

Abstract
Understanding the origins of normal and pathological behavior is one of the most exciting opportunities in contemporary biomedical research. There is increasing evidence that, in addition to DNA sequence and the environment, epigenetic modifications of DNA and histone proteins may contribute to complex phenotypes. Inherited and/or acquired epigenetic factors are partially stable and have regulatory roles in numerous genomic activities, thus making epigenetics a promising research path in etiological studies of psychiatric disease. In this article, we review recent epigenetic studies examining the brain and other tissues, including those from individuals affected with schizophrenia and bipolar disorder. We also highlight heuristic aspects of the epigenetic theory of psychiatric disease and discuss the future directions of psychiatric epigenetics.
Keywords: Epigenetics, DNA methylation, histone modifications, schizophrenia, bipolar disorder, epigenome-wide association study
Psychiatry meets epigenetics
Psychiatric disorders are characterized by severely debilitating behavioral abnormalities that often persist over a life time and are resistant to medical or psychotherapeutic interventions. Here we focus on two major psychiatric diseases – schizophrenia (SCZ) and bipolar disorder (BPD) - that have received the greatest attention in molecular biological studies. SCZ and BPD are severe forms of mental illness each causing 1% of global disability [1]. SCZ is characterized by delusions, disturbances in reasoning, hallucinations and withdrawal from social activity. The disease usually affects people in late adolescence or early twenties. BPD is characterized by recurrent mania or hypomania and depressive episodes that impair overall function and quality of life. Patients with SCZ and BPD, diseases collectively called major psychosis, have a suicide risk that is significantly higher than those with any other illness [1]. The net result of major psychosis is significant distress, disability and societal burden.
Both SCZ and BPD are complex diseases that, like cancer or diabetes, aggregate in families but do not segregate in a mendelian manner. It has generally been assumed that psychiatric diseases are caused by combinations of genetic (DNA) risk polymorphisms or mutations that interact with hazardous environmental factors [2]. Over the past three decades, numerous genetic association and linkage studies have been performed to understand major psychosis, but they have resulted in poor replication and risk alleles with small effect sizes. Difficulties in identifying candidate genes implicated in psychiatric disease have traditionally been explained by locus- and allelic- heterogeneity, imprecisely defined clinical phenotypes, epistasis and the interaction of genes with non-genetic factors [3]. Though often stated, the contributions of these potential confounding factors have not yet been proven. The history of epidemiological studies is even longer than the molecular genetic one. In general, epidemiological studies are confounded by complex cause and effect relationships, the unclear mechanism by which non-shared environmental factors mediate disease risk and an inability to reconcile the “heritable” component embedded within what appears to be an environmental domain [4]. Althoguh pre-/peri-natal complications, socioeconomic status, urban living and negative life events have sometimes demonstrated correlation with major psychosis, none have led to the identification of specific causal risk factors [5]. Thus, despite significant effort, limited progress has been made in understanding the genetic and environmental basis of SCZ and BPD. In this article, we propose that epigenetic mechanisms may operate in the etiopathogenesis of major psychosis, with a contribution that may extend and potentially exceed that of the DNA sequence and environmental factors.
Epigenetic mechanisms are a heritable and dynamic means of regulating various genomic functions, including gene expression, through covalent modifications of DNA and histones. Although DNA and histone modifications are often investigated separately, crosstalk exists between the different types of marks, thus allowing epigenetic modifications to act in concert [6]. In contrast to the DNA sequence, the epigenome shows considerable heterogeneity within the tissues of an organism, differing even between brain regions [7], and acquires changes in response to environmental cues [8]. Research on the temporal and spatial dynamics of epigenetic regulation has recently gained momentum (Box 1).
Box 1. Temporal and spatial dimensions of the dynamic epigenome.
To coordinate changes in transcriptional regimes through the life cycle of an organism, epigenetic changes must be temporally dynamic. Organismal age is one axis where epigenomic dynamics are most evident. In population studies of human disease, cases and controls must be matched for age. From the epigenetic point of view, “age” is a complex characteristic that, for the lack of more specific information, serves as a proxy for effects of developmental programs, changes in hormonal milieu, environmental events, cellular senescence and stochasticity, among other factors. The overall density of modified cytosines decreases with age, an effect observed at the level of the epigenome in white blood cells in adults [79]. However, the relationship of DNA methylation with age is not strictly linear, as children exhibit a threefold to fourfold higher rate of methylation change compared with adults [80]. Similarly, in the prefrontal cortex of the brain, the methylome is most dynamic during embryogenesis, with gradual stabilization after birth and during aging [81]. Some [82], but not all [83], twin studies have found an age-dependent divergence in twin epigenomes; older monozygotic twins exhibited greater differences in overall density and in genomic distribution of DNA methylation and histone acetylation compared with younger twins. There is also evidence for an age-dependent increase in variability at individual loci (or age-specific heteroscedasticity) (Oh and Petronis, unpublished).
Bidirectional or cyclical epigenetic changes regulating transcriptional regimes have also been detected. Strand-specific cycling in methylation/demethylation paralleled the transcriptional cycling of the pS2/TFF1 gene promoter upon activation by estrogens [84]. Addition and removal of methyl groups from DNA also affected the sequential recruitment of chromatin-remodeling proteins involved in cycles of transcriptional activation [84]. Epigenetic machinery also modulates transcriptional programs in response to circadian cues [85].
The spatial organization of the epigenome is another area of recent interest. The genome contains specific regions with dense DNA methylation, which impact the three-dimensional nuclear organization of the genome as well as nucleosomal structure [86, 87]. Separately, the epigenome is likely itself organized into multi-locus functional “modules”, similar to the transcriptional networks it regulates. A recent study found fragmentation in the network architecture of the methylome from brains of patients diagnosed with psychosis, relative to that from control brains [88]. Although the precise implications of these changes are unclear, one can speculate that changes in modular network structures may be related to disturbances in distinct areas of high epigenetic activity in the nucleus.
