

Abstract
The ability of female sex steroids to regulate tissue function has long been appreciated, however their role in the regulation of striated muscle function has received considerably less attention. The purpose of this symposium review is to document recent evidence indicating the role female sex steroids have in defining the functional characteristics of striated muscle. The presentations provide substantial evidence indicating that estrogens are critical to the physiological and metabolic regulation of striated muscle, thus when considering women’s health issues striated muscle must included as an important target tissue along with other classically thought of estrogen sensitive tissues.
Keywords: estrogens, muscle, metabolism, cardiac, sex, exercise
Introduction
In 1990 the National Institutes of Health (NIH) formed the Office of Research on Women’s Health within the NIH Directors office. One of the major thrusts of this office is to improve the health of women through biomedical and behavioral research on the roles of sex (biological characteristics of being female or male) and gender (social influences based on sex) in health and disease. At this time, it been recognized that we have a poor understanding of the biological differences that exist between men and women. Although, significant progress has been made in understanding the contributions of female sex steroids to the function of traditional thought of estrogen sensitive tissues (i.e. breast tissue) at this time we have a poor understanding of the effects of estrogens on striated muscle tissue function. Evidence is accumulating demonstrating that estrogens play a critical role in the physiological and metabolic function of striated muscle. However, at this time we have yet to define the mechanisms by which estrogens regulate these parameters within striated muscle.
The purpose of this review is to summarize the presentations of participants in a symposium presented at the National American College of Sports Medicine meeting in Denver, CO in 2011. The symposium was entitled the ‘Novel Mechanistic Insights Into the Role of Female Sex Steroids in Regulating Physiological and Metabolic Function of Striated Muscle.’ The purpose of the symposium was to prevent novel and recent findings concerning the role of female sex steroids in the physiological and metabolic regulation of skeletal and cardiac muscle. In the following paragraphs each speaker will present a short review of their symposium presentation. Dr. Dawn Lowe will review recent data highlighting the role of estrogens in the regulation of skeletal muscle contractility. Dr. Paige Geiger will discuss the role of the estrogen receptor in the regulation of skeletal muscle glucose dynamics. Dr. Espen Spangenburg will review data concerning the effects of reduced estrogen function on muscle lipid metabolism. Finally, Dr. Lesile Leinwand will present new insights into the consideration of sex within the cardiac tissue.
Interactive effects of estradiol and physical activity on muscle contractile function
A primary function of skeletal muscle is to generate force. While much is known about the influence of androgens on muscle force, comparatively little is known about the female counterpart, estrogens. What is known is that muscle fibers contain estrogen receptors, the α estrogen receptor in muscle is responsive to changes in circulating estrogens (3), and the expression of that receptor is greater following exercise training. These observations imply that estrogen receptors and their ligand, estrogens, have a role in skeletal muscle function but the mechanism(s) by which this occurs is not understood.
Clinical studies on muscle force generation, i.e., strength, in women provide some insight on functional consequences of estrogens in skeletal muscle. In a frequently-cited paper (53), Phillips and coworkers showed that women lost strength at an earlier age then men, and that the loss began at the age of menopause. Furthermore, they showed that strength was preserved in post-menopausal women who were on an estrogen-based hormone therapy. Their report that women on hormone therapy were stronger has not been supported by all studies. To address the discrepancies in the literature, we completed a rigorous systematic review and meta-analyses on studies that compared muscle strength in postmenopausal women who were and were not on hormone therapy (24). Twenty three studies fit our criteria and the collective data on nearly 10,000 women showed that hormone therapy provided a small benefit to strength (effect size=0.23). Five studies reported size of the muscle that was tested allowing for the evaluation of specific force. A larger effect size of 0.45 was realized indicating that estrogens may be important for specific force. Similar meta-analyses were conducted on data from rodent studies in which force generation in estrogen-replete and –deficient animals were compared. The main finding was that estradiol had a large effect on specific force (effect size=0.66), equating to rodents with circulating estradiol having ~7% greater normalized strength than those that were estrogen-deficient. Thus, the collective literature on humans and rodents show that the presence of estrogens beneficially influences the ability of skeletal muscle to generate force and appears to function in a qualitative as opposed to quantitative manner. In other words, unlike androgens that affect force production by promoting primarily protein synthesis and hypertrophy (i.e., the quantity of muscle), it appears that estrogens influence muscle strength by improving muscle’s intrinsic ability to generate force (i.e., the quality of muscle). Potential anabolic or anti-catabolic effects of estrogens should not be disregarded, however, as recently shown in a study of postmenopausal monozygotic twins who were discordant for hormone therapy, with those on hormone therapy having greater thigh muscle area and power (59).
It had been hypothesized that ovarian hormones influence muscle strength by directly affecting contractile proteins (53), and two studies have addressed that idea using permeabilized muscle fibers. Permeabilized fibers do not have intact nerve or membranes so excitation-coupling mechanisms are bypassed and contractile responses directly reflect contractile protein function. In support of the hypothesis that hormones affect contractile proteins, permeabilized fibers from rat soleus muscles generated 20% lower specific force 10–14 weeks following ovariectomy compared with fibers from control rats (71). In contrast, permeabilized fibers from women who were and were not on hormone therapy showed no difference in specific force (72). We have used electron paramagnetic spectroscopy paired with site-specific spin labeling to investigate force production at the molecular level of myosin and actin in muscles from mice with altered levels of estrogen. We showed that the loss of estrogens, specifically 17 β-estradiol (Table 1), detrimentally affects strong-binding myosin (47,48). These results directly implicate contractile proteins in estrogenic effects on skeletal muscle’s force generating capacity.
Table 1.
