

Abstract
Autophagy is a catabolic process that allows cellular macromolecules to be broken down and recycled as metabolic precursors. The influence of non-coding microRNAs (miRNAs) in autophagy has not been explored in colon cancer. In this study, we discover a novel mechanism of autophagy regulated by hsa-miR-502-5p (miR-502) by suppression of Rab1B, a critical mediator of autophagy. A number of other miR-502 suppressed mRNA targets (e.g. DHODH) are also identified by microarray analysis. Ectopic expression of miR-502 inhibited autophagy, colon cancer cell growth, and cell cycle progression of colon cancer cells in vitro. miR-502 also inhibited in vivo colon cancer growth in a mouse tumor xenografts model. In addition, the expression of miR-502 was regulated by p53 via a negative feedback regulatory mechanism. The expression of miR-502 was down-regulated in colon cancer patient specimens compared to the paired normal control samples. These results suggest that miR-502 may function as a potential tumor suppressor and therefore be a novel candidate for developing miR-502 based therapeutic strategies.
Keywords: miR-502, autophagy, p53, colon cancer
Introduction
Colon cancer is one of the most common types of cancer and causes 655,000 deaths per year worldwide (1). In the United States, colorectal cancer ranks the second in cancer-related mortality (2), and it is estimated that more than 49,000 colorectal cancer patients will succumb to the disease in 2011 according to the National Cancer Institute. There has been steady and significant improvement in patient survival in the past decade due to early detection and improved treatment strategies (2).
In the past 10 years, small regulatory RNAs have gained enormous interest in colon cancer research. miRNAs are a class of non-coding RNA molecules, 18–25 nucleotides in length, that regulate the expression of their target genes by translational arrest or mRNA cleavage mostly via direct interacting with the 3’-UTRs of the target mRNAs (3, 4). Base pairing between at least six consecutive nucleotides within the 5’-seed of the miRNA with the target site on the mRNA is reported to be a minimum requirement for the miRNA-mRNA interaction (5). miRNAs have been found to regulate many cellular processes including apoptosis (6–9), differentiation (4, 10, 11) and cell proliferation (6, 11–13).
p53, one of the major tumor suppressor genes, has been found to be deleted and mutated in more than 50 percent of the colon cancer cases (14–16). It regulates its target gene expression either as a transcriptional activator or suppressor (17). It can also act as an RNA-binding protein to modulate gene expression at the post-transcriptional level (18, 19). In 2006, our laboratory first identified putative p53-binding sites in nearly 40% of miRNA promoter regions (20). Some of these miRNAs (e.g. miR-215, miR-192, miR-34s) were directly activated by p53 to impact cell proliferation, cell cycle control and chemoresistance (20–31), providing p53 with a greater flexibility to control cell cycle and cell death.
Moreover, p53 also functions as a key regulator of autophagy (32–35), a catabolic pathway for degradation and recycling of proteins and cellular organelles, which has been shown to be dysregulated in cancers (36, 37). Under prolonged nutrition starvation, p53 sustained active autophagic flux, which was beneficial for colon cancer cell survival (35). In addition, Rab1B, a small GTPase from Ras super family, has been demonstrated to modulate autophagic activity in Hela cells through the regulation of autophagosome formation (38). Since translational control provides cells with acute response to growth stress, we reasoned that some of the p53 mediated miRNAs may play important roles in autophagy. We discovered that Rab1B was predicted to be the direct target of hsa-miR-502-5p (miR-502), which was among the top 10 miRNA candidates containing potential p53-binding sites in their promoter regions (20). Currently little is known about the regulatory function of miR-502 in colon cancer and its impact on autophagy.
In this study, we investigated the mechanisms and impact of miR-502 in colon cancer. We systematically discovered miR-502 mediated targets via microarray analysis and experimentally validated that the repressions of Rab1B and dihydroorotate dehydrogenase (DHODH) were directly regulated by miR-502. We further demonstrated that ectopic expression of miR-502 in colon cancer cells interrupted the autophagic flux, and such impact was also linked directly to the p53 status. The expression of miR-502 and p53 was mediated through a negative feedback loop. In addition, miR-502 inhibited colon cancer cell growth and triggered cell cycle arrest in vitro. We administered miR-502 precursor to a mouse colon cancer tumor xenograft model and further demonstrated that it can suppress colon tumor growth in vivo. To demonstrate the clinical relevance, we quantified the expression of miR-502 and Rab1B from paired normal and colorectal tumor specimens and discovered that the expression of miR-502 was down-regulated and Rab1B was upregulated in colorectal tumor tissues.
Results
Rab1B is a direct target of miR-502 in colon cancer cells
Rab1B has been shown to regulate vesicle trafficking at multiple stages and directly impact autophagy (39, 40), and it was found to be overexpressed in liver cancer (41). We have identified a putative miR-502 binding site at position 1160 to 1166 (GCAAGGA) in 3’-UTR of Rab1B mRNA using TargetScan analysis (Figure 1A). To experimentally confirm that the expression of Rab1B is a direct target of miR-502, we transfected colon cancer cell lines, HCT116 and SW480, with either negative control miRNA or miR-502 precursors, and quantified the expression of Rab1B at both protein and mRNA levels by western blot analysis and real-time qRT-PCR analysis, respectively. Our results revealed that miR-502 specifically suppressed the protein expression of Rab1B in both cell lines (Figure 1B) and reduced the Rab1B mRNA transcript level (Figure 1C) compared to the controls. To further confirm the direct binding of miR-502 to the 3’-UTR of Rab1B mRNA, we cloned full-length Rab1B 3’-UTR and inserted it to the downstream of a firefly luciferase gene in a luciferase reporter vector, which was then transfected into colon cancer HCT116 cells together with miR-502 or negative control miRNA. miR-502 significantly inhibited the luciferase activity compared to the negative control miRNA (Figure 1D), suggesting that miR-502 was able to bind directly to the 3’-UTR of Rab1B mRNA to suppress translation. In addition, miR-502 failed to inhibit the luciferase activity of the reporter vector containing Rab1B 3’UTR with two point mutations in miR-502 binding site (GCCCGGA) (Figure 1A, D). Based on these results, we conclude that miR-502 directly regulates Rab1B expression in colon cancer cell lines.