The rationale for epigenetic and epigenomic explorations into psychiatric diseases is based on two sets of findings. First, rapidly growing evidence from basic research indicates that epigenetic regulation underlies normal cognition, and that cognitive dysfunction occurs upon epigenetic misregulation. Second, epigenetics research is providing new insights into heritable and non-heritable components of complex psychiatric disease. Putative epigenetic misregulation is consistent with the various clinical and epidemiological features of psychiatric diseases, such as discordance of identical twins, sex- and parent-of-origin effects, coincidence between disease onset and the time of major hormonal changes in the organism and major fluctuations in clinical course. Below, we discuss how epigenetics influences brain function and describe the general features of epigenetics relevant to psychiatric disease.
Epigenetics is a new frontier in neurobiology
Epigenetic factors can influence genomic activities in the brain to produce long-term changes in synaptic signaling, organization and morphology, which in turn underlie cognitive function [9]. For example, the phosphorylated form of MeCP2, an MBD protein that binds to methylated DNA and regulates transcription, binds broadly throughout the genome, affecting chromatin state, dendritic and synaptic development and hippocampus-dependent memory [10, 11]. In mice, hippocampal neuronal activity induces active DNA demethylation or de novo methylation [12], and targeted knockouts of DNA de novo methyltransferases cause learning and memory impairments [13]. DNA methylation has also been implicated in the maintenance of long-term memories, as pharmacological inhibition of DNA methylation abolishes remote memories [9, 14]. Finally, the recent rediscovery of DNA hydroxymethylation may uncover epigenetic mechanisms unique to brain function (Box 2). These findings indicate the importance of covalent DNA modifications in mediating synaptic plasticity and cognitive functions, both of which are disturbed in psychiatric illness.
Box 2. The many flavors of DNA modification.
Covalent modifications of DNA occur at cytosine residues, typically in the context of CG dinucleotides, although a recent study has shown an abundance of non-CG modifications in the adult mouse brain [89]. In addition to the well-known form of mammalian DNA modification, methylated cytosine (5-mC), a second modification, hydroxymethylation (5-hmC) of the CpG dinucleotide, was recently rediscovered [90, 91]. 5-hmC was first detected in the rat brain 40 years ago [92], but did not attract any interest for decades. 5-hmC studies have been accompanied by the recognition that this modification has a different genomic distribution than that of 5-mC [93]. The role of 5-hmC in brain function is a topic of active research. This base accounts for about 40% of modified cytosines in neurons, increases in the brain with postnatal age and is produced in response to neuronal activity [94]. 5-hmC is an intermediate in active DNA demethylation, a phenomenon that, while long suspected, is only recently being mechanistically understood [94]. Active demethylation occurs in a number of contexts, including learning and memory [13]. Further studies will determine if there are additional regulatory functions of 5-hmC that are independent from that of 5-mC. The rediscovery of 5-hmC has created an opportunity to further resolve the epigenomic landscape in various cellular contexts, and identify the respective contributions of each modification to neuronal function and cognitive processes [9]. In addition to 5-mC and 5-hmC, the Tet (ten eleven translocation) proteins can catalyze 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC) from 5-mC; however, the density of these modifications is much smaller than those of 5-mC and 5-hmC [95].
Changes in histone modifications can also influence long-term memory formation by altering chromatin accessibility and the transcription of genes relevant to learning and memory. Memory formation and the associated enhancements in synaptic transmission are accompanied by increases in histone acetylation, which promote an active chromatin state [15]. Some types of histone methylation are also required for normal cognitive function [16]. Conversely, a neuronal increase in histone deacetylase activity, which promotes chromatin compaction, results in reduced synaptic plasticity and impairs memory [17]. Pharmacological inhibition of histone deacetylases augments memory formation [17, 18], further suggesting that histone (de)acetylation regulates this process. Mutations in CBP, a transcriptional regulator with histone acetyltransferase activity, produce cognitive disturbances related to Rubinstein-Taybi syndrome, a disease characterized by short stature, mental retardation and distinct physical anomalies [19]. Overall, these studies demonstrate that misregulation of epigenetic modifications and of their regulatory enzymes is capable of orchestrating prominent deficits in neuronal plasticity and cognitive function, abnormalities relevant to many psychiatric disorders.
Epigenetic perspective on the “missing” heritability, ephemeral environment and non-mendelian features of major psychosis
The second group of arguments supporting epigenetic applications in psychiatric research – and in other complex diseases – is a potential reinterpretation of the “DNA + environment” paradigm of disease causation. Twin and family studies have demonstrated a genetic influence in all psychiatric diseases, with heritability reaching 80% in SCZ and BPD [20]. Despite high heritability estimates, common genetic risk factors mapped in genome-wide association studies (GWAS) [21] and rare DNA mutations, including copy-number variants (CNVs) [22, 23], have explained only a small fraction of the total inherited risk of major psychosis. The term ‘missing heritability’ refers to the discrepancy between epidemiological heritability estimates and the proportion of phenotypic variation explained by DNA sequence differences [24].
The concept of missing heritability has been discussed in over a hundred articles, and several theories have been used to explain the small risk effects detected in GWAS. It has been suggested that the missing heritability is related to large numbers of rare variants, or that the heritability is not missing, but is “hidden” across linkage disequilibrium blocks, each containing numerous weakly-contributing genetic risk factors [25]. Another possibility is that some additive heritability estimates may be inflated by epistasis and gene-environment interactions, detection of which in GWAS is not trivial, resulting in “phantom” heritability [26]. Such models, though scientifically sound, would require studies with hundreds of thousands of samples, and the translational benefit of this knowledge is not obvious. In major psychosis, the unaccounted heritability is so large that one is tempted to believe that other models and mechanisms of disease heritability may be involved. In this regard, epigenetic heritability offers an interesting opportunity.
Traditionally, “epi-genetic” has been perceived as DNA- and histone-modification marks that were faithfully transferred across a limited number of mitotic divisions in differentiated somatic cells, but that were erased and reset between generations. Transmission of parent-specific epigenetic signatures was thought to be impossible due to reprogramming during gametogenesis and early development. However, transgenerational inheritance of natural- and pharmacologically-induced epimutations has been well documented in animal models [reviewed in 27], indicating that some epigenetic marks can indeed survive the two reprogramming events. Studies in twins have found that monozygotic twins (derived from the same zygote) are epigenetically more similar than dizygotic twins (derived from two different zygotes) [28, 29], which is consistent with the inheritance of epigenetic marks from gametes. In this view, epigenetic contributions to heritability in twins would be mediated by a partial epigenetic stability in somatic cells of a single generation, while epigenetic contributions to transgenerational heritability would additionally require epigenetic stability during gametogenesis. Thus, transgenerational- and twin-based epigenetic inheritance may account for some of the missing heritability.