Glossary of Terms
| Estrogen Receptor (ER): a member of the nuclear hormone receptor family that is activated by estrogens. Primarily exists in two forms: alpha (α) and beta (β), which are expressed across numerous tissues throughout the body. |
| 17α-estradiol: Most concentrated form of circulating estrogen. It should be noted it is not the only form of circulating estrogen. |
| Glucose transporter 4 (GLUT4): GLUT4 is the insulin-contraction regulated glucose transporter found in striated muscle and responsible for glucose transport into the cell. |
| Propylpyrazoletriyl (PPT): Specific ERα agonist |
| Akt: a serine/threonine kinase that plays a key role in insulin cell signaling. Also known as protein kinase B (PKB). |
| AMP-activated protein kinase (AMPK): A hetreotrimeric enzyme that regulates intracellular metabolic systems through intrinsic energy sensing capabilities |
| WAT: white adipose tissue |
| Perlipin1 (PLIN1): A lipid droplet coating protein found mainly in mature adipocytes. PLIN1 plays a supportive role in the storage and hydrolysis of triacylglyercol. |
| Lipid droplet (LD): cellular organelle responsible for storage of neutral lipids. |
| Adipose tissue glycerol lipase (ATGL): A key lipase responsible for the initial step in triglyceride hydrolysis. |
| Comparative Gene Identification-58 (CGI-58): A member of the alpha/beta hydrolase family of proteins that is often localized on the lipid droplet. |
| Cluster of Differentiation 36/Fatty acid translocase (CD36/FAT): A long chain fatty acid transporter found on the sarcolemma of striated muscle. |
| G-protein coupled estrogen receptor (GPR30/GPER): An estrogen sensitive G-protein coupled receptor that is involved in rapid non-genomic signaling. |
| Estrogen response element (ERE): Specific DNA sequences that are the site of recognition for the alpha and beta forms of the estrogen receptor. |
| Peroxisome proliferator-activated receptor gamma (PPARγ): A member of the nuclear receptor family that regulates genes involved in fatty acid storage and glucose metabolism. |
| Histone Deacteylases (HDAC): a class of enzyme that removes acetyl groups from lysine amino acids on histones that encase genomic DNA. |
A consequence of manipulating estrogens in rodents is that behavior, most notably physical activity, is altered. As summarized by Lightfoot (37), female rodents are more active than males with the sex hormones estrogens and testosterone explaining most of the difference. The effect is mediated largely through the α estrogen receptor in the activity centers of the brain, requiring aromatization of testosterone in males, and likely involving dopaminergic systems. This effect is robust as demonstrated by C57BL female mice given running wheels in their home cages. For example, we showed that distance ran dropped by as much as 90% when ovarian hormones were removed and impressively, running distance quickly and completely rebounded with estradiol replacement (23). Similar to wheel running, cage activities such as ambulation distance, jumping, and rearing are significantly lower in ovariectomized mice compared to ovariectomized mice treated with estradiol (25). In that study, the difference in these cage activities equated to the hormone-deficient mice being one hour less physically active per day, which theoretically could cause a loss of muscle strength.
To determine if estrogens affect skeletal muscle and myosin force generation directly or indirectly (through alterations in physical activity), we conducted two studies in which mouse and muscle activity were equalized by hindlimb suspension and muscle denervation interventions, respectively (25). In both studies, C57BL mice that were deficient in ovarian hormones were compared to those that had intact ovaries or were treated with estradiol. There were main effects of estradiol on maximal isometric tetanic force (Po), with force normalized to physiological cross-sectional area (specific Po) and contractile protein (normalized Po) being ~20% greater in soleus muscles from mice with estradiol compared to those that were hormone-deficient. These results were irrespective of whether or not the muscle was unloaded or denervated (i.e., there were no significant interactions between estrogen status and activity). The findings clearly demonstrate that estradiol favorably impacts muscle force generation independent of mouse or muscle activity.
The converse question can also be asked. That is, in female mice, to what extent does physical activity affect force generation independent of estradiol? Data to address this question lies within a paper we published reporting bone-muscle functional relationships in female C57BLmice (70). Soleus muscles were studied from cohorts of mice that underwent sham or ovariectomy surgeries and voluntarily wheel ran or were sedentary for 30 or 60 days. As previously reported, soleus muscle maximal isometric tetanic force was greater in sham-operated than ovariectomized mice, and this occurred irrespective of activity (Figure 1, top). Force generation was also greater in mice that ran than those that were sedentary, irrespective of ovarian hormone status (Figure 1, top). The running-induced strength gain was due to protein accretion and represents a quantitative effect on the muscle (Figure 1, middle). Thus, when soleus muscle force was normalized to protein content, the qualitative effect on muscle force generation from estradiol is evident (Figure 1, bottom). Again, no statistically significant interactions between ovarian hormones and activity were detected for any of these variables. This coupled with the inference that estradiol and exercise improve force generation by different underlying mechanisms, qualitative versus quantitative, infers that the effects could be additive and warrants further investigation.
Figure 1.
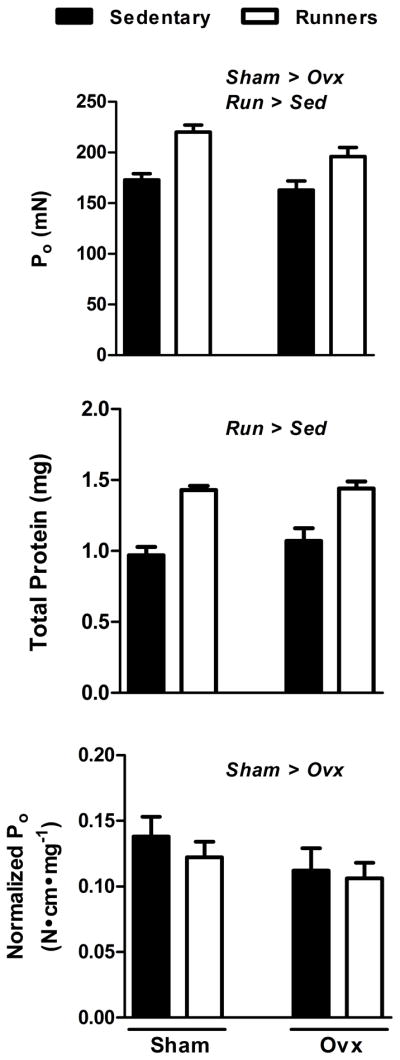
Ovarian hormones and wheel running for 30 d independently benefit soleus muscle force generation. Top; Maximal isometric tetanic force (Po) is greater in mice with intact ovaries and circulating estrogens (Sham) compared to ovariectomized mice lacking estrogens for 30 d (Ovx). Po is also greater in mice that wheel ran for 30 d (Run) compared to mice that did not have wheels, that is, mice that were sedentary (Sed). Middle; Total protein is greater in soleus muscles from Run than Sed mice, illustrating a quantitative enhancement in contractility by physical activity. Bottom; Soleus muscle maximal isometric tetanic force normalized to protein content (Normalized Po) is greater in Sham than Ovx mice, demonstrating a qualitative improvement in contractility by estrogens. Data were analyzed by 2-way ANOVA with significant main effects (P<0.05) indicated above the bars.
In summary, the female sex hormone, estradiol, favorably affects skeletal muscle contractility via a qualitative mechanism. This effect is likely at the level of the contractile proteins. In female mice, the benefits of estradiol on muscle strength is independent of physical activity. Physical activity also impacts skeletal muscle force generation in mice, independent of estrogen status, and does so in a quantitative manner by protein accretion.