Figure 1. Rab1B is the direct target of miR502.

(A) A putative miR-502 binding site exists in the 3’-UTR of Rab1B mRNA and two point mutations were generated in the binding site. Ectopic expression of miR-502 or siRNAs against Rab1B (siRab1B) in HCT116 and SW480 cells decreased Rab1B protein levels by (B) Western blot analysis and (C) mRNA level by real-time qRT-PCR. (D) Transfection of miR-502 inhibited firefly luciferase activity of pMIR-REPORT-3UTRRab1B (wt) and such inhibition was absent with mutations in the miR-502 binding site (mut). The negative miRNA was used as the negative control in all experiments. The impact of miR-502 and siRab1B on Rab1B expression was normalized and compared to those of negative miRNA (n=3, p < 0.001).
In addition to Rab1B, we systematically identified miR-502 mediated targets via microarray expression analysis by comparing mRNA profiles between the control and miR-502 transfected cell lines. We have listed 11 top candidate mRNA targets cross validated using TargetScan analysis (Supplementary Table 1). We further experimentally validated that the expression of DHODH, with a predictive miR-502 binding site at the position 245 to 251 in 3’-UTR of its mRNA (Supplementary Figure 1A), was directly regulated by miR-502. Ectopic expression of miR-502 precursor in HCT116 cells led to significant decreases in DHODH mRNA and protein levels compared to negative miRNA transfection (Supplementary Figure 1B, C). Moreover, miR-502 inhibited the luciferase activity of reporter vector expressing DHODH 3’UTR (Supplementary Figure 1D). These results indicate that DHODH is a direct target of miR-502.
miR-502 and p53 form a negative feedback loop in colon cancer cells
Previous studies from our laboratory have shown that a number of miRNAs (e.g. miR-34, miR-192, miR-194, miR-215, miR-200c, miR-26a) had potential p53 binding sites in their putative promoter regions (20), and miR-502 was among the top listed candidates. By using a bioinformatics approach, we identified a putative p53 binding site at 2472 bp upstream of miR-502 (sequence TCACATGCCT…GAGCATGTTT) (20), indicating a miR-502 level may be affected by p53. We used a pair of HCT116 cell lines with wild-type p53 (HCT116 p53+) and null p53 (HCT116 p53−) to investigate the relationship between p53 and miR-502. To induce p53 expression, we treated HCT116 (p53+/+) cell with 10µM nutlin-3 (a specific p53 inducer) for 24 hours, which has been shown to effectively elevate p53 levels in HCT116 cells (42). The increased level of p53 after the treatment was confirmed by the Western blot analysis (Figure 2B). Total cellular RNAs were extracted from the control and treated cells, and the expression of endogenous miR-502 was quantified via real-time qRT-PCR analysis. Interestingly, we discovered that nutlin-3 treatment triggered down-regulation of miR-502 to 52.4% of non-treated cells (Figure 2C), indicating p53 functioned as a transcriptional repressor of miR-502. To further investigate the repressive function of p53 on miR-502, we cloned the 2.5Kb gene fragment (containing p53 binding site) upstream of the miR-502 coding sequence and inserted it upstream of SV40 promoter of pGL3-promoter vector (Figure 2D). We discovered that nutlin treatment triggered a decrease of in approximately 63.3% in luciferase activity compared to the untreated control HCT116 (p53+/+) cells. In contrast, such an inhibitory effect was absent in HCT116 (p53−/−) cells (Figure 2D), suggesting that elevated level of p53 function as a repressor for miR-502 transcription.
Figure 2. Negative feedback loop existed between miR-502 and p53.

(A) Ectopic expression of miR-502 in HCT116 cells decreased protein levels of p53 and p21 by western blot analysis. (B) Treating HCT116 (p53+/+) cells with 10µM nutlin-3 for 24 hours induced elevated protein levels of p53 and (C) decreased levels of endogenous miR-502 by real-time qRT-PCR (n=3, p < 0.001). (D) A 2.5 Kb gene fragment containing putative p53 binding site at 2472 bp upstream of miR-502 coding sequence was inserted into pGL3-promoter vector. Nutlin-3 treatment inhibited the luciferase activity in HCT116 (p53+/+) cells but not in HCT116 (p53−/−) cells as compared to the negative control (NC) (n=3, p < 0.001).
Furthermore, we discovered that the ectopic overexpression of miR-502 in HCT116 (p53+/+) cells decreased the level of p53 protein expression (Figure 2A) without significant changes in p53 mRNA levels (Supplementary Figure 2A). Luciferase activity with a luciferase reporter containing full-length p53 3’-UTR (Supplementary Figure 2B) did not change with elevated miR-502, suggesting that the suppression of p53 was an indirect effect of miR-502. Moreover we examined the level of MDM2, the major mediator for p53 ubiquitination and degradation, but did not find any significant change by overexpression of miR-502 (Supplementary Figure 2C). These results suggest that the suppression of p53 by miR-502 is indirect. In addition, we also found that miR-502 decreased the expression levels of p21 in both HCT116 (p53+/+) and HCT116 (p53−/−) colon cancer cells (Figure 2A), suggesting that the inhibition of p21 by miR-502 was independent of p53.
miR-502 suppressed autophagy in colon cancer cells
Based on the inhibitory effects of miR-502 on p53 expression and the suppression of Rab1B, which was demonstrated to impact autophagosome in Hela cells (38), we reasoned that miR-502 may have an impact on autophagy. We investigated the effects of miR-502 on autophagy in HCT116 (p53+/+) and HCT116 (p53−/−) cells by quantifying the level of lipidated MAP1B-light chain 3 (LC3B-II) and p62 after the cells were induced by nutrient starvation (EBSS) to trigger autophagy for two and 24 hours. To monitor the active turnover of autophagosomes, we treated cells with lysosomal inhibitor Bafilomycin A1 (BafA1) in the presence of EBSS, and deduced autophagic flux between the levels of LC3B-II in the presence and absence of BafA1.