Separately, epigenetics may shed new light on the non-heritable (i.e. environmental) factors contributing to SCZ and BPD. There is increasing experimental evidence that environmental agents, including dietary factors, environmental chemicals and rearing environment, can alter the epigenetic status of specific genes and genomes [reviewed in 8, 30]. Therefore, in principle, epigenetic approaches can measure the effects of the environment on a molecular level. Epigenetic marks also exhibit stochastic mitotic instability, which are epigenetic changes that are not induced by measureable environmental stimuli [31, 32]. Stochastically-induced epigenetic misregulation has the potential to affect disease susceptibility, an effect that would have traditionally been misattributed to environmental contribution [5].
From the epigenetic point of view, “inherited” and “acquired” marks need not be independent in origin, as they are in the traditional “genes + environment” paradigm. Environmental factors may impact epigenetic regulation and thereby become heritable, an idea that provides new ways to conceptualize and conduct molecular analyses of gene-environment interactions [reviewed in 27].
Finally, epigenetic misregulation is also consistent with various epidemiological, clinical and molecular puzzles in psychiatric diseases, including discordance of identical twins, sexual dimorphism in incidence and severity, parent-of-origin effects, fluctuating clinical course, decline of clinical symptoms in aging SCZ and BPD patients and the non-decreasing incidence of SCZ despite the significantly-reduced reproductive fitness of the affected individuals [33, 34].
In summary, although there is no direct evidence to date for the causal role of epigenetic factors in psychiatric disease, numerous observations support the heuristic potential of the epigenetic theory of major psychiatric disease as a viable alternative to the existing research program in complex non-mendelian genetics and biology. These arguments warrant a dedicated search for epigenetic risk factors in psychiatric disease.
The short (half a decade) history of epigenetic studies of SCZ and BPD
Both SCZ and BPD have been examined for disease-associated changes in DNA methylation [35]. Initial studies investigated methylation in candidate genes such as reelin [36, 37], sex-determining region Y (SRY)-box 10 [38], forkhead box P2 [39], and serotonin receptor 1A [40], using postmortem brains and peripheral blood samples. However, the findings from these studies were not always replicated [41]. The first epigenome-wide study characterizing DNA methylation in major psychosis surveyed 12,000 GC-rich regions, including CpG islands, in the prefrontal cortex of the brain [42]. This study identified several dozen sites with DNA methylation alterations, some of which were sex-specific. Loci with significant epigenetic differences between affected individuals and controls contained genes involved in brain development and neurotransmitter pathways, which had previously been associated with major psychosis [42]. One of the identified genes, the HLA complex group 9 gene (HCG9), maps to chromosome 6p21.33, which also exhibited an association with psychosis in GWAS [21]. DNA methylation patterns in HCG9 were separately analyzed in approximately 1400 DNA samples from major psychosis patients and controls using bisulfite conversion coupled with pyrosequencing [43]. BPD patients exhibited a lower degree of HCG9 methylation in several tissues, including in the post-mortem prefrontal cortex and sperm cells from living individuals [43]. The DNA methylation change in HCG9 across many tissues suggests the occurrence of an epimutation that is inherited or acquired before tissue differentiation in embryogenesis.
One powerful experimental design in epigenetic studies is the comparison of discordant monozygotic (MZ) twins that are naturally matched for DNA sequence and demographic variables. To date, the most comprehensive twin study examined blood samples from 22 twin pairs discordant for SCZ or BPD and interrogated over 27,000 CpG dinucleotides using microarrays [44]. Among all genes with disease-associated changes, those involved in neuronal development and pathways implicated in neurological disease were statistically overrepresented. Another study examined X-chromosome inactivation patterns in blood and/or buccal cells from 63 female MZ twin pairs concordant or discordant for BPD or SCZ, alongside healthy twin controls [45]. Discordant BPD twins showed greater differences in the methylation of maternal and paternal X-alleles compared to concordant twin pairs [45]. This result suggests that differential skewing of X-chromosome inactivation may contribute to MZ twin discordance in BPD, further supporting a role for genes on the X chromosome in mood disorders.
Perturbations of DNA methylation in major psychosis may also result from the abnormal activity of DNA methyltransferases (DNMT) or changes in the levels of methyl-group donors and co-factors affecting DNA methylation. DNMT genes have been reported to be upregulated in the prefrontal cortex of SCZ and BPD patients [46, 47]. In addition, several studies have detected differences in the amounts of S-adenosyl methionine (SAM), a methyl donor, and other molecules part of the one-carbon metabolism cycle (i.e. homocysteine, folate) in patients with major psychosis [48, 49].
Differences in patterns of histone modifications have also been found in major psychosis. While histone modifications are less frequently studied in the post-mortem human brain, owing to postmortem autolysis of the marks, histone methylation may be less vulnerable to this phenomenon [50]. A study measuring levels of histone 3 lysine 4 trimethylation (H3K4me3), a mark associated with an open chromatin state and active transcription, found reduced levels of this mark at the glutamate decarboxylase 1 (GAD1) locus in the female SCZ prefrontal cortex [51]. The GAD1 protein synthesizes the neurotransmitter GABA; dysregulation of GABAergic neurotransmission has been implicated in the pathophysiology of SCZ, and reduced expression of GAD1 has frequently been observed in the post-mortem prefrontal cortex and hippocampus of affected individuals [52]. Accordingly, decreases in H3K4me3 were accompanied by deficits in GAD1 mRNA expression in the prefrontal cortex of SCZ patients [51]. A different study that measured several histone marks, including methylation, acetylation and phosphorylation, in the prefrontal cortex of patients with SCZ, did not find any overall significant differences, although a subset of eight schizophrenia patients had a 30% increase in the histone methylation mark H3-(methyl)arginine 17 (H3meR17), a sign of closed chromatin, in comparison to control subjects [53]. Elevations in the repressive chromatin mark H3K9me2 (histone 3 lysine 9 dimethylation) have also been observed in lymphocytes of living schizophrenia patients, and higher levels of H3K9me2 were correlated with an earlier age of disease onset [54]. Furthermore, enzymes that catalyze post-translational histone modifications may be dysregulated in major psychosis. Overexpression of histone deacetylase 1 (HDAC1) has been reported in the post-mortem prefrontal cortex of patients with SCZ [55]. Inverse correlations between the expression of GAD1 and HDAC1, -3 and -4 have also been observed in the brain of individuals with major psychosis [55, 56]. Together, these studies suggest that disturbances in histone regulatory enzymes and histone patterns in the brain mediate transcriptional changes that may contribute to the risk for psychotic disorders.