Estrogen receptors and the modulation of skeletal muscle glucose uptake
Evidence from both human and rodent studies demonstrates the ability of estrogens to modify glucose homeostasis. Premenopausal women have increased insulin sensitivity compared with age-matched men (11). Compared to postmenopausal women, premenopausal women are also less likely to develop insulin resistance and have higher levels of GLUT4, the protein responsible for insulin-stimulated glucose uptake in skeletal muscle. In contrast, following menopause a significant decline in insulin sensitivity occurs along with a corresponding increase in fat mass (11,38). Estrogen replacement has been shown to ameliorate the increased risk for type 2 diabetes in postmenopausal women and improve whole body (38,44,54) and skeletal muscle glucose metabolism (54). In animal models, insulin sensitivity and glucose metabolism are impaired following ovariectomy (35) and aromatase knockout mice, which lack the ability to synthesize estrogen hormones, are insulin resistant.
The physiological actions of estrogens are mediated by two receptors, estrogen receptor (ER) α and ERβ (Table 1). A number of studies suggest that ERα is more highly expressed in insulin sensitive tissues (3,55) and increased adiposity occurs in humans and mice as a result of decreased ERα activation (29). While mice with global knockout of ERα exhibit impaired glucose tolerance and skeletal muscle insulin resistance (8,29), OB/OB mice given the ERα specific agonist propylpyrazoletriyl (PPT) demonstrated improved glucose tolerance and insulin sensitivity (43). These findings established ERα as a positive mediator of glucose metabolism. The ERα knockout has been associated with hepatic insulin resistance, and also skeletal muscle insulin resistance (8). A study by Ribas et al. (55) attributed decreased glucose tolerance in ERα knockout mice primarily to impaired insulin action in skeletal muscle. In addition, activation of ERα via the specific agonist propylpyrazoletriyl (PPT) for 3 days resulted in increased insulin-stimulated skeletal muscle glucose uptake (22), further highlighting the muscle-specific actions of ERα.
Increased skeletal muscle glucose uptake as a result of ERα activation occurs at least in part by stimulation of the insulin signaling pathway. In vivo administration of PPT for 3 days results in increased phosphorylation of Akt (Table 1) and insulin-stimulated phospho-Akt substrate (PAS) in soleus and EDL muscles (22). Estrogen and selective activation of ERα via PPT have been shown to potentiate the insulin signaling pathway and increase glucose transport in adipocytes in culture (50) and AMPK (Table 1) (14) in C2C12 muscle cells. Activation of ERα with PPT results in increased pAMPK in soleus and EDL muscles (22) and ERα-KO mice demonstrate decreased pAMPK in skeletal muscle (55). Furthermore, skeletal muscle simulated with estrogen in vivo and in vitro can increase AMPK activation (14,57) with a recent study showing estrogen-induced AMPK activation is mediated by ERα (58). Together, these findings suggest that ERα acts3 as a positive modulator of both insulin signaling and AMPK activation in skeletal muscle.
The ERs may also be involved in modulation of GLUT4 (Table 1) transcription; however, current results are conflicting. In the absence of ERα, GLUT4 is decreased in the gastrocnemius muscle of male ERα knock-out (ERα-KO) mice (4). In contrast, Ribas et al. (55) did not observe a decrease in GLUT4 in the quadriceps or soleus muscle in female ERα-KO mice. Activation of ERα via PPT resulted in increased GLUT4 in the EDL (fast-twitch) but not in the soleus (slow-twitch), suggesting that the ability of ERα to regulate GLUT4 may be fiber type specific (22). It is also possible that expression patterns of both ERα and ERβ are critical to determining tissue specific effects of estrogens.
ERα and ERβ maintain a complex inter-regulatory relationship that varies with the target tissue. For example, previous studies have shown that activation of ERβ can oppose the action of ERα with a resulting negative impact on glucose metabolism (4,45). As estrogens activate3 both ERs, estrogen administration could have variable effects depending on the tissue or physiological model examined. In the first study to test the effects of estrogen on rodent skeletal muscle in vitro, acute incubations in estrogen for 10 minutes did not increase insulin-stimulated glucose transport in soleus muscle (58). Similarly, three days of estrogen treatment in vivo also had no effect on insulin-stimulated glucose uptake in soleus or EDL muscles (22). The lack of an acute effect of estrogen on skeletal muscle glucose uptake could be due to ERβ activation offsetting any stimulation of ERα via estrogen. Importantly, it is unknown if aging or metabolic disease alters the ratio of ERα to ERβ in skeletal muscle, thereby changing the tissue response to estrogen and estrogen receptor modulators. Therefore, while important functional information has been gained using ER knockout models, future studies need to focus on the physiological changes in ERα and ERβ expression and activation, and the impact of these changes on metabolic function.
In summary, the ability of estrogens to positively mediate glucose uptake in skeletal muscle may depend on the expression and activation pattern of estrogen receptors. ERα activation can result in an increase in skeletal muscle glucose uptake through a combination of insulin-dependent and –independent pathways. As a result, it may be possible to harness the beneficial effects of estrogens, and mitigate the negative effects, by specifically targeting ERs pharmacologically. The expression and activation patterns of ERs in response to diet, age and disease should be the focus of future investigations on this topic.
Reductions in circulating estrogens contributes to a unique metabolic profile in skeletal muscle
In human and animals, any reduction in estrogen function leads to a significant increase in visceral adiposity. For example, surgical ovariectomy (32,75,76) or genetic ablation of the estrogen receptor all result in substantial gains in visceral fat mass (29). Significant increases in visceral fat mass are associated with increased risk of developing peripheral insulin resistance and overall metabolic dysfunction (5). It has been documented that women transitioning into menopause or being treating for estrogen positive cancers via estrogen receptor antagonists results in an increased risk for development of the metabolic syndrome. In addition, numerous women undergo surgical hysterectomy/ovariectomy for clinical reasons, which also results in increased likelihood of developing the metabolic syndrome. We have previously found that surgical ovariectomy (OVX) in mice results in substantial gains in visceral fat mass of ~200% within 8 weeks of the surgery (32,75,76). These increases in fat mass are mediated by increases in adipocyte size without an increase in adipocyte number (data unpublished, Spangenburg et al.). It is often assumed that these increases in fat mass are the result of the onset of hyperphagia in the OVX mouse model, however previous studies have clearly shown that hyperphagia is specific to the rat model and does not develop in the mouse (31,74). The data from our group and that of others suggest that the OVX mouse model suffers from reductions in physical activity levels rather than increases in eating behavior (23,32,75). In humans, there are few data examining changes in eating behavior or physical activity with the onset of reduced estrogen function so it is unclear if either the OVX rat or OVX mouse are more appropriate for mimicking the human condition.