Consistent with the previous report (35), HCT116 (p53−/−) cells showed significantly stalled autophagic flux compared to HCT116 (p53+/+) cells after 24-hour EBSS treatment (Figure 3D) but not after 2-hour treatment (Figure 4B). Two hours after nutrient deprivation, ectopic expression of miR-502 in HCT116 (p53+/+) showed elevated levels of LC3B-II with reduced accumulation of autophagosomes compared to the negative controls (Figure 3A). This effect was more prominent after 24-hour treatment (Figure 3C). After calculating autophagic flux, we discovered that the overexpression of miR-502 impaired the autophagosome recycling at 67.8% and 30.5% of negative control at two and 24 hours, respectively (Figure 3B, D). Similar impairment of miR-502 on autophagy was also observed in HCT116 (p53−/−) cells (44.6% and 54.9% of negative control after 2-hour and 24-hour starvation, respectively, Figure 3). In addition to LC3B, the levels of p62 showed a trend similar to that of miR-502 transfected cells with less accumulated autophagosomes after autophagy induction for 2 hours and 24 hours (Figure 3A, C). These results suggest that miR-502 regulated autophagy through multiple mediators such as p53 and Rab1B in colon cancer cells. This is highly consistent with the function of Rab1B in autophagy of previous studies (38).
Figure 3. miR-502 inhibited autophagy in HCT116 cells.

Forty-eight hours after transfection of miR-502 and negative miRNA, HCT116 (p53+/+) and HCT116 (p53−/−) cells were treated with EBSS and lysosomal inhibitor BafA1. Western blot analysis of LC3B and p62 levels was performed after two hours (A) and 24 hours (C) and the densitometry of LC3B-II normalized to GAPDH is labeled under each lane. The fold changes in autophagic flux were shown in (B) and (D), respectively (n=3).
Figure 4. miR-502 inhibited colon cancer cell growth and induced cell cycle arrest partially through Rab1B.

HCT116 (p53+/+) (A), HCT116 (p53−/−) (B) and SW480 cells (C) were transfected with miR-502 or siRab1B, and cell numbers were measured with WST-1 assay at days 1, 3 and 5. Cell cycle analysis was performed to determine the impact of miR-502 and siRab1B. The representative flow cytometry pattern was shown in (D) and the G1/S and G2/S ratios were shown in (E) (n=3).
Overexpression of miR-502 inhibited colon cancer cell growth in vitro
After ectopic expression of miR-502 in HCT116 and SW480 cells, we quantified the colon cancer cell proliferation by WST-1 assay. The cell proliferation was significantly inhibited by miR-502 compared to negative control miRNA (Figure 4A, C). At day 5, the number of miR-502 transfected HCT116 and SW480 cells were 33.2% and 23.9% of negative controls respectively. We further analyzed cell cycle control by flow cytometry and found that miR-502 overexpression increased cells in both G1 and G2 phases with a decrease in S phase (Figure 4D). The G1/S and G2/S ratios (Figure 4E) indicated that miR-502 induced cell cycle arrest at both G1 and G2 checkpoints.
We further demonstrated that ectopic expression of miR-502 suppressed colon cancer growth. However, the growth inhibitory affect of miR-502 was contributed in part, by the suppression of Rab1B as siRab1B can only account for a portion of the growth inhibitory effect. We demonstrated that the suppression of Rab1B contributed to growth arrest of HCT116 (p53−/−) (Figure 4A, B) by a siRNA knockdown based approach. It is quite likely that other targets (e.g. DHODH) of miR-502 are involved in regulating cell proliferation.
It is well known that p53 and p21 serve as an essential regulator of cell cycle control. However, in this case, miR-502 reduced both p53 and p21 expression, suggesting that the cell cycle control was mediated independent of p53 and p21 status. We have provided evidence that the inhibition of Rab1B contributed to the G2 cell cycle arrest (Figure 4E). The G1 arrest was likely due to the suppression of DHODH by miR-502, as it has been demonstrated previously that DHODH regulated cell cycle at the G1 phase (43).
Inhibition of colon cancer tumor growth by miR-502 in vivo
To access the impact of miR-502 on colon tumor growth in vivo, we further investigated the role of miR-502 in colon cancer progression in mouse colon cancer xenografts models. We subcutaneously inoculated 2.5×106 HCT116 (p53+/+) cells in NOD/SCID mice, and delivered miR-502 precursor, negative miRNA or siPORTamine only (vehicle control) into the tumors when they reached 100 – 150 mm3 at day 14. The miRNAs were administered at 3-day intervals for a total of three times before the tumors were collected at day 24. We followed tumor growth and found that the administration of negative miRNA did not affect tumor growth compared to the vehicle control group (data not shown). However, miR-502 treatment began to affect tumor growth as early as three days after the first injection (Figure 5A). miR-502-treated tumors were significantly smaller than the control tumors (Figure 5B, C), and their average volume was approximately 76.4% of the control group at day 24 (Figure 5A), indicating miR-502 has a potential as a novel adjuvant therapeutic agent for future therapeutic development. In addition, we quantified miR-502 expression levels in tumor xenografts at day 24, and found that the miR-502 injected tumors had nearly 17 fold increase of miR-502 than the negative control group (Figure 5D), suggesting the observed inhibitory effect on tumor growth was due to overexpression of miR-502.
Figure 5. miR-502 inhibited colon tumor growth in vivo.

