

SUMMARY
Neuromuscular junction (NMJ) formation requires precise interaction between motoneurons and muscle fibers. LRP4 is a receptor of agrin that is thought to act incis to stimulate MuSK in muscle fibers for postsynaptic differentiation. Here we dissected the roles of LRP4 in muscle fibers and motoneurons in NMJ formation by cell-specific mutation. Studies of muscle-specific mutants suggest that LRP4 is involved in deciding where to form AChR clusters in muscle fibers, postsynaptic differentiation, and axon terminal development. LRP4 in HEK293 cells increased synapsin or SV2 puncta in contacting axons of co-cultured neurons, suggesting a synaptogenic function. Analysis of LRP4 muscle and motoneuron double mutants and mechanistic studies suggest that NMJ formation may also be regulated by LRP4 in motoneurons, which could serve as agrin’s receptor in trans to induce AChR clusters. These observations uncovered distinct roles of LRP4 in motoneurons and muscles in NMJ development.
INTRODUCTION
Brain functions are made possible by synapses, contacts formed between neurons or between a neuron and a target cell. The neuromuscular junction (NMJ) is a cholinergic synapse between motoneurons and skeletal muscle fibers that has most, if not all, features characteristic of a chemical synapse in the brain. Because of its simplicity, high spatial resolution and accessibility, the NMJ has served as an informative model of synaptogenesis (Sanes and Lichtman, 1999, 2001; Wu et al., 2010). Its development requires the precise coordination between presynaptic motoneurons and postsynaptic muscle fibers. Mechanisms by which motoneurons instruct postsynaptic differentiation are better characterized whereas relatively little is known about retrograde signals from the muscle fibers. Agrin is a nerve-derived organizer of postsynaptic differentiation during NMJ formation (McMahan, 1990). It stimulates AChR cluster formation in myotubes in culture (Ferns et al., 1993; Nitkin et al., 1987) and mice lacking agrin do not form the NMJ (Gautam et al., 1996). MuSK is a receptor tyrosine kinase that is essential for agrin-induced clustering and for NMJ formation in vivo (DeChiara et al., 1996; Glass et al., 1996; Kim and Burden, 2008; Lin et al., 2001; Yang et al., 2001). However, agrin and MuSK do not directly interact (Glass et al., 1996); rather, MuSK activation by agrin requires LRP4, a member of the LDL receptor family (Kim et al., 2008; Zhang et al., 2008).
LRP4 is a single-transmembrane protein that possesses a large extracellular domain with multiple LDLR repeats, EGF-like and β-propeller repeats; a transmembrane domain; and a short C-terminal region without an identifiable catalytic motif (Johnson et al., 2005; Lu et al., 2007; Tian et al., 2006; Yamaguchi et al., 2006). Mice lacking LRP4 die at birth and do not form the NMJ, indicating a critical role in NMJ formation (Weatherbee et al., 2006). Evidence suggests that agrin binds to LRP4 and is necessary and sufficient to enable agrin signaling (Kim et al., 2008; Zhang et al., 2008). It also interacts with MuSK and this interaction is increased in response to agrin. Recent studies of the crystal structure of an agrin-LRP4 complex suggest that monomeric agrin interacts with LRP4 to form a binary complex, which promotes the synergistic formation of a tetramer crucial for agrin-induced AChR clustering (Zong et al., 2012). These observations support a working hypothesis that agrin binds to LRP4 in muscle cells, which acts in cis to interact and activate MuSK to initiate signaling necessary for postsynaptic differentiation (Kim et al., 2008; Wu et al., 2010; Zhang et al., 2008; Zhang et al., 2011).
To further investigate how LRP4 regulates NMJ formation, we generated and characterized mutant mice that lack LRP4 specifically in muscle cells or motoneurons or both cells. Remarkably, HSA-LRP4−/− mice, where LRP4 is specifically ablated in muscle cells survived at birth and formed primitive NMJs, unlike LRP4 null mutant mice, suggesting that a role of LRP4 in motoneurons or other cells in NMJ formation in the absence of muscle LRP4. Severe morphological and functional deficits were observed in motor nerve terminals in HSA-LRP4−/− mice, indicating a critical role of muscle LRP4 for presynaptic differentiation. These hypotheses were further tested in mutant mice that lacked LRP4 in motoneurons or in both muscle fibers and motoneurons. Results revealed distinct functions of LRP4 in muscle fibers and in motoneurons in NMJ formation and maintenance, and suggest that LRP4 of motoneurons was able to serve as agrin’s receptor intrans to stimulate MuSK-dependent AChR clustering.
RESULTS
Aberrant NMJ Formation in the Absence of Muscle LRP4
Genetic rescues demonstrated that LRP4 in muscle cells is sufficient to initiate signaling for NMJ formation (data not shown) (Gomez and Burden, 2011). To further investigate the role of muscle LRP4, we generated LRP4f/f mice (see methods and Figure S1A for details) and crossed them with HSA-Cre mice which express the Cre gene under the control of HSA promoter. Cre expression in this line is active at E9.5 in myotomal regions of somites (Brennan and Hardeman, 1993; Crawford et al., 2000; Jaworski and Burden, 2006; Li et al., 2008; Miniou et al., 1999; Muscat and Kedes, 1987; Schwander et al., 2003) and is detectable in almost all muscle fibers in P0 mice (Li et al., 2008). Both mRNA and protein levels of LRP4 in resulting HSA-Cre;LRP4f/f (or HSA-LRP4−/−) mice were significantly reduced, compared to control LRP4f/f mice. The reduction was specific for muscles and was not observed in other tissues including spinal cords (Figures S1C and S1D). Residual LRP4 detected in the “muscle” preparation may be due to contamination by nerve terminals, blood vessel and Schwann cells in muscles, as observed previously (Li et al., 2008) (see below). Remarkably, HSA-LRP4−/− pups were viable at birth and apparently able to breathe and suck milk. A majority of HSA-LRP4−/− pups did not die until P15 (Figure S1E). These results were unexpected because the ablation of critical genes including agrin, MuSK, rapsyn, Dok-7, as well as LRP4 prevents NMJ formation and thus leads to neonatal lethality (DeChiara et al., 1996; Gautam et al., 1996; Gautam et al., 1995; Glass et al., 1996; Okada et al., 2006; Weatherbee et al., 2006).
The neonatal survival of HSA-LRP4−/− mice suggest that NMJs may form in the absence of LRP4 in muscle fibers. To test this hypothesis, HSA-LRP4−/− diaphragms were whole-mount stained for AChR and phrenic nerve terminals. Indeed AChR clusters were observed in HSA-LRP4−/− diaphragms (Figures 1A–1C). However, compared to those in control LRP4f/f diaphragms, the clusters in HSA-LRP4−/− mice were abnormal with the following characteristics. First, they were distributed in a wider area in the middle of muscle fibers. The endplate band width increased from 166 ± 28 μm in control to 806 ± 103 μm (P < 0.01, n = 5) in P0 as well as P10 HSA-LRP4−/− mice (Figures 1A–1D), suggesting a role of muscle LRP4 in restricting AChR clusters in the central region. Second, the clusters appeared elongated in morphology. In control mice, 93.9% of the clusters were between 10–30 μm in length; however, in HSA-LRP4−/− mice, cluster length ranged from 5 to > 40 μm (Figures 1A–1C, and 1E). The average size of AChR clusters was reduced (Figure 1F). Moreover, the clusters distributed along nerve terminals were consistently smaller (Figure 1C), suggesting that motor terminals were unable to induce normal clusters. Third, we quantified AChR clusters in 1-mm segments of left ventral diaphragms, to include clusters distributed outside the central area. The number was increased from 588 ± 96 in control to 708 ± 89 in HSA-LRP4−/− diaphragms (P=0.03, n = 4) (Figures 1A–1C, and 1G). The average number of AChR clusters per muscle fiber increased from 1.30 ± 0.45 in control to 1.59 ± 0.72 in HSA-LRP4−/− mutants (P< 0.05, n=37) (Figures S2A and S2B). These results suggest formation of abnormal ectopic AChR clusters in HSA-LRP4−/− mice. Finally, the AChR density at HSA-LRP4−/− NMJs was reduced because the amplitudes of miniature endplate potentials (mEPPs) were smaller than those in control mice (Figures 2A–2C). These results indicate that AChR clusters formed in mutant mice were immature with altered number, size, and AChR density. Together, these results indicate that the formation of the immature clusters is not dependent on LRP4 in muscles, but LRP4 from a non-muscle source, likely motoneurons. LRP4 in muscle cells, however, appeared to be necessary for AChR cluster restriction in the central region and AChR cluster maturation (see below).