Using the OVX mouse model, we have previously shown that with the increases in fat mass, there is a subsequent loss of regulatory control over lipolytic process leading to spill over of free fatty acids into other tissues (75). The OVX animals exhibit significant increases in unstimulated serum glycerol and NEFA compared to the SHAM animals. This increase in NEFA levels in OVX animals was associated with an upregulation of adipose tissue glycerol lipoase (ATGL, Table 1) protein and an increase in the functional interaction between ATGL and comparative gene idenfication-58 (CGI-58, Table 1) in the white adipose tissue (WAT, Table 1) in the visceral region compared to the SHAM animals. CGI-58 increases the catalytic activity of ATGL, thus these data would suggest that increased activation of ATGL likely contributes to the increased basal lipolysis in the OVX animals. In addition, we also found decreases in perilipin (PLIN1, Table 1) protein content in the WAT of the OVX mice compared to the SHAM mice. PLIN1 is a lipid droplet (LD) coating protein that plays both a plays a protective role in preventing unstimulated lipolysis and facilitates activated lipolysis (40). In genetic ablation models of PLIN1 there is a significant increase in basal lipolysis (40), suggesting that the decreases in PLIN1 in our model likely contributes to the increased basal or unstimulated lipolysis in the OVX animals compared to the intact SHAM group. Our data suggest that the loss of regulatory control of WAT lipolytic function is a consequence of reduced PLIN1 content and increased activation of ATGL, which could contribute to increased risk of developing lipotoxicity due to peripheral tissue being exposed to higher concentrations of circulating lipid. Indeed, we recently demonstrated that hepatic tissue is a likely target of this spillover due to the anatomical relationship of the liver with visceral tissue via the portal circulation (32). Further, since estrogen receptor is known to regulate mitochondrial function in skeletal muscle and due to the ability of skeletal muscle to store lipid, skeletal muscle is a likely target to be affected by the increase in circulating NEFA in the OVX animals.
Here, we discuss further evidence that this relationship also extends to skeletal muscle, in that we have found evidence of increasing lipid droplet formation in skeletal muscle fibers from OVX mice compared to SHAM mice (see Fig 2, quantification not shown). Although skeletal muscle is capable of storing significant amounts of triacylglycerol (TAG), there is a known association of insulin resistance with an increasing frequency of LD in skeletal muscle in sedentary individuals. In collaboration, with Dr A. Bonen we have determined that there is a significant upregulation of the major long chain fatty acid tranporters (i.e. CD36/FAT and FABPpm (Table 1)) in skeletal muscle from the OVX mice compared to the SHAM mice (unpublished observations, Spangenburg et al), which would suggest that increased exposure of muscle to high levels of circulating lipid would result in increased lipid storage. Thus, it is likely that increase in LD frequency is a result of enhanced lipid movement into the muscle cell, which could contribute to the development of insulin resistance.
Figure 2.

Lipid droplet staining frequency is higher in single muscle fibers isolated from OVX mice compared to SHAM mice. LD are stained with BODIPY (493/503) (green) as previously described (65).
Our data suggest that metabolic mechanisms within skeletal muscle that regulate some aspect of lipid metabolism are potentially dysfunctional under conditions of reduced estrogen function. Using a metabolic profiling approach of the skeletal muscle from the OVX mice compared to the SHAM mice in collaboration with Dr(s) Debbie Muoio and Tim Koves we determined that there was a significant reduction in the long chain acylcarnitines and short chain acylcarnitines (unpublished data, Spangenburg et al.). These results suggests that in the skeletal muscle from the OVX mouse, under conditions of increased fatty acid flux there maybe an inability to transport long chain fatty acids into the mitochondria thus encouraging ectopic lipid deposition. Indeed, Campbell et al previously demonstrated a reduction in carnitine palmitoyltransferase I (CPT-1) activity, which is the primary mitochondrial transporter of long chain fatty acids, in skeletal muscle from OVX rats compared to SHAM rats (9). These data suggest that estrogens influence lipid entry dynamics into the mitochondria through some undefined influence on CPT-1. Although, the ovary secretes a number of different hormones it is likely that this effect is specific to estrogens since skeletal muscle fatty acid oxidation is significantly reduced in the estrogen receptor alpha knock out mice (56). Further, estrogens appear to have a positive effect on mitochondria function across a number of tissues (33). These data would suggest that reductions in estrogen function result in increased lipid accumulation in skeletal muscle due to poor lipid entry into the mitochondria.
Thus, our data coupled with others suggest that reductions or attenuations of estrogen function leads to increases in visceral fat mass coupled with increased fatty acid release by the WAT. Simultaneously, due to the reduced estrogen function there are potential losses in mitochondrial function leading to poor lipid utilization occurring in the skeletal muscle. Thus, skeletal muscle from OVX mice that are exposed to increased levels of fatty acids is at increased risk of developing of lipotoxicity. These data suggest that decreases in estrogen function in results in multiple metabolic defects that occur across a variety of tissues that all could contribute to increased risk of developing the metabolic syndrome. Thus, in females estrogens play a key role in maintaining metabolic function across a number of tissues and any intervention that results in inhibition of estrogen function must be considered carefully to account for increased risk of developing the metabolic syndrome.
Roles of estrogen in males and female hearts
Estrogen is important in both males and females but its actions in each sex may be different. For example, men with coronary artery disease given high doses of estrogen have increased incidence of myocardial infarction (MI) and pulmonary embolism (1). While many studies show that estrogen can be cardioprotective in females, there are a number of genetic animal models of cardiovascular disease where it is not (19,51). There are also examples of estrogen acting oppositely in males vs. females. Estrogen induces collagen expression in male cardiac fibroblasts but represses it in female cardiac fibroblasts (52). We and others have begun to study adult male and female cardiac myocytes and some aspects of their biology are, not surprisingly, distinct. For example, female adult rat ventricular myocytes (ARVMs) are protected more than males against H2O2-mediated oxidative stress, possibly due to higher basal activation of Akt in female cells (69).