(A) 2.5 × 106 HCT116 (p53+/+) cells were subcutaneously injected into NOD/SCID mice. On days 14, 17 and 20 (indicated by arrows), miR-502 precursor (n=6) and negative miRNA (n=5) conjugated with siPORTamine reagent were delivered into tumors, and tumor sizes were measured during the treatment until day 24 when mice were sacrificed. (B) Representative images of mice bearing HCT116 tumors and (C) gross morphology of tumors after dissection at day 24 were shown. Arrows indicate the subcutaneous tumors. Scale bar: 2 cm (B) and 1 cm (C). (D) Expression level of miR-502 in tumor xenografts was quantified by real-time qRT-PCR and miR-502 injected tumors had significantly higher levels of miR-502 than the negative controls (n=3, p < 0.05).
miR-502 was down-regulated in human colorectal tumor tissues
To directly demonstrate the clinical relevance of miR-502 and its targets expression, we profiled miR-502, Rab1B and DHODH levels in paired human colorectal tumor specimens and normal controls. The expression of miR-502 was quantified from total RNA samples and the expression was normalized to the internal control RNU44. The expression of miR-502 was significantly reduced in colorectal tumor tissues compared to the normal controls (Figure 6A). This suggests that the reduced levels of miR-502 may be associated with the colorectal tumor disease development. Similarly, the expression levels of Rab1B and DHODH were normalized to the internal control GAPDH. In contrast Rab1B was significantly up-regulated in CRC tumor tissue than normal tissue (Figure 6B), which was opposite to the expression pattern of miR-502. However, no significant difference was found in DHODH expression levels between normal and tumor tissue (Supplementary Figure 3).
Figure 6. miR-502 was down-regulated in CRC clinical samples.

Twenty-three pairs of tumor and normal tissue from CRC patients were used, and miR-502 and Rab1B levels were quantified by real-time qRT-PCR and normalized to the internal control RNU44 and GAPDH respectively. The miR-502 and Rab1B levels in tumor tissues were compared to corresponding normal tissues.
Based on our results, a model was proposed for the potential roles of miR-502 in colon cancer (Figure 7). miR-502 inhibits autophagy via the suppression of Rab1B. miR-502 suppresses the expression of Rab1B and DHODH to influence tumor growth and cell cycle control. miR-502 and p53 form a negative feedback loop to regulate autophagy. The impact of autophagy was dependent on Rab1B and p53 status. Additional targets mediated by miR-502 may also contribute to colon cancer growth and cell cycle.
Figure 7. Proposed model of miR-502 function in CRC development and progression.
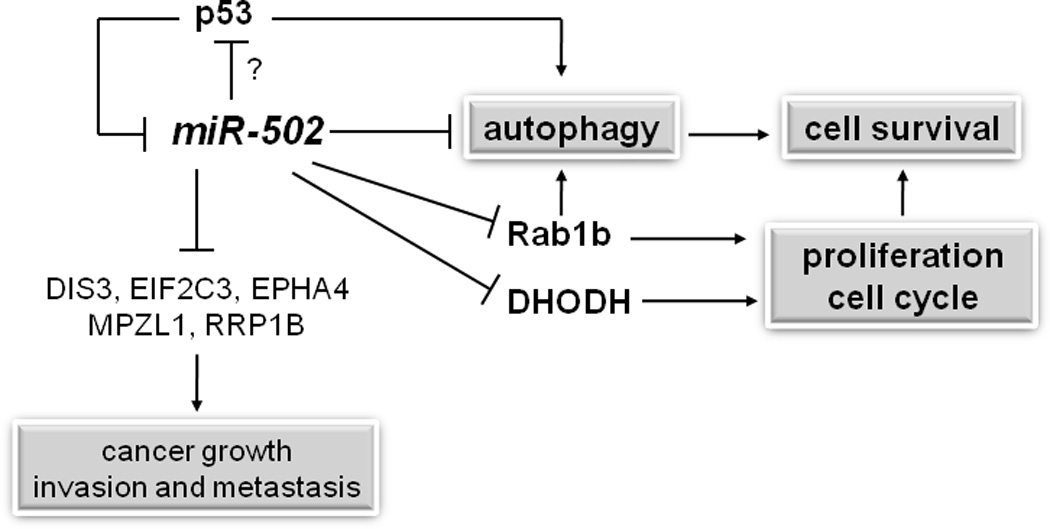
The targets of miR-502 in bold fonts were confirmed in this study.
Discussion
There is mounting evidence to link autophagy and cancer (37, 44). It has been demonstrated extensively that autophagy mediates tumor survival by supplying nutrients to stressed cancer cells. Therefore, an anti-autophagy approach may offer a therapeutic strategy for treating cancer (45–47). Moreover, it has been demonstrated that inhibition of autophagy by 3-methyladenine or siRNA against Atg7 enhances the apoptosis induced by 5-FU treatment in colorectal cancer cell lines in vitro and in vivo (48). Contrary to this, other studies have shown an anticancer role in autophagy (32, 37, 49, 50).
In this study, we demonstrated that miR-502 suppresses autophagy with reduced tumor growth in colon cancer cells. We discovered a novel mechanism of autophagy mediated by miR-502 by suppressing the critical target Rab1B. We showed that miR-502 impaired the autophagy flux in HCT116 cells, implying that the inhibitory effect on colon cancer cell growth by miR-502 may be partially due to the interrupted autophagy. We also demonstrated that miR-502 suppressed p53 mediated autophagy via a negative feedback mechanism. miR-502 also suppressed p53 independent autophagy in an HCT116 (p53−/−) cell line. More than 50 percent of human colorectal cancer cases have mutant or deleted p53. However, nearly half of the human colon tumors contain wild type p53. It has been shown that wild type p53 promotes autophagy and provides a survival advantage to colon cancer cells under chronic exposure to nutrient deprivation (35). In our study, we showed that miR-502 reduced tumor cell survival by decreasing p53 levels in HCT116 (p53+/+) cells and inhibited autophagy flux in HCT116 (p53+/+). These results indicate that miR-502 has a tumor suppressive function by abolishing p53 dependent autophagy, which has been shown to prolong colon cancer cell survival (35). In addition, miR-502 also reduced autophagy in HCT116 (p53−/−) cells, suggesting a broader impact on autophagy to suppress tumor growth. We confirmed Rab1B as one of the direct targets for miR-502, and it has been reported to participate in autophagy by regulating autophagosome formation in Hela cells (38). Another target of miR-502 is AP2B1 (Supplementary Table 1), which was reported to regulate vesicle trafficking during autophagy (51).