Epigenetic misregulation has also been associated with depression and drug addiction, which are common co-morbid conditions with SCZ and BPD [57, 58]. Changes in the expression of the de novo DNA methyltransferase gene DNMT3a in the brain reward system of adult mice regulated addictive behaviors and depressive-like phenotypes [59]. Histone deacetylases and the histone methyltransferase G9a also modulated preferences for addictive substances, stress vulnerability and depression-like behaviors in adult animals [60–62]. Chronic administration of the widely-used anti-depressant imipramine reversed many of the histone methylation and gene expression abnormalities induced by defeat stress [63], and increased histone acetylation at brain-derived neurotrophic factor (BDNF), a gene important in mediating depressive responses following stress [64].
Finally, a number of psychotropic medications have been shown to produce epigenetic changes in the brain (Box 3), although it is still unclear if the therapeutic effects are a direct consequence of these epigenetic alterations. Together, all the above studies underscore the widespread impact of and prominent role for epigenetic factors in a number of psychiatric diseases.
Box 3. The tools of epigenomic profiling.
Epigenomic assays involve the selective enrichment of DNA containing the epigenetic mark(s) in question, followed by the position-specific quantification of enriched sequences. Modifications are enriched using DNA methylation-sensitive restriction enzymes, antibodies or chemical approaches. Two popular methods for epigenomic screens are microarrays and direct DNA sequencing. Sequencing offers the advantage of single base pair resolution and, unlike most arrays, is not limited to certain genomic loci (probes). This method is being successfully used to generate “reference epigenomic atlases” for individual cell types [96], including the full human DNA methylome [97]. However, microarrays still offer some advantages for mapping disease epimutations. Relative to sequencing, the library preparation and computational costs of arrays are still significantly lower, making the latter cost-effective for populational studies, which require hundreds (if not thousands) of samples. The cost of sequencing-based approaches is directly proportional to the fold coverage, which may differ dramatically for each specific type of epigenetic modification. The rule is to increase the number of unique reads covering a particular region until saturation occurs, that is when further sequencing fails to discover additional regions (“peaks”) above background [98]. In histone modification studies, the general recommendation for sequencing depth is 20 million aligned reads per replicate, at a 36 base read length (http://www.roadmapepigenomics.org/), but lower sequencing depths may be sufficient for more localized, rare histone modifications (e.g. H3K4me2, H3K4me3) [99]. In bisulfite-based DNA modification studies, the peak criterion does not apply due to the abundance of these modifications and required sequencing depth may be as high as >50 reads on average per locus [100] to account for the large degree of intra- and inter-individual variation of DNA modification. To offset the costs of next-generation sequencing, one strategy has been to interrogate a subset of the epigenome (i.e. reduced representation bisulfite sequencing or enrichment by antibody). In general, microarray and direct sequencing methods each presents a tradeoff in cost, genome coverage and accuracy. Given the rapidly falling costs of direct sequencing, this method will likely replace microarrays as the main assay for population-based epigenomic profiling in the next five years.
Future directions and challenges for identification of epimutations in major psychosis
As discussed above, epigenetics and epigenetic models of disease causation have numerous characteristics that distinguish them from genetic studies, and accordingly, the up-and-coming wave of epigenome-wide association studies (or “EWAS”) is being accompanied by thoughts on theoretical and experimental considerations for these studies [65]. Here we focus on epigenomic research strategies that are directly relevant to the brain.
The instability of the epigenetic code is a double-edged sword, in that the very diversity and changeability of the mechanisms that could explain facets of disease etiology presents an experimental challenge for their identification. Epimutations may exhibit variations in their genomic span and distribution, the types of epigenetic modifications affected and the number of tissues carrying the epimutation. This variability will influence the considerations of sample size, sample type and epigenomic assays required to reliably identify disease-associated epimutations (Figure 1). A hypothetical example of “low-hanging fruit” is an inherited long-ranging epimutation that affects both DNA and histones; detection of such an epimutation would require relatively few samples, lower-resolution mapping techniques and easily-accessible human tissues, such as blood. Such broad somatic epimutations have been detected in cancers; gene silencing of a 4-Mb band of chromosome 2q.14.2 in colorectal cancer was related to three regions with DNA hypermethylation, the largest of which spanned nearly 1Mb [66]. By contrast, a “point epimutation”, i.e. an epigenetic change occurring in a limited genomic region and tissue/cell type, may not be identified without assaying hundreds of diseased tissue samples, and still may be difficult to detect without a priori knowledge (i.e. of the affected brain region) to help narrow the search. The sheer complexity of the epigenome may limit the success of certain experimental designs and techniques, particularly in the detection of small “private” epimutations (epigenetic changes in very short regions and mapping to different genes in different patients).
Figure 1. Features of epimutations likely vary in complexity, necessitating sophisticated experimental designs and possibly explaining why some designs fail.