Figure 1. Aberrant NMJ Formation in HSA-LRP4−/− Mice.

(A) Abnormal NMJ morphology in HSA-LRP4−/− mice. P0 control and mutant diaphragms were stained whole-mount with α-BTX (red) to label AChR clusters and with NF/synaptophysin antibodies (Syn) to label nerve terminals, which was visualized by Alexa Fluor 488-conjugated goat anti-rabbit antibody (green). Shown were the left, ventral areas of hemi-diaphragms. Arrow, primary nerve branches; arrowheads, secondary nerve branches. M, medial; L, lateral; V, ventral.
(B) Wider endplate areas in P0 and P10 HSA-LRP4−/− muscles, which were counterstained with phalloidin. Dashed lines indicate clusters-enriched regions.
(C) Increased secondary nerve branches (arrowhead) and over-shot axon terminals (empty arrowheads). Arrows, primary branches; dashed lines, boundary of cluster-rich regions. S, sensory or autonomic nerve axons; L, lateral.
(D–J) Compared to controls, HSA-LRP4−/− mice showed increased band width (**, P<0.01; n=5, t-test) (D), increased variability in cluster sizes (calculated using LSM Image Browser software, Zeiss) (E); decreased size of all clusters (F), increased number of AChR clusters (G); no difference in the distances between primary nerve branches and the muscle midlines (H); increased number of secondary/intramuscular branches (I); and increased length of secondary branches (J). Data are shown as mean ± SEM. *p < 0.05; **p < 0.01 (n = 4–8; t test). Please also see Figures S1 and S2.
Figure 2. Compromised NMJ Transmission in HSA-LRP4−/− Mice.

mEPPs were recorded in P0 (A–E) and P15 (F–J) control and HSA-LRP4−/− muscles. A, F: representative mEPP traces; B, G: Cumulative probability plot of mEPP amplitude distribution; C, H: Reduced mEPP amplitudes in P0 HSA-LRP4−/− mice; D, I: Cumulative probability plot of mEPP frequency distribution; E, J: Reduced mEPP frequencies in HSA-LRP4−/− mice. *p < 0.05; **p < 0.01 (mean ± SEM, n = 5 for P0; n = 4 for P15; t test). Black, control mice; red, HSA-LRP4−/− mice.
LRP4 in Muscle for Motor Nerve Terminal Development
Motor nerve terminal differentiation is impaired in LRP4mitt null mice. The terminals failed to stop in the central region of muscle fibers, and instead arborized extensively as if to search for AChR clusters which do not form at all in LRP4mitt null mutant mice (Weatherbee et al., 2006). These results suggest that LRP4 is critical for motor nerve terminal differentiation. Muscle rescue experiments suggested a role of muscle LRP4 in this event (data not shown) (Gomez and Burden, 2011). However, loss-of-function evidence is lacking, which is critical because LRP4 in motoneurons may also regulate NMJ formation (see below). Gain-of-function studies were unable to dissect exact roles of muscle LRP4 in motor terminal navigation and differentiation. In particular, the relationship between AChR clusters and arborized terminals in the absence of LRP4 could not be investigated because LRP4mitt null mice do not form AChR clusters.
In HSA-LRP4−/− mutant mice, primary nerve branches were located in the central region of muscle fibers, as in control mice (Figures 1A, and 1H), indicating proper nerve navigation in the absence of muscle LRP4. However, the secondary or intramuscular branches were increased remarkably in number from 55 ± 5.5 in control to 74 ± 8.8 in HSA-LRP4−/− mutant mice (Figures 1A and 1I) and in length from 49.7 ± 15.3 μm in control to 170 ± 89.4 μm in HSA-LRP4−/− mutant mice (P<0.01, n=5) (Figures 1C, 1J and Table S1). In addition, they formed tertiary and quaternary branches that effectively increased the number of nerve terminals in HSA-LRP4−/− diaphragms (Figures 1A and 1C). These phenotypes qualitatively resembled those in LRP4mitt null mice, indicating a critical role of LRP4 in muscles, not motor neurons, in presynaptic differentiation. Intriguingly, motor axons in HSA-LRP4−/− mice did not end with AChR clusters, unlike those in controls where terminals associated with clusters. Rather, the axons ignored or by-passed the clusters, to “over-shoot” towards the periphery of muscle fibers where AChR clusters were absent (Figure 1C, arrows). These observations indicate that muscle, but not neuronal, LRP4 is necessary for a stop signal to motor axons, and suggest that such stop signal is not mediated by homophilic interaction between LRP4 in muscles and motoneurons.
In LRP4f/+ control littermates, BTX staining showed almost complete registration to synaptophysin (Figure S2C, arrows). In contrast, however, the area stained by both BTX and synaptophysin was reduced from 51.9 ± 10.0% in control to 22.9 ± 15.2% at HSA-LRP4−/− NMJs (Figure S2D) although every AChR cluster was positive for synaptophysin. In agreement, mEPP frequency was reduced by 53.3% at HSA-LRP4−/− NMJs (0.42 ± 0.09/min in HSA-LRP4−/− versus 0.90 ± 0.11/min in control littermates) (Figures 2A, 2D, and 2E), indicating compromised vesicle release in the absence of muscle LRP4. Amplitudes of endplate potentials (EPPs) were decreased from 22.2 ± 2.17 mV in control to 6.75 ± 2.22 mV in HSA-LRP4−/− mice (p<0.01, control n=5, and HSA-LRP4−/− n=4), suggesting a reduction in quantum contents. During development in wild type mice, mEPP amplitudes decreased from 2.30 ± 0.23 mV at P0 to 1.63 ± 0.10 mV at P15 NMJs (Figures 2C and 2H) whereas the frequencies increased during this time (from 0.90 ± 0.11/min at P0 to 27.0 ± 3.1/min at P15) (Figures 2E and 2J), in agreement with previous reports (Kelly, 1978). In HSA-LRP4−/− mice, however, mEPP frequencies were unable to increase during this time and were only 2.8% of control littermate at P15 when most mutant mice died (Figures 2E and 2J). Notice that mEPP amplitudes in HSA-LRP4−/− mice between P0 and P15 were similar (Figures 2C and 2H). These results indicate in the absence of muscle LRP4, the development or maturation of presynaptic terminals is more severely impaired than postsynaptic counterparts.
Next we examined NMJ structures in control and HSA-LRP4−/− mice by electron microscopic analysis. In control or LRP4loxp/+ NMJs, axon terminals were filled with synaptic vesicles, some of which were docked on electron-dense active zones (Figure 3A). Quantitatively, nerve terminal numbers at P0 decreased from 3.16 ± 0.75 in control to 1.5 ± 0.54 in HSA-LRP4−/− mice and at P15, from 4.0 ± 0.89 in control to 1.40 ± 0.55 in HSA-LRP4−/− mice respectively (P<0.01, n=6) (Figure 3B). The number of active zones was decreased from 2.17 ± 0.75 and 3.0 ± 0.89 in control to 1.0 ± 0.63 and 0.33 ± 0.52 in mutant mice at P0 and P15, respectively (P<0.05 in P0 and P<0.01 in P15, n=6) (Figure 3C). The density of synaptic vesicles within 0.04-μm2 areas surrounding active zones or adjacent to terminals was decreased in both P0 and P15 HSA-LRP4−/− mice (5.67 ± 1.0 in control to 1.67 ± 1.73 in mutants and 9.78 ± 1.20 in control to 3.67 ± 2.12 in mutants, respectively) (P<0.01, n=9) (Figure 3D) although the sizes or diameters of synaptic vesicles were similar between control and HSA-LRP4−/− mice (Figure 3E). Muscle LRP4 mutation seems to have little effect on the synaptic cleft width (Figure 3F); however, postjunctional folds, which were occasional at P0 and prominent at P15, were decreased (Figure 3A), as indicated by reduction in postsynaptic membrane length from 9.93 ± 2.57 μm in control to 6.19 ± 1.26 μm in HSA-LRP4−/− mice at P0 (P<0.05, n=6) and from 30.2 ± 8.51 μm in control to 15.9 ± 6.90 μm in HSA-LRP4−/− mice at P15 (P<0.01, n=8) (Figure 3G). These results provide anatomical evidence for NMJ deficits in HSA-LRP4−/− mice, in agreement with impaired neurotransmission revealed by electrophysiological recording.