In addition to autophagy, we found overexpression of miR-502 induced cell cycle arrest both at G1 and G2 checkpoints, which was more prominent in HCT116 cells with wild type p53. Activation of p53 and p21 was required for cell cycle control (52, 53). However, miR-502 mediated cell cycle arrest was independent of p53 and p21, as both of them were reduced by miR-502. These results suggest that other cell cycle control genes were involved in this process. We investigated the effects of suppression of Rab1B on cell cycle control and found that inhibition of Rab1B led to cell cycle arrest at the G2 checkpoint in the absence of p53, suggesting the inhibitory function of miR-502 at the G2 checkpoint was dependent, in part, on Rab1B. Another target candidate for cell cycle arrest is DHODH, which has been shown to promote melanoma cell growth in vitro and in mouse xenografts (43, 54). The DHODH inhibitor, leflunomide, can induce cell cycle arrest at the G1 checkpoint by regulating expression levels of cyclin D2 and pRb (43). We confirmed that DHODH was a direct target of miR-502 as overexpression of miR-502 led to a down-regulation of DHODH (Supplementary Figure 1). The suppression of DHODH may contribute to the cell cycle arrest at the G1 checkpoint by miR-502. Taken together, our findings show that miR-502 has a broad impact on cell cycle regulation through multiple targets including Rab1B and DHODH.
The expression of miR-502 was reduced in colorectal tumor specimens compared to paired normal controls, suggesting a clinical relevance of miR-502 in colorectal cancer development. In addition to Rab1B, we have identified other potential miR-502 mediated targets and pathways. Many of these (e.g. DHODH) contribute to tumor growth and cell cycle control (Supplementary Table 1), suggesting miR-502 as a broad suppressive miRNA for colon tumor growth. We reason that miR-502 may have potential as a novel therapeutic strategy for treating colorectal cancer patients, as miRNAs and their inhibitors have been tested in multiple cancer animal models based on their intrinsic tumor-suppressive and oncogenic functions. For example, miR-143 and miR-145 have been shown to be down-regulated in colorectal cancer (55–57), and ectopic expression of miR-143 or miR-145 suppressed tumor cell proliferation and triggered apoptosis of tumor cells in vitro and in vivo (58–61). In the mouse colon cancer xenograft models, miR-145 delivered systemically or locally to the tumors led to decreased tumor burden (62). Similarly, we found that local delivery of miR-502 to colon cancer xenografts led to reduced tumor growth compared to negative controls. Consistent with our findings, leflunomide, the inhibitor of DHODH, is able to reduce the tumor burden, inhibit transcriptional elongation of genes essential for melanoma growth, and enhance the chemosensitivity of melanoma cells to chemotherapeutic drugs (43, 54). These results suggest that miR-502 can be a potential candidate for new therapeutic developments.
The relationship between miR-502 and p53 is also an important feature, as several recent studies demonstrated a clear significance of such a regulatory relationship between p53 and miRNAs in various cancer types (63–65). In 2006, we first systematically analyzed the miRNA profiles in colon cancer cell lines HCT116 with either wild type p53 or null-p53, and identified putative p53-binding sites in nearly 40% of miRNA promoter regions (e.g. miR-34s, miR-192, miR-194, miR-215, miR-200c, miR-26) (20). miR-34a, miR-192, miR-215 and miR-26a have been extensively studied in colon cancer with a positive feedback mechanism. These miRNAs are down-regulated in colorectal tumor tissue, and ectopic expression of these miRNAs induced cell cycle arrest and growth (20–24, 26–30). In contrast, in this study we discovered a novel negative feedback mechanism between the p53 and miR-502. Such negative regulatory mechanism has been recently documented that another transcription factor, c-myc, not only can activate certain miRNAs, but also suppress a number of miRNA expressions (66). Elevated levels of p53 decreased miR-502 level in HCT116 (p53+/+) cells, and overexpression of miR-502 decreased p53 levels, suggesting that p53 functioned as a transcriptional repressor for miR-502. As it has been well investigated that p53 can function as a transcriptional suppressor to down-regulate gene expression (17, 67, 68), we have also ruled out that the reduction of p53 by miR-502 was not due to the changes in MDM2 expression. The exact detailed mechanism remained to be fully investigated in future studies.
In summary, we identified a novel mechanism of autophagy mediated by miR-502 via suppressing the expression of Rab1B. miR-502 reduces autophagy in both p53 wild type and p53 null colon cancer cell lines, suggesting a broad impact on autophagy in colon cancer. miR-502 suppresses the colon tumor growth both in vitro and in vivo. It has been demonstrated that the suppression of autophagy will have a synergy with 5-FU treatment in colon cancer (48). As a result, miR-502 based therapeutic strategies may offer a new treatment option for treating colorectal cancer.
Materials and Methods
Cell lines and reagents
The human CRC cell lines HCT116 (p53+/+) and HCT116 (p53−/−) were kindly provided by Professor Bert Vogelstein (The Johns Hopkins University), and maintained in McCoy’s 5A medium (Gibco Laboratories). Another human colon cancer line SW480 (mutated p53) was purchased from the American Type Culture Collection (ATCC), and was maintained in L15 medium (Gibco Laboratories). All the media were supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich). Nutlin-3 was purchased from Sigma-Aldrich.
Clinical colorectal cancer samples
Twenty-three colorectal cancer specimens were selected from patients who underwent surgical resection of primary tumors at the Department of Visceral and Transplantation Surgery, University of Ulm, Germany. Patient consent forms were obtained from each patient according to institutional policies. Each patient sample contained a pair of snap-frozen specimens from normal colorectal mucosa and tumor. The expression level of miR-502, Rab1b and DHODH in the tumors was compared to that of the paired normal tissue.
miRNA and siRNA transfection
HCT116 cells and SW480 cells were plated in 6-well plates at 2×105 and 4×105 per well, respectively. Twenty-four hours after plating, 100 pmole of miR-502 precursor (Ambion) or siRNAs against Rab1B (Invitrogen) were transfected to the cells with oligofectamine (Invitrogen) following manufacture protocol. Negative miRNA (Ambion) or scramble siRNA (Invitrogen) was also transfected as negative controls.