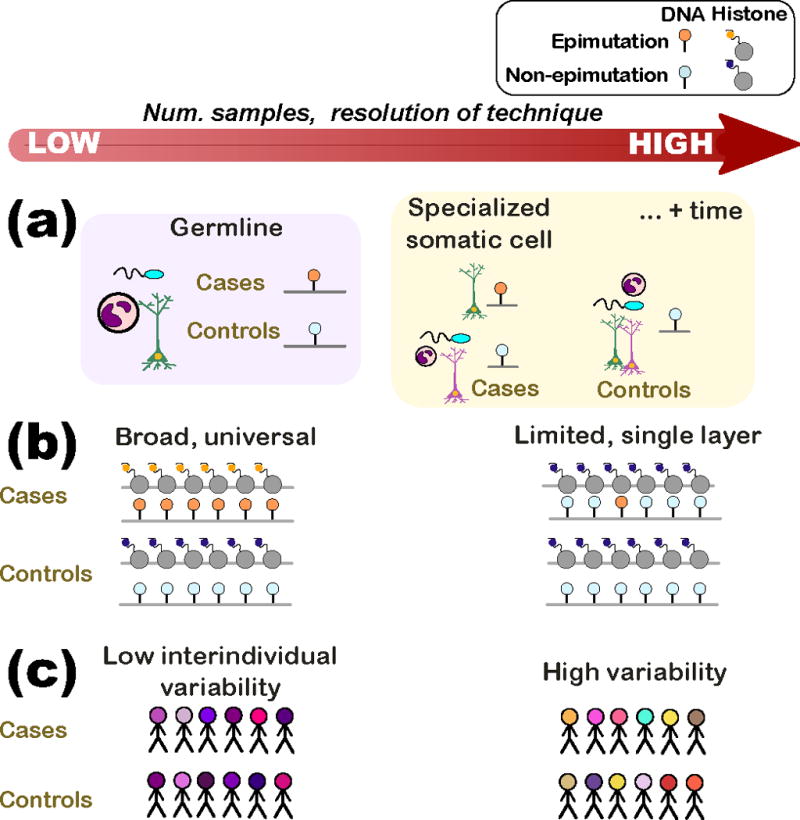
(a) An epimutation may be inherited (germline) or may be acquired during the individual’s lifetime. In the former case (purple box), an epimutation would be consistently present in all cell types of the affected individuals. In the latter (orange box), the epimutation may only be found in particular cell types of the affected individual or, still more complex, in a specialized cell type following a particular developmental stage (e.g. after puberty).
(b) Epimutations may vary in the stretch of epigenome affected and in the number of epigenetic layers where the mutation is evident. (Left) An epimutation that spans a large stretch of the epigenome (similar to a translocation) and multiple epigenetic layers would be detected by a low-resolution technique and several assays. (Right) At the other extreme, an epimutation that affects a single locus (similar to a point mutation) in a single layer would require single base-pair resolution techniques and would not be found in assays investigating other epigenetic modifications.
(c) The interindividual variability in the epigenome surrounding the epimutation also affects sample size and technique resolution. (Left) Regions of the epigenome that have low interindividual variability (e.g. conserved regulatory domains) require fewer samples than in cases where the epimutation is in a region of high variability (i.e. regulatory region affected by lifestyle choices). There is currently little understanding of the population variability of the epigenome.
A different consideration in experimental design is the resolution of causal versus correlative epimutations. The majority of reports that associate epigenetic abnormalities with psychiatric disease do not ascertain if the association is causal, or if it is a consequence of disease, its treatment or other disease-related confounds (i.e. recreational drug use). Several approaches may help differentiate causal and non-causal associations. First, an epimutation identified in multiple tissues of affected individuals – including the brain – would favor (but not prove) a causal relationship. However, causality cannot be rejected if brain epimutations are not detected in non-brain tissues. Causality may be further supported if disease-specific epimutations are detected in the germline (sperm) of affected individuals or of their fathers. Second, DNA- and histone-modification differences between psychiatric patients and controls can be further analyzed prospectively in the offspring born to affected parents. About 10% of such offspring will develop SCZ or BPD when they reach the critical age of susceptibility [67], suggesting that they likely carry inherited disease risk factors. Third, animals with induced epimutations can be used to monitor the dynamics of epigenetic regulation during embryogenesis, post-natal development and aging, in parallel with behavioral assessments. Epimutations could either be induced globally (by altering the activity of epigenetic regulators) or in a locus-specific manner (i.e. by using hybrid DNA methyltransferases [68] and non-coding RNA-based approaches [69]).
Some challenges posed to the epigenomic study of disease will be mitigated by technological advancements. Technological trends for epigenetic assays have advanced from analyzing selected loci (i.e. small-scale bisulfite mapping) to unbiased searches of the epigenome (i.e. epigenetic modification-specific antibodies coupled with microarrays or next-generation sequencing), using continually smaller starting populations of cells [70] (Box 3). Ideally, sample choice would be narrowed by an understanding of the basic biology underpinning disease symptoms (i.e. choosing tissues based on studies in animals or cell lines [71, 72]), or by the selection of major cell populations (i.e. neurons, glia), leading to experimental designs that are more economical and easier to interpret. This refinement is of particular relevance in the brain, which is heterogenous in cell populations as well as in regional epigenetic signatures [7, 73].
Yet another strategy could combine epigenomic association maps with those from genetic studies (GWAS+EWAS) to mine genetic-epigenetic interactions in disease. Allele-specific DNA methylation (ASM) [reviewed in 74] is a well-documented phenomenon that likely reflects tissue-specific cis regulatory influences of DNA polymorphisms on epigenetic status. ASM may help identify causal genetic polymorphisms within a genetic linkage disequilibrium (LD) block that contains multiple disease-associated SNPs, and/or uncover the molecular mechanisms of disease. Notably, genetic-epigenetic interactions may not be limited to the unidirectional effects of ASM, and epigenetic factors may also contribute to DNA sequence variation. The presence of a 5′ methyl group increases mutability of a cytosine base [75], and knockouts of DNMT1 exhibit a higher DNA mutation rate [76]. The human brain contains 100 billion neurons and 10X more glial cells [77]; epigenetically-induced mutations in a small population of cells, particularly during early neurodevelopmental stages, could affect predisposition to psychiatric disorders. In addition, epigenetic factors seem to play a role in the origin of de novo CNVs. The regions of the sperm genome displaying the lowest (bottom 1%) density of DNA methylation had 10X more structural rearrangements compared to the genome-wide average [78]. In addition, rare CNVs occurring in the genomes of individuals diagnosed with SCZ and BPD were significantly more concentrated within such hypomethylated regions. For example, BPD-specific deletions measured in sperm were significantly enriched (> 2-fold) in regions with the lowest methylation levels compared to the control-specific deletions [78]. Future studies on the epigenetic regulation of gene expression and other genomic functions within CNVs will be valuable, as this regulation may either compensate or further aggravate the effect of changed copy number. In summary, unraveling the bi-directional DNA-epigenetic interactions in psychiatric disease, in addition to investigating each separately, could provide a better understanding of the nature of gene misregulation in psychiatric disorders.