Figure 3. Aberrant NMJ Structures in HSA-LRP4−/− Mice.

(A) Representative electron micrographic images of NMJs in P0 and P15 LRP4loxP/+ control and HSA-LRP4−/− mice. Asterisks, active zones; JF, junctional folds; SBL, synaptic basal lamina; N, nerve terminals; M, muscle fibers; SC, Schwann cells. (B–G) Quantitative data were shown for nerve terminal numbers (B), active zone number per nerve terminal (C), synaptic vesicle density (D), synaptic vesicle diameter (E), synaptic cleft width (F), and postsynaptic membrane length (G). Data are shown as mean ± SEM. *p < 0.05; **p < 0.01 (n = 6, t test).
LRP4 in Motoneurons is Dispensable for NMJ Formation
The finding that NMJs formed in HSA-LRP4−/− mice, but not in LRP4mitt null mice suggests a role of LRP4 in motoneurons in NMJ formation. Indeed, LRP4 is a ubiquitous protein, present in various tissues including the spinal cord and brain, in addition to skeletal muscles (Lu et al., 2007; Weatherbee et al., 2006) (Figure S1D) (see below). Is LRP4 in motoneurons required for NMJ formation? To address this question, we generated motoneuron-specific LRP4 mutant mice, HB9-LRP4−/−, by crossing HB9-Cre mice with floxed LRP4 mice. HB9 is a motoneuron-specific transcription factor critical for motoneuron differentiation (Arber et al., 1999; Thaler et al., 1999). HB9-Cre mice express Cre specifically in motoneurons at E9.5 (Arber et al., 1999) and have been used to study proteins in motor neuron development and motoneuron proteins in NMJ formation (Arber et al., 1999; Bolis et al., 2005; Li et al., 1999; Yang et al., 2001). In agreement, levels of LRP4 protein and mRNA were reduced in the spinal cord of HB9-LRP4−/− mice, compared to controls (Figures S3A–S3D). A mild, but significant reduction in LRP4 was also observed in HB9-LRP4−/− muscles, suggesting that LRP4 is present in motor nerves and terminals in muscles. However, HB9-LRP4−/− mice were viable at birth, showed no difference, compared to controls, in ability to breathe and suck milk and mobility, and survived as long as more than 1 year after birth (data not shown). Whole-mount staining of P0 diaphragms indicated that NMJ morphology in HB9-LRP4−/− mice was similar to that of control littermates (Figure S3E). No difference was observed in primary branch localization, the number and size of secondary branches, and AChR clusters, the band width of clusters, as well as AChE distribution (Figures S3F–S3J) (data not shown). Electrophysiological characterization failed to reveal any difference either in the frequency and amplitudes of mEPPs (Figures S3K, S3M, and S3N), or in EPP amplitudes (Figures S3L and S3O) between HB9-LRP4−/− and control muscles, indicating normal neuromuscular transmission. These observations demonstrate that LRP4 in motoneurons is not required for NMJ formation or function when LRP4 is available in muscle fibers.
Double Mutation of LRP4 in Both Muscles and Motoneurons Generates Similar Phenotypes of LRP4 Null Mutation
The observation that HSA-LRP4−/− mice form AChR clusters, many of which were innervated (Figures 1A, 1C, and S2C), suggests that LRP4 from non-muscle cells could be critical. Considering the intimate, direct interaction between motor nerve terminals and muscle fibers, we hypothesized that LRP4 in motoneurons may be involved. Yet HB9-LRP4−/− showed no deficit in NMJ formation or function (Figure S3). Alternatively, AChR clusters in HSA-LRP4−/− mice may result from incomplete or mosaic ablation of the LRP4 gene in muscles. Previous work from several groups including ours indicates that in HSA-Cre mice, Cre is expressed in most, if not all, muscle fibers when crossed with Rosa reporter mice (Escher et al., 2005; Jaworski and Burden, 2006; Li et al., 2008). Moreover, when β-catenin is ablated in muscle cells using HSA-Cre, mutant mice die immediately after birth without functional NMJs (Li et al., 2008).
We further generated double knockout mice – HSA-Cre;HB9-Cre;LRP4f/f (HSA/HB9-LRP4−/−) – where the LRP4 gene is ablated in both motoneurons and muscles. Remarkably, HSA/HB9-LRP4−/− pups died soon after birth with cyanosis. AChR cluster formation was severely impaired (Figure 4) as clusters were almost undetectable in HSA/HB9-LRP4−/− diaphragms, except occasional smaller, weak clusters. Their size was only 9.6% of that in LRP4loxP/+ control and 34.9% of that in HSA-LRP4−/− (Table S1). Quantitatively, the number of AChR clusters in double KO pups was reduced by 95.5%, compared to LRP4loxP/+ controls, and by 96.2%, compared to HSA-LRP4−/− pups (588 ± 95.9 per mm2 in controls, 707 ± 89.2 per mm2 in HSA-LRP4−/−, and 26.7 ± 15.8 per mm2 in HSA/HB9-LRP4−/− pups) (Table S1). These results demonstrate that the ablation of LRP4 in motoneurons further impairs AChR clustering in HSA-LRP4−/− mice, and identifies a role of motoneuron LRP4 in AChR clustering (Figure 4F). As observed in LRP4mitt null mice, aneural AChR clusters were almost undetectable in diaphragms of E13.5 HSA/HB9-LRP4−/− embryos, indicating of impaired prepatterning of muscle fibers (Figure S4A).
Figure 4. Motoneuron-muscle Double Mutant Mice Exhibit Similar NMJ Deficits as LRP4mitt Mutant Mice.

(A) Abnormal NMJ morphology in HSA/HB9-LRP4−/− mice. Diaphragms (P0) were stained whole mount as in Figure 1. M, medial; L, lateral; V, ventral; asterisk, phrenic nerves; arrows, primary nerve branches; arrowheads, secondary or tertiary nerve branches.
(B) Reduced AChR clusters and extensively arborized axon terminals in HSA/HB9-LRP4−/− mice. Arrows, primary nerve branches; arrowheads, secondary or tertiary nerve branches or AChR clusters.
(C) Reduction in AChR clusters in HSA/HB9-LRP4−/− diaphragms. *p < 0.05; ###p < 0.001 (mean ± SEM, n = 4, two-way ANOVA).
(D) Increased length of secondary branches in HSA-LRP4−/−, HSA/HB9-LRP4−/−, and LRP4mitt muscles. **p < 0.01; #p < 0.05 (mean ± SEM, n = 5, two-way ANOVA).
(E) Increased number of tertiary and quaternary branches in HSA/HB9-LRP4−/− and LRP4mitt muscles, compared to HSA-LRP4−/− samples. **p < 0.01; ##p < 0.01 (mean ± SEM, n = 4, two-way ANOVA).
(F) Diagrams summarizing NMJ morphological phenotypes of control and respective mutant mice. Green, nerve; red, AChR clusters; blue ovals, nuclei.
Please also see Figures S3 and S4 and Table S1.
Regulation of Presynaptic Differentiation by Motoneuron and Muscle LRP4
The presynaptic deficits in HSA/HB9-LRP4−/− mice also resemble those in LRP4mitt null as the number and length of secondary and tertiary branches of these two genotypes were similar (Table S1) (Figures 4A and 4B). However, compared to HSA-LRP4−/−, secondary branches were significantly longer in HSA/HB9-LRP4−/− and LRP4mitt null mice, indicating a role of motoneuron LRP4 in nerve terminal differentiation. This notion was supported by increased number of tertiary and quaternary branches in HSA/HB9-LRP4−/− and LRP4mitt mice (Figure 4E). Moreover, motor nerve terminals appeared to be fragmented in diaphragms of both HSA/HB9-LRP4−/− mice and LRP4mitt null mice. In contrast, such discontinuous intumescence of nerve terminals was not observed in HSA-LRP4−/− muscles (Figures S4B and S4C). LRP4 null mutation or conditional mutation (in muscles or in both muscles and motoneurons) had little effect on the number and distribution of motoneurons (Figures S4D and S4E), suggesting that LRP4 controls neuron differentiation, but not survival.