Estrogen receptor biology in the cardiac myocyte
ERs have been targets of drug discovery and laboratory investigation because of their importance in health and disease. Estrogen’s interactions with its receptors are central to many of its actions (Figure 3). ERα and ERβ can form homodimers or heterodimers. In addition, there are multiple alternatively spliced isoforms; particularly of ERβ. There is also a more recently described G-protein coupled ER, termed GPR30 or GPER (Table 1) (46) which may be responsible for the rapid, non-genomic effects of estrogen. There is a null mouse line which lacks GPER and this mouse has impaired cardiac function, but in males only (15). However, activation of GPER pharmacologically is cardioprotecitve in both male and female rats (17). Through its receptors estrogen can have both genomic and non-genomic effects. Genomic effects are typical ligand/nuclear receptor binding to estrogen responsive elements (EREs, Table 1) and for ERα, activating genes via AP-1 sites (36). Non-genomic effects involve activation of multiple signal transduction pathways that can be mediated by membrane-associated ERs. ERs a and β are both expressed in male and female cardiac myocytes and the 2 sexes express equivalent levels of each receptors in the heart (39). ERs are important in both sexes, as evidenced by the fact that both sexes of ERα null mice are infertile (41). Cardiac phenotypes of ERα and β total null mice are not straightforward, in part because of their systemic traits. For example, ERα null mice have 5–20 fold higher levels of circulating estrogen and testosterone and exhibit metabolic syndrome and obesity (8,29). ERβ null animals are fertile but they are chronically hypoxic and have high blood pressure (49). ERs also have ligand-independent activity most likely originating from those ERs located at the membrane that can alter growth factor signaling pathways (27). ERα and ERβ show different subcellular localization (39) which likely points to distinct cellular functions. All of this speaks to the need for conditional null alleles in cardiac myocytes to address the role of these receptors in the myocardium. In this section, we will not explore the biology of estrogen-related receptors or androgens and their receptors although we acknowledge their importance.
Figure 3.

Estrogen (E2) and genistein (Gen) induce multiple types of cell signaling in cardiac tissue that include genomic (mins to hours) and non-genomic (rapid; secs to mins) induction of estrogen receptors. TF=transcription factor; GF=growth factor; Trxn=transcription; G/E=gen or E2; P=phosphorylation.
Soy phytoestrogen endocrine disruption
The soy phytoestrogen, genistein, has become a topic of debate as it is defined as an endocrine disruptor (30) but is also thought to have beneficial properties in the setting of stopping cancer cell growth (62). We hypothesize that the properties of genistein that may make it effective in inhibiting tumor cell growth are the same properties that can make it harmful to the heart. The concern of the public derives from genistein’s widespread prevalence in the environment, including foods and beverages. Human consumption of soy products is growing, including infant formula and phytoestrogen supplements. Also, most laboratory rodents eat soy-based diets and have 9–10μM concentrations of genistein in their serum (7). Thus they are subject to genistein’s potent physiological effects. For example, dietary phytoestrogens partially correct the ovarian phenotypes in aromatase null (estrogen-free) mice (6). Genistein binds to both ERs (but binds ERβ more tightly (67)) and activates genes with EREs (61). In addition to estrogenic effects, genistein is a potent broad spectrum TKI (2). It is also an activating ligand of PPAR (Table 1) which can be harmful in the heart (64). We and others have shown that genistein has significant effects on gene expression and cardiac function in a mouse model of HCM and in exercise of wild type mice; this effect is more potent in males than females (42). Further, we have shown that genistein inhibits different RTKs in male and female cardiac myocytes.
Receptor tyrosine kinases and tyrosine kinase inhibition
There are numerous RTKs in the human genome and they regulate many important cell processes. RTK mutations and aberrant activation of their intracellular signaling pathways cause many diseases. Ligand-induced dimerization or oligomerization results in activation of the tyrosine kinase domain which, in turn, undergoes autophosphorylation, increasing catalytic activity and phosphorylation of other parts of the protein (63). While RTKs are the targets of many small molecule anti-cancer drugs, the selectivity of these TKIs is highly variable (28). One emerging problem with TKIs is that many of them are associated with cardiotoxicity in 20–40% of patients that can lead to heart failure (12). We have also shown that genistein has a very similar pattern of RTK inhibition to some chemotherapeutic drugs with high cardiotoxicity (18).
Histone Deacetylases (HDACs) and ERα
Cardiac adaptation and chromatin remodeling are tightly linked, and HDAC (Table 1) activity is involved in both processes, consistent with a molecular link between these biological phenomena. There are 4 classes of HDACs which catalyze directly or indirectly the removal of acetyl groups on lysines in both histones and non-histone proteins. HDACs play important roles in cardiac biology (43). Broad HDAC inhibition has been shown to attenuate pathologic cardiac growth (34), likely by inhibiting autophagy (10). The histone acetylase p300 acetylates ERα which stabilizes it and increases its transcriptional activity (73). Class IIa HDACs 5 and 9 deacetylate ERα and also repress its transcriptional activity through direct binding when estrogen is present (68). In the absence of HDACs 5 or 9, ERα is significantly upregulated and activated (68). Of interest, HDAC 5 and 9 null female mice show improved recovery from myocardial infarction but male null mice have worsened survival (68). Dysregulation of HDACs has been shown to occur in a variety of diseases, particularly cancer, and a focus of drug discovery is the development of specific HDAC inhibitors.
The regression of pathological cardiac hypertrophy
There are several clinical settings during which the regression of hypertrophic growth occurs, such as treatment of hypertension and bariatric surgery but a large number of people do not show regression (see 26). There may be sex differences in this process; one study demonstrated that post-surgical regression of cardiac hypertrophy due to aortic stenosis is greater in females than males (52). In our model of cardiac atrophy due to cancer, females are protected in an ER-dependent manner while E2 administration to males worsens their atrophy (13). Several animal models for the regression of pathologic growth exist. For example, the hypertrophy seen after treatment with isoproterenol (Iso) for 7 days regresses within 7–14 days after agonist removal (60). Also, there have been several studies of aortic banding and removal of banding (20,66). These authors found that gene expression profiles for banding are quite distinct from de-banding but sex has not been studied. (54,66). The cessation of exercise and the end of pregnancy also involve the regression of hypertrophic growth; but they are understudied. For instance, there is only one report about postpartum cardiac signaling (21). The proteasome and autophagy have a clear role in hypertrophy; for example, in a mouse model of pressure overload, proteasome activity is induced and proteasome inhibition blocks hypertrophy (16). HDAC inhibition attenuates pathologic cardiac hypertrophy by suppressing autophagy (10).