Concluding remarks
The coming decade will likely see a large assortment of epigenomic assays of disease that have increased resolution, sample size and scope of analysis. It is possible that epigenetic analysis in psychiatric disease will be inundated by the complexities of epigenetic maps, yielding small and non-replicable findings. However, the optimistic outlook is that this research will identify new molecular mechanisms to explain features of non-mendelian biology and transform our understanding of the molecular basis of psychiatric disease. Importantly, epigenetic studies may additionally identify novel therapeutic targets and strategies for the treatment of psychiatric disorders (Box 4).
Box 4. Epigenetics of psychiatric treatment and epigenetic therapy.
The therapeutic action of current medications for psychotic disorders may occur via epigenetic mechanisms. Clozapine is an atypical antipsychotic that is widely used to treat SCZ and other psychiatric illnesses. Treatment with clozapine has been associated with an elevation in H3K4me3 at the promoter of GAD1 (which encodes the enzyme that synthesizes the neurotransmitter GABA) in the prefrontal cortex of SCZ patients and in the mouse cortex [51]. The mood stabilizing effect of valproate, a drug routinely used to treat BPD, may be mediated by its ability to promote chromatin accessibility through HDAC inhibition, and thereby increase histone acetylation [101]. Indeed, a dose-dependent increase in histone acetylation in response to valproate was observed in lymphocyte nuclear protein extracts from patients affected with major psychosis [102]. In targeted DNA modification studies, clozapine and sulpiride (another atypical antipsychotic) both caused dose-dependent increases in cortical and striatal demethylation of the hypermethylated reelin and GAD67 promoters in the mouse brain [103]; similar to histone acetylation, DNA demethylation at a gene promoter increases chromatin accessibility. These studies show that antipsychotics and mood stabilizing agents are capable of promoting epigenetic modifications associated with an active transcriptional state at disease-relevant loci, suggesting new molecular mechanisms of antipsychotic efficacy.
The epigenetic and epigenomic studies of psychiatric disease bring the promise of reversing disease-causing epimutations, even in adulthood. Of particular interest are the opportunities of targeted corrections of epigenetic misregulation. In theory, two technologies can be used in the development of future epigenetic therapies [reviewed in 104]. The first is based on zinc-finger proteins (ZFPs), which recognize specific DNA sequences and bind to short stretches of DNA (typically 9–18 base pairs) [68]. ZFPs can be used to carry out a variety of cellular activities when combined with different protein domains. An epigenetic problem may be resolved if an epigenetically misregulated gene is exposed to a gene-specific ZFP that is attached to the appropriate histone- or DNA-modifying enzyme. Another promising technology is based on small interfering RNAs (siRNAs), which target and cleave mRNA [69]. RNA interference may modify local chromatin structure and promote heterochromatin assembly and gene silencing. Although the potential of epigenetic treatments and personalized medicine is starting to be explored, to a large extent clinical translation will depend on the success in identifying targets for epigenetic therapy.
Acknowledgments
This work was supported by the Canadian Institutes for Health and Research (186007), the National Institutes of Health (MH074127; MH088413), and the Krembil Foundation to AP. VL is supported by a Canadian Institutes of Health Research fellowship. AP is Senior Fellow, Ontario Mental Health Foundation, and Tapscott Chair in Schizophrenia Studies, University of Toronto.
Glossary
- Copy number variation
is present when DNA sections (larger than 1 kb) show differences in copy number between individuals. These rare structural variations in the genome can result in the duplication, deletion, or disruption of gene copies
- DNA methyltransferases (DNMTs)
enzymes that establish and maintain DNA methylation, using methyl-group donor compounds or cofactors. Main mammalian DNMTs are DNMT1, which maintains methylation state across DNA replication, and DNMT3a, DNMT3b which perform de novo methylation
- DNA modifications
covalent modifications of mammalian DNA occur via the methylation (5-mC) or hydroxymethylation (5-hmC) of cytosine, typically in the context of the CpG dinucleotide. More recently, other DNA modifications involved in DNA demethylation (5-carboxylcytosine, 5-formylcytosine) have also been identified. It is important to note that the conventional methods used for mapping of 5-mC, such as bisulfite-sequencing and methylation-sensitive restriction enzyme-based approaches, do not differentiate it from 5-hmC. We use the term “DNA methylation” to be consistent with primary publications; however, the more correct term “DNA modification” is used in the rest of the article
- Epigenome
the genome-wide distribution of epigenetic marks, including covalent DNA modifications and post-translational histone modifications, that influences genomic functions without altering the DNA sequence
- Genome-wide association study (GWAS)
study that maps common DNA polymorphisms in cases and matched controls with an aim to identify causal genetic variants
- Histone acetyltransferase (HAT) and deacetylase (HDAC)
HATs are enzymes that transfer acetyl groups to specific positions on histone tails, promoting an “open” chromatin state and transcriptional activation. HDACs remove these acetyl groups, resulting in a “closed” chromatin state and transcriptional repression
- Histone modifications
post-translational modifications of the N-terminal “tails” of histone proteins, which serve as a major mode of epigenetic regulation. These modifications include acetylation, phosphorylation, methylation, sumoylation, ubiquitination and ADP-ribosylation
- Linkage Disequilibrium
the non-random occurrence of a combination of alleles or genetic markers in a population
- Monozygotic (MZ) twins
two individual organisms that originated from the same zygote and are therefore genetically identical or very similar. The epigenetic profiling of MZ twins discordant for disease is a unique experimental design as it eliminates the genetic-, age-, and sex- differences from consideration
- Next-generation sequencing
an umbrella term for a collection of methods used to directly sequence nucleotides (DNA, RNA). Next-generation technologies can be applied in a variety of contexts, including whole-genome sequencing, profiling RNA (mRNAs and other small and non-coding RNAs), examining DNA binding proteins and chromatin structures, and assessing DNA modification patterns
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Collins PY, et al. Grand challenges in global mental health. Nature. 2011;475:27–30. doi: 10.1038/475027a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sullivan PF, et al. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 3.Burmeister M, et al. Psychiatric genetics: progress amid controversy. Nat Rev Genet. 2008;9:527–540. doi: 10.1038/nrg2381. [DOI] [PubMed] [Google Scholar]