To investigate how muscle LRP4 regulates presynaptic differentiation, we tested if LRP4 could be synaptogenic using an established co-culture assay (Biederer et al., 2002; Fogel et al., 2011; Graf et al., 2004; Scheiffele et al., 2000). HEK293 were transfected with EGFP alone (control) or together with LRP4 and co-cultured with cortical neurons. Cells were stained for synapsin and SV2, both markers for presynaptic differentiation. Synapsin or SV2 puncta in axons were not altered by HEK293 cells expressing EGFP alone (Figures 5A and 5D). In contrast, the puncta in axon segments in contact with HEK293 cells expressing LRP4 were increased. Quantitatively, the numbers of positive HEK293 cells (i.e., those associated with synapsin or SV2 puncta) were increased in the co-culture with cells expressing LRP4, compared to those expressing EGFP alone (Figures 5B and 5E). The intensity of synapsin and SV2 puncta overlapping with LRP4-expressing cells was higher than that with control cells (Figures 5C and 5F). These results demonstrate that LRP4 may have synaptogenic activity to induce or promote presynaptic differentiation. Together, these observations indicate distinct functions of LRP4 in muscle and in motoneurons for presynaptic differentiation.
Figure 5. Expression of LRP4 in HEK293 Cells Promotes Presynaptic Development in Contacting Axons.

(A and D) HEK293 cells transfected with EGFP alone or together with Flag-LRP4 (1:10) were cocultured with cortical neurons for 36 hr and immunostained with anti-synapsin (A) or SV2 (D) antibodies (red). Cell nuclei were counterstained with DAPI (blue). Arrows, synapsin or SV2 punctas in axons in contact with HEK293 cells; arrowheads, axons in contact with EGFP-labeled HEK293 cells.
(B and E) Number of HEK293 cells contacting axons with synapsin or SV2 punctas.
(C and F) Total integrated intensity of synapsin or SV2 puncta. Data are shown as mean ± SEM. *p < 0.05, n = 3 experiments with 30–50 HEK293 cells per experiment.
Soluble Complex of the Extracellular Domain of LRP4 and Agrin is Sufficient to Stimulate MuSK and Induce AChR Clustering
How would LRP4 in motoneurons regulate postsynaptic differentiation? Transmembrane proteins of the LDLR family could undergo proteolytic cleavage at the extracellular domain to release diffusible ecto-domain (Carter, 2007; Selvais et al., 2011; von Arnim et al., 2005; Willnow et al., 1996). We wondered whether the extracellular domain of LRP4 (ecto-LRP4) could be cleaved by similar mechanisms and the soluble ecto-LRP4 may serve as agrin receptor. Earlier we showed that ecto-LRP4 is able to bind to agrin in solution (Zhang et al., 2008); however, it is unknown the soluble binary complex is sufficient to activate MuSK and/or induce AChR clusters. HEK293 cells do not express LRP4 and thus do not respond to agrin even after transfection with MuSK (Zhang et al., 2008) (Figure 6, lanes 5 and 6). Co-transfection with full length LRP4 enabled HEK293 cells to respond to agrin, with increased MuSK tyrosine phosphorylation (Figure 6, lanes 1 and 2), in agreement with our previous study (Zhang et al., 2008). Intriguingly, stimulation with agrin together with ecto-LRP4 was also able to elicit tyrosine phosphorylation of MuSK in HEK293 cells that were transfected only with MuSK (Figure 6, lanes 3 and 4). These results demonstrate that the soluble complex of ecto-LRP4 and agrin is sufficient to stimulate MuSK, in agreement with a recent report (Zhang et al., 2011).
Figure 6. Soluble ecto-LRP4 is Sufficient to Activate MuSK and Induce AChR Clusters in Muscle Cells.

(A) Ecto-LRP4 enabled agrin activation of MuSK in HEK293 cells. Cells were transfected with Flag-MuSK alone (lanes 3 to 6) or with Flag-LRP4 (lanes 1 and 2). Transfected cells were treated with agrin alone or together with ecto-LRP4 for 2 hr. Flag-MuSK was immunoprecipitated with anti-Flag (M2) antibody and examined for tyrosine phosphorylation by western blotting with anti-phosphotyrosine antibody 4G10. Immunoprecipitates and lysates were blotted with anti-Flag (M2) antibody to demonstrate equal amounts of MuSK in precipitates or lysates. Expression of transfected Flag-LRP4 was shown by blotting lysates with anti-LRP4 (ECD) antibody. IP, immunoprecipitation; IB, immunoblotting; MW, molecular weight; kD, kilodalton.
(B) Quantitative analysis of data in A. **, P<0.01; #, P<0.05; ††, P<0.01 (mean ± SEM, n=3, two-way ANOVA).
(C) Ecto-LRP4 and agrin were sufficient to induce AChR clusters in LRP4-deficient myotubes. Young C2C12 myotubes were transfected with scrambled miRNA (Scr-miRNA, white bars) or LRP4-specific miRNA (miLRP4-1062, red bars) and were stimulated with or without agrin in the presence or absence of soluble ecto-LRP4. AChR clusters were visualized by staining with α-BTX in myotubes expressing GFP that was encoded by the miRNA parental vector. Arrows, AChR clusters. Representative images of experiments that were repeated at least three times with similar results are shown.
(D) Quantitative analysis of data in C. AChR clusters larger than 4 μm were scored. **, P<0.01; ##, P<0.01; ††, P<0.01 (mean ± SEM, n=3, two-way ANOVA).
Next, we determined if the agrin-ecto-LRP4 complex is sufficient to induce AChR clusters in muscle cells. C2C12 myoblasts were transfected with miLRP4-1062 or scrambled control miRNA and resulting transfected myotubes were identified by GFP that is expressed by the miRNA vector. LRP4 knockdown inhibits agrin induction of AChR clusters in miLRP4-1062-transfected myotubes, as observed before (Zhang et al., 2008). Treatment of myotubes with ecto-LRP4, in the absence of agrin, had no effect on basal, indicating that ecto-LRP4 is unable to serve as ligand for MuSK without agrin. It had no effect on agrin-induced clusters in control myotubes which express wild type LRP4. Remarkably, in the presence of agrin, ecto-LRP4 increased the number of agrin-induced AChR clusters in miLRP4-1062-transfected myotubes (compared to that in the absence of ecto-LRP4) (Figures 6C and 6D). This result indicates that soluble ecto-LRP4 is sufficient to serve as a receptor for agrin to initiate pathways for AChR clustering.
Inhibition of Extracellular Cleavage of LRP4 Reduced AChR Clusters in HSA-LRP4−/− Mice
To identify the protease(s) that cleave LRP4, HEK293 cells were transfected with Flag-LRP4 and ecto-LRP4. A Flag-tagged LRP4 fragment was detected in the conditioned media of transfected cells, at the molecular weight of 180kD, similar to that of Flag-ecto-LRP4 (Figure 7B, left lane). This result suggests that LRP4 could be released into the cultured media by proteolytic shedding in the extracellular juxtamembrane domain (Figure 7A, red arrow; Figure 7B). Interestingly, treatment of GM6001, an inhibitor of MMP, but not β-secretase inhibitor IV, significantly reduced the amount of Flag-tagged soluble LRP4 in the medium (Figures 7B and 7C), suggesting possible involvement of MMPs in generating ecto-LRP4, in agreement with a recent report (Dietrich et al., 2010).
Figure 7. MMP-mediated LRP4 Proteolytic Cleavage is Required for Immature AChR Clusters in HSA-LRP4−/− Mice.

(A) Schematic diagram of LRP4’s structure. LRP4 (ECD) monoclonal antibody recognizes the epitope localized between amino acid residues 26 and 350.
(B) MPP inhibition reduced the amount of soluble ecto-LRP4 in conditioned media. HEK293 cells were transfected with Flag-ecto-LRP4 (left lane, as control) or Flag-LRP4 (right three lanes). Transfected cells were rinsed and cultured in fresh media in the presence of vehicle (control) or GM6001, a general MMP inhibitor, or β-secretase inhibitor IV (both at 10 μM) for another 24 hr. Conditioned media were analyzed for soluble ecto-LRP4 by anti-LRP4 (ECD) antibody. Cell lysates were also probed with Flag (M2) and α-Tubulin antibodies to indicate equal amounts of proteins.
(C) Quantitative analysis of results in B. **, P<0.01 (mean ± SEM, n=3, two-way ANOVA).
(D) MMP inhibition reduced AChR clusters in HSA-LRP4−/−, but not in LRP4loxP/+, muscles. Pregnant females were injected with GM6001 (100 mg/kg, i.p.) or vehicle DMSO three times at gestation day 13.5, 15.5 and 17.5, respectively. Diaphragms of P0 mice were stained whole-mount as described in Figure 1. Arrows, primary nerve branches; arrowheads, secondary nerve branches. Insets, images of AChR clusters in the central region. M, medial; L, lateral; V, ventral.