Cell proliferation assay
Twenty-four hours after transfection, HCT116 and SW480 cells were seeded in 96-well plates at 2,000 or 4,000 cells per well, respectively. The cell proliferation assay was performed on days 1, 3 and 5 by incubating 10 µl WST-1 (Roche Applied Science) in the culture medium for one hour and reading the absorbance at 450 and 630 nm (27, 69). The cell proliferation rate was calculated by subtracting the absorbance at 450 nm from the obsorbance at 630 nm.
Cell cycle analysis
Thirty-six hours after transfection, cells were harvested and resuspended at 0.5 to 1×106 cells/ml in modified Krishan buffer with 0.02 mg/ml RNase H (Invitrogen) and 0.05 mg/ml propidium iodide (PI, Sigma-Aldrich) (70, 71). The samples were analyzed by flow cytometry and results were calculated with Modfit LT™ software.
RNA extraction and real-time quantitative RT-PCR (qRT-PCR) analysis of mRNA and miRNA
Total RNA, including miRNAs, was isolated from clinical specimens, mouse xenografts and cell lines 24 hours after transfection or treatment by using the TRIzol reagent (Invitrogen) following manufacturer protocol. All reagents for real-time qRT-PCR were ordered from Applied Biosystems. 1µg of RNA was used as a template for cDNA synthesis with high capacity cDNA synthesis kit and random primers. Then the cDNA templates were mixed with gene-specific primers for Rab1B, DHODH and internal control GAPDH and Taqman 2x universal PCR master mix. Applied Biosystems 7500 Real-Time PCR machine was used for q-RT-PCR and programmed as: 50°C, 2 minutes; 95°C, 10 minutes; 95°C, 15 seconds; 60°C, 1 minute and the latter three steps were repeated for 40 cycles. For quantification of miR-502, 10ng of RNA was used as a template and cDNA was synthesized with miRNA-specific primers. Similarly as mRNA qRT-PCR, the miR-502 level was analyzed with its specific primers and internal control RNU44. Fluorescent signals from each sample were collected at the endpoint of every cycle, and the expression level of genes and miR-502 was calculated by ΔCT values based on the internal controls, normalized to the control group and plotted as relative value (RQ).
Western blot
The protein samples were collected 48 hours after transfection with RIPA buffer (Sigma-Aldrich). Equal amounts of samples were loaded to SDS-PAGE, then transferred to polyvinylidene difluoride (PVDF) membrane. The membranes were blocked with 5% non-fat milk in TBS/0.5% Tween-20 (TBS-T) at room temperature for one hour, then probed with mouse anti-human p53 antibody (1:1000, DAKO), mouse anti-human p21 antibody (1:1000, Cell Signaling), mouse anti-human GAPDH antibody (1:1000, Santa Cruz), mouse anti-human α-tubulin antibody (1:1000, Millipore), rabbit anti-human LC3B antibody (1:1000, Cell Signaling), rabbit anti-human Rab1B antibody (1:500, Santa Cruz), rabbit anti-human p62 antibody (1:1000, Sigma Aldrich) or rabbit anti-human DHODH antibody (1:200, Santa Cruz) overnight at 4°C. Goat anti-rabbit and anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson Immunoresearch) were used at 1:5000. HRP activity was detected with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and visualized in an UVP Bioimaging system. Expression levels were quantified using ImageJ software, and normalized to loading controls.
Luciferase assay
The full length of 3’UTR of Rab1B was cloned from human genomic DNA and inserted into pMIR-REPORT plasmid (Invitrogen). Twenty-four hours before transfection, 1.5×104 cells were plated in a 96-well plate. 10 pmole of miR-502 or negative miRNA was transfected into cells together with 100ng of pMIR-REPORT-3UTRRab1B and 1ng of Renilla luciferase plasmid pRL-SV40 (Promega) by DharmaFect Duo (Dharmacon). Luciferase assay was performed 24 hours after transfection by the dual-luciferase reporter assay system (Promega). For each sample, firefly luciferase activity was normalized to Renilla luciferase activity and the inhibition of miR-502 on Rab1B 3’-UTR was normalized to the control miRNA.
The 2.5 Kb gene fragment upstream of miR-502 coding sequence was cloned from human genomic DNA and inserted into pGL3-promoter plasmid (Promega). Twenty-four hours before transfection, 1.5×104 cells were plated in a 96-well plate. 100ng of pGL3-promoter-miR502 and 1ng of pRL-SV40 was transfected by DharmaFect Duo. Twenty-four hours after transfection, cells were treated with 10µM nutlin-3 for an additional 24 hours. Luciferase assay was performed with the dual-luciferase reporter assay system.
Autophagy assay
For western blot analysis of autophagy, cells were treated with Earle's Balanced Salt Solution (EBSS) for two or 24 hours at 48 hours after transfection. Protein samples were then collected for western blot analysis of LC3B and p62. 100nM Bafilomycin A1 (BafA1, Santa Cruz) was used as the lysosomal inhibitor. The protein level of LC3B-II and p62 was measured and normalized to GAPDH level by Image J. The autophagy flux was calculated as the ratio between LC3B-II with BafA1 and without BafA1.
Tumor implantation
Ten-twelve week old NOD/SCID mice (Jackson Laboratories) were used for tumor implantation. All animal procedures were approved by the Stony Brook University Institutional Animal Care and Use Committee (IACUC). Mice were bred in-house under maximum isolation conditions on a 12:12 hour light:dark cycle with food ad libitum. The tumor implantation and miRNA injection protocol was modified from Trang et al (72). The mice were anesthetized by isoflurane inhalation. 2.5 × 106 HCT116 (wild type p53) cells resuspended in 100 µl PBS were injected into lower back areas of the mice. The tumor size was measured by a caliper as length × width2/2. When tumor sizes reach 100 – 150 mm3 at day 14 post-injection, the mice were randomly assigned into two groups. 6.25µg miR-502 precursor or negative miRNA (Ambion) complexed with 1.6µl siPORTAmine transfection reagent (Ambion) in 50 µl PBS was injected to the tumors every three days. The procedures was repeated three times. The mice were euthanized on day 24 post-injection by CO2 inhalation, and tumors were dissected out for future evaluation.