- 4.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 5.Oh G, Petronis A. Environmental studies of schizophrenia through the prism of epigenetics. Schizophrenia bulletin. 2008;34:1122–1129. doi: 10.1093/schbul/sbn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 7.Gregg C, et al. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science. 2010;329:643–648. doi: 10.1126/science.1190830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2011;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 9.Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70:813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skene PJ, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, et al. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat Neurosci. 2011;14:1001–1008. doi: 10.1038/nn.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo JU, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng J, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nature neuroscience. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller CA, et al. Cortical DNA methylation maintains remote memory. Nature neuroscience. 2010;13:664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan Z, et al. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- 16.Schaefer A, et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron. 2009;64:678–691. doi: 10.1016/j.neuron.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan JS, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levenson JM, et al. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- 19.Alarcon JM, et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 20.Cardno AG, et al. Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch Gen Psychiatry. 1999;56:162–168. doi: 10.1001/archpsyc.56.2.162. [DOI] [PubMed] [Google Scholar]
- 21.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malhotra D, Sebat J. CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheng G, et al. Differences in the Circuitry-Based Association of Copy Numbers and Gene Expression Between the Hippocampi of Patients With Schizophrenia and the Hippocampi of Patients With Bipolar Disorder. Arch Gen Psychiatry. 2012 doi: 10.1001/archgenpsychiatry.2011.1882. [DOI] [PubMed] [Google Scholar]
- 24.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 25.Visscher PM, et al. Evidence-based psychiatric genetics, AKA the false dichotomy between common and rare variant hypotheses. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.65. [DOI] [PubMed] [Google Scholar]
- 26.Zuk O, et al. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daxinger L, Whitelaw E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet. 2012 doi: 10.1038/nrg3188. [DOI] [PubMed] [Google Scholar]
- 28.Kaminsky ZA, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–245. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 29.Ollikainen M, et al. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Human molecular genetics. 2010;19:4176–4188. doi: 10.1093/hmg/ddq336. [DOI] [PubMed] [Google Scholar]
- 30.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nature reviews Genetics. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ushijima T, et al. Fidelity of the methylation pattern and its variation in the genome. Genome Res. 2003;13:868–874. doi: 10.1101/gr.969603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1757–1764. doi: 10.1073/pnas.0906183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petronis A, et al. Schizophrenia: an epigenetic puzzle? Schizophr Bull. 1999;25:639–655. doi: 10.1093/oxfordjournals.schbul.a033408. [DOI] [PubMed] [Google Scholar]
- 34.Petronis A. Human morbid genetics revisited: relevance of epigenetics. Trends in genetics : TIG. 2001;17:142–146. doi: 10.1016/s0168-9525(00)02213-7. [DOI] [PubMed] [Google Scholar]
- 35.Pidsley R, Mill J. Epigenetic studies of psychosis: current findings, methodological approaches, and implications for postmortem research. Biol Psychiatry. 2011;69:146–156. doi: 10.1016/j.biopsych.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 36.Grayson DR, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura Y, et al. Epigenetic aberration of the human REELIN gene in psychiatric disorders. Mol Psychiatry. 2007;12:519, 593–600. doi: 10.1038/sj.mp.4002014. [DOI] [PubMed] [Google Scholar]
- 38.Iwamoto K, et al. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. J Neurosci. 2005;25:5376–5381. doi: 10.1523/JNEUROSCI.0766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tolosa A, et al. FOXP2 gene and language impairment in schizophrenia: association and epigenetic studies. BMC medical genetics. 2010;11:114. doi: 10.1186/1471-2350-11-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carrard A, et al. Increased DNA methylation status of the serotonin receptor 5HTR1A gene promoter in schizophrenia and bipolar disorder. J Affect Disord. 2011;132:450–453. doi: 10.1016/j.jad.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 41.Tochigi M, et al. Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol Psychiatry. 2008;63:530–533. doi: 10.1016/j.biopsych.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 42.Mill J, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaminsky Z, et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.64. [DOI] [PubMed] [Google Scholar]
- 44.Dempster EL, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–4796. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosa A, et al. Differential methylation of the X-chromosome is a possible source of discordance for bipolar disorder female monozygotic twins. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. 2008;147B:459–462. doi: 10.1002/ajmg.b.30616. [DOI] [PubMed] [Google Scholar]
- 46.Veldic M, et al. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc Natl Acad Sci U S A. 2005;102:2152–2157. doi: 10.1073/pnas.0409665102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhubi A, et al. An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophr Res. 2009;111:115–122. doi: 10.1016/j.schres.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guidotti A, et al. S-adenosyl methionine and DNA methyltransferase-1 mRNA overexpression in psychosis. Neuroreport. 2007;18:57–60. doi: 10.1097/WNR.0b013e32800fefd7. [DOI] [PubMed] [Google Scholar]
- 49.Kale A, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175:47–53. doi: 10.1016/j.psychres.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 50.Huang HS, et al. Chromatin immunoprecipitation in postmortem brain. Journal of neuroscience methods. 2006;156:284–292. doi: 10.1016/j.jneumeth.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 51.Huang HS, et al. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27:11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torrey EF, et al. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry. 2005;57:252–260. doi: 10.1016/j.biopsych.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 53.Akbarian S, et al. Chromatin alterations associated with down-regulated metabolic gene expression in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry. 2005;62:829–840. doi: 10.1001/archpsyc.62.8.829. [DOI] [PubMed] [Google Scholar]
