaudin JF, Joubert C, Lectard B, et al. (2009) Molecular analysis of the Ink4a/Rb1-Arf/Tp53 pathways in radon-induced rat lung tumors. Lung Cancer 63: 348–353. [DOI] [PubMed] [Google Scholar]
- 70. Katsuda K, Kataoka M, Uno F, Murakami T, Kondo T, et al. (2002) Activation of caspase-3 and cleavage of Rb are associated with p16-mediated apoptosis in human non-small cell lung cancer cells. Oncogene 21: 2108–2113. [DOI] [PubMed] [Google Scholar]
- 71. Gregorc V, Darwish S, Ludovini V, Pistola L, De Angelis V, et al. (2003) The clinical relevance of Bcl-2, Rb and p53 expression in advanced non-small cell lung cancer. Lung Cancer 42: 275–281. [DOI] [PubMed] [Google Scholar]
- 72. Dacic S, Kelly L, Shuai Y, Nikiforova MN (2010) miRNA expression profiling of lung adenocarcinomas: correlation with mutational status. Mod Pathol 23: 1577–1582. [DOI] [PubMed] [Google Scholar]
- 73. Garofalo M, Quintavalle C, Di Leva G, Zanca C, Romano G, et al. (2008) MicroRNA signatures of TRAIL resistance in human non-small cell lung cancer. Oncogene 27: 3845–3855. [DOI] [PubMed] [Google Scholar]
- 74. Yu SL, Chen HY, Chang GC, Chen CY, Chen HW, et al. (2008) MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 13: 48–57. [DOI] [PubMed] [Google Scholar]
- 75. Wang G, Mao W, Zheng S (2008) MicroRNA-183 regulates Ezrin expression in lung cancer cells. FEBS Lett 582: 3663–3668. [DOI] [PubMed] [Google Scholar]
- 76. Cho WC, Chow AS, Au JS (2009) Restoration of tumour suppressor hsa-miR-145 inhibits cancer cell growth in lung adenocarcinoma patients with epidermal growth factor receptor mutation. Eur J Cancer 45: 2197–2206. [DOI] [PubMed] [Google Scholar]
- 77. Bandi N, Zbinden S, Gugger M, Arnold M, Kocher V, et al. (2009) miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res 69: 5553–5559. [DOI] [PubMed] [Google Scholar]
- 78. Webster RJ, Giles KM, Price KJ, Zhang PM, Mattick JS, et al. (2009) Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J Biol Chem 284: 5731–5741. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The 4,381 Gene Ontology (GO) sets of mRNA. Each line describes a gene set. The first field contains the Gene Ontology (GO) term name, the second field contains the number of mRNAs in the set, and the remaining fields list the mRNAs in the set.
(TXT)
The 4,381 Gene Ontology (GO) sets of microRNA. Each line describes a microRNA set. The first field contains the Gene Ontology (GO) term name, the second field contains the number of microRNAs in the set, and the remaining fields list the microRNAs in the set.
(TXT)
The 4,381 Gene Ontology (GO) sets of methylation. Each line describes a gene set. The first field contains the Gene Ontology (GO) term name, the second field contains the number of genes in the set, and the remaining fields list the genes in the set.
(TXT)
The microRNA - target gene pairs that were predicted by at least three tools.
(XLSX)
The methylation, microRNA and mRNA dysfunction groups of the 4,381 Gene Ontology (GO) gene sets.
(XLSX)
The top 20 dysfunctional gene sets in lung cancer.
(PDF)
The high frequency genes and microRNAs.
(XLSX)