(E) Quantitative analysis of AChR clusters in D. *, P<0.05 (mean ± SEM, n=3, t-test). Please also see Figures S5 and S6.
Ecto-LRP4 was detectable in motor nerves as well as skeletal muscles (Figures S5A and S5B). The amount of LRP4 in synapse-rich regions appeared higher than that in non-synapse regions of skeletal muscles. To study if LRP4 cleavage is involved in NMJ formation, GM6001 was injected into pregnant females, and NMJs were analyzed in new born pups of indicated genotypes. It had little effect on NMJ formation in LRP4loxP/+ control mice (582 ± 31.8/mm2 in GM6001-injected and 589 ± 39.6/mm2 in DMSO-injected mice; n=3, P=0.81). This result was in agreement with the finding of normal NMJs in HB9-LRP4−/− mice (i.e., motoneuron LRP4 is not critical when muscle LRP4 is available) and suggested that the majority of muscle LRP4 functions in cis as agrin receptor. However, the number of primitive AChR clusters was significantly reduced in GM6001-injected HSA-LRP4−/− mice (134 ± 34.2 /mm2), compared to DMSO-injected mice (644 ± 52.1/mm2) (n = 3, P < 0.01) (Figures 7D and 7E). These results could support the hypothesis that ecto-LRP4 from motoneurons may serve as an agrin receptor in trans for MuSK activation in muscle fibers.
DISCUSSION
This study confirms that LRP4 in muscles serves as an obligate receptor for agrin, and is necessary and sufficient to mediate agrin signaling in NMJ formation and maturation. It reveals functions of LRP4 in NMJ formation. Muscle LRP4 appears to restrict AChR clusters in the middle region of muscle fibers, directs a stop signal for axon terminals, and is critical for presynaptic differentiation. On the other hand, LRP4 in motoneurons has at least two functions. It promotes the formation of immature AChR clusters that are sufficient to prevent neonatal lethality. This effect appears to be mediated by ecto-LRP4 from motoneurons that serves as agrin’s receptor in trans to initiate agrin signaling in muscles. Moreover, motoneuron LRP4 is also necessary for axon terminal differentiation and well-being.
Muscle LRP4 for Presynaptic Differentiation
In LRP4mitt null mutant mice, motor axons fail to terminate in the middle region of muscle fibers; instead, they arborized extensively as if to search for AChR clusters which do not form at all in LRP4mitt null mutant mice (Weatherbee et al., 2006). These phenotypes were duplicated in HSA-LRP4−/− mice, indicating that presynaptic deficits are caused by the lack of LRP4 in muscles, but not in motoneurons. In addition to extensive arborization, axon terminals contained fewer synaptic vesicles and active zones. These results suggest that muscle LRP4 may direct a retrograde mechanism for presynaptic differentiation. The extensive terminal arborization in LRP4 or MuSK null mutants was thought to be a compensatory response of motoneurons to look for AChR clusters the mutant mice fail to form. Intriguingly, axons in HSA-LRP4−/− mice appeared to ignore primitive AChR clusters and extend to outside of the already-widened cluster-rich areas. These observations suggest that the LRP4-dependent stop signal may not be retained in AChR clusters.
How muscle LRP4 directs presynaptic differentiation remains unclear. Intriguingly, our in vitro study suggests that LRP4 of HEK293 cells may have synaptogenic activity for cortical neurons. Having a large extracellular domain, LRP4 is able to interact with LRP4 of another cell in a homophilic manner (Kim et al., 2008). However, this mechanism is not supported by lack of NMJ deficits in HB9-LRP4−/− mice (Figure S3). Whether muscle LRP4 may organize presynaptic differentiation via direct interaction with a receptor on motoneurons demands further investigation. Of note, such a cell contact-dependent mechanism may be more feasible for developing NMJs, but less for mature NMJs whose synaptic clefts could be as large as 100 nm in distance (Sanes and Lichtman, 1999).
It is worth pointing out that AChR clusters that are formed in the absence of muscle LRP4 are primitive: varying in size and being distributed in a wider central region (Figure 1). Reduced mEPP amplitudes suggest that they are impaired in function (Figure 2). Moreover, junctional folds were reduced in HSA-LRP4−/− NMJs. These deficits plus the presynaptic deficits described above demonstrate that LRP4 in muscles plays an unequivocal role in postsynaptic differentiation. They also raise a possibility that the presynaptic phenotypes in HSA-LRP4−/− mice may be secondary to neuromuscular deficits as in agrin and MuSK mutant mice (DeChiara et al., 1996; Gautam et al., 1996; Glass et al., 1996). This mechanism and the possible impaired synaptogenic activity are not mutually exclusive and worthy further investigation. Moreover, the presynaptic deficits of LRP4 null or HSA-LRP4−/− mice are different from those in muscle-specific β-catenin mutant mice (Li et al., 2008), suggesting complexity of retrograde mechanisms.
Motoneuron LRP4 for Postsynaptic Differentiation
The findings that primitive AChR clusters are formed in HSA-LRP4−/− mice, but abolished by additional ablation of motoneuron LRP4 (i.e., in HSA/HB9-LRP4−/− mice) suggest a role of motoneuron LRP4 in NMJ formation. Because HB9-LRP4−/− mice are viable with normal NMJs, motoneuron LRP4 is dispensable when LRP4 is available from muscles. We speculate that LRP4 is so critical for NMJ formation and/or maintenance that a safe-guard mechanism is created in case of abnormal expression or function of LRP4 in muscles or motoneurons under pathological conditions.
Our data suggest that LRP4 in motoneurons may serve as a receptor of agrin in trans. It is possible that the extracellular domain of LRP4 may hang like a chandelier from motoneurons and interacts with agrin and MuSK. It is unclear, however, whether the extracellular region is able to reach MuSK on muscle cells. Alternatively, it is cleaved to release ecto-LRP4 which binds to agrin to activate MuSK. The latter model is supported by the following evidence. First, ecto-LRP4 could serve as a receptor for agrin to stimulate MuSK or AChR clustering. Second, ecto-LRP4 was detected in motor nerves and appeared enriched in the synapse-rich region (Figure S5B). Third, inhibition of MMP attenuated the formation of primitive AChR clusters in HSA-LRP4−/− mice. These observations suggest that motoneuron LRP4 may function as a receptor for agrin in trans to activate MuSK in muscle cells for postsynaptic differentiation. In this case, the soluble agrin-ecto-LRP4 complex could be considered as a “coligand” for the kinase. It is worthwhile to notice that ecto-LRP4 does not stimulate AChR clustering by itself (i.e., without agrin); and in the presence of full-length LRP4 in muscle, it does not further increase agrin’s effect, suggesting that ecto-LRP4 acts via a similar mechanism of full-length LRP4 in muscles. MMP3 mutation was shown to impair the NMJ (VanSaun et al., 2003) and one of its substrate is agrin (VanSaun and Werle, 2000). It remains unknown whether MMP3 also cleaves LRP4. However, GM6001 is an inhibitor of multiple MMPs and thus may alter cleavage of various substrates.
Why would motoneuron LRP4 be unable to form normal NMJs (in the absence of muscle LRP4)? First, the amounts of LRP4 contributed by motoneurons at the NMJ may be limited and is insufficient to initiate necessary signaling thresholds in muscle cells. This hypothesis is supported by western blot analysis of motoneuron- and/or muscle-specific LRP4 knockout muscles. As shown in Figure S6, 63.5% of LRP4 in muscles was derived from expression in muscle fibers, 19.4% from motor nerve terminals, and the residual 17.1% from Schwann cells or blood vessels. Moreover, heterozygous muscle-specific mutant mice (HSA-LRP4+/−) did not show NMJ deficits because they appeared to express more than 60% of LRP4 (Figures S5C–S5F). Likewise, MMP inhibition impaired NMJ formation in HSA-LRP4−/− mice, but not control mice (Figures 7D and 7E). Second, LRP4 receptor function in cis in muscles may be more efficient than the trans-receptor. Third, in addition to function as a receptor of agrin, LRP4 in muscles appears to serve as a scaffold for the MuSK signaling complex. AChR was shown to associate with MuSK and rapsyn (Apel et al., 1995; Fuhrer et al., 1997; Moransard et al., 2003). This interaction was abolished or attenuated in muscle-specific, double, and null LRP4 mutant muscles, but not in motoneuron-specific mutant muscles (Figures S7A and S7B). Finally, motoneuron LRP4 may be unable to generate the retrograde signals for presynaptic differentiation.