Overall Conclusions
It is clear that female sex steroids have both powerful and diverse effects on the striated muscle function, which help to define the phenotypic characteristics of the cell. Importantly, loss of estrogenic function also contributes to alterations in muscle function that when extrapolated to the human would be predicted to contribute to increased risk of various chronic conditions including sarcopenia, type 2 diabetes, metabolic syndrome and cardiovascular disease. Collectively, our data and the work of others provide key insights which indicate that striated muscle should be considered an important target for female sex steroids and deserve important consideration in women’s health initiatives. In addition, the data clearly demonstrate that estrogens provide a critical signaling avenue for striated muscle that defines its overall function, which may be important for considering the use of estrogen therapy or the inability to use estrogen therapy. Specifically, recent and somewhat controversial evidence has suggested estrogen therapy may exacerbate the development of certain diseases in women. Perhaps exercise training is the solution, in that a number of the conditions that result from loss estrogenic function are alleviated or attenuated by exercise training. Thus, it is critical that future studies consider biological effect of sex within their experimental design to help elucidate the role of female sex steroids and their interaction with exercise in defining the striated muscle function.
Acknowledgments
All of the authors would like to acknowledge the contribution of their lab members who made the presentation of these data and ideas possible and in particular to thank Krsiten Barthel for Figure 3. In addition, we apologize for not being able to acknowledge all of the outstanding work in this area due to space limitations. Work presented by Dr. Paige Geiger was supported was supported by NIH AG031575 and grant number P20 RR016475 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health NIH. Work presented by Dr. Leslie Leinwand was supported by NIH HL50560. Work presented by Dr. Dawn Lowe was supported by NIH grants AG031743 and AG036827.
Footnotes
No conflicts of interest to declare for each author.
The results of the present do not constitute endorsement by ACSM.
References
- 1.The Coronary Drug Project. Initial findings leading to modifications of its research protocol. JAMA. 1970;214(7):1303–1313. [PubMed] [Google Scholar]
- 2.Akiyama T, Ishida J, Nakagawa S, et al. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262(12):5592–5595. [PubMed] [Google Scholar]
- 3.Baltgalvis KA, Greising SM, Warren GL, Lowe DA. Estrogen regulates estrogen receptors and antioxidant gene expression in mouse skeletal muscle. PloS one. 2010;5(4):e10164. doi: 10.1371/journal.pone.0010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barros RP, Machado UF, Warner M, Gustafsson JA. Muscle GLUT4 regulation by estrogen receptors ERbeta and ERalpha. Proc Natl Acad Sci U S A. 2006;103(5):1605–1608. doi: 10.1073/pnas.0510391103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergman RN, Kim SP, Catalano KJ, et al. Why visceral fat is bad: mechanisms of the metabolic syndrome. Obesity (Silver Spring) 2006;14 (Suppl 1):16S–19S. doi: 10.1038/oby.2006.277. [DOI] [PubMed] [Google Scholar]
- 6.Britt KL, Simpson ER, Findlay JK. Effects of phytoestrogens on the ovarian and pituitary phenotypes of estrogen-deficient female aromatase knockout mice. Menopause. 2005;12(2):174–185. doi: 10.1097/00042192-200512020-00012. [DOI] [PubMed] [Google Scholar]
- 7.Brown NM, Setchell KD. Animal models impacted by phytoestrogens in commercial chow: implications for pathways influenced by hormones. Lab Invest. 2001;81(5):735–747. doi: 10.1038/labinvest.3780282. [DOI] [PubMed] [Google Scholar]
- 8.Bryzgalova G, Gao H, Ahren B, et al. Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia. 2006;49(3):588–597. doi: 10.1007/s00125-005-0105-3. [DOI] [PubMed] [Google Scholar]
- 9.Campbell SE, Febbraio MA. Effect of ovarian hormones on mitochondrial enzyme activity in the fat oxidation pathway of skeletal muscle. Am J Physiol Endocrinol Metab. 2001;281(4):E803–808. doi: 10.1152/ajpendo.2001.281.4.E803. [DOI] [PubMed] [Google Scholar]
- 10.Cao DJ, Wang ZV, Battiprolu PK, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108(10):4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr MC. The emergence of the metabolic syndrome with menopause. J Clin Endocrinol Metab. 2003;88(6):2404–2411. doi: 10.1210/jc.2003-030242. [DOI] [PubMed] [Google Scholar]
- 12.Cheng H, Force T. Why do kinase inhibitors cause cardiotoxicity and what can be done about it? Prog Cardiovasc Dis. 2010;53(2):114–120. doi: 10.1016/j.pcad.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Cosper PF, Leinwand LA. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 2011;71(5):1710–1720. doi: 10.1158/0008-5472.CAN-10-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Eon TM, Rogers NH, Stancheva ZS, Greenberg AS. Estradiol and the estradiol metabolite, 2-hydroxyestradiol, activate AMP-activated protein kinase in C2C12 myotubes. Obesity (Silver Spring, Md. 2008;16(6):1284–1288. doi: 10.1038/oby.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delbeck M, Golz S, Vonk R, et al. Impaired left-ventricular cardiac function in male GPR30-deficient mice. Mol Med Report. 2011;4(1):37–40. doi: 10.3892/mmr.2010.402. [DOI] [PubMed] [Google Scholar]
- 16.Depre C, Wang Q, Yan L, et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114(17):1821–1828. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- 17.Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol. 2009;297(5):H1806–1813. doi: 10.1152/ajpheart.00283.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Lorenzo G, Autorino R, Bruni G, et al. Cardiovascular toxicity following sunitinib therapy in metastatic renal cell carcinoma: a multicenter analysis. Ann Oncol. 2009;20(9):1535–1542. doi: 10.1093/annonc/mdp025. [DOI] [PubMed] [Google Scholar]
- 19.Gao XM, Agrotis A, Autelitano DJ, et al. Sex hormones and cardiomyopathic phenotype induced by cardiac beta 2-adrenergic receptor overexpression. Endocrinology. 2003;144(9):4097–4105. doi: 10.1210/en.2002-0214. [DOI] [PubMed] [Google Scholar]
- 20.Gao XM, Kiriazis H, Moore XL, et al. Regression of pressure overload-induced left ventricular hypertrophy in mice. Am J Physiol Heart Circ Physiol. 2005;288(6):H2702–2707. doi: 10.1152/ajpheart.00836.2004. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez AM, Osorio JC, Manlhiot C, et al. Hypertrophy signaling during peripartum cardiac remodeling. Am J Physiol Heart Circ Physiol. 2007;293(5):H3008–3013. doi: 10.1152/ajpheart.00401.2007. [DOI] [PubMed] [Google Scholar]