Statistical analysis
All experiments were repeated at least three times. All statistical analyses were performed with Graphpad Prism software. The statistical significance between two groups was determined using Student’s t-test (paired t-test for clinical samples, and unpaired t-test for all other samples). For comparison of more than two groups, one-way ANOVA followed by a Bonferroni-Dunn test was used. Data were expressed as mean ± standard error of the mean (SEM). The statistical significance is either described in figure legends, or indicated with asterisks (*). *=P <0.05; **=P<0.01; ***=P< 0.001.
Supplementary Material
Acknowledgements
We appreciate the critical review by Ms. Sonya R. Lorrain. We thank Dr. Stella E. Tsirka for technical support in animal experiments. This study was supported in part by Stony Brook University Translational Research Laboratory Start-up Fund (J. Ju), R01CA155019 (J. Ju) and R33CA147966 (J. Ju).
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Alwan A. World Health Organization. Disaster Med Public Health Prep. 2007;1:7–8. doi: 10.1097/DMP.0b013e3180676d32. [DOI] [PubMed] [Google Scholar]
- 2.Hegde SR, Sun W, Lynch JP. Systemic and targeted therapy for advanced colon cancer. Expert Rev Gastroenterol Hepatol. 2008;2:135–149. doi: 10.1586/17474124.2.1.135. [DOI] [PubMed] [Google Scholar]
- 3.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 4.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 5.Gunaratne PH, Creighton CJ, Watson M, Tennakoon JB. Large-scale integration of MicroRNA and gene expression data for identification of enriched microRNA-mRNA associations in biological systems. Methods Mol Biol. 2010;667:297–315. doi: 10.1007/978-1-60761-811-9_20. [DOI] [PubMed] [Google Scholar]
- 6.Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- 7.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Research. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 8.Ghodgaonkar MM, Shah RG, Kandan-Kulangara F, Affar EB, Qi HH, Wiemer E, et al. Abrogation of DNA vector-based RNAi during apoptosis in mammalian cells due to caspase-mediated cleavage and inactivation of Dicer-1. Cell Death Differ. 2009;16:858–868. doi: 10.1038/cdd.2009.15. [DOI] [PubMed] [Google Scholar]
- 9.Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2006;94:776–780. doi: 10.1038/sj.bjc.6603023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang F. Small RNAs in mammalian germline: Tiny for immortal. Differentiation. 79:141–146. doi: 10.1016/j.diff.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Navarro F, Lieberman J. Small RNAs guide hematopoietic cell differentiation and function. J Immunol. 184:5939–5947. doi: 10.4049/jimmunol.0902567. [DOI] [PubMed] [Google Scholar]
- 12.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67:7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 14.Scott N, Sagar P, Stewart J, Blair GE, Dixon MF, Quirke P. p53 in colorectal cancer: clinicopathological correlation and prognostic significance. Br J Cancer. 1991;63:317–319. doi: 10.1038/bjc.1991.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crawford LV, Pim DC, Lamb P. The cellular protein p53 in human tumours. Mol Biol Med. 1984;2:261–272. [PubMed] [Google Scholar]
- 16.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 17.Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, et al. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000;14:981–993. [PMC free article] [PubMed] [Google Scholar]
- 18.Miller SJ, Suthiphongchai T, Zambetti GP, Ewen ME. p53 binds selectively to the 5' untranslated region of cdk4, an RNA element necessary and sufficient for transforming growth factor beta- and p53-mediated translational inhibition of cdk4. Mol Cell Biol. 2000;20:8420–8431. doi: 10.1128/mcb.20.22.8420-8431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu L, Minden MD, Benchimol S. Translational regulation of human p53 gene expression. EMBO J. 1996;15:4392–4401. [PMC free article] [PubMed] [Google Scholar]
- 20.Xi Y, Shalgi R, Fodstad O, Pilpel Y, Ju J. Differentially regulated micro-RNAs and actively translated messenger RNA transcripts by tumor suppressor p53 in colon cancer. Clin Cancer Res. 2006;12:2014–2024. doi: 10.1158/1078-0432.CCR-05-1853. [DOI] [PubMed] [Google Scholar]
- 21.Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A. 2007;104:15472–15477. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 25.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song B, Wang Y, Kudo K, Gavin EJ, Xi Y, Ju J. miR-192 Regulates dihydrofolate reductase and cellular proliferation through the p53-microRNA circuit. Clin Cancer Res. 2008;14:8080–8086. doi: 10.1158/1078-0432.CCR-08-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song B, Wang Y, Titmus MA, Botchkina G, Formentini A, Kornmann M, et al. Molecular mechanism of chemoresistance by miR-215 in osteosarcoma and colon cancer cells. Mol Cancer. 2010;9:96. doi: 10.1186/1476-4598-9-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karaayvaz M, Pal T, Song B, Zhang C, Georgakopoulos P, Mehmood S, et al. Prognostic Significance of miR-215 in Colon Cancer. Clin Colorectal Cancer. 2011 doi: 10.1016/j.clcc.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braun CJ, Zhang X, Savelyeva I, Wolff S, Moll UM, Schepeler T, et al. p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res. 2008;68:10094–10104. doi: 10.1158/0008-5472.CAN-08-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Georges SA, Biery MC, Kim SY, Schelter JM, Guo J, Chang AN, et al. Coordinated regulation of cell cycle transcripts by p53-Inducible microRNAs, miR-192 and miR-215. Cancer Res. 2008;68:10105–10112. doi: 10.1158/0008-5472.CAN-08-1846. [DOI] [PubMed] [Google Scholar]
- 31.Xi Y, Formentini A, Chien M, Weir DB, Russo JJ, Ju J, et al. Prognostic Values of microRNAs in Colorectal Cancer. Biomark Insights. 2006;2:113–121. [PMC free article] [PubMed] [Google Scholar]