Coordinated Efforts of Motoneuron and Muscle LRP4 in Synapse Formation
Results of this study highlight distinct functions of motoneuron and muscle LRP4 for NMJ formation and maintenance (Figure 8). In a working model, motoneuron LRP4 contributes to formation of primitive AChR clusters and initial postsynaptic differentiation. This effect is likely mediated by ecto-LRP4, released by extracellular cleavage, which acts as agrin’s receptor in trans to stimulate AChR clustering. Motoneuron LRP4 is also necessary for well-being of motor axons. On the other hand, muscle LRP4 plays a dominant role in NMJ formation. It instructs where nerve terminals stop and where muscle fibers form AChR clusters, is essential for NMJ maturation, and regulates presynaptic differentiation. These observations may have an implication for understanding how LRP4-like “receptors” work in other contexts including CNS synapse formation and plasticity.
Figure 8. A Working Model for LRP4 at the NMJ.
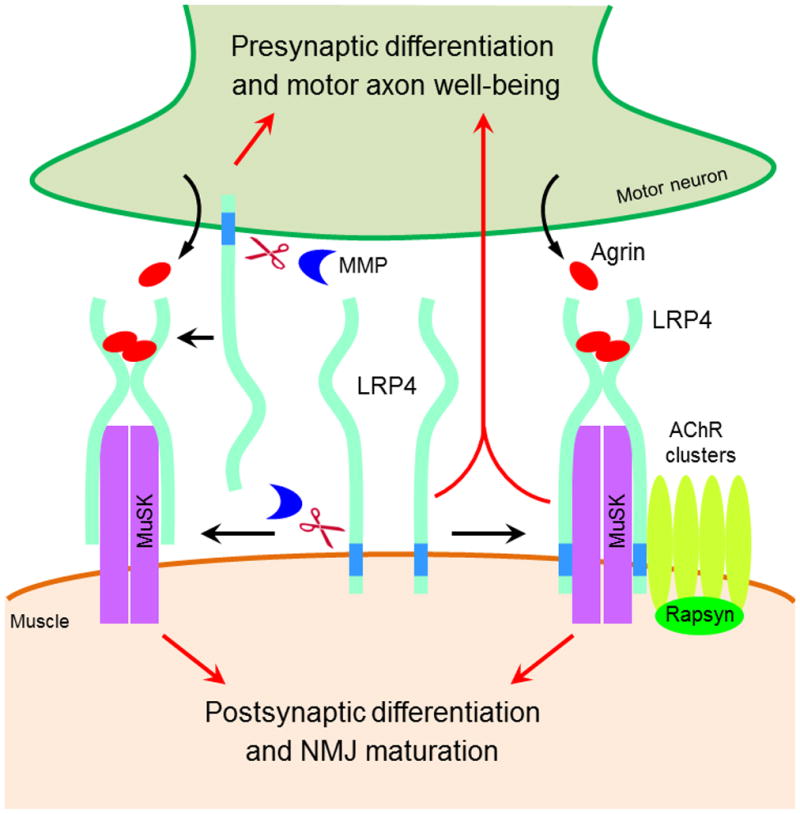
LRP4 in muscles serves as an obligate receptor for agrin, which is necessary and sufficient to mediate postsynaptic differentiation and NMJ maturation. In addition, it directs axons about where to stop, restricts AChR clusters in the middle region of muscle fibers, and regulates presynaptic differentiation. On the other hand, LRP4 in motoneurons undergoes extracellular cleavage to release ecto-LRP4, which acts as agrin’s receptor in trans to stimulate AChR clustering. Motoneuron LRP4 is also necessary for differentiation and well-being of motor axons. Please also see Figure S7.
EXPERIMENTAL PROCEDURES
Reagents, Antibodies, and Constructs
Chemicals were purchased from Sigma-Aldrich Company unless otherwise indicated. Alexa Fluor 350 phalloidin (A22281; 1:200 for staining) and Alexa Fluor 594 α-bungarotoxin (BTX) (B-13423; 1:3000 for staining) were purchased from Invitrogen (Carlsbad, CA). Matrix metalloproteinase (MMP) inhibitor N-[(2R)-2(hydroxamideocarbonylmethyl)-4-methylpantanoyl]-L-tryptophan methylamide (GM6001) (364206) and β-Secretase inhibitor IV (565788) were purchased from Calbiochem (EMD Millipore, Billerica, MA). Information of antibodies was as follows: neurofilament (Millipore, Billerica, MA) (AB1991; 1:1000 for staining); synaptophysin (Dako, Carpinteria, CA) (A0010; 1:2000 for staining); LRP4 (ECD) clone N207/27 (UC Davis/NIH NeuroMab Facility, Davis, CA) (75–221; 1:1000 for western blotting); Flag (M2) (Sigma-Aldrich, St. Louis, MO) (F3165; 1:2000 for western blotting); 4G10 (Millipore, Billerica, MA) (05-1050X; 1:2000 for western blotting); GFP (Abcam, Cambridge, MA) (ab6556; 1:2000 for western blotting); β-III-Tubulin (Covance, Richmond, CA) (MMS-435P; 1:1000 for immunostaining); HB9 antibodies (C-terminal 307–403, gift from Dr. Samuel Pfaff; 1:4000 for staining) (Thaler et al., 1999); synapsin (Santa Cruz, CA) (sc-20780; 1:500 for immunostaining); SV2 (Developmental Studies Hybridoma Bank, Iowa City, IA) (1:1000 for immunostaining); α-Tubulin (Santa Cruz, CA) (sc-23948; 1:3000 for western blotting); β-actin (Novus, Littleton, CO) (NB600-501; 1:3000 for western blotting); and Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, Carlsbad, CA) (A-11034, 1:1000 for staining). Polyclonal horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (32260), goat anti-mouse IgG (32230) and goat anti-rat IgG (31470) secondary antibodies were purchased from Pierce (Thermo Scientific, Rockford, IL) (1:3000 for western blotting). Anti-MuSK, anti-rapsyn and anti-AChRα antibodies were described previously (1:1000 for western blotting) (Luo et al., 2002).
Original agrin and MuSK constructs were gifts from Dr. Zach Hall, and original LRP4 constructs were gifts from Dr. Tatsuo Suzuki. Flag-MuSK was generated as previously described (Zhang et al., 2008). To generate Flag-LRP4 and Flag-ecto-LRP4, full length LRP4 and ecto-LRP4 cDNA was amplified by PCR from original LRP4 construct and subcloned into HindIII/BglII sites in pFlag-CMV1 downstream of an artificial signal peptide sequence and a Flag epitope. LRP4-miRNA construct miLRP4-1062 was generated using the BLOCK-It Pol II miR RNAi Expression Vector Kit (K4936-00, Invitrogen, Carlsbad, CA) which has been previously described and verified to be most potent in inhibiting LRP4 expression (Zhang et al., 2008). For all the constructs, the authenticity was verified by DNA sequencing in Eurofins MWG Operon (Huntsville, AL).
Generation of LRP4 Floxed Mice, Crossing and Genotyping
LRP4loxP mice, where the exon 1 of the LRP4 gene was flanked by loxP sites, were generated as described in Supplemental Information. They were crossed with HSA-Cre and HB9-Cre transgenic mice to generate muscle or motoneuron specific knockout (KO) as well as double knockout (dKO) LRP4 mutant mice. The mutant mice were generated on a BL6/129 mixed background and backcrossed into C57BL/5J mice. Crosses generated the expected Mendelian numbers of each genotype and genotyping procedures were described in Supplemental Information. Mice were housed in a room with a 12 hr light/dark cycle with ad libitum access to water and rodent chow diet (Diet 1/4″ 7097, Harlan Teklad, Madison, WI). Experiments with animals were approved by Institutional Animal Care and Use Committee of the Georgia Health Sciences University.
Light Microscopic and Electrophysiological Analysis of NMJs
Whole-mount staining of diaphragms, quantitative analysis of NMJs, single muscle fiber assay, and electrophysiological recording were performed as previously described with modification (Dong et al., 2006; Li et al., 2008) (see Supplemental Information for details).