- 22.Gorres BK, Bomhoff GL, Morris JK, Geiger PC. In vivo stimulation of oestrogen receptor alpha increases insulin-stimulated skeletal muscle glucose uptake. The Journal of physiology. 2011;589(Pt 8):2041–2054. doi: 10.1113/jphysiol.2010.199018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorzek JF, Hendrickson KC, Forstner JP, et al. Estradiol and tamoxifen reverse ovariectomy-induced physical inactivity in mice. Medicine and science in sports and exercise. 2007;39(2):248–256. doi: 10.1249/01.mss.0000241649.15006.b8. [DOI] [PubMed] [Google Scholar]
- 24.Greising SM, Baltgalvis KA, Lowe DA, Warren GL. Hormone therapy and skeletal muscle strength: a meta-analysis. The journals of gerontology Series A, Biological sciences and medical sciences. 2009;64(10):1071–1081. doi: 10.1093/gerona/glp082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greising SM, Baltgalvis KA, Kosir AM, et al. Estradiol’s beneficial effect on murine muscle function is independent of muscle activity. J Appl Physiol. 2011;110(1):109–115. doi: 10.1152/japplphysiol.00852.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Habib GB, Mann DL, Zoghbi WA. Normalization of cardiac structure and function after regression of cardiac hypertrophy. Am Heart J. 1994;128(2):333–343. doi: 10.1016/0002-8703(94)90487-1. [DOI] [PubMed] [Google Scholar]
- 27.Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276(40):36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 28.Hasinoff BB. The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol Appl Pharmacol. 2010;244(2):190–195. doi: 10.1016/j.taap.2009.12.032. [DOI] [PubMed] [Google Scholar]
- 29.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A. 2000;97(23):12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henley DV, Korach KS. Physiological effects and mechanisms of action of endocrine disrupting chemicals that alter estrogen signaling. Hormones (Athens) 2010;9(3):191–205. doi: 10.14310/horm.2002.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isken F, Pfeiffer AF, Nogueiras R, et al. Deficiency of glucose-dependent insulinotropic polypeptide receptor prevents ovariectomy-induced obesity in mice. Am J Physiol Endocrinol Metab. 2008;295(2):E350–355. doi: 10.1152/ajpendo.00008.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson KC, Wohlers LM, Valencia AP, et al. Wheel running prevents the accumulation of monounsaturated fatty acids in the liver of ovariectomized mice by attenuating changes in SCD-1 content. Appl Physiol Nutr Metab. 2011;36(6):798–810. doi: 10.1139/h11-099. [DOI] [PubMed] [Google Scholar]
- 33.Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008;105(6):1342–1351. doi: 10.1002/jcb.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kong Y, Tannous P, Lu G, et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation. 2006;113(22):2579–2588. doi: 10.1161/CIRCULATIONAHA.106.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumagai S, Holmang A, Bjorntorp P. The effects of oestrogen and progesterone on insulin sensitivity in female rats. Acta physiologica Scandinavica. 1993;149(1):91–97. doi: 10.1111/j.1748-1716.1993.tb09596.x. [DOI] [PubMed] [Google Scholar]
- 36.Kushner PJ, Agard DA, Greene GL, et al. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74(5):311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 37.Lightfoot JT. Sex hormones’ regulation of rodent physical activity: a review. International journal of biological sciences. 2008;4(3):126–132. doi: 10.7150/ijbs.4.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindheim SR, Buchanan TA, Duffy DM, et al. Comparison of estimates of insulin sensitivity in pre- and postmenopausal women using the insulin tolerance test and the frequently sampled intravenous glucose tolerance test. J Soc Gynecol Investig. 1994;1(2):150–154. doi: 10.1177/107155769400100210. [DOI] [PubMed] [Google Scholar]
- 39.Lizotte E, Grandy SA, Tremblay A, Allen BG, Fiset C. Expression, distribution and regulation of sex steroid hormone receptors in mouse heart. Cell Physiol Biochem. 2009;23(1–3):75–86. doi: 10.1159/000204096. [DOI] [PubMed] [Google Scholar]
- 40.Londos C, Brasaemle DL, Schultz CJ, et al. On the control of lipolysis in adipocytes. Ann N Y Acad Sci. 1999;892:155–168. doi: 10.1111/j.1749-6632.1999.tb07794.x. [DOI] [PubMed] [Google Scholar]
- 41.Lubahn DB, et al. Alteration of repruductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA. 1993;90(23):11162–11166. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luczak ED, Barthel KK, Stauffer BL, et al. Remodeling the cardiac transcriptional landscape with diet. Physiol Genomics. 2011;43(12):772–780. doi: 10.1152/physiolgenomics.00237.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lundholm L, Bryzgalova G, Gao H, et al. The estrogen receptor α-selective agonist propyl pyrazole triol improves glucose tolerance in ob/ob mice; potential molecular mechanisms. The Journal of endocrinology. 2008;199(2):275–286. doi: 10.1530/JOE-08-0192e. [DOI] [PubMed] [Google Scholar]
- 44.Margolis KL, Bonds DE, Rodabough RJ, et al. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women’s Health Initiative Hormone Trial. Diabetologia. 2004;47(7):1175–1187. doi: 10.1007/s00125-004-1448-x. [DOI] [PubMed] [Google Scholar]
- 45.Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Interv. 2003;3(5):281–292. doi: 10.1124/mi.3.5.281. [DOI] [PubMed] [Google Scholar]
- 46.Meyer MR, Prossnitz ER, Barton M. The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function. Vascul Pharmacol. 2011;55(1–3):17–25. doi: 10.1016/j.vph.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moran AL, Warren GL, Lowe DA. Removal of ovarian hormones from mature mice detrimentally affects muscle contractile function and myosin structural distribution. J Appl Physiol. 2006;100(2):548–559. doi: 10.1152/japplphysiol.01029.2005. [DOI] [PubMed] [Google Scholar]
- 48.Moran AL, Nelson SA, Landisch RM, Warren GL, Lowe DA. Estradiol replacement reverses ovariectomy-induced muscle contractile and myosin dysfunction in mature female mice. J Appl Physiol. 2007;102(4):1387–1393. doi: 10.1152/japplphysiol.01305.2006. [DOI] [PubMed] [Google Scholar]
- 49.Morani A, Barros RP, Imamov O, et al. Lung dysfunction causes systemic hypoxia in estrogen receptor beta knockout (ERbeta−/−) mice. Proc Natl Acad Sci U S A. 2006;103(18):7165–7169. doi: 10.1073/pnas.0602194103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagira K, Sasaoka T, Wada T, et al. Altered subcellular distribution of estrogen receptor alpha is implicated in estradiol-induced dual regulation of insulin signaling in 3T3-L1 adipocytes. Endocrinology. 2006;147(2):1020–1028. doi: 10.1210/en.2005-0825. [DOI] [PubMed] [Google Scholar]