- 32.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tasdemir E, Maiuri MC, Orhon I, Kepp O, Morselli E, Criollo A, et al. p53 represses autophagy in a cell cycle-dependent fashion. Cell Cycle. 2008;7:3006–3011. doi: 10.4161/cc.7.19.6702. [DOI] [PubMed] [Google Scholar]
- 34.Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M, et al. A dual role of p53 in the control of autophagy. Autophagy. 2008;4:810–814. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- 35.Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A. 2010;107:18511–18516. doi: 10.1073/pnas.1006124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis. 2009;14:376–391. doi: 10.1007/s10495-008-0307-5. [DOI] [PubMed] [Google Scholar]
- 37.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zoppino FC, Militello RD, Slavin I, Alvarez C, Colombo MI. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic. 2010;11:1246–1261. doi: 10.1111/j.1600-0854.2010.01086.x. [DOI] [PubMed] [Google Scholar]
- 39.Plutner H, Cox AD, Pind S, Khosravi-Far R, Bourne JR, Schwaninger R, et al. Rab1b regulates vesicular transport between the endoplasmic reticulum and successive Golgi compartments. J Cell Biol. 1991;115:31–43. doi: 10.1083/jcb.115.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 41.He H, Dai F, Yu L, She X, Zhao Y, Jiang J, et al. Identification and characterization of nine novel human small GTPases showing variable expressions in liver cancer tissues. Gene Expr. 2002;10:231–242. doi: 10.3727/000000002783992406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, et al. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem. 2004;279:53015–53022. doi: 10.1074/jbc.M410233200. [DOI] [PubMed] [Google Scholar]
- 43.Baumann P, Mandl-Weber S, Volkl A, Adam C, Bumeder I, Oduncu F, et al. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Mol Cancer Ther. 2009;8:366–375. doi: 10.1158/1535-7163.MCT-08-0664. [DOI] [PubMed] [Google Scholar]
- 44.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 48.Li J, Hou N, Faried A, Tsutsumi S, Kuwano H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur J Cancer. 2010;46:1900–1909. doi: 10.1016/j.ejca.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 49.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 51.Hirst J, Bright NA, Rous B, Robinson MS. Characterization of a fourth adaptor-related protein complex. Mol Biol Cell. 1999;10:2787–2802. doi: 10.1091/mbc.10.8.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010;22:1003–1012. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471:518–522. doi: 10.1038/nature09882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akao Y, Nakagawa Y, Naoe T. MicroRNA-143 and-145 in colon cancer. DNA Cell Biol. 2007;26:311–320. doi: 10.1089/dna.2006.0550. [DOI] [PubMed] [Google Scholar]
- 56.Michael MZ, SM OC, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–891. [PubMed] [Google Scholar]
- 57.Slaby O, Svoboda M, Fabian P, Smerdova T, Knoflickova D, Bednarikova M, et al. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology. 2007;72:397–402. doi: 10.1159/000113489. [DOI] [PubMed] [Google Scholar]
- 58.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi B, Sepp-Lorenzino L, Prisco M, Linsley P, deAngelis T, Baserga R. Micro RNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. J Biol Chem. 2007;282:32582–32590. doi: 10.1074/jbc.M702806200. [DOI] [PubMed] [Google Scholar]
- 60.Akao Y, Nakagawa Y, Iio A, Naoe T. Role of microRNA-143 in Fas-mediated apoptosis in human T-cell leukemia Jurkat cells. Leuk Res. 2009;33:1530–1538. doi: 10.1016/j.leukres.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 61.Akao Y, Nakagawa Y, Hirata I, Iio A, Itoh T, Kojima K, et al. Role of anti-oncomirs miR-143 and-145 in human colorectal tumors. Cancer Gene Ther. 2010;17:398–408. doi: 10.1038/cgt.2009.88. [DOI] [PubMed] [Google Scholar]
- 62.Ibrahim AF, Weirauch U, Thomas M, Grunweller A, Hartmann RK, Aigner A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011 doi: 10.1158/0008-5472.CAN-10-4645. [DOI] [PubMed] [Google Scholar]
- 63.Shin S, Lee EM, Cha HJ, Bae S, Jung JH, Lee SM, et al. MicroRNAs that respond to histone deacetylase inhibitor SAHA and p53 in HCT116 human colon carcinoma cells. Int J Oncol. 2009;35:1343–1352. doi: 10.3892/ijo_00000452. [DOI] [PubMed] [Google Scholar]
- 64.Shin S, Cha HJ, Lee EM, Jung JH, Lee SJ, Park IC, et al. MicroRNAs are significantly influenced by p53 and radiation in HCT116 human colon carcinoma cells. Int J Oncol. 2009;34:1645–1652. doi: 10.3892/ijo_00000295. [DOI] [PubMed] [Google Scholar]
- 65.Zhou J, Zhou Y, Yin B, Hao W, Zhao L, Ju W, et al. 5-Fluorouracil and oxaliplatin modify the expression profiles of microRNAs in human colon cancer cells in vitro. Oncol Rep. 2010;23:121–128. [PubMed] [Google Scholar]
- 66.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang X, Milyavsky M, Shats I, Erez N, Goldfinger N, Rotter V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene. 2004;23:5759–5769. doi: 10.1038/sj.onc.1207706. [DOI] [PubMed] [Google Scholar]
- 68.Wang X, Wu X, Wang C, Zhang W, Ouyang Y, Yu Y, et al. Transcriptional suppression of breast cancer resistance protein (BCRP) by wild-type p53 through the NF-kappaB pathway in MCF-7 cells. FEBS Lett. 2010;584:3392–3397. doi: 10.1016/j.febslet.2010.06.033. [DOI] [PubMed] [Google Scholar]
- 69.Song B, Wang Y, Xi Y, Kudo K, Bruheim S, Botchkina GI, et al. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene. 2009;28:4065–4074. doi: 10.1038/onc.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krishan A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol. 1975;66:188–193. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dressler LG, Seamer LC, Owens MA, Clark GM, McGuire WL. DNA flow cytometry and prognostic factors in 1331 frozen breast cancer specimens. Cancer. 1988;61:420–427. doi: 10.1002/1097-0142(19880201)61:3<420::aid-cncr2820610303>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 72.Trang P, Medina PP, Wiggins JF, Ruffino L, Kelnar K, Omotola M, et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2010;29:1580–1587. doi: 10.1038/onc.2009.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.