Electron Microscopic Analysis
Electron microscopic studies were carried as described previously (Wu 2012, in press). Briefly, entire diaphragms (P0 and P15) were isolated and fixed in 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate buffer for 1 hr at 25°C and 4°C overnight. Synaptic segments of muscles were isolated and fixed in sodium cacodylate-buffered (pH 7.3) 1% osmium tetroxide for 1 hr at 25°C. After washing three times with phosphate buffer, 10 min each, synaptic segments were dehydrated through a series of ethanol steps (30%, 50%, 70%, 80%, 90%, 100%), rinsed with 100% propylene oxide three times, embedded in plastic resin (EM-bed 812, EM Sciences, Fort Washington, PA), and subjected to serial thick-sectioning (1–2 μm). Some sections will be stained with 1% toluidine blue for light microscopic identification of phrenic nerves. Adjacent sections were cut into ultra-thin sections, mounted on 200 mesh unsupported copper grids and stained with uranyl acetate (3% in 50% methanol) and lead citrate (2.6% lead nitrate and 3.5% sodium citrate, pH 12.0). Electron micrographs were taken by a JEOL 100CXII operated at 80 KeV. Micrographs were digitized and morphometric analysis was performed using National Institutes of Health (NIH) Image J software, as described previously (An et al., 2010; Reddy et al., 2003).
qRT-PCR and Biochemical Analysis
qRT-PCR was performed as described previously (Liu et al., 2011). Pull-down assays and western blotting were carried as described previously (Luo et al., 2008). Details were provided in Supplemental Information.
In vitro AChR Clustering Assay
AChR clusters in cultured myotubes were assayed as described previously (Luo et al., 2008; Luo et al., 2002; Zhang et al., 2008). Details are provided in Supplemental Information.
Imaging of Motoneurons in Spinal Cords
Spinal cords between C3 and C5 were dissected from P0 mice, fixed in 4% PFA in PBS (pH 7.3) overnight, immersed in 0.1 M phosphate buffer (pH 7.3) containing 30% sucrose for 24 hr, and embedded in OCT compound (Tissue-Tek) (Sakura Finetek, Torrance, CA). Cross-sections (14 μm) were stained by hematoxylin and eosin or with anti-HB9 antibodies as described previously (Arber et al., 1999). For quantification, HB9-positive motor neurons from hemiventral columns in every fourth section were counted by individuals blind to genotypes.
Neuron-HEK293 Cells Coculture Assay
Cortical neurons were prepared from Sprague Dawley rat embryos (E18) and cultured in the neurobasal medium (21103-049; Invitrogen) supplemented with 1× B27 (17504-044; Invitrogen), and 1× penicillin-streptomycin (30-003-CI; Cellgro) as described previously (Ting et al., 2011). Neuron-HEK293 cells coculture assays were set up as previously described (Biederer et al., 2002; Fogel et al., 2011; Graf et al., 2004; Scheiffele et al., 2000). Briefly, HEK293 cells in 60 mm culture dishes were cotransfected with 8 μg Flag-LRP4 and pEGFPC1 (BD Biosciences Clontech) vector (10:1) or pEGFPC1 alone using Lipofectamine 2000 (11668-019; Invitrogen, Carlsbad, CA). Twenty-four hours later, 60,000 HEK293 cells were resuspended and cocultured with primary cortical neurons (DIV 7). Neurons had been seeded on coverslips (12-545-84; Fisher Scientific) which were coated with Ploy-L-Lysine (P2636; Sigma-Aldrich) in 12-well plates at 30,000 cells/well. After 24–48 hr of coculture, cells were fixed with 4% paraformaldehyde and stained for synapsin or SV2. For quantification, HEK293 cells contacting axons with synapsin or SV2 punctas were accounted as positive (Umemori and Sanes, 2008). Integrated puncta intensity was quantified as described previously (Graf et al., 2004). The intensity of synapsin or SV2 staining in neurites contacting HEK293 cells was subtracted with off-cell background, and normalized to synapsin or SV2 intensity in neurites in cell-free regions (also subtracted with off-cell background).
Statistical Analysis
Statistics were computed using Prism 5.0 (GraphPad, La Jolla, CA) software. Survival curves were analyzed first all genotypes and, if significant, reanalyzed for the experimental pair using the Log-Rank (Mantel-Cox) test. All data were presented as mean ± standard error of the mean (SEM) and analyzed using Student’s t-test or two-way ANOVA analysis, wherever appropriate. Graphs were also generated by Prism5.0. Difference were considered significant at P value <0.05.
Supplementary Material
Acknowledgments
We are grateful to Dr. J. Melki, Dr. S. Arber, and Dr. L.A. Niswander for valuable mouse lines; Dr. S. Pfaff, Dr. R. Rotundo, Dr. Z. Hall, and Dr. T. Suzuki for valuable reagents; and Dr. Chien-Ping Ko for advice on EM analysis. We thank members of the Mei and Xiong laboratories for discussion. This work was supported in part by grants from National Institutes of Health (NS040480 and NS056415, L.M. and W.C.X.) and Muscular Dystrophy Association (L.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An MC, Lin W, Yang J, Dominguez B, Padgett D, Sugiura Y, Aryal P, Gould TW, Oppenheim RW, Hester ME, et al. Acetylcholine negatively regulates development of the neuromuscular junction through distinct cellular mechanisms. Proc Natl Acad Sci U S A. 2010;107:10702–10707. doi: 10.1073/pnas.1004956107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel ED, Roberds SL, Campbell KP, Merlie JP. Rapsyn may function as a link between the acetylcholine receptor and the agrin-binding dystrophin-associated glycoprotein complex. Neuron. 1995;15:115–126. doi: 10.1016/0896-6273(95)90069-1. [DOI] [PubMed] [Google Scholar]
- Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, Sockanathan S. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron. 1999;23:659–674. doi: 10.1016/s0896-6273(01)80026-x. [DOI] [PubMed] [Google Scholar]
- Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Sudhof TC. SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science. 2002;297:1525–1531. doi: 10.1126/science.1072356. [DOI] [PubMed] [Google Scholar]
- Bolis A, Coviello S, Bussini S, Dina G, Pardini C, Previtali SC, Malaguti M, Morana P, Del Carro U, Feltri ML, et al. Loss of Mtmr2 phosphatase in Schwann cells but not in motor neurons causes Charcot-Marie-Tooth type 4B1 neuropathy with myelin outfoldings. J Neurosci. 2005;25:8567–8577. doi: 10.1523/JNEUROSCI.2493-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan KJ, Hardeman EC. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J Biol Chem. 1993;268:719–725. [PubMed] [Google Scholar]
- Carter CJ. Convergence of genes implicated in Alzheimer’s disease on the cerebral cholesterol shuttle: APP, cholesterol, lipoproteins, and atherosclerosis. Neurochem Int. 2007;50:12–38. doi: 10.1016/j.neuint.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol. 2000;150:1399–1410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Dietrich MF, van der Weyden L, Prosser HM, Bradley A, Herz J, Adams DJ. Ectodomains of the LDL receptor-related proteins LRP1b and LRP4 have anchorage independent functions in vivo. PLoS One. 2010;5:e9960. doi: 10.1371/journal.pone.0009960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XP, Li XM, Gao TM, Zhang EE, Feng GS, Xiong WC, Mei L. Shp2 is dispensable in the formation and maintenance of the neuromuscular junction. Neurosignals. 2006;15:53–63. doi: 10.1159/000094484. [DOI] [PubMed] [Google Scholar]
- Escher P, Lacazette E, Courtet M, Blindenbacher A, Landmann L, Bezakova G, Lloyd KC, Mueller U, Brenner HR. Synapses form in skeletal muscles lacking neuregulin receptors. Science. 2005;308:1920–1923. doi: 10.1126/science.1108258. [DOI] [PubMed] [Google Scholar]
- Ferns MJ, Campanelli JT, Hoch W, Scheller RH, Hall Z. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 1993;11:491–502. doi: 10.1016/0896-6273(93)90153-i. [DOI] [PubMed] [Google Scholar]