- 54.Gavin DP, et al. Dimethylated lysine 9 of histone 3 is elevated in schizophrenia and exhibits a divergent response to histone deacetylase inhibitors in lymphocyte cultures. Journal of psychiatry & neuroscience : JPN. 2009;34:232–237. [PMC free article] [PubMed] [Google Scholar]
- 55.Sharma RP, et al. Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the National Brain Databank microarray collection. Schizophr Res. 2008;98:111–117. doi: 10.1016/j.schres.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benes FM, et al. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc Natl Acad Sci U S A. 2007;104:10164–10169. doi: 10.1073/pnas.0703806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Callaghan RC, et al. Methamphetamine Use and Schizophrenia: A Population-Based Cohort Study in California. Am J Psychiatry. 2011 doi: 10.1176/appi.ajp.2011.10070937. [DOI] [PubMed] [Google Scholar]
- 58.Beaulieu S, et al. The Canadian Network for Mood and Anxiety Treatments (CANMAT) task force recommendations for the management of patients with mood disorders and comorbid substance use disorders. Annals of clinical psychiatry : official journal of the American Academy of Clinical Psychiatrists. 2012;24:38–55. [PubMed] [Google Scholar]
- 59.LaPlant Q, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maze I, et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Covington HE, 3rd, et al. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011;71:656–670. doi: 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Renthal W, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 63.Wilkinson MB, et al. Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J Neurosci. 2009;29:7820–7832. doi: 10.1523/JNEUROSCI.0932-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsankova NM, et al. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 65.Rakyan VK, et al. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clark SJ. Action at a distance: epigenetic silencing of large chromosomal regions in carcinogenesis. Hum Mol Genet. 2007;16(Spec No 1):R88–95. doi: 10.1093/hmg/ddm051. [DOI] [PubMed] [Google Scholar]
- 67.Lichtenstein P, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu GL, Bestor TH. Cytosine methylation targetted to pre-determined sequences. Nat Genet. 1997;17:376–378. doi: 10.1038/ng1297-376. [DOI] [PubMed] [Google Scholar]
- 69.Saxe JP, Lin H. Small noncoding RNAs in the germline. Cold Spring Harb Perspect Biol. 3:a002717. doi: 10.1101/cshperspect.a002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adli M, Bernstein BE. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat Protoc. 6:1656–1668. doi: 10.1038/nprot.2011.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weaver IC, et al. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dolmetsch R, Geschwind DH. The human brain in a dish: the promise of iPSC-derived neurons. Cell. 2011;145:831–834. doi: 10.1016/j.cell.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gibbs JR, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tycko B. Allele-specific DNA methylation: beyond imprinting. Hum Mol Genet. 19:R210–220. doi: 10.1093/hmg/ddq376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pfeifer GP. Mutagenesis at methylated CpG sequences. Current topics in microbiology and immunology. 2006;301:259–281. doi: 10.1007/3-540-31390-7_10. [DOI] [PubMed] [Google Scholar]
- 76.Chen RZ, et al. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 77.Herculano-Houzel S. The human brain in numbers: a linearly scaled-up primate brain. Frontiers in human neuroscience. 2009;3:31. doi: 10.3389/neuro.09.031.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li J, et al. Genomic hypomethylation in the human germline associates with structural mutability in the human genome. PLoS Genetics. doi: 10.1371/journal.pgen.1002692. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fuke C, et al. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Annals of human genetics. 2004;68:196–204. doi: 10.1046/j.1529-8817.2004.00081.x. [DOI] [PubMed] [Google Scholar]
- 80.Alisch RS, et al. Age-associated DNA methylation in pediatric populations. Genome Res. 2012 doi: 10.1101/gr.125187.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Numata S, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet. 2012;90:260–272. doi: 10.1016/j.ajhg.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fraga MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heijmans BT, et al. Heritable rather than age-related environmental and stochastic factors dominate variation in DNA methylation of the human IGF2/H19 locus. Human molecular genetics. 2007;16:547–554. doi: 10.1093/hmg/ddm010. [DOI] [PubMed] [Google Scholar]
- 84.Metivier R, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 85.Masri S, Sassone-Corsi P. Plasticity and specificity of the circadian epigenome. Nat Neurosci. 2010;13:1324–1329. doi: 10.1038/nn.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fraser P, Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature. 2007;447:413–417. doi: 10.1038/nature05916. [DOI] [PubMed] [Google Scholar]
- 87.Gilbert N, et al. DNA methylation affects nuclear organization, histone modifications, and linker histone binding but not chromatin compaction. The Journal of cell biology. 2007;177:401–411. doi: 10.1083/jcb.200607133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mill J, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xie W, et al. Base-Resolution Analyses of Sequence and Parent-of-Origin Dependent DNA Methylation in the Mouse Genome. Cell. 2012;148:816–831. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Penn NW, et al. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem J. 1972;126:781–790. doi: 10.1042/bj1260781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Szulwach KE, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guo JU, et al. Hydroxylation of 5-Methylcytosine by TET1 Promotes Active DNA Demethylation in the Adult Brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bernstein BE, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nature biotechnology. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harris RA, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nature biotechnology. 2010;28:1097–1105. doi: 10.1038/nbt.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Koch CM, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome research. 2007;17:691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Petronis A, et al. Monozygotic twins exhibit numerous epigenetic differences: clues to twin discordance? Schizophr Bull. 2003;29:169–178. doi: 10.1093/oxfordjournals.schbul.a006988. [DOI] [PubMed] [Google Scholar]
- 101.Phiel CJ, et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 102.Sharma RP, et al. Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: preliminary results from a clinical population. Schizophr Res. 2006;88:227–231. doi: 10.1016/j.schres.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 103.Dong E, et al. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci U S A. 2008;105:13614–13619. doi: 10.1073/pnas.0805493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ptak C, Petronis A. Epigenetics and complex disease: from etiology to new therapeutics. Annu Rev Pharmacol Toxicol. 2008;48:257–276. doi: 10.1146/annurev.pharmtox.48.113006.094731. [DOI] [PubMed] [Google Scholar]