- 51.O’Connell TD, Ishizaka S, Nakamura A, et al. The alpha(1A/C)- and alpha(1B)-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J Clin Invest. 2003;111(11):1783–1791. doi: 10.1172/JCI16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petrov G, Regitz-Zagrosek V, Lehmkuhl E, et al. Regression of myocardial hypertrophy after aortic valve replacement: faster in women? Circulation. 2010;122(11 Suppl):S23–28. doi: 10.1161/CIRCULATIONAHA.109.927764. [DOI] [PubMed] [Google Scholar]
- 53.Phillips SK, Rook KM, Siddle NC, Bruce SA, Woledge RC. Muscle weakness in women occurs at an earlier age than in men, but strength is preserved by hormone replacement therapy. Clin Sci (Lond) 1993;84(1):95–98. doi: 10.1042/cs0840095. [DOI] [PubMed] [Google Scholar]
- 54.Riant E, Waget A, Cogo H, et al. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. 2009;150(5):2109–2117. doi: 10.1210/en.2008-0971. [DOI] [PubMed] [Google Scholar]
- 55.Ribas V, Nguyen MT, Henstridge DC, et al. Impaired Oxidative Metabolism and Inflammation are Associated with Insulin Resistance in ERα–Deficient Mice. American journal of physiology. 2009;298:E304–319. doi: 10.1152/ajpendo.00504.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rogers NH, Perfield JW, 2nd, Strissel KJ, Obin MS, Greenberg AS. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology. 2009;150(5):2161–2168. doi: 10.1210/en.2008-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rogers NH, Witczak CA, Hirshman MF, Goodyear LJ, Greenberg AS. Estradiol stimulates Akt, AMP-activated protein kinase (AMPK) and TBC1D1/4, but not glucose uptake in rat soleus. Biochem Biophys Res Commun. 2009;382(4):646–650. doi: 10.1016/j.bbrc.2009.02.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ronkainen PH, Kovanen V, Alen M, et al. Postmenopausal hormone replacement therapy modifies skeletal muscle composition and function: a study with monozygotic twin pairs. J Appl Physiol. 2009;107(1):25–33. doi: 10.1152/japplphysiol.91518.2008. [DOI] [PubMed] [Google Scholar]
- 59.Saadane N, Alpert L, Chalifour LE. Expression of immediate early genes, GATA-4, and Nkx-2. 5 in adrenergic-induced cardiac hypertrophy and during regression in adult mice. Br J Pharmacol. 1999;127(5):1165–1176. doi: 10.1038/sj.bjp.0702676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shanle EK, Xu W. Endocrine disrupting chemicals targeting estrogen receptor signaling: identification and mechanisms of action. Chem Res Toxicol. 2011;24(1):6–19. doi: 10.1021/tx100231n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shanmugam MK, Kannaiyan R, Sethi G. Targeting cell signaling and apoptotic pathways by dietary agents: role in the prevention and treatment of cancer. Nutr Cancer. 2011;63(2):161–173. doi: 10.1080/01635581.2011.523502. [DOI] [PubMed] [Google Scholar]
- 62.Shrivastava A, Radziejewski C, Campbell E, et al. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. 1997;1(1):25–34. doi: 10.1016/s1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- 63.Son NH, Park TS, Yamashita H, et al. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117(10):2791–2801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spangenburg EE, Pratt SJ, Wohlers LM, Lovering RM. Use of BODIPY (493/503) to visualize intramuscular lipid droplets in skeletal muscle. Journal of biomedicine & biotechnology. 2011;2011:598358. doi: 10.1155/2011/598358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stansfield WE, Charles PC, Tang RH, et al. Regression of pressure-induced left ventricular hypertrophy is characterized by a distinct gene expression profile. J Thorac Cardiovasc Surg. 2009;137(1):232–238. 238e231–238. doi: 10.1016/j.jtcvs.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Swedenborg E, Power KA, Cai W, Pongratz I, Ruegg J. Regulation of estrogen receptor beta activity and implications in health and disease. Cell Mol Life Sci. 2009;66(24):3873–3894. doi: 10.1007/s00018-009-0118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Rooij E, Fielitz J, Sutherland LB, et al. Myocyte enhancer factor 2 and class II histone deacetylases control a gender-specific pathway of cardioprotection mediated by the estrogen receptor. Circ Res. 2010;106(1):155–165. doi: 10.1161/CIRCRESAHA.109.207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang F, He Q, Sun Y, Dai X, Yang XP. Female adult mouse cardiomyocytes are protected against oxidative stress. Hypertension. 2010;55(5):1172–1178. doi: 10.1161/HYPERTENSIONAHA.110.150839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Warren GL, Moran AL, Hogan HA, et al. Voluntary run training but not estradiol deficiency alters the tibial bone-soleus muscle functional relationship in mice. American journal of physiology Regulatory, integrative and comparative physiology. 2007;293(5):R2015–2026. doi: 10.1152/ajpregu.00569.2007. [DOI] [PubMed] [Google Scholar]
- 70.Wattanapermpool J, Reiser PJ. Differential effects of ovariectomy on calcium activation of cardiac and soleus myofilaments. The American journal of physiology. 1999;277(2 Pt 2):H467–473. doi: 10.1152/ajpheart.1999.277.2.H467. [DOI] [PubMed] [Google Scholar]
- 71.Widrick JJ, Maddalozzo GF, Lewis D, et al. Morphological and functional characteristics of skeletal muscle fibers from hormone-replaced and nonreplaced postmenopausal women. The journals of gerontology Series A, Biological sciences and medical sciences. 2003;58(1):3–10. doi: 10.1093/gerona/58.1.b3. [DOI] [PubMed] [Google Scholar]
- 72.Wilson BJ, Tremblay AM, Deblois G, Sylvain-Drolet G, Giguere V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol Endocrinol. 2010;24(7):1349–1358. doi: 10.1210/me.2009-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Witte MM, Resuehr D, Chandler AR, Mehle AK, Overton JM. Female mice and rats exhibit species-specific metabolic and behavioral responses to ovariectomy. Gen Comp Endocrinol. 166(3):520–528. doi: 10.1016/j.ygcen.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wohlers LM, Spangenburg EE. 17beta-estradiol supplementation attenuates ovariectomy-induced increases in ATGL signaling and reduced perilipin expression in visceral adipose tissue. J Cell Biochem. 2010;110(2):420–427. doi: 10.1002/jcb.22553. [DOI] [PubMed] [Google Scholar]
- 75.Wohlers LM, Jackson KC, Spangenburg EE. Lipolytic signaling in response to acute exercise is altered in female mice following ovariectomy. J Cell Biochem. 2011;112(12):3675–3684. doi: 10.1002/jcb.23302. [DOI] [PMC free article] [PubMed] [Google Scholar]