- Fogel AI, Stagi M, Perez de Arce K, Biederer T. Lateral assembly of the immunoglobulin protein SynCAM 1 controls its adhesive function and instructs synapse formation. EMBO J. 2011;30:4728–4738. doi: 10.1038/emboj.2011.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer C, Sugiyama JE, Taylor RG, Hall ZW. Association of muscle-specific kinase MuSK with the acetylcholine receptor in mammalian muscle. EMBO J. 1997;16:4951–4960. doi: 10.1093/emboj/16.16.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, et al. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Gomez AM, Burden SJ. The extracellular region of Lrp4 is sufficient to mediate neuromuscular synapse formation. Dev Dyn. 2011;240:2626–2633. doi: 10.1002/dvdy.22772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski A, Burden SJ. Neuromuscular synapse formation in mice lacking motor neuron- and skeletal muscle-derived Neuregulin-1. J Neurosci. 2006;26:655–661. doi: 10.1523/JNEUROSCI.4506-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EB, Hammer RE, Herz J. Abnormal development of the apical ectodermal ridge and polysyndactyly in Megf7-deficient mice. Hum Mol Genet. 2005;14:3523–3538. doi: 10.1093/hmg/ddi381. [DOI] [PubMed] [Google Scholar]
- Kelly SS. The effect of age on neuromuscular transmission. J Physiol. 1978;274:51–62. doi: 10.1113/jphysiol.1978.sp012133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Burden SJ. MuSK controls where motor axons grow and form synapses. Nat Neurosci. 2008;11:19–27. doi: 10.1038/nn2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Arber S, Jessell TM, Edlund H. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet. 1999;23:67–70. doi: 10.1038/12669. [DOI] [PubMed] [Google Scholar]
- Li XM, Dong XP, Luo SW, Zhang B, Lee DH, Ting AK, Neiswender H, Kim CH, Carpenter-Hyland E, Gao TM, et al. Retrograde regulation of motoneuron differentiation by muscle beta-catenin. Nat Neurosci. 2008;11:262–268. doi: 10.1038/nn2053. [DOI] [PubMed] [Google Scholar]
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- Liu X, Bates R, Yin DM, Shen C, Wang F, Su N, Kirov SA, Luo Y, Wang JZ, Xiong WC, et al. Specific regulation of NRG1 isoform expression by neuronal activity. J Neurosci. 2011;31:8491–8501. doi: 10.1523/JNEUROSCI.5317-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Tian QB, Endo S, Suzuki T. A role for LRP4 in neuronal cell viability is related to apoE-binding. Brain Res. 2007;1177:19–28. doi: 10.1016/j.brainres.2007.08.035. [DOI] [PubMed] [Google Scholar]
- Luo S, Zhang B, Dong XP, Tao Y, Ting A, Zhou Z, Meixiong J, Luo J, Chiu FC, Xiong WC, et al. HSP90 beta regulates rapsyn turnover and subsequent AChR cluster formation and maintenance. Neuron. 2008;60:97–110. doi: 10.1016/j.neuron.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Wang Q, Zhou J, Wang J, Liu M, He X, Wynshaw-Boris A, Xiong W, Lu B, Mei L. Regulation of AChR Clustering by Dishevelled Interacting with MuSK and PAK1. Neuron. 2002;35:489–505. doi: 10.1016/s0896-6273(02)00783-3. [DOI] [PubMed] [Google Scholar]
- McMahan UJ. The agrin hypothesis. [Review] Cold Spring Harb Symp Quant Biol. 1990;55:407–418. doi: 10.1101/sqb.1990.055.01.041. [DOI] [PubMed] [Google Scholar]
- Miniou P, Tiziano D, Frugier T, Roblot N, Le Meur M, Melki J. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 1999;27:e27. doi: 10.1093/nar/27.19.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moransard M, Borges LS, Willmann R, Marangi PA, Brenner HR, Ferns MJ, Fuhrer C. Agrin regulates rapsyn interaction with surface acetylcholine receptors, and this underlies cytoskeletal anchoring and clustering. J Biol Chem. 2003;278:7350–7359. doi: 10.1074/jbc.M210865200. [DOI] [PubMed] [Google Scholar]
- Muscat GE, Kedes L. Multiple 5′-flanking regions of the human alpha-skeletal actin gene synergistically modulate muscle-specific expression. Mol Cell Biol. 1987;7:4089–4099. doi: 10.1128/mcb.7.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitkin RM, Smith MA, Magill C, Fallon JR, Yao YM, Wallace BG, McMahan UJ. Identification of agrin, a synaptic organizing protein from Torpedo electric organ. J Cell Biol. 1987;105:2471–2478. doi: 10.1083/jcb.105.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, Kubo S, Shiraishi H, Eguchi K, Motomura M, et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–1805. doi: 10.1126/science.1127142. [DOI] [PubMed] [Google Scholar]
- Reddy LV, Koirala S, Sugiura Y, Herrera AA, Ko CP. Glial cells maintain synaptic structure and function and promote development of the neuromuscular junction in vivo. Neuron. 2003;40:563–580. doi: 10.1016/s0896-6273(03)00682-2. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Schwander M, Leu M, Stumm M, Dorchies OM, Ruegg UT, Schittny J, Muller U. Beta1 integrins regulate myoblast fusion and sarcomere assembly. Dev Cell. 2003;4:673–685. doi: 10.1016/s1534-5807(03)00118-7. [DOI] [PubMed] [Google Scholar]
- Selvais C, D’Auria L, Tyteca D, Perrot G, Lemoine P, Troeberg L, Dedieu S, Noel A, Nagase H, Henriet P, et al. Cell cholesterol modulates metalloproteinase-dependent shedding of low-density lipoprotein receptor-related protein-1 (LRP-1) and clearance function. FASEB J. 2011;25:2770–2781. doi: 10.1096/fj.10-169508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, Pfaff SL. Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron. 1999;23:675–687. doi: 10.1016/s0896-6273(01)80027-1. [DOI] [PubMed] [Google Scholar]
- Tian QB, Suzuki T, Yamauchi T, Sakagami H, Yoshimura Y, Miyazawa S, Nakayama K, Saitoh F, Zhang JP, Lu Y, et al. Interaction of LDL receptor-related protein 4 (LRP4) with postsynaptic scaffold proteins via its C-terminal PDZ domain-binding motif, and its regulation by Ca/calmodulin-dependent protein kinase II. Eur J Neurosci. 2006;23:2864–2876. doi: 10.1111/j.1460-9568.2006.04846.x. [DOI] [PubMed] [Google Scholar]
- Ting AK, Chen Y, Wen L, Yin DM, Shen C, Tao Y, Liu X, Xiong WC, Mei L. Neuregulin 1 promotes excitatory synapse development and function in GABAergic interneurons. J Neurosci. 2011;31:15–25. doi: 10.1523/JNEUROSCI.2538-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemori H, Sanes JR. Signal regulatory proteins (SIRPS) are secreted presynaptic organizing molecules. J Biol Chem. 2008;283:34053–34061. doi: 10.1074/jbc.M805729200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanSaun M, Herrera AA, Werle MJ. Structural alterations at the neuromuscular junctions of matrix metalloproteinase 3 null mutant mice. J Neurocytol. 2003;32:1129–1142. doi: 10.1023/B:NEUR.0000021907.68461.9c. [DOI] [PubMed] [Google Scholar]
- VanSaun M, Werle MJ. Matrix metalloproteinase-3 removes agrin from synaptic basal lamina. J Neurobiol. 2000;43:140–149. [PubMed] [Google Scholar]
- von Arnim CA, Kinoshita A, Peltan ID, Tangredi MM, Herl L, Lee BM, Spoelgen R, Hshieh TT, Ranganathan S, Battey FD, et al. The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J Biol Chem. 2005;280:17777–17785. doi: 10.1074/jbc.M414248200. [DOI] [PubMed] [Google Scholar]
- Weatherbee SD, Anderson KV, Niswander LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
- Willnow TE, Moehring JM, Inocencio NM, Moehring TJ, Herz J. The low-density-lipoprotein receptor-related protein (LRP) is processed by furin in vivo and in vitro. Biochem J. 1996;313(Pt 1):71–76. doi: 10.1042/bj3130071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lu Y, Barik A, Joseph A, Taketo M, Xiong WC, Mei L. Beta-catenin gain-of-function in muscles impairs neuromuscular junction formation. Development. 2012;139:2392–2404. doi: 10.1242/dev.080705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi YL, Tanaka SS, Kasa M, Yasuda K, Tam PP, Matsui Y. Expression of low density lipoprotein receptor-related protein 4 (Lrp4) gene in the mouse germ cells. Gene Expr Patterns. 2006;6:607–612. doi: 10.1016/j.modgep.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, Birchmeier C, Burden SJ. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 2001;30:399–410. doi: 10.1016/s0896-6273(01)00287-2. [DOI] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Coldefy AS, Hubbard SR, Burden SJ. Agrin binds to the N-terminal region of Lrp4 and stimulates association between Lrp4 and the first Ig-like domain in MuSK. J Biol Chem. 2011;286:40624–40630. doi: 10.1074/jbc.M111.279307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Y, Zhang B, Gu S, Lee K, Zhou J, Yao G, Figueiredo D, Perry K, Mei L, Jin R. Structural basis of agrin-LRP4-MuSK signaling. Genes Dev. 2012;26:247–258. doi: 10.1101/gad.180